
Hedgehog Morphogens Act as Growth Factors Critical to Pre- and Postnatal Cardiac Development and Maturation: How Primary Cilia Mediate Their Signal Transduction


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
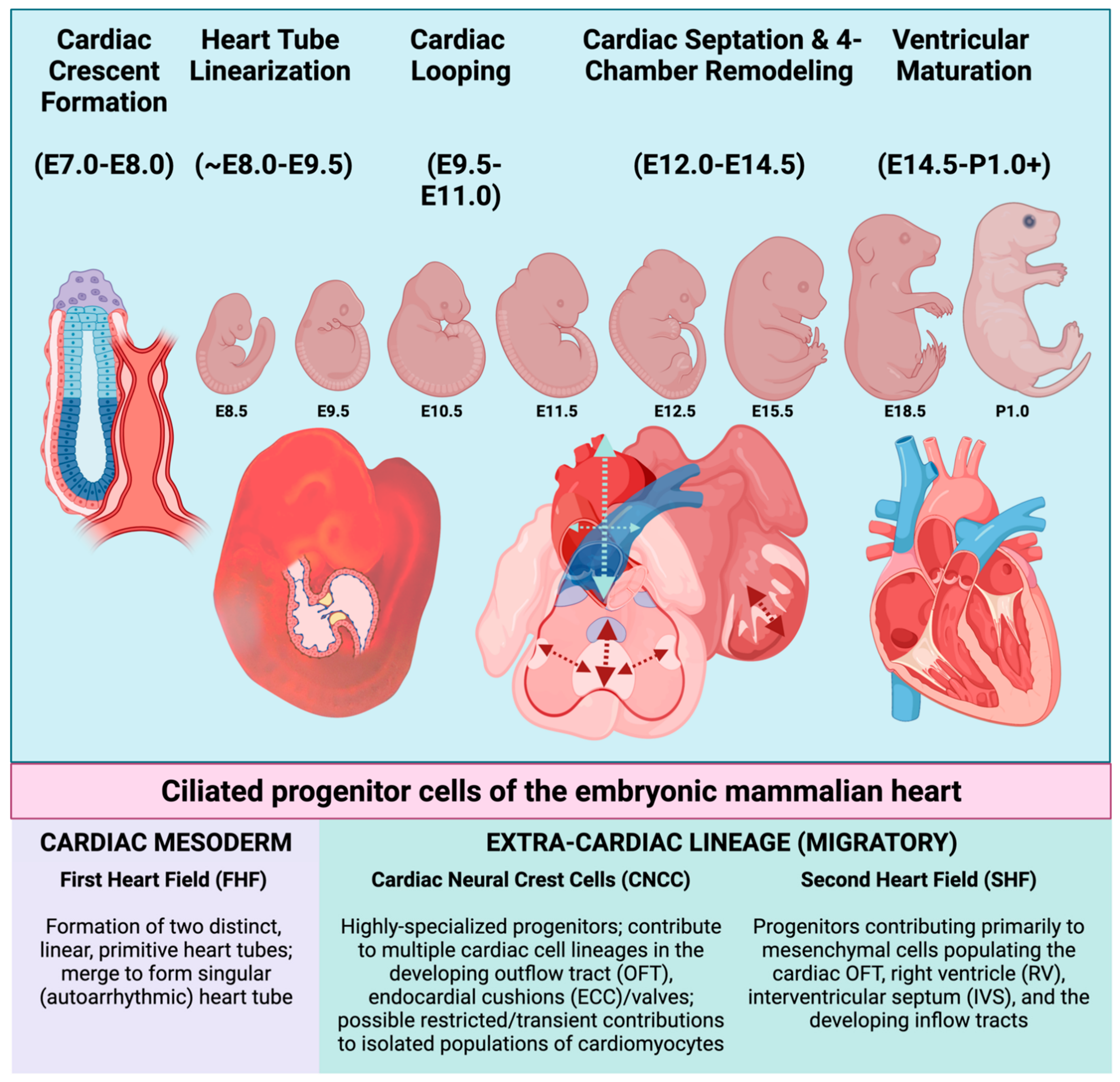
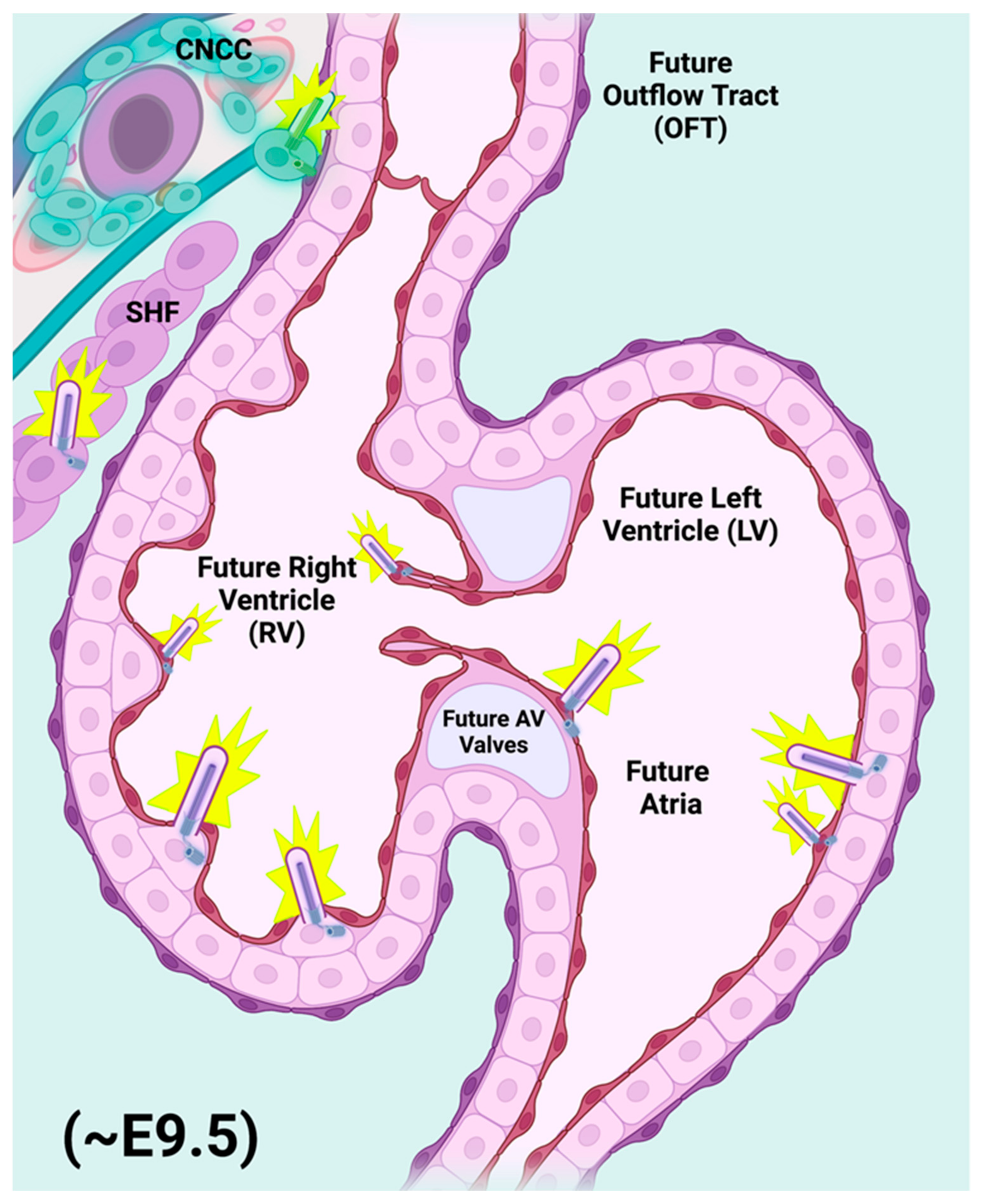
2. Primary Cilia and Their Function in the Development of Specific Cardiac Structures
2.1. Primary Cilia within the Heart: Distribution and Phenotypic Consequences Attendant with Ciliary Dysfunction
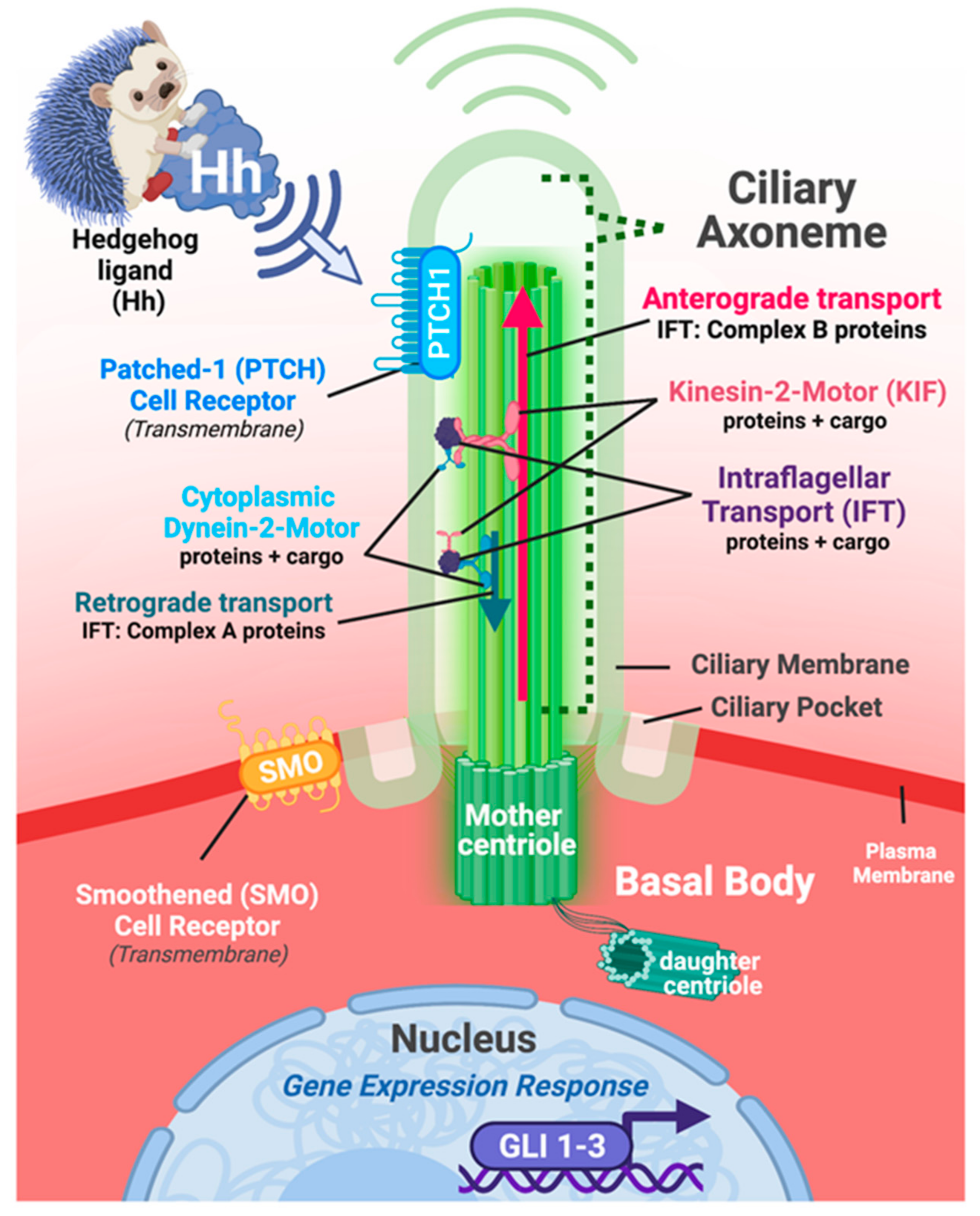
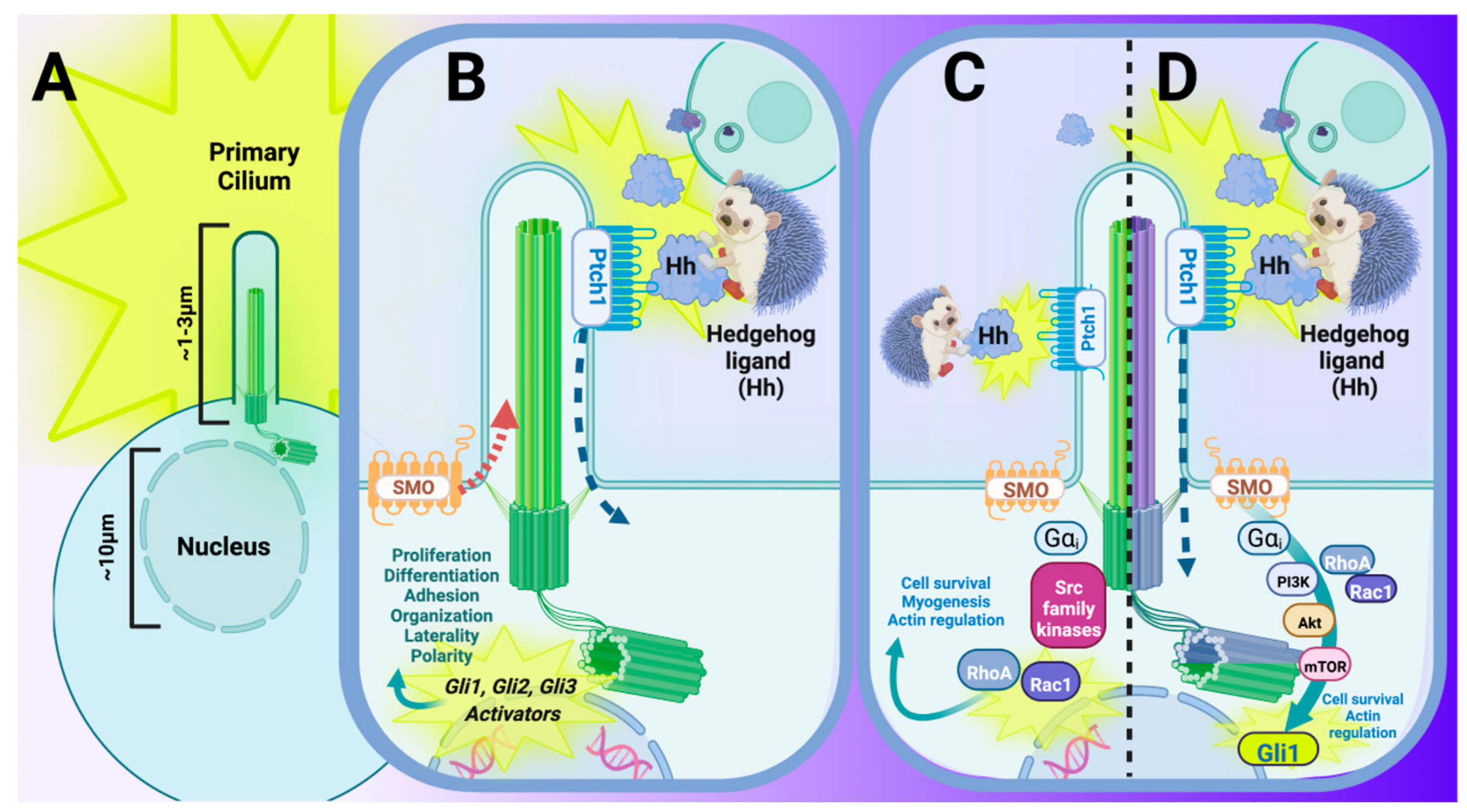
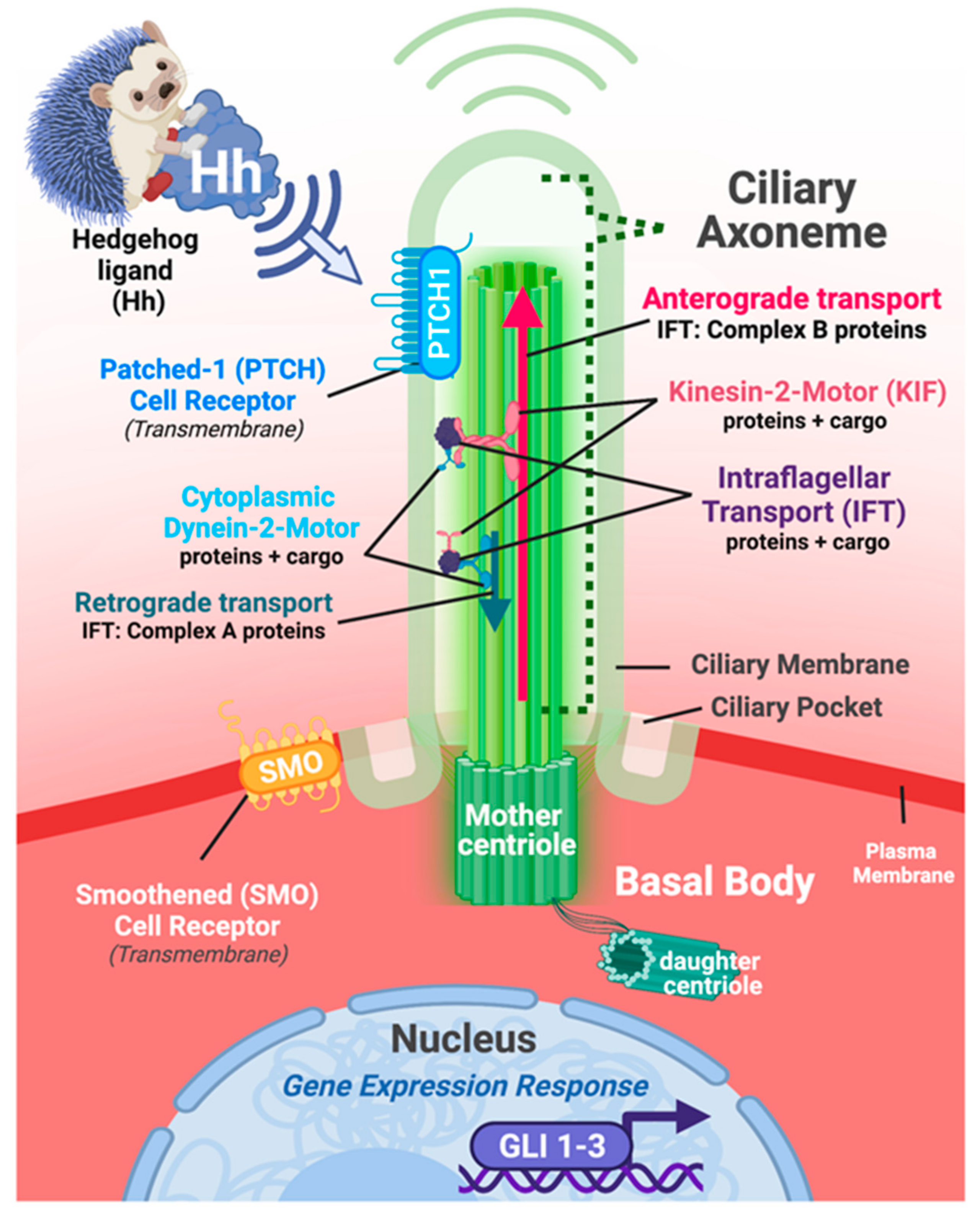
2.2. Primary Cilia and Hedgehog Signaling: A Long-Standing History
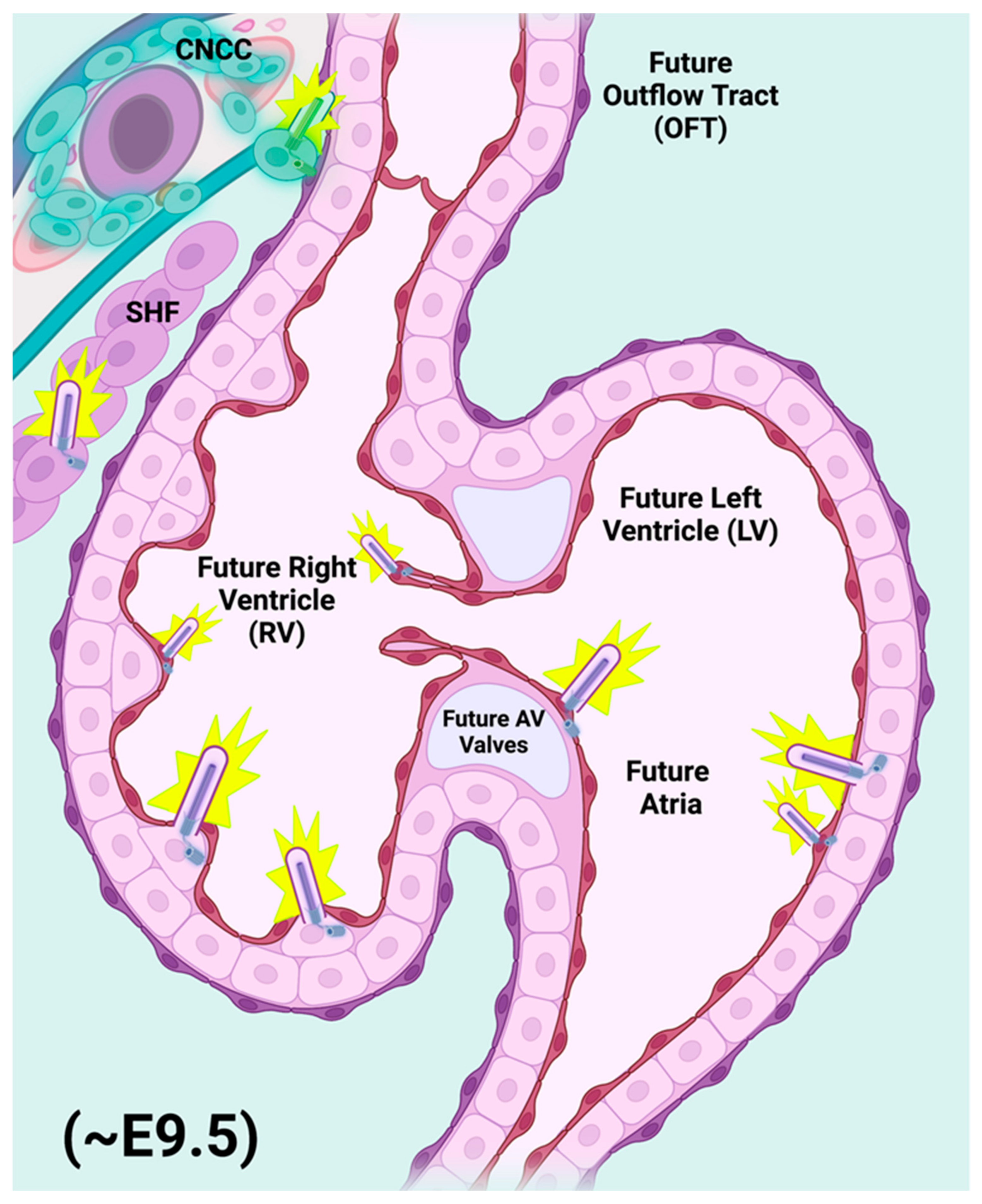
2.3. Cilia of the Embryonic Node
2.4. Primary Cilia of the Migratory CNCC Population
2.5. Primary Cilia of the Endocardial Cushions
2.6. Primary Cilia of the Atrio-Ventricular Septa
2.6.1. Atrial Septation
2.6.2. Ventricular Septation
2.7. Primary Cilia of Cardiac Fibrocytes
2.8. Primary Cilia of the Endocardium
2.9. Primary Cilia of the Ventricular Myocardium
3. The Multiple Roles That Hedgehog Signaling Plays in Cardiac Development and Function
3.1. Hedgehog Signaling and Progenitor Cells of Mammalian Heart Development
3.2. Hedgehog Signaling in Pre- and Postnatal Cardiomyocytes
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rash, J.E.; Shay, J.W.; Biesele, J.J. Cilia in Cardiac Differentiation. J. Ultrasructure Res. 1969, 29, 470–484. [Google Scholar] [CrossRef]
- Gabriel, G.C.; Young, C.B.; Lo, C.W. Role of Cilia in the Pathogenesis of Congenital Heart Disease. Semin. Cell Dev. Biol. 2021, 110, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Djenoune, L.; Berg, K.; Brueckner, M.; Yuan, S. A Change of Heart: New Roles for Cilia in Cardiac Development and Disease. Nat. Rev. Cardiol. 2022, 19, 211–227. [Google Scholar] [CrossRef] [PubMed]
- Slack, J.M.W.; Dale, L. Essential Developmental Biology, 4th ed.; John Wiley & Sons: Hoboken, NJ, USA, 2021. [Google Scholar]
- Meilhac, S.M.; Lescroart, F.; Blanpain, C.D.; Buckingham, M.E. Cardiac Cell Lineages That Form the Heart. Cold Spring Harb. Perspect. Med. 2014, 4, a013888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buckingham, M.; Meilhac, S.; Zaffran, S. Building the Mammalian Heart from Two Sources of Myocardial Cells. Nat. Rev. Genet. 2005, 6, 826–835. [Google Scholar] [CrossRef] [PubMed]
- Goetz, S.C.; Anderson, K.V. The Primary Cilium: A Signalling Centre during Vertebrate Development. Nat. Rev. Genet. 2010, 11, 331–344. [Google Scholar] [CrossRef] [PubMed]
- Wheway, G.; Nazlamova, L.; Hancock, J.T. Signaling through the Primary Cilium. Front. Cell Dev. Biol. 2018, 6, 8. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Fisher, R.L.; Bowser, S.S.; Pentecost, B.T.; Sui, H. Three-Dimensional Architecture of Epithelial Primary Cilia. Proc. Natl. Acad. Sci. USA 2019, 116, 9370–9379. [Google Scholar] [CrossRef] [Green Version]
- Hoyer-Fender, S. Primary and Motile Cilia: Their Ultrastructure and Ciliogenesis. In Cilia and Nervous System Development and Function; Tucker, K.L., Caspary, T., Eds.; Springer: Dordrecht, Netherlands, 2013; pp. 1–53. [Google Scholar]
- Scholey, J.M. Intraflagellar Transport. Annu. Rev. Cell Dev. Biol. 2003, 19, 423–443. [Google Scholar] [CrossRef] [Green Version]
- Haycraft, C.J.; Zhang, Q.; Song, B.; Jackson, W.S.; Detloff, P.J.; Serra, R.; Yoder, B.K. Intraflagellar Transport Is Essential for Endochondral Bone Formation. Development 2007, 134, 307–316. [Google Scholar] [CrossRef] [Green Version]
- Koefoed, K.; Veland, I.R.; Bang, L.; Lars, P.; Larsen, A.; Christensen, S.T.; Pedersen, L.B.; Larsen, L.A.; Christensen, S.T. Cilia and Coordination of Signaling Networks during Heart Development. Organogenesis 2014, 10, 108–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reiter, J.F.; Leroux, M.R. Genes and Molecular Pathways Underpinning Ciliopathies. Nat. Rev. Mol. Cell Biol. 2017, 18, 533–547. [Google Scholar] [CrossRef] [PubMed]
- Li, L.X.; Li, X. Epigenetically Mediated Ciliogenesis and Cell Cycle Regulation, and Their Translational Potential. Cells 2021, 10, 1662. [Google Scholar] [CrossRef] [PubMed]
- Mirvis, M.; Siemers, K.A.; Nelson, W.J.; Stearns, T.P. Primary Cilium Loss in Mammalian Cells Occurs Predominantly by Whole-Cilium Shedding. PLoS Biol. 2019, 17, e3000381. [Google Scholar] [CrossRef] [Green Version]
- Zimmermann, K.W. Beiträge Zur Kenntniss Einiger Drüsen Und Epithelien. Arch. Für Mikrosk. Anat. 1898, 52, 552–706. [Google Scholar] [CrossRef]
- Luck, D.J.L. Genetic and Biochemical Dissection of the Eucaryotic Flagellum. J. Cell Biol. 1984, 98, 789. [Google Scholar] [CrossRef] [Green Version]
- Pazour, G.J.; Dickert, B.L.; Vucica, Y.; Seeley, E.S.; Rosenbaum, J.L.; Witman, G.B.; Cole, D.G. Chlamydomonas IFT 88 and Its Mouse Homologue, Polycystic Kidney Disease Gene Tg 737, Are Required for Assembly of Cilia and Flagella. J. Cell Biol. 2000, 151, 709–718. [Google Scholar] [CrossRef]
- Badano, J.L.; Mitsuma, N.; Beales, P.L.; Katsanis, N. The Ciliopathies: An Emerging Class of Human Genetic Disorders. Annu. Rev. Genom. Hum. Genet. 2006, 7, 125–148. [Google Scholar] [CrossRef] [Green Version]
- Adams, M.; Smith, U.M.; Logan, C.V.; Johnson, C.A. Recent Advances in the Molecular Pathology, Cell Biology and Genetics of Ciliopathies. J. Med. Genet. 2008, 45, 257–267. [Google Scholar] [CrossRef]
- Snell, W.J.; Pan, J.; Wang, Q. Cilia and Flagella Revealed: From Flagellar Assembly in Chlamydomonas to Human Obesity Disorders. Cell 2004, 117, 693–697. [Google Scholar] [CrossRef] [Green Version]
- Wann, A.K.T.; Zuo, N.; Haycraft, C.J.; Jensen, C.G.; Poole, C.A.; McGlashan, S.R.; Knight, M.M. Primary Cilia Mediate Mechanotransduction through Control of ATP-Induced Ca2+ Signaling in Compressed Chondrocytes. FASEB J. 2012, 26, 1663–1671. [Google Scholar] [CrossRef] [Green Version]
- Menzl, I.; Lebeau, L.; Pandey, R.; Hassounah, N.B.; Li, F.W.; Nagle, R.; Weihs, K.; McDermott, K.M. Loss of Primary Cilia Occurs Early in Breast Cancer Development. Cilia 2014, 3, 7. [Google Scholar] [CrossRef] [Green Version]
- Jenks, A.D.; Vyse, S.; Wong, J.P.; Kostaras, E.; Keller, D.; Burgoyne, T.; Shoemark, A.; Tsalikis, A.; de la Roche, M.; Michaelis, M.; et al. Primary Cilia Mediate Diverse Kinase Inhibitor Resistance Mechanisms in Cancer. Cell Rep. 2018, 23, 3042–3055. [Google Scholar] [CrossRef]
- Han, Y.G.; Kim, H.J.; Dlugosz, A.A.; Ellison, D.W.; Gilbertson, R.J.; Alvarez-Buylla, A. Dual and Opposing Roles of Primary Cilia in Medulloblastoma Development. Nat. Med. 2009, 15, 1062–1065. [Google Scholar] [CrossRef] [Green Version]
- Manasek, F.J. Embryonic Development of the Heart. I. A Light and Electron Microscopic Study of Myocardial Development in the Early Chick Embryo. J. Morphol. 1968, 125, 329–365. [Google Scholar] [CrossRef]
- Van Der Heiden, K.; Groenendijk, B.C.W.; Hierck, B.P.; Hogers, B.; Koerten, H.K.; Mommaas, A.M.; Gittenberger-de Groot, A.C.; Poelmann, R.E. Monocilia on Chicken Embryonic Endocardium in Low Shear Stress Areas. Dev. Dyn. 2006, 235, 19–28. [Google Scholar] [CrossRef]
- Toomer, K.A.; Yu, M.; Fulmer, D.; Guo, L.; Moore, K.S.; Moore, R.; Ka’la, D.D.; Glover, J.; Peterson, N.; Ramos-Ortiz, S.; et al. Primary Cilia Defects Causing Mitral Valve Prolapse. Sci. Transl. Med. 2019, 11, eaax0290. [Google Scholar] [CrossRef] [Green Version]
- Diguet, N.; Le Garrec, J.-F.F.; Lucchesi, T.; Meilhac, S.M. Imaging and Analyzing Primary Cilia in Cardiac Cells. Methods Cell Biol. 2015, 127, 55–73. [Google Scholar] [CrossRef]
- Willaredt, M.A.; Gorgas, K.; Gardner, H.A.R.R.; Tucker, K.L. Multiple Essential Roles for Primary Cilia in Heart Development. Cilia 2012, 1, 23. [Google Scholar] [CrossRef] [Green Version]
- Slough, J.; Cooney, L.; Brueckner, M. Monocilia in the Embryonic Mouse Heart Suggest a Direct Role for Cilia in Cardiac Morphogenesis. Dev. Dyn. 2008, 237, 2304–2314. [Google Scholar] [CrossRef]
- Praetorius, H.A.; Spring, K.R. Bending the MDCK Cell Primary Cilium Increases Intracellular Calcium. J. Membr. Biol. 2001, 184, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Hofherr, A.; Köttgen, M. Polycystic Kidney Disease: Cilia and Mechanosensation Revisited. Nat. Rev. Nephrol. 2016, 12, 318–319. [Google Scholar] [CrossRef] [PubMed]
- Delling, M.; Indzhykulian, A.A.; Liu, X.; Li, Y.; Xie, T.; Corey, D.P.; Clapham, D.E. Primary Cilia Are Not Calcium-Responsive Mechanosensors. Nature 2016, 531, 656–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egorova, A.D.; Khedoe, P.P.S.J.; Goumans, M.J.T.H.; Yoder, B.K.; Nauli, S.M.; Ten Dijke, P.; Poelmann, R.E.; Hierck, B.P. Lack of Primary Cilia Primes Shear-Induced Endothelial-to-Mesenchymal Transition. Circ. Res. 2011, 108, 1093–1101. [Google Scholar] [CrossRef] [Green Version]
- Ma, M. Cilia and Polycystic Kidney Disease. Semin. Cell Dev. Biol. 2021, 110, 139–148. [Google Scholar] [CrossRef]
- Wu, G.; Markowitz, G.S.; Li, L.; D’Agati, V.D.; Factor, S.M.; Geng, L.; Tibara, S.; Tuchman, J.; Cai, Y.; Park, J.H.; et al. Cardiac Defects and Renal Failure in Mice with Targeted Mutations in Pkd2. Nat. Genet. 2000, 24, 75–78. [Google Scholar] [CrossRef]
- Boulter, C.; Mulroy, S.; Webb, S.; Fleming, S.; Brindle, K.; Sandford, R. Cardiovascular, Skeletal, and Renal Defects in Mice with a Targeted Disruption of the Pkd1 Gene. Proc. Natl. Acad. Sci. USA 2001, 98, 12174–12179. [Google Scholar] [CrossRef] [Green Version]
- Clement, C.A.; Kristensen, S.G.; Møllgård, K.; Pazour, G.J.; Yoder, B.K.; Larsen, L.A.; Christensen, S.T.; Møllgard, K.; Pazour, G.J.; Yoder, B.K.; et al. The Primary Cilium Coordinates Early Cardiogenesis and Hedgehog Signaling in Cardiomyocyte Differentiation. J. Cell Sci. 2009, 122 Pt 17, 3070–3082. [Google Scholar] [CrossRef] [Green Version]
- Günthel, M.; Barnett, P.; Christoffels, V.M. Development, Proliferation, and Growth of the Mammalian Heart. Mol. Ther. 2018, 26, 1599–1609. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Klena, N.T.; Gabriel, G.C.; Liu, X.; Kim, A.J.; Lemke, K.; Chen, Y.; Chatterjee, B.; Devine, W.; Damerla, R.R.; et al. Global Genetic Analysis in Mice Unveils Central Role for Cilia in Congenital Heart Disease. Nature 2015, 521, 520–524. [Google Scholar] [CrossRef] [Green Version]
- Caspary, T.; Larkins, C.E.; Anderson, K.V. The Graded Response to Sonic Hedgehog Depends on Cilia Architecture. Dev. Cell 2007, 12, 767–778. [Google Scholar] [CrossRef] [Green Version]
- Nüsslein-Volhard, C.; Wieschaus, E. Mutations Affecting Segment Number and Polarity in Drosophila. Nature 1980, 287, 795–801. [Google Scholar] [CrossRef]
- Pathi, S.; Pagan-Westphal, S.; Baker, D.P.; Garber, E.A.; Rayhorn, P.; Bumcrot, D.; Tabin, C.J.; Blake Pepinsky, R.; Williams, K.P. Comparative Biological Responses to Human Sonic, Indian, and Desert Hedgehog. Mech. Dev. 2001, 106, 107–117. [Google Scholar] [CrossRef]
- Tsukui, T.; Capdevila, J.; Tamura, K.; Ruiz-Lozano, P.; Rodriguez-Esteban, C.; Yonei-Tamura, S.; Magallón, J.; Chandraratna, R.A.S.; Chien, K.; Blumberg, B.; et al. Multiple Left-Right Asymmetry Defects in Shh-/- Mutant Mice Unveil a Convergence of the Shh and Retinoic Acid Pathways in the Control of Lefty-1. Proc. Natl. Acad. Sci. USA 1999, 96, 11376–11381. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.M.; Ramalho-Santos, M.; McMahon, A.P. Smoothened Mutants Reveal Redundant Roles for Shh and Ihh Signaling Including Regulation of L/R Asymmetry by the Mouse Node. Cell 2001, 105, 781–792. [Google Scholar] [CrossRef] [Green Version]
- Bonnafe, E.; Touka, M.; AitLounis, A.; Baas, D.; Barras, E.; Ucla, C.; Moreau, A.; Flamant, F.; Dubruille, R.; Couble, P.; et al. The Transcription Factor RFX3 Directs Nodal Cilium Development and Left-Right Asymmetry Specification. Mol. Cell. Biol. 2004, 24, 4417–4427. [Google Scholar] [CrossRef] [Green Version]
- Francis, R.J.B.; Christopher, A.; Devine, W.A.; Ostrowski, L.; Lo, C. Congenital Heart Disease and the Specification of Left-Right Asymmetry. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H2102–H2111. [Google Scholar] [CrossRef] [Green Version]
- Parmantier, E.; Lynn, B.; Lawson, D.; Turmaine, M.; Namini, S.S.; Chakrabarti, L.; McMahon, A.P.; Jessen, K.R.; Mirsky, R. Schwann Cell-Derived Desert Hedgehog Controls the Development of Peripheral Nerve Sheaths. Neuron 1999, 23, 713–724. [Google Scholar] [CrossRef] [Green Version]
- Whitfield, J.F. The Solitary (Primary) Cilium—A Mechanosensory Toggle Switch in Bone and Cartilage Cells. Cell. Signal. 2008, 20, 1019–1024. [Google Scholar] [CrossRef]
- Wang, Z.; Wann, A.K.T.; Thompson, C.L.; Hassen, A.; Wang, W.; Knight, M.M. IFT88 Influences Chondrocyte Actin Organization and Biomechanics. Osteoarthr. Cartil. 2016, 24, 544–554. [Google Scholar] [CrossRef] [Green Version]
- Rohatgi, R.; Milenkovic, L.; Scott, M.P. Patched1 Regulates Hedgehog Signaling at the Primary Cilium. Science 2007, 317, 372–376. [Google Scholar] [CrossRef] [Green Version]
- Corbit, K.C.; Aanstad, P.; Singla, V.; Norman, A.R.; Stainier, D.Y.R.; Reiter, J.F. Vertebrate Smoothened Functions at the Primary Cilium. Nature 2005, 437, 1018–1021. [Google Scholar] [CrossRef]
- Stecca, B.; Ruiz I Altaba, A. Context-Dependent Regulation of the GLI Code in Cancer by HEDGEHOG and Non-HEDGEHOG Signals. J. Mol. Cell Biol. 2010, 2, 84. [Google Scholar] [CrossRef] [Green Version]
- Echelard, Y.; Epstein, D.J.; St-Jacques, B.; Shen, L.; Mohler, J.; McMahon, J.A.; McMahon, A.P. Sonic Hedgehog, a Member of a Family of Putative Signaling Molecules, Is Implicated in the Regulation of CNS Polarity. Cell 1993, 75, 1417–1430. [Google Scholar] [CrossRef]
- Chen, M.-H.H.; Wilson, C.W.; Li, Y.-J.J.; Law, K.K.L.; Lu, C.-S.S.; Gacayan, R.; Zhang, X.; Hui, C.-C.C.; Chuang, P.-T.T.; King, K.; et al. Cilium-Independent Regulation of Gli Protein Function by Sufu in Hedgehog Signaling Is Evolutionarily Conserved. Genes Dev. 2009, 23, 1910–1928. [Google Scholar] [CrossRef] [Green Version]
- Alexander, S.P.H.; Kelly, E.; Mathie, A.; Peters, J.A.; Veale, E.L.; Armstrong, J.F.; Faccenda, E.; Harding, S.D.; Pawson, A.J.; Sharman, J.L.; et al. THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: G protein-coupled receptors. Br. J. Pharmacol. 2019, 176 (Suppl. S1), S21–S141. [Google Scholar] [CrossRef] [Green Version]
- Brennan, D.; Chen, X.; Cheng, L.; Mahoney, M.; Riobo, N.A. Noncanonical Hedgehog Signaling. Vitam. Horm. 2012, 88, 55–72. [Google Scholar] [CrossRef] [Green Version]
- Akhshi, T.; Shannon, R.; Trimble, W.S. The Complex Web of Canonical and Non-Canonical Hedgehog Signaling. BioEssays 2022, 44, e2100183. [Google Scholar] [CrossRef]
- Jeong, M.H.; Leem, Y.E.; Kim, H.J.; Kang, K.; Cho, H.; Kang, J.S. A Shh Coreceptor Cdo Is Required for Efficient Cardiomyogenesis of Pluripotent Stem Cells. J. Mol. Cell. Cardiol. 2016, 93, 57–66. [Google Scholar] [CrossRef]
- Tenzen, T.; Allen, B.L.; Cole, F.; Kang, J.S.; Krauss, R.S.; McMahon, A.P. The Cell Surface Membrane Proteins Cdo and Boc Are Components and Targets of the Hedgehog Signaling Pathway and Feedback Network in Mice. Dev. Cell 2006, 10, 647–656. [Google Scholar] [CrossRef] [Green Version]
- Allen, B.L.; Tenzen, T.; McMahon, A.P. The Hedgehog-Binding Proteins Gas1 and Cdo Cooperate to Positively Regulate Shh Signaling during Mouse Development. Genes Dev. 2007, 21, 1244–1257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinelli, D.C.; Fan, C.M. Gas1 Extends the Range of Hedgehog Action by Facilitating Its Signaling. Genes Dev. 2007, 21, 1231–1243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sigafoos, A.N.; Paradise, B.D.; Fernandez-Zapico, M.E.; Stecca, B. Hedgehog/GLI Signaling Pathway: Transduction, Regulation, and Implications for Disease. Cancers 2021, 13, 3410. [Google Scholar] [CrossRef] [PubMed]
- Polizio, A.H.; Chinchilla, P.; Chen, X.; Manning, D.R.; Riobo, N.A. Sonic Hedgehog Activates the GTPases Rac1 and RhoA in a Gli-Independent Manner via Coupling of Smoothened to Gi Proteins. Sci Signal. 2011, 4, pt7. [Google Scholar] [CrossRef] [Green Version]
- Pietrobono, S.; Gagliardi, S.; Stecca, B. Non-Canonical Hedgehog Signaling Pathway in Cancer: Activation of GLI Transcription Factors beyond Smoothened. Front. Genet. 2019, 10, 556. [Google Scholar] [CrossRef] [Green Version]
- Kojima, Y.; Tam, O.H.; Tam, P.P.L. Timing of Developmental Events in the Early Mouse Embryo. Semin. Cell Dev. Biol. 2014, 34, 65–75. [Google Scholar] [CrossRef]
- Antony, D.; Brunner, H.G.; Schmidts, M. Ciliary Dyneins and Dynein Related Ciliopathies. Cells 2021, 10, 1885. [Google Scholar] [CrossRef]
- Wislet, S.; Vandervelden, G.; Rogister, B. From Neural Crest Development to Cancer and Vice Versa: How P75NTR and (Pro)Neurotrophins Could Act on Cell Migration and Invasion? Front. Mol. Neurosci. 2018, 11, 244. [Google Scholar] [CrossRef]
- Lumiaho, A.; Miettinen, R.; Niemitukia, L.; Laitinen, T.; Rantala, A.; Lampainen, E.; Laakso, M.; Hartikainen, J. Mitral Valve Prolapse and Mitral Regurgitation Are Common in Patients With Polycystic Kidney Disease Type 1. Am. J. Kidney Dis. 2001, 38, 1208–1216. [Google Scholar] [CrossRef]
- Durst, R.; Sauls, K.; Peal, D.S.; DeVlaming, A.; Toomer, K.; Leyne, M.; Salani, M.; Talkowski, M.E.; Brand, H.; Perrocheau, M.; et al. Mutations in DCHS1 Cause Mitral Valve Prolapse. Nature 2015, 525, 109–113. [Google Scholar] [CrossRef]
- Ecile Dau, C.; Fliegauf, M.; Omran, H.; Schlensog, M.; Dahl, E.; Van Roeyen, C.R.; Kriz, W.; Moeller, M.J.; Braun, G.S. The Atypical Cadherin Dachsous1 Localizes to the Base of the Ciliary Apparatus in Airway Epithelia. Biochem. Biophys. Res. Commun. 2016, 473, 1177–1184. [Google Scholar] [CrossRef]
- Kyndt, F.; Gueffet, J.P.; Probst, V.; Jaafar, P.; Legendre, A.; Le Bouffant, F.; Toquet, C.; Roy, E.; McGregor, L.; Lynch, S.A.; et al. Mutations in the Gene Encoding Filamin A as a Cause for Familial Cardiac Valvular Dystrophy. Circulation 2007, 115, 40–49. [Google Scholar] [CrossRef] [Green Version]
- Adams, M.; Simms, R.J.; Abdelhamed, Z.; Dawe, H.R.; Szymanska, K.; Logan, C.V.; Wheway, G.; Pitt, E.; Gull, K.; Knowles, M.A.; et al. A Meckelin-Filamin A Interaction Mediates Ciliogenesis. Hum. Mol. Genet. 2012, 21, 1272–1286. [Google Scholar] [CrossRef]
- Wang, C.; Low, W.C.; Liu, A.; Wang, B. Centrosomal Protein DZIP1 Regulates Hedgehog Signaling by Promoting Cytoplasmic Retention of Transcription Factor GLI3 and Affecting Ciliogenesis. J. Biol. Chem. 2013, 288, 29518–29529. [Google Scholar] [CrossRef] [Green Version]
- Fulmer, D.; Toomer, K.A.; Glover, J.; Guo, L.; Moore, K.; Moore, R.; Stairley, R.; Gensemer, C.; Abrol, S.; Rumph, M.K.; et al. Desert Hedgehog-Primary Cilia Cross Talk Shapes Mitral Valve Tissue by Organizing Smooth Muscle Actin. Dev. Biol. 2020, 463, 26–38. [Google Scholar] [CrossRef]
- Toomer, K.A.; Fulmer, D.; Guo, L.; Drohan, A.; Peterson, N.; Swanson, P.; Brooks, B.; Mukherjee, R.; Body, S.; Lipschutz, J.H.; et al. A Role for Primary Cilia in Aortic Valve Development and Disease. Dev. Dyn. 2017, 246, 625–634. [Google Scholar] [CrossRef] [Green Version]
- Karp, N.; Grosse-Wortmann, L.; Bowdin, S. Severe Aortic Stenosis, Bicuspid Aortic Valve and Atrial Septal Defect in a Child with Joubert Syndrome and Related Disorders (JSRD)—A Case Report and Review of Congenital Heart Defects Reported in the Human Ciliopathies. Eur. J. Med. Genet. 2012, 55, 605–610. [Google Scholar] [CrossRef]
- Fulmer, D.; Toomer, K.; Guo, L.; Moore, K.; Glover, J.; Moore, R.; Stairley, R.; Lobo, G.; Zuo, X.; Dang, Y.; et al. Defects in the Exocyst-Cilia Machinery Cause Bicuspid Aortic Valve Disease and Aortic Stenosis. Circulation 2019, 140, 1331–1341. [Google Scholar] [CrossRef]
- Hoffmann, A.D.; Peterson, M.A.; Friedland-Little, J.M.; Anderson, S.A.; Moskowitz, I.P. Sonic Hedgehog Is Required in Pulmonary Endoderm for Atrial Septation. Development 2009, 136, 1761–1770. [Google Scholar] [CrossRef] [Green Version]
- Smoak, I.W.; Byrd, N.A.; Abu-issa, R.; Goddeeris, M.M.; Anderson, R.; Morris, J.; Yamamura, K.; Klingensmith, J.; Meyers, E.N. Sonic Hedgehog Is Required for Cardiac Outflow Tract and Neural Crest Cell Development. Dev. Biol. 2005, 283, 357–372. [Google Scholar] [CrossRef] [Green Version]
- Burnicka-Turek, O.; Steimle, J.D.; Huang, W.; Felker, L.; Kamp, A.; Kweon, J.; Peterson, M.; Reeves, R.H.; Maslen, C.L.; Gruber, P.J.; et al. Cilia Gene Mutations Cause Atrioventricular Septal Defects by Multiple Mechanisms. Hum. Mol. Genet. 2016, 25, 3011–3028. [Google Scholar] [CrossRef]
- Klena, N.T.; Gibbs, B.C.; Lo, C.W. Cilia and Ciliopathies in Congenital Heart Disease. Cold Spring Harb. Perspect. Biol. 2017, 9, a028266. [Google Scholar] [CrossRef]
- Vierkotten, J.; Dildrop, R.; Peters, T.; Wang, B.; Rüther, U. Ftm Is a Novel Basal Body Protein of Cilia Involved in Shh Signalling. Development 2007, 134, 2569–2577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerhardt, C.; Lier, J.M.; Kuschel, S.; Rü, U.; Rüther, U. The Ciliary Protein Ftm Is Required for Ventricular Wall and Septal Development. PLoS ONE 2013, 8, e57545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villalobos, E.; Criollo, A.; Schiattarella, G.G.; Altamirano, F.; French, K.M.; May, H.I.; Jiang, N.; Nguyen, N.U.N.; Romero, D.; Roa, J.C.; et al. Fibroblast Primary Cilia Are Required for Cardiac Fibrosis. Circulation 2019, 139, 2342–2357. [Google Scholar] [CrossRef] [PubMed]
- Saint-jean, L.; Barkas, N.; Harmelink, C.; Tompkins, K.L.; Oakey, R.J.; Baldwin, H.S. Myocardial Differentiation Is Dependent upon Endocardial Signaling during Early Cardiogenesis in Vitro. Development 2019, 146, dev172619. [Google Scholar] [CrossRef] [Green Version]
- Pucéat, M. Embryological Origin of the Endocardium and Derived Valve Progenitor Cells: From Developmental Biology to Stem Cell-Based Valve Repair. Biochim. Biophys. Acta—Mol. Cell Res. 2013, 1833, 917–922. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.; Lin, C.; Chen, C.; Zhou, B.; Chang, C. Partitioning the Heart: Mechanisms of Cardiac Septation and Valve Development. Development 2012, 3299, 3277–3299. [Google Scholar] [CrossRef] [Green Version]
- Artap, S.; Manderfield, L.J.; Smith, C.L.; Poleshko, A.; Aghajanian, H.; See, K.; Li, L.; Jain, R.; Epstein, J.A. Endocardial Hippo Signaling Regulates Myocardial Growth and Cardiogenesis. Dev. Biol. 2018, 440, 22–30. [Google Scholar] [CrossRef]
- Samsa, L.A.; Yang, B.; Liu, J. Embryonic Cardiac Chamber Maturation: Trabeculation, Conduction, and Cardiomyocyte Proliferation. Am. J. Med. Genet. Part C Semin. Med. Genet. 2013, 163, 157–168. [Google Scholar] [CrossRef] [Green Version]
- Samsa, L.A.; Givens, C.; Tzima, E.; Stainier, D.Y.R.R.; Qian, L.; Liu, J. Cardiac Contraction Activates Endocardial Notch Signaling to Modulate Chamber Maturation in Zebrafish. Development 2015, 142, 4080–4091. [Google Scholar] [CrossRef] [Green Version]
- Pentassuglia, L.; Sawyer, D.B. The Role of Neuregulin-1β/ErbB Signaling in the Heart. Exp. Cell Res. 2009, 315, 627–637. [Google Scholar] [CrossRef] [Green Version]
- Wheatley, D.N.; Wang, A.M.; Strugnell, G.E. Expression of Primary Cilia in Mammalian Cells. Cell Biol. Int. 1996, 20, 73–81. [Google Scholar] [CrossRef]
- Hua, K.; Ferland, R.J. Fixation Methods Can Differentially Affect Ciliary Protein Immunolabeling. Cilia 2017, 6, 5. [Google Scholar] [CrossRef] [Green Version]
- Bell, P.D.; Fitzgibbon, W.; Sas, K.; Stenbit, A.E.; Amria, M.; Houston, A.; Reichert, R.; Gilley, S.; Siegal, G.P.; Bissler, J.; et al. Loss of Primary Cilia Upregulates Renal Hypertrophic Signaling and Promotes Cystogenesis. J. Am. Soc. Nephrol. 2011, 22, 839–848. [Google Scholar] [CrossRef] [Green Version]
- Nauli, S.M.; Jin, X.; AbouAlaiwi, W.A.; El-Jouni, W.; Su, X.; Zhou, J. Non-Motile Primary Cilia as Fluid Shear Stress Mechanosensors. Methods Enzymol. 2013, 525, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Pala, R.; Jamal, M.; Alshammari, Q.; Nauli, S.M. The Roles of Primary Cilia in Cardiovascular Diseases. Cells 2018, 7, 233. [Google Scholar] [CrossRef] [Green Version]
- Kaur, S.; McGlashan, S.R.; Ward, M.-L. Evidence of Primary Cilia in the Developing Rat Heart. Cilia 2018, 7, 4. [Google Scholar] [CrossRef] [Green Version]
- Sedmera, D.; Pexieder, T.; Hu, N.; Clark, E.B. A Quantitative Study of the Ventricular Myoarchitecture in the Stage 21- 29 Chick Embryo Following Decreased Loading. Eur. J. Morphol. 1998, 36, 105–119. [Google Scholar] [CrossRef]
- Goenezen, S.; Rennie, M.Y.; Rugonyi, S. Biomechanics of Early Cardiac Development. Biomech. Model. Mechanobiol. 2013, 11, 1187–1204. [Google Scholar] [CrossRef] [Green Version]
- Paige, S.L.; Plonowska, K.; Xu, A.; Wu, S.M. Molecular Regulation of Cardiomyocyte Differentiation. Circ. Res. 2015, 116, 341–353. [Google Scholar] [CrossRef] [Green Version]
- Ma, P.; Gu, S.; Karunamuni, G.H.; Jenkins, M.W.; Watanabe, M.; Rollins, A.M. Cardiac Neural Crest Ablation Results in Early Endocardial Cushion and Hemodynamic Flow Abnormalities. Am. J. Physiol.—Hear. Circ. Physiol. 2016, 311, H1150–H1159. [Google Scholar] [CrossRef] [Green Version]
- Lindsey, S.E.; Butcher, J.T.; Yalcin, H.C. Mechanical Regulation of Cardiac Development. Front. Physiol. 2014, 5, 318. [Google Scholar] [CrossRef] [Green Version]
- Vestergaard, M.L.; Grubb, S.; Koefoed, K.; Anderson-Jenkins, Z.; Grunnet-Lauridsen, K.; Calloe, K.; Clausen, C.; Christensen, S.T.; Møllgård, K.; Andersen, C.Y. Human Embryonic Stem Cell-Derived Cardiomyocytes Self-Arrange with Areas of Different Subtypes during Differentiation. Stem Cells Dev. 2017, 26, 1566–1577. [Google Scholar] [CrossRef]
- Grubb, S.; Vestergaard, M.L.; Andersen, A.S.; Rasmussen, K.K.; Mamsen, L.S.; Tuckute, G.; Grunnet-Lauridsen, K.; Møllgård, K.; Ernst, E.; Christensen, S.T.; et al. Comparison of Cultured Human Cardiomyocyte Clusters Obtained from Embryos/Fetuses or Derived from Human Embryonic Stem Cells. Stem Cells Dev. 2019, 28, 608–619. [Google Scholar] [CrossRef]
- Robbins, D.J.; Fei, D.L.; Riobo, N.A. The Hedgehog Signal Transduction Network. Sci. Signal. 2012, 5, re6. [Google Scholar] [CrossRef] [Green Version]
- Martí, E.; Takada, R.; Bumcrot, D.A.; Sasaki, H.; McMahon, A.P. Distribution of Sonic Hedgehog Peptides in the Developing Chick and Mouse Embryo. Development 1995, 121, 2537–2547. [Google Scholar] [CrossRef]
- Epstein, J.; Li, J.; Lang, D.; Chen, F.; Brown, C.B.; Jin, F.; Lu, M.M.; Thomas, M.; Liu, E.; Wessels, A.; et al. Migration of Cardiac Neural Crest Cells in Splotch Embryos. Development 2000, 127, 1869–1878. [Google Scholar] [CrossRef]
- Goddeeris, M.M.; Schwartz, R.; Klingensmith, J.; Meyers, E.N. Independent Requirements for Hedgehog Signaling by Both the Anterior Heart Field and Neural Crest Cells for Outflow Tract Development. Development 2007, 134, 1593–1604. [Google Scholar] [CrossRef] [Green Version]
- Le, T.L.; Sribudiani, Y.; Dong, X.; Huber, C.; Kois, C.; Baujat, G.; Gordon, C.T.; Mayne, V.; Galmiche, L.; Serre, V.; et al. Bi-Allelic Variations of SMO in Humans Cause a Broad Spectrum of Developmental Anomalies Due to Abnormal Hedgehog Signaling. Am. J. Hum. Genet. 2020, 106, 779–792. [Google Scholar] [CrossRef]
- Meilhac, S.M.; Buckingham, M.E. The Deployment of Cell Lineages That Form the Mammalian Heart. Nat. Rev. Cardiol. 2018, 15, 705–724. [Google Scholar] [CrossRef] [PubMed]
- Dyer, L.A.; Kirby, M.L. Sonic Hedgehog Maintains Proliferation in Secondary Heart Field Progenitors and Is Required for Normal Arterial Pole Formation. Dev. Biol. 2009, 330, 305–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christ, A.; Marczenke, M.; Willnow, T.E. LRP2 Controls Sonic Hedgehog-Dependent Differentiation of Cardiac Progenitor Cells during Outflow Tract Formation. Hum. Mol. Genet. 2020, 29, 3183–3196. [Google Scholar] [CrossRef] [PubMed]
- Briggs, L.E.; Burns, T.A.; Lockhart, M.M.; Phelps, A.L.; Van den Hoff, M.J.B.; Wessels, A. Wnt/β-Catenin and Sonic Hedgehog Pathways Interact in the Regulation of the Development of the Dorsal Mesenchymal Protrusion. Dev. Dyn. 2016, 245, 103–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dyer, L.A.; Makadia, F.A.; Scott, A.; Pegram, K.; Hutson, M.R.; Kirby, M.L. BMP Signaling Modulates Hedgehog-Induced Secondary Heart Field Proliferation. Dev. Biol. 2010, 348, 167–176. [Google Scholar] [CrossRef] [Green Version]
- Goddeeris, M.M.; Rho, S.; Petiet, A.; Davenport, C.L.; Johnson, G.A.; Meyers, E.N.; Klingensmith, J.; Goddeeris, M.M. Intracardiac Septation Requires Hedgehog-Dependent Cellular Contributions from Outside the Heart. Development 2008, 135, 1887–1895. [Google Scholar] [CrossRef] [Green Version]
- Tang, W.; Martik, M.L.; Li, Y.; Bronner, M.E. Cardiac Neural Crest Contributes to Cardiomyocytes in Amniotes and Heart Regeneration in Zebrafish. eLife 2019, 8, e47929. [Google Scholar] [CrossRef]
- Abdul-Wajid, S.; Demarest, B.L.; Yost, H.J. Loss of Embryonic Neural Crest Derived Cardiomyocytes Causes Adult Onset Hypertrophic Cardiomyopathy in Zebrafish. Nat. Commun. 2018, 9, 4603. [Google Scholar] [CrossRef] [Green Version]
- Bookman, D.E.; Redmond, M.E.; Waldo, K.; Davis, H.; Kirby, M.L. Effect of Neural Crest Ablation on Development of the Heart and Arch Arteries in the Chick. Am. J. Anat. 1987, 180, 332–341. [Google Scholar] [CrossRef]
- Kelly, R.G.; Buckingham, M.E.; Moorman, A.F. Heart Fields and Cardiac Morphogenesis. Cold Spring Harb. Perspect. Med. 2014, 4, a015750. [Google Scholar] [CrossRef] [Green Version]
- Cheng, L.; Al-Owais, M.; Covarrubias, M.L.; Koch, W.J.; Manning, D.R.; Peers, C.; Riobo-Del Galdo, N.A. Coupling of Smoothened to Inhibitory G Proteins Reduces Voltage-Gated K+ Currents in Cardiomyocytes and Prolongs Cardiac Action Potential Duration. J. Biol. Chem. 2018, 293, 11022–11032. [Google Scholar] [CrossRef] [Green Version]
- Torrisi, F.; Alberghina, C.; Lo Furno, D.; Zappalà, A.; Valable, S.; Li Volti, G.; Tibullo, D.; Vicario, N.; Parenti, R. Connexin 43 and Sonic Hedgehog Pathway Interplay in Glioblastoma Cell Proliferation and Migration. Biology 2021, 10, 767. [Google Scholar] [CrossRef]
- Dobrowolski, R.; Hertig, G.; Lechner, H.; Wörsdörfer, P.; Wulf, V.; Dicke, N.; Eckert, D.; Bauer, R.; Schorle, H.; Willecke, K. Loss of Connexin43-Mediated Gap Junctional Coupling in the Mesenchyme of Limb Buds Leads to Altered Expression of Morphogens in Mice. Hum. Mol. Genet. 2009, 18, 2899–2911. [Google Scholar] [CrossRef] [Green Version]
- Diagbouga, M.R.; Morel, S.; Cayron, A.F.; Haemmerli, J.; Georges, M.; Hierck, B.P.; Allémann, E.; Lemeille, S.; Bijlenga, P.; Kwak, B.R. Primary Cilia Control Endothelial Permeability by Regulating Expression and Location of Junction Proteins. Cardiovasc. Res. 2022, 118, 1583–1596. [Google Scholar] [CrossRef]
- Jeong, M.-H.; Kim, H.-J.; Pyun, J.-H.; Choi, K.-S.; Lee, D.I.; Solhjoo, S.; O’rourke, B.; Tomaselli, G.F.; Jeong, D.S.; Cho, H.; et al. Cdon Deficiency Causes Cardiac Remodeling through Hyperactivation of WNT/β-Catenin Signaling. Proc. Natl. Acad. Sci. USA 2017, 114, E1345–E1354. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Lu, P.; Zhao, D.; Sheng, J. Targeting the Hedgehog Signaling Pathway for Cardiac Repair and Regeneration. Herz 2017, 42, 662–668. [Google Scholar] [CrossRef]
- Paulis, L.; Fauconnier, J.; Cazorla, O.; Thireau, J.; Soleti, R.; Vidal, B.; Ouillé, A.; Bartholome, M.; Bideaux, P.; Roubille, F.; et al. Activation of Sonic Hedgehog Signaling in Ventricular Cardiomyocytes Exerts Cardioprotection against Ischemia Reperfusion Injuries. Sci. Rep. 2015, 5, 7983. [Google Scholar] [CrossRef] [Green Version]
- Kawagishi, H.; Xiong, J.; Rovira, I.I.; Pan, H.; Yan, Y.; Fleischmann, B.K.; Yamada, M.; Finkel, T. Sonic Hedgehog Signaling Regulates the Mammalian Cardiac Regenerative Response. J. Mol. Cell. Cardiol. 2018, 123, 180–184. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fitzsimons, L.A.; Brewer, V.L.; Tucker, K.L. Hedgehog Morphogens Act as Growth Factors Critical to Pre- and Postnatal Cardiac Development and Maturation: How Primary Cilia Mediate Their Signal Transduction. Cells 2022, 11, 1879. https://doi.org/10.3390/cells11121879
Fitzsimons LA, Brewer VL, Tucker KL. Hedgehog Morphogens Act as Growth Factors Critical to Pre- and Postnatal Cardiac Development and Maturation: How Primary Cilia Mediate Their Signal Transduction. Cells. 2022; 11(12):1879. https://doi.org/10.3390/cells11121879
Chicago/Turabian StyleFitzsimons, Lindsey A., Victoria L. Brewer, and Kerry L. Tucker. 2022. "Hedgehog Morphogens Act as Growth Factors Critical to Pre- and Postnatal Cardiac Development and Maturation: How Primary Cilia Mediate Their Signal Transduction" Cells 11, no. 12: 1879. https://doi.org/10.3390/cells11121879
APA StyleFitzsimons, L. A., Brewer, V. L., & Tucker, K. L. (2022). Hedgehog Morphogens Act as Growth Factors Critical to Pre- and Postnatal Cardiac Development and Maturation: How Primary Cilia Mediate Their Signal Transduction. Cells, 11(12), 1879. https://doi.org/10.3390/cells11121879