
Immunogenicity of Novel AAV Capsids for Retinal Gene Therapy


 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Human Blood Donors
2.2. AAV Vectors
2.3. Determination of Capsid Titers of AAV2 Preparations
2.4. Antibody Responses to AAV2 Capsids
2.5. Neutralizing Antibody Assay
2.6. In Vitro Stimulation of PBMCs
2.7. Cytokine Bioplex Assay
2.8. AAV-Induced Expansion of Cell Populations and Intracellular Cytokine Expression
2.9. Statistics
3. Results
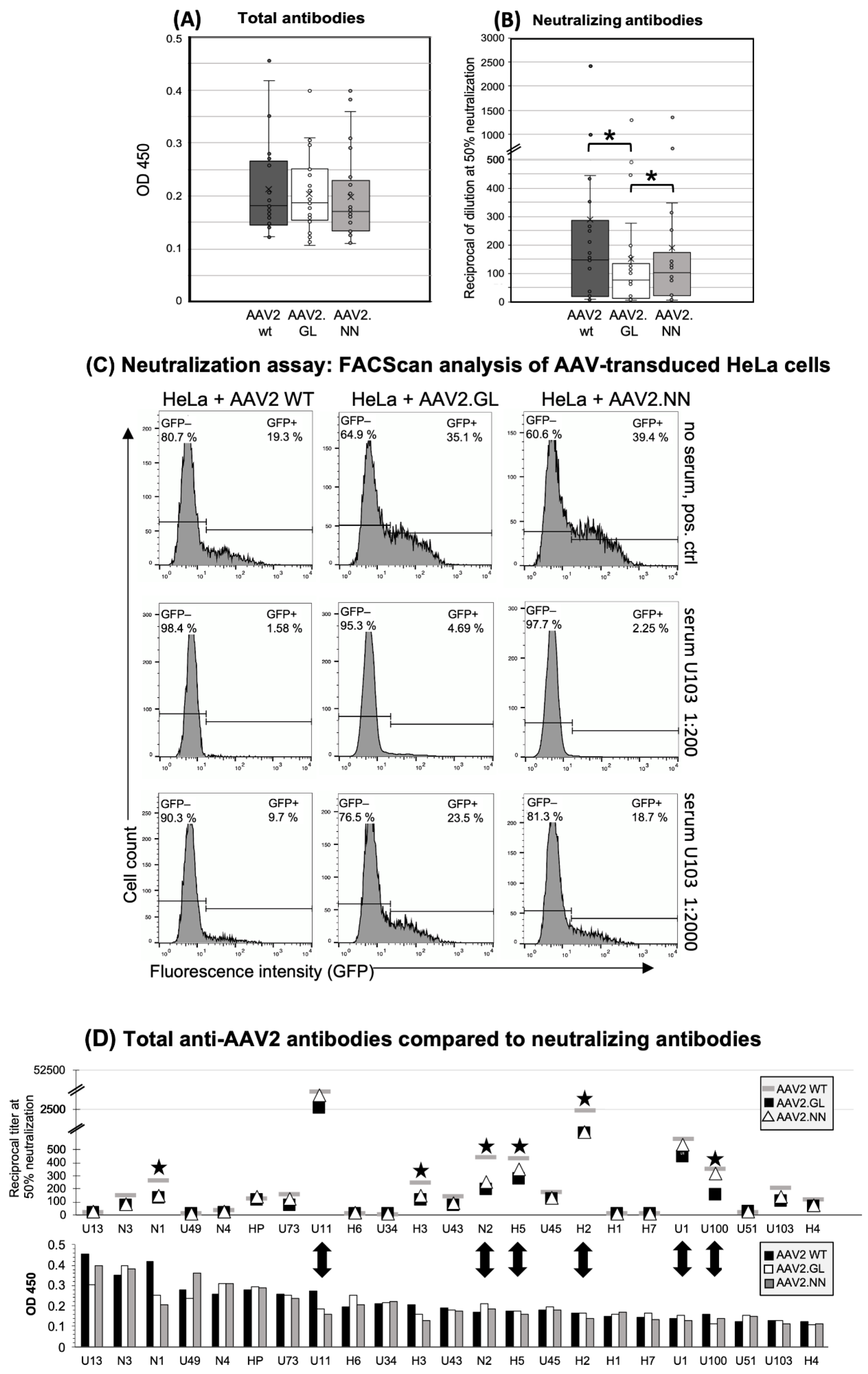
3.1. Similar Total Antibody Binding Abilities for AAV2 WT, AAV2.GL, and AAV2.NN
3.2. Decreased Neutralizing Antibody Binding to the AAV2.GL Mutant
3.3. AAV-Induced Proliferation of Innate Cells and IFN-Beta Production after in Vitro Stimulation
3.4. Secretion of Cytokines and Chemokines Triggered In Vitro by AAV2 Variants
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, D.; Tai, P.W.L.; Gao, G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat. Rev. Drug Discov. 2019, 18, 358–378. [Google Scholar] [CrossRef] [PubMed]
- Keeler, A.M.; Flotte, T.R. Recombinant Adeno-Associated Virus Gene Therapy in Light of Luxturna (and Zolgensma and Glybera): Where Are We, and How Did We Get Here? Annu. Rev. Virol. 2019, 6, 601–621. [Google Scholar] [CrossRef] [PubMed]
- Kuzmin, D.A.; Shutova, M.V.; Johnston, N.R.; Smith, O.P.; Fedorin, V.V.; Kukushkin, Y.S.; van der Loo, J.C.M.; Johnstone, E.C. The clinical landscape for AAV gene therapies. Nat. Rev. Drug Discov. 2021, 20, 173–174. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A. In vivo tissue-tropism of adeno-associated viral vectors. Curr. Opin. Virol. 2016, 21, 75–80. [Google Scholar] [CrossRef] [Green Version]
- Grimm, D.; Büning, H. Small but Increasingly Mighty: Latest Advances in AAV Vector Research, Design, and Evolution. Hum. Gene Ther. 2017, 28, 1075–1086. [Google Scholar] [CrossRef]
- Verdera, H.C.; Kuranda, K.; Mingozzi, F. AAV Vector Immunogenicity in Humans: A Long Journey to Successful Gene Transfer. Mol. Ther. 2020, 28, 723–746. [Google Scholar] [CrossRef]
- Dudek, A.M.; Zabaleta, N.; Zinn, E.; Pillay, S.; Zengel, J.; Porter, C.; Franceschini, J.S.; Estelien, R.; Carette, J.E.; Zhou, G.L.; et al. GPR108 Is a Highly Conserved AAV Entry Factor. Mol. Ther. 2020, 28, 367–381. [Google Scholar] [CrossRef]
- Büning, H.; Srivastava, A. Capsid Modifications for Targeting and Improving the Efficacy of AAV Vectors. Mol. Ther. Methods Clin. Dev. 2019, 12, 248–265. [Google Scholar] [CrossRef]
- Li, C.; Samulski, R.J. Engineering adeno-associated virus vectors for gene therapy. Nat. Rev. Genet. 2020, 21, 255–272. [Google Scholar] [CrossRef]
- Barnes, C.; Scheideler, O.; Schaffer, D. Engineering the AAV capsid to evade immune responses. Curr. Opin. Biotechnol. 2019, 60, 99–103. [Google Scholar] [CrossRef]
- Hamilton, B.A.; Wright, J.F. Challenges Posed by Immune Responses to AAV Vectors: Addressing Root Causes. Front. Immunol. 2021, 12, 675897. [Google Scholar] [CrossRef]
- Weber, T. Anti-AAV Antibodies in AAV Gene Therapy: Current Challenges and Possible Solutions. Front. Immunol. 2021, 12, 658399. [Google Scholar] [CrossRef]
- Ronzitti, G.; Gross, D.-A.; Mingozzi, F. Human Immune Responses to Adeno-Associated Virus (AAV) Vectors. Front. Immunol. 2020, 11, 670. [Google Scholar] [CrossRef]
- Shieh, P.B.; Bonnemann, C.G.; Muller-Felber, W.; Blaschek, A.; Dowling, J.J.; Kuntz, N.L.; Seferian, A.M. Re: "Moving Forward After Two Deaths in a Gene Therapy Trial of Myotubular Myopathy" by Wilson and Flotte. Hum. Gene Ther. 2020, 31, 787. [Google Scholar] [CrossRef]
- Ross, M.; Ofri, R. The future of retinal gene therapy: Evolving from subretinal to intravitreal vector delivery. Neural Regen. Res. 2021, 16, 1751–1759. [Google Scholar] [CrossRef]
- Cheng, S.Y.; Punzo, C. Ocular Inflammation with Anti-Vascular Endothelial Growth Factor Treatments. Hum. Gene 2021, 32, 639–641. [Google Scholar] [CrossRef]
- Mishra, A.; Vijayasarathy, C.; Cukras, C.A.; Wiley, H.E.; Sen, H.N.; Zeng, Y.; Wei, L.L.; Sieving, P.A. Immune function in X-linked retinoschisis subjects in an AAV8-RS1 phase I/IIa gene therapy trial. Mol. Ther. 2021, 29, 2030–2040. [Google Scholar] [CrossRef]
- Newman, N.J.; Yu-Wai-Man, P.; Carelli, V.; Moster, M.L.; Biousse, V.; Vignal-Clermont, C.; Sergott, R.C.; Klopstock, T.; Sadun, A.A.; Barboni, P.; et al. Efficacy and Safety of Intravitreal Gene Therapy for Leber Hereditary Optic Neuropathy Treated within 6 Months of Disease Onset. Ophthalmology 2021, 128, 649–660. [Google Scholar] [CrossRef]
- Bucher, K.; Rodríguez-Bocanegra, E.; Dauletbekov, D.; Fischer, M.D. Immune responses to retinal gene therapy using adeno-associated viral vectors—Implications for treatment success and safety. Prog. Retin. Eye Res. 2021, 83, 100915. [Google Scholar] [CrossRef]
- Keino, H.; Horie, S.; Sugita, S. Immune Privilege and Eye-Derived T-Regulatory Cells. J. Immunol. Res. 2018, 2018, 1679197. [Google Scholar] [CrossRef]
- Daya, S.; Berns, K.I. Gene therapy using adeno-associated virus vectors. Clin. Microbiol. Rev. 2008, 21, 583–593. [Google Scholar] [CrossRef] [Green Version]
- Mével, M.; Bouzelha, M.; Leray, A.; Pacouret, S.; Guilbaud, M.; Penaud-Budloo, M.; Alvarez-Dorta, D.; Dubreil, L.; Gouin, S.G.; Combal, J.P.; et al. Chemical modification of the adeno-associated virus capsid to improve gene delivery. Chem. Sci. 2020, 11, 1122–1131. [Google Scholar] [CrossRef] [Green Version]
- Pavlou, M.; Schön, C.; Occelli, L.M.; Rossi, A.; Meumann, N.; Boyd, R.F.; Bartoe, J.T.; Siedlecki, J.; Gerhardt, M.J.; Babutzka, S.; et al. Novel AAV capsids for intravitreal gene therapy of photoreceptor disorders. Embo Mol. Med. 2021, 13, e13392. [Google Scholar] [CrossRef]
- Becirovic, E.; Böhm, S.; Nguyen, O.N.P.; Riedmayr, L.M.; Hammelmann, V.; Schön, C.; Butz, E.S.; Wahl-Schott, C.; Biel, M.; Michalakis, S. AAV Vectors for FRET-Based Analysis of Protein-Protein Interactions in Photoreceptor Outer Segments. Front. Neurosci. 2016, 10, 356. [Google Scholar] [CrossRef] [Green Version]
- Kaufmann, U.; Diedrichs-Mohring, M.; Wildner, G. Dynamics of intraocular IFN-gamma, IL-17 and IL-10-producing cell populations during relapsing and monophasic rat experimental autoimmune uveitis. PLoS One 2012, 7, e49008. [Google Scholar] [CrossRef]
- Chan, Y.K.; Dick, A.D.; Hall, S.M.; Langmann, T.; Scribner, C.L.; Mansfield, B.C. Inflammation in Viral Vector-Mediated Ocular Gene Therapy: A Review and Report from a Workshop Hosted by the Foundation Fighting Blindness, 9/2020. Transl. Vis Sci. Technol. 2021, 10, 3. [Google Scholar] [CrossRef]
- Moskalenko, M.; Chen, L.; van Roey, M.; Donahue, B.A.; Snyder, R.O.; McArthur, J.G.; Patel, S.D. Epitope mapping of human anti-adeno-associated virus type 2 neutralizing antibodies: Implications for gene therapy and virus structure. J. Virol. 2000, 74, 1761–1766. [Google Scholar] [CrossRef] [Green Version]
- Tseng, Y.S.; Gurda, B.L.; Chipman, P.; McKenna, R.; Afione, S.; Chiorini, J.A.; Muzyczka, N.; Olson, N.H.; Baker, T.S.; Kleinschmidt, J.; et al. Adeno-associated virus serotype 1 (AAV1)- and AAV5-antibody complex structures reveal evolutionary commonalities in parvovirus antigenic reactivity. J. Virol. 2015, 89, 1794–1808. [Google Scholar] [CrossRef] [Green Version]
- McCraw, D.M.; O’Donnell, J.K.; Taylor, K.A.; Stagg, S.M.; Chapman, M.S. Structure of adeno-associated virus-2 in complex with neutralizing monoclonal antibody A20. Virology 2012, 431, 40–49. [Google Scholar] [CrossRef] [Green Version]
- Gurda, B.L.; DiMattia, M.A.; Miller, E.B.; Bennett, A.; McKenna, R.; Weichert, W.S.; Nelson, C.D.; Chen, W.J.; Muzyczka, N.; Olson, N.H.; et al. Capsid antibodies to different adeno-associated virus serotypes bind common regions. J. Virol. 2013, 87, 9111–9124. [Google Scholar] [CrossRef] [Green Version]
- Perabo, L.; Endell, J.; King, S.; Lux, K.; Goldnau, D.; Hallek, M.; Büning, H. Combinatorial engineering of a gene therapy vector: Directed evolution of adeno-associated virus. J. Gene Med. 2006, 8, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Maersch, S.; Huber, A.; Büning, H.; Hallek, M.; Perabo, L. Optimization of stealth adeno-associated virus vectors by randomization of immunogenic epitopes. Virology 2010, 397, 167–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, T. Antibody Interactions with Rhinovirus: Lessons for Mechanisms of Neutralization and the Role of Immunity in Viral Evolution. Curr. Top. Microbiol. Immunol. 2001, 260, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Huttner, N.A.; Girod, A.; Perabo, L.; Edbauer, D.; Kleinschmidt, J.A.; Büning, H.; Hallek, M. Genetic modifications of the adeno-associated virus type 2 capsid reduce the affinity and the neutralizing effects of human serum antibodies. Gene 2003, 10, 2139–2147. [Google Scholar] [CrossRef] [Green Version]
- Maheshri, N.; Koerber, J.T.; Kaspar, B.K.; Schaffer, D.V. Directed evolution of adeno-associated virus yields enhanced gene delivery vectors. Nat. Biotechnol. 2006, 24, 198–204. [Google Scholar] [CrossRef]
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373, 871–876. [Google Scholar] [CrossRef]
- Chirmule, N.; Propert, K.J.; Magosin, S.A.; Qian, Y.; Qian, R.; Wilson, J.M. Immune responses to adenovirus and adeno-associated virus in humans. Gene Ther. 1999, 6, 1574–1583. [Google Scholar] [CrossRef] [Green Version]
- Calcedo, R.; Wilson, J. Humoral Immune Response to AAV. Front. Immunol. 2013, 4. [Google Scholar] [CrossRef] [Green Version]
- Antia, A.; Ahmed, H.; Handel, A.; Carlson, N.E.; Amanna, I.J.; Antia, R.; Slifka, M. Heterogeneity and longevity of antibody memory to viruses and vaccines. Plos Biol. 2018, 16, e2006601. [Google Scholar] [CrossRef]
- Borghans, J.; Ribeiro, R.M. The maths of memory. eLife 2017, 6, e26754. [Google Scholar] [CrossRef] [Green Version]
- Hernandez Yosbani, J.; Wang, J.; Kearns William, G.; Loiler, S.; Poirier, A.; Flotte Terence, R. Latent Adeno-Associated Virus Infection Elicits Humoral but Not Cell-Mediated Immune Responses in a Nonhuman Primate Model. J. Virol. 1999, 73, 8549–8558. [Google Scholar] [CrossRef] [Green Version]
- Zaiss, A.-K.; Liu, Q.; Bowen, G.P.; Wong, N.C.W.; Bartlett, J.S.; Muruve, D.A. Differential activation of innate immune responses by adenovirus and adeno-associated virus vectors. J. Virol. 2002, 76, 4580–4590. [Google Scholar] [CrossRef] [Green Version]
- Allan, R.S.; Waithman, J.; Bedoui, S.; Jones, C.M.; Villadangos, J.A.; Zhan, Y.; Lew, A.M.; Shortman, K.; Heath, W.R.; Carbone, F.R. Migratory Dendritic Cells Transfer Antigen to a Lymph Node-Resident Dendritic Cell Population for Efficient CTL Priming. Immunity 2006, 25, 153–162. [Google Scholar] [CrossRef] [Green Version]
- Le Borgne, M.; Etchart, N.; Goubier, A.; Lira, S.A.; Sirard, J.C.; van Rooijen, N.; Caux, C.; Aït-Yahia, S.; Vicari, A.; Kaiserlian, D.; et al. Dendritic Cells Rapidly Recruited into Epithelial Tissues via CCR6/CCL20 Are Responsible for CD8+ T Cell Crosspriming In Vivo. Immunity 2006, 24, 191–201. [Google Scholar] [CrossRef]
- Granelli-Piperno, A.; Golebiowska, A.; Trumpfheller, C.; Siegal, F.P.; Steinman, R.M. HIV-1-infected monocyte-derived dendritic cells do not undergo maturation but can elicit IL-10 production and T cell regulation. Proc. Natl. Acad. Sci. USA 2004, 101, 7669. [Google Scholar] [CrossRef] [Green Version]
- Kuranda, K.; Jean-Alphonse, P.; Leborgne, C.; Hardet, R.; Collaud, F.; Marmier, S.; Costa Verdera, H.; Ronzitti, G.; Veron, P.; Mingozzi, F. Exposure to wild-type AAV drives distinct capsid immunity profiles in humans. J. Clin. Investig. 2018, 128, 5267–5279. [Google Scholar] [CrossRef]
- Oliveira-Nascimento, L.; Massari, P.; Wetzler, L.M. The Role of TLR2 in Infection and Immunity. Front. Immunol. 2012, 3, 79. [Google Scholar] [CrossRef] [Green Version]
- Shepardson, K.M.; Schwarz, B.; Larson, K.; Morton, R.V.; Avera, J.; McCoy, K.; Caffrey, A.; Harmsen, A.; Douglas, T.; Rynda-Apple, A.; et al. Induction of Antiviral Immune Response through Recognition of the Repeating Subunit Pattern of Viral Capsids Is Toll-Like Receptor 2 Dependent. mBio 2017, 8, e01356-17. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Martínez, S.; Cancino-Díaz, M.E.; Jiménez-Zamudio, L.; García-Latorre, E.; Cancino-Díaz, J.C. TLRs and NODs mRNA expression pattern in healthy mouse eye. Br. J. Ophthalmol. 2005, 89, 904. [Google Scholar] [CrossRef] [Green Version]
- Tummala, G.; Crain, A.; Rowlan, J.; Pepple, K.L. Characterization of Gene Therapy Associated Uveitis Following Intravitreal Adeno-Associated Virus Injection in Mice. Investig. Ophthalmol. Vis. Sci. 2021, 62, 41. [Google Scholar] [CrossRef]
- Hösel, M.; Broxtermann, M.; Janicki, H.; Esser, K.; Arzberger, S.; Hartmann, P.; Gillen, S.; Kleeff, J.; Stabenow, D.; Odenthal, M.; et al. Toll-like receptor 2-mediated innate immune response in human nonparenchymal liver cells toward adeno-associated viral vectors. Hepatology 2012, 55, 287–297. [Google Scholar] [CrossRef]
- Melchjorsen, J.; Sørensen, L.N.; Paludan, S.R. Expression and function of chemokines during viral infections: From molecular mechanisms to in vivo function. J. Leukoc. Biol. 2003, 74, 331–343. [Google Scholar] [CrossRef]
- Michael, B.D.; Bricio-Moreno, L.; Sorensen, E.W.; Miyabe, Y.; Lian, J.; Solomon, T.; Kurt-Jones, E.A.; Luster, A.D. Astrocyte- and Neuron-Derived CXCL1 Drives Neutrophil Transmigration and Blood-Brain Barrier Permeability in Viral Encephalitis. Cell Rep. 2020, 32, 108150. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Donor | Gender | Age | P/S |
|---|---|---|---|---|
| 1. | H1 | female | 60 | P/S |
| 2. | H2 | female | 41 | P/S |
| 3. | H3 | male | 43 | P/S |
| 4. | H4 | female | 28 | P/S |
| 5. | H5 | female | 60 | P/S |
| 6. | H6 | male | 45 | P/S |
| 7. | H7 | male | 60 | P/ |
| 8. | HP | male pool | 24 | S |
| 9. | N1 | female | 58 | S |
| 10. | N2 | male | 57 | S |
| 11. | N3 | female | 49 | S |
| 12. | N4 | female | 61 | S |
| 13. | U1 | male | 36 | S |
| 14. | U11 | male | 47 | S |
| 15. | U13 | male | 37 | S |
| 16. | U34 | male | 27 | S |
| 17. | U43 | female | 30 | S |
| 18. | U45 | female | 29 | S |
| 19. | U49 | male | 33 | S |
| 20. | U51 | male | 25 | S |
| 21. | U73 | female | 11 | S |
| 22. | U100 | male | 44 | S |
| 23. | U103 | female | 21 | S |
| 24. | C1 | male | 31 | P |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gehrke, M.; Diedrichs-Möhring, M.; Bogedein, J.; Büning, H.; Michalakis, S.; Wildner, G. Immunogenicity of Novel AAV Capsids for Retinal Gene Therapy. Cells 2022, 11, 1881. https://doi.org/10.3390/cells11121881
Gehrke M, Diedrichs-Möhring M, Bogedein J, Büning H, Michalakis S, Wildner G. Immunogenicity of Novel AAV Capsids for Retinal Gene Therapy. Cells. 2022; 11(12):1881. https://doi.org/10.3390/cells11121881
Chicago/Turabian StyleGehrke, Miranda, Maria Diedrichs-Möhring, Jacqueline Bogedein, Hildegard Büning, Stylianos Michalakis, and Gerhild Wildner. 2022. "Immunogenicity of Novel AAV Capsids for Retinal Gene Therapy" Cells 11, no. 12: 1881. https://doi.org/10.3390/cells11121881
APA StyleGehrke, M., Diedrichs-Möhring, M., Bogedein, J., Büning, H., Michalakis, S., & Wildner, G. (2022). Immunogenicity of Novel AAV Capsids for Retinal Gene Therapy. Cells, 11(12), 1881. https://doi.org/10.3390/cells11121881