
NADPH Oxidase (NOX) Targeting in Diabetes: A Special Emphasis on Pancreatic β-Cell Dysfunction


{kind=link}
{kind=link}
Abstract
:1. Introduction
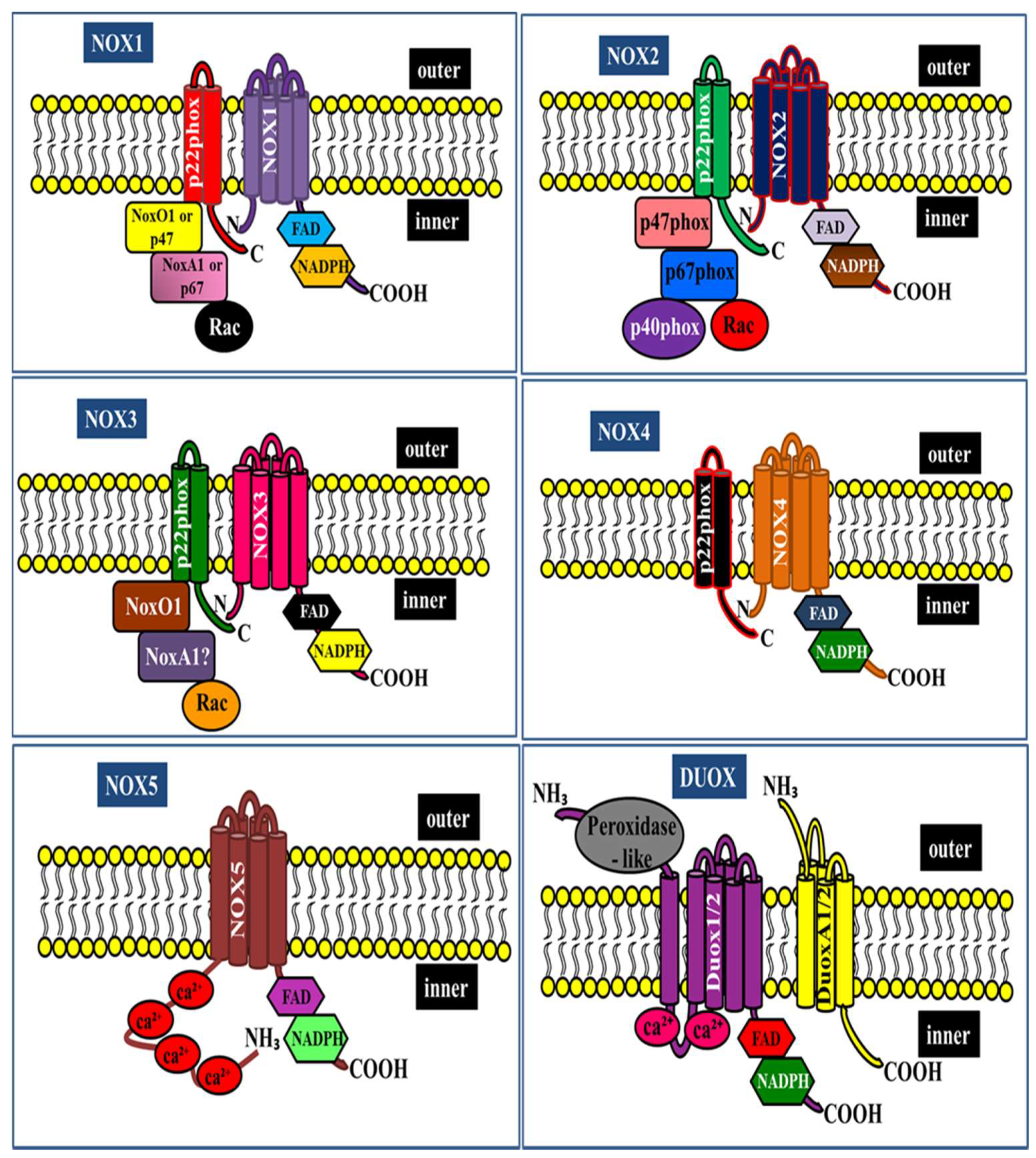
2. NADPH Oxidase Isoforms: An Overview
3. The Role of NADPH Oxidase in Insulin Secretion
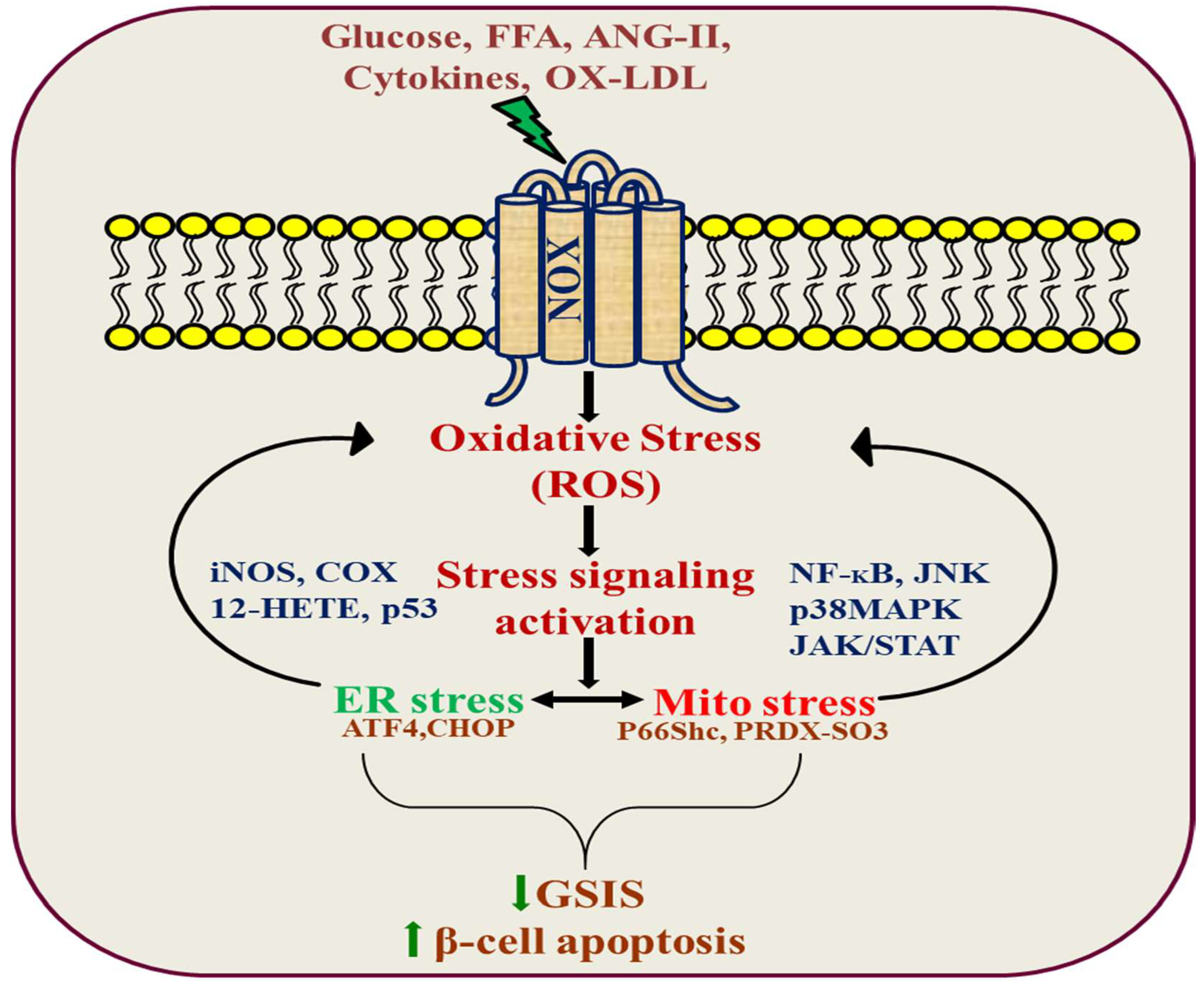
4. The Role of NADPH Oxidase in Hyperglycemia-Induced β-Cell Dysfunction
5. The Role of NADPH Oxidase in β-Cell Dysfunction Caused by Lipotoxicity
6. NADPH Oxidase-Targeted Therapeutic Agents
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kahn, S.E. The relative contributions of insulin resistance and beta-cell dysfunction to the pathophysiology of Type 2 diabetes. Diabetologia 2003, 46, 3–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robertson, R.P.; Harmon, J.; Tran, P.O.; Poitout, V. Beta-cell glucose toxicity, lipotoxicity, and chronic oxidative stress in type 2 diabetes. Diabetes 2004, 53, S119–S124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cnop, M.; Welsh, N.; Jonas, J.C.; Jorns, A.; Lenzen, S.; Eizirik, D.L. Mechanisms of pancreatic beta-cell death in type 1 and type 2 diabetes: Many differences, few similarities. Diabetes 2005, 54, S97–S107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weir, G.C. Glucolipotoxicity, beta-Cells, and Diabetes: The Emperor Has No Clothes. Diabetes 2020, 69, 273–278. [Google Scholar] [CrossRef]
- Prentki, M.; Peyot, M.L.; Masiello, P.; Madiraju, S.R.M. Nutrient-Induced Metabolic Stress, Adaptation, Detoxification, and Toxicity in the Pancreatic beta-Cell. Diabetes 2020, 69, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Cerf, M.E. Beta Cell Physiological Dynamics and Dysfunctional Transitions in Response to Islet Inflammation in Obesity and Diabetes. Metabolites 2020, 10, 452. [Google Scholar] [CrossRef]
- Schrenzel, J.; Serrander, L.; Banfi, B.; Nusse, O.; Fouyouzi, R.; Lew, D.P.; Demaurex, N.; Krause, K.H. Electron currents generated by the human phagocyte NADPH oxidase. Nature 1998, 392, 734–737. [Google Scholar] [CrossRef]
- Babior, B.M.; Lambeth, J.D.; Nauseef, W. The neutrophil NADPH oxidase. Arch. Biochem. Biophys. 2002, 397, 342–344. [Google Scholar] [CrossRef] [PubMed]
- Guichard, C.; Pedruzzi, E.; Dewas, C.; Fay, M.; Pouzet, C.; Bens, M.; Vandewalle, A.; Ogier-Denis, E.; Gougerot-Pocidalo, M.A.; Elbim, C. Interleukin-8-induced priming of neutrophil oxidative burst requires sequential recruitment of NADPH oxidase components into lipid rafts. J. Biol. Chem. 2005, 280, 37021–37032. [Google Scholar] [CrossRef] [Green Version]
- Groemping, Y.; Rittinger, K. Activation and assembly of the NADPH oxidase: A structural perspective. Biochem. J. 2005, 386, 401–416. [Google Scholar] [CrossRef] [Green Version]
- Sumimoto, H.; Miyano, K.; Takeya, R. Molecular composition and regulation of the Nox family NAD(P)H oxidases. Biochem. Biophys. Res. Commun. 2005, 338, 677–686. [Google Scholar] [CrossRef]
- Brown, D.I.; Griendling, K.K. Nox proteins in signal transduction. Free Radic. Biol. Med. 2009, 47, 1239–1253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bedard, K.; Lardy, B.; Krause, K.H. NOX family NADPH oxidases: Not just in mammals. Biochimie 2007, 89, 1107–1112. [Google Scholar] [CrossRef] [PubMed]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Lambeth, J.D. Nox enzymes, ROS, and chronic disease: An example of antagonistic pleiotropy. Free Radic. Biol. Med. 2007, 43, 332–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banfi, B.; Clark, R.A.; Steger, K.; Krause, K.H. Two novel proteins activate superoxide generation by the NADPH oxidase NOX1. J. Biol. Chem. 2003, 278, 3510–3513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banfi, B.; Tirone, F.; Durussel, I.; Knisz, J.; Moskwa, P.; Molnar, G.Z.; Krause, K.H.; Cox, J.A. Mechanism of Ca2+ activation of the NADPH oxidase 5 (NOX5). J. Biol. Chem. 2004, 279, 18583–18591. [Google Scholar] [CrossRef] [Green Version]
- Ameziane-El-Hassani, R.; Morand, S.; Boucher, J.L.; Frapart, Y.M.; Apostolou, D.; Agnandji, D.; Gnidehou, S.; Ohayon, R.; Noel-Hudson, M.S.; Francon, J.; et al. Dual oxidase-2 has an intrinsic Ca2+-dependent H2O2-generating activity. J. Biol. Chem. 2005, 280, 30046–30054. [Google Scholar] [CrossRef] [Green Version]
- Ashcroft, F.M.; Harrison, D.E.; Ashcroft, S.J. Glucose induces closure of single potassium channels in isolated rat pancreatic beta-cells. Nature 1984, 312, 446–448. [Google Scholar] [CrossRef]
- Jitrapakdee, S.; Wutthisathapornchai, A.; Wallace, J.C.; MacDonald, M.J. Regulation of insulin secretion: Role of mitochondrial signalling. Diabetologia 2010, 53, 1019–1032. [Google Scholar] [CrossRef] [Green Version]
- Pi, J.; Bai, Y.; Zhang, Q.; Wong, V.; Floering, L.M.; Daniel, K.; Reece, J.M.; Deeney, J.T.; Andersen, M.E.; Corkey, B.E.; et al. Reactive oxygen species as a signal in glucose-stimulated insulin secretion. Diabetes 2007, 56, 1783–1791. [Google Scholar] [CrossRef] [Green Version]
- Saadeh, M.; Ferrante, T.C.; Kane, A.; Shirihai, O.; Corkey, B.E.; Deeney, J.T. Reactive oxygen species stimulate insulin secretion in rat pancreatic islets: Studies using mono-oleoyl-glycerol. PLoS ONE 2012, 7, e30200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliveira, H.R.; Verlengia, R.; Carvalho, C.R.; Britto, L.R.; Curi, R.; Carpinelli, A.R. Pancreatic beta-cells express phagocyte-like NAD(P)H oxidase. Diabetes 2003, 52, 1457–1463. [Google Scholar] [CrossRef] [Green Version]
- Uchizono, Y.; Takeya, R.; Iwase, M.; Sasaki, N.; Oku, M.; Imoto, H.; Iida, M.; Sumimoto, H. Expression of isoforms of NADPH oxidase components in rat pancreatic islets. Life Sci. 2006, 80, 133–139. [Google Scholar] [CrossRef]
- Morgan, D.; Oliveira-Emilio, H.R.; Keane, D.; Hirata, A.E.; Santos da Rocha, M.; Bordin, S.; Curi, R.; Newsholme, P.; Carpinelli, A.R. Glucose, palmitate and pro-inflammatory cytokines modulate production and activity of a phagocyte-like NADPH oxidase in rat pancreatic islets and a clonal beta cell line. Diabetologia 2007, 50, 359–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imoto, H.; Sasaki, N.; Iwase, M.; Nakamura, U.; Oku, M.; Sonoki, K.; Uchizono, Y.; Iida, M. Impaired insulin secretion by diphenyleneiodium associated with perturbation of cytosolic Ca2+ dynamics in pancreatic beta-cells. Endocrinology 2008, 149, 5391–5400. [Google Scholar] [CrossRef] [Green Version]
- Weir, E.K.; Wyatt, C.N.; Reeve, H.L.; Huang, J.; Archer, S.L.; Peers, C. Diphenyleneiodonium inhibits both potassium and calcium currents in isolated pulmonary artery smooth muscle cells. J. Appl. Physiol. 1994, 76, 2611–2615. [Google Scholar] [CrossRef] [PubMed]
- Morgan, D.; Rebelato, E.; Abdulkader, F.; Graciano, M.F.; Oliveira-Emilio, H.R.; Hirata, A.E.; Rocha, M.S.; Bordin, S.; Curi, R.; Carpinelli, A.R. Association of NAD(P)H oxidase with glucose-induced insulin secretion by pancreatic beta-cells. Endocrinology 2009, 150, 2197–2201. [Google Scholar] [CrossRef]
- Bengis-Garber, C.; Gruener, N. Protein kinase A downregulates the phosphorylation of p47 phox in human neutrophils: A possible pathway for inhibition of the respiratory burst. Cell. Signal. 1996, 8, 291–296. [Google Scholar] [CrossRef]
- Pyne, N.J.; Furman, B.L. Cyclic nucleotide phosphodiesterases in pancreatic islets. Diabetologia 2003, 46, 1179–1189. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Xiaokaiti, Y.; Yang, J.; Pan, J.; Li, Z.; Luria, V.; Li, Y.; Song, G.; Zhu, X.; Zhang, H.T.; et al. Inhibition of phosphodiesterase 2 reverses gp91phox oxidase-mediated depression- and anxiety-like behavior. Neuropharmacology 2018, 143, 176–185. [Google Scholar] [CrossRef]
- Bensellam, M.; Laybutt, D.R.; Jonas, J.C. The molecular mechanisms of pancreatic beta-cell glucotoxicity: Recent findings and future research directions. Mol. Cell. Endocrinol. 2012, 364, 1–27. [Google Scholar] [CrossRef]
- Jonas, J.C.; Bensellam, M.; Duprez, J.; Elouil, H.; Guiot, Y.; Pascal, S.M. Glucose regulation of islet stress responses and beta-cell failure in type 2 diabetes. Diabetes Obes. Metab. 2009, 11, 65–81. [Google Scholar] [CrossRef]
- Tang, C.; Koulajian, K.; Schuiki, I.; Zhang, L.; Desai, T.; Ivovic, A.; Wang, P.; Robson-Doucette, C.; Wheeler, M.B.; Minassian, B.; et al. Glucose-induced beta cell dysfunction in vivo in rats: Link between oxidative stress and endoplasmic reticulum stress. Diabetologia 2012, 55, 1366–1379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newsholme, P.; Morgan, D.; Rebelato, E.; Oliveira-Emilio, H.C.; Procopio, J.; Curi, R.; Carpinelli, A. Insights into the critical role of NADPH oxidase(s) in the normal and dysregulated pancreatic beta cell. Diabetologia 2009, 52, 2489–2498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Syed, I.; Kyathanahalli, C.N.; Jayaram, B.; Govind, S.; Rhodes, C.J.; Kowluru, R.A.; Kowluru, A. Increased phagocyte-like NADPH oxidase and ROS generation in type 2 diabetic ZDF rat and human islets: Role of Rac1-JNK1/2 signaling pathway in mitochondrial dysregulation in the diabetic islet. Diabetes 2011, 60, 2843–2852. [Google Scholar] [CrossRef] [Green Version]
- Abo, A.; Pick, E.; Hall, A.; Totty, N.; Teahan, C.G.; Segal, A.W. Activation of the NADPH oxidase involves the small GTP-binding protein p21rac1. Nature 1991, 353, 668–670. [Google Scholar] [CrossRef] [PubMed]
- Abo, A.; Webb, M.R.; Grogan, A.; Segal, A.W. Activation of NADPH oxidase involves the dissociation of p21rac from its inhibitory GDP/GTP exchange protein (rhoGDI) followed by its translocation to the plasma membrane. Biochem. J. 1994, 298, 585–591. [Google Scholar] [CrossRef] [Green Version]
- Sarfstein, R.; Gorzalczany, Y.; Mizrahi, A.; Berdichevsky, Y.; Molshanski-Mor, S.; Weinbaum, C.; Hirshberg, M.; Dagher, M.C.; Pick, E. Dual role of Rac in the assembly of NADPH oxidase, tethering to the membrane and activation of p67phox: A study based on mutagenesis of p67phox-Rac1 chimeras. J. Biol. Chem. 2004, 279, 16007–16016. [Google Scholar] [CrossRef] [Green Version]
- Chi, X.; Wang, S.; Huang, Y.; Stamnes, M.; Chen, J.L. Roles of rho GTPases in intracellular transport and cellular transformation. Int. J. Mol. Sci. 2013, 14, 7089–7108. [Google Scholar] [CrossRef]
- Asahara, S.; Shibutani, Y.; Teruyama, K.; Inoue, H.Y.; Kawada, Y.; Etoh, H.; Matsuda, T.; Kimura-Koyanagi, M.; Hashimoto, N.; Sakahara, M.; et al. Ras-related C3 botulinum toxin substrate 1 (RAC1) regulates glucose-stimulated insulin secretion via modulation of F-actin. Diabetologia 2013, 56, 1088–1097. [Google Scholar] [CrossRef] [Green Version]
- Marei, H.; Carpy, A.; Macek, B.; Malliri, A. Proteomic analysis of Rac1 signaling regulation by guanine nucleotide exchange factors. Cell Cycle 2016, 15, 1961–1974. [Google Scholar] [CrossRef] [Green Version]
- Marei, H.; Malliri, A. GEFs: Dual regulation of Rac1 signaling. Small GTPases 2017, 8, 90–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kowluru, A. Small G proteins in islet beta-cell function. Endocr. Rev. 2010, 31, 52–78. [Google Scholar] [CrossRef] [Green Version]
- Dang, P.M.; Cross, A.R.; Babior, B.M. Assembly of the neutrophil respiratory burst oxidase: A direct interaction between p67PHOX and cytochrome b558. Proc. Natl. Acad. Sci. USA 2001, 98, 3001–3005. [Google Scholar] [CrossRef] [Green Version]
- Dang, P.M.; Johnson, J.L.; Babior, B.M. Binding of nicotinamide adenine dinucleotide phosphate to the tetratricopeptide repeat domains at the N-terminus of p67PHOX, a subunit of the leukocyte nicotinamide adenine dinucleotide phosphate oxidase. Biochemistry 2000, 39, 3069–3075. [Google Scholar] [CrossRef]
- Inoguchi, T.; Li, P.; Umeda, F.; Yu, H.Y.; Kakimoto, M.; Imamura, M.; Aoki, T.; Etoh, T.; Hashimoto, T.; Naruse, M.; et al. High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C—Dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes 2000, 49, 1939–1945. [Google Scholar] [CrossRef] [Green Version]
- Xiang, F.L.; Lu, X.; Strutt, B.; Hill, D.J.; Feng, Q. NOX2 deficiency protects against streptozotocin-induced beta-cell destruction and development of diabetes in mice. Diabetes 2010, 59, 2603–2611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Souza, A.H.; Santos, L.R.B.; Roma, L.P.; Bensellam, M.; Carpinelli, A.R.; Jonas, J.C. NADPH oxidase-2 does not contribute to beta-cell glucotoxicity in cultured pancreatic islets from C57BL/6J mice. Mol. Cell. Endocrinol. 2017, 439, 354–362. [Google Scholar] [CrossRef] [PubMed]
- Anvari, E.; Wikstrom, P.; Walum, E.; Welsh, N. The novel NADPH oxidase 4 inhibitor GLX351322 counteracts glucose intolerance in high-fat diet-treated C57BL/6 mice. Free Radic. Res. 2015, 49, 1308–1318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Elksnis, A.; Wikstrom, P.; Walum, E.; Welsh, N.; Carlsson, P.O. The novel NADPH oxidase 4 selective inhibitor GLX7013114 counteracts human islet cell death in vitro. PLoS ONE 2018, 13, e0204271. [Google Scholar] [CrossRef]
- Lastra, G.; Manrique, C. The expanding role of oxidative stress, renin angiotensin system, and beta-cell dysfunction in the cardiometabolic syndrome and Type 2 diabetes mellitus. Antioxid. Redox Signal. 2007, 9, 943–954. [Google Scholar] [CrossRef]
- Lupi, R.; Del Guerra, S.; Bugliani, M.; Boggi, U.; Mosca, F.; Torri, S.; Del Prato, S.; Marchetti, P. The direct effects of the angiotensin-converting enzyme inhibitors, zofenoprilat and enalaprilat, on isolated human pancreatic islets. Eur. J. Endocrinol. 2006, 154, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Chu, K.Y.; Lau, T.; Carlsson, P.O.; Leung, P.S. Angiotensin II type 1 receptor blockade improves beta-cell function and glucose tolerance in a mouse model of type 2 diabetes. Diabetes 2006, 55, 367–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sauter, N.S.; Thienel, C.; Plutino, Y.; Kampe, K.; Dror, E.; Traub, S.; Timper, K.; Bedat, B.; Pattou, F.; Kerr-Conte, J.; et al. Angiotensin II induces interleukin-1beta-mediated islet inflammation and beta-cell dysfunction independently of vasoconstrictive effects. Diabetes 2015, 64, 1273–1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, K.Y.; Leung, P.S. Angiotensin II Type 1 receptor antagonism mediates uncoupling protein 2-driven oxidative stress and ameliorates pancreatic islet beta-cell function in young Type 2 diabetic mice. Antioxid. Redox Signal. 2007, 9, 869–878. [Google Scholar] [CrossRef] [PubMed]
- Weaver, J.R.; Grzesik, W.; Taylor-Fishwick, D.A. Inhibition of NADPH oxidase-1 preserves beta cell function. Diabetologia 2015, 58, 113–121. [Google Scholar] [CrossRef]
- Weaver, J.R.; Taylor-Fishwick, D.A. Regulation of NOX-1 expression in beta cells: A positive feedback loop involving the Src-kinase signaling pathway. Mol. Cell. Endocrinol. 2013, 369, 35–41. [Google Scholar] [CrossRef]
- Weaver, J.; Taylor-Fishwick, D.A. Relationship of NADPH Oxidase-1 expression to beta cell dysfunction induced by inflammatory cytokines. Biochem. Biophys. Res. Commun. 2017, 485, 290–294. [Google Scholar] [CrossRef]
- Weaver, J.R.; Holman, T.R.; Imai, Y.; Jadhav, A.; Kenyon, V.; Maloney, D.J.; Nadler, J.L.; Rai, G.; Simeonov, A.; Taylor-Fishwick, D.A. Integration of pro-inflammatory cytokines, 12-lipoxygenase and NOX-1 in pancreatic islet beta cell dysfunction. Mol. Cell. Endocrinol. 2012, 358, 88–95. [Google Scholar] [CrossRef]
- Kowluru, A. Oxidative Stress in Cytokine-Induced Dysfunction of the Pancreatic Beta Cell: Known Knowns and Known Unknowns. Metabolites 2020, 10, 480. [Google Scholar] [CrossRef]
- Michalska, M.; Wolf, G.; Walther, R.; Newsholme, P. Effects of pharmacological inhibition of NADPH oxidase or iNOS on pro-inflammatory cytokine, palmitic acid or H2O2-induced mouse islet or clonal pancreatic beta-cell dysfunction. Biosci. Rep. 2010, 30, 445–453. [Google Scholar] [CrossRef] [Green Version]
- Mohammed, A.M.; Syeda, K.; Hadden, T.; Kowluru, A. Upregulation of phagocyte-like NADPH oxidase by cytokines in pancreatic beta-cells: Attenuation of oxidative and nitrosative stress by 2-bromopalmitate. Biochem. Pharmacol. 2013, 85, 109–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subasinghe, W.; Syed, I.; Kowluru, A. Phagocyte-like NADPH oxidase promotes cytokine-induced mitochondrial dysfunction in pancreatic beta-cells: Evidence for regulation by Rac1. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 300, R12–R20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Briaud, I.; Harmon, J.S.; Kelpe, C.L.; Segu, V.B.; Poitout, V. Lipotoxicity of the pancreatic beta-cell is associated with glucose-dependent esterification of fatty acids into neutral lipids. Diabetes 2001, 50, 315–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haber, E.P.; Ximenes, H.M.; Procopio, J.; Carvalho, C.R.; Curi, R.; Carpinelli, A.R. Pleiotropic effects of fatty acids on pancreatic beta-cells. J. Cell. Physiol. 2003, 194, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Prentki, M.; Nolan, C.J. Islet beta cell failure in type 2 diabetes. J. Clin. Investig. 2006, 116, 1802–1812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Unger, R.H.; Zhou, Y.T. Lipotoxicity of beta-cells in obesity and in other causes of fatty acid spillover. Diabetes 2001, 50, S118–S121. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.W.; Moon, J.S.; Seo, Y.J.; Park, S.Y.; Kim, J.Y.; Yoon, J.S.; Lee, I.K.; Lee, H.W.; Won, K.C. Inhibition of fatty acid translocase cluster determinant 36 (CD36), stimulated by hyperglycemia, prevents glucotoxicity in INS-1 cells. Biochem. Biophys. Res. Commun. 2012, 420, 462–466. [Google Scholar] [CrossRef]
- Wallin, T.; Ma, Z.; Ogata, H.; Jorgensen, I.H.; Iezzi, M.; Wang, H.; Wollheim, C.B.; Bjorklund, A. Facilitation of fatty acid uptake by CD36 in insulin-producing cells reduces fatty-acid-induced insulin secretion and glucose regulation of fatty acid oxidation. Biochim. Biophys. Acta 2010, 1801, 191–197. [Google Scholar] [CrossRef] [Green Version]
- Elumalai, S.; Karunakaran, U.; Lee, I.K.; Moon, J.S.; Won, K.C. Rac1-NADPH oxidase signaling promotes CD36 activation under glucotoxic conditions in pancreatic beta cells. Redox Biol. 2017, 11, 126–134. [Google Scholar] [CrossRef] [Green Version]
- Gharib, M.; Tao, H.; Fungwe, T.V.; Hajri, T. Cluster Differentiating 36 (CD36) Deficiency Attenuates Obesity-Associated Oxidative Stress in the Heart. PLoS ONE 2016, 11, e0155611. [Google Scholar] [CrossRef] [PubMed]
- Karunakaran, U.; Elumalai, S.; Moon, J.S.; Won, K.C. CD36 dependent redoxosomes promotes ceramide-mediated pancreatic beta-cell failure via p66Shc activation. Free Radic. Biol. Med. 2019, 134, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Hirosumi, J.; Tuncman, G.; Chang, L.; Gorgun, C.Z.; Uysal, K.T.; Maeda, K.; Karin, M.; Hotamisligil, G.S. A central role for JNK in obesity and insulin resistance. Nature 2002, 420, 333–336. [Google Scholar] [CrossRef]
- Holzer, R.G.; Park, E.J.; Li, N.; Tran, H.; Chen, M.; Choi, C.; Solinas, G.; Karin, M. Saturated fatty acids induce c-Src clustering within membrane subdomains, leading to JNK activation. Cell 2011, 147, 173–184. [Google Scholar] [CrossRef] [Green Version]
- Kant, S.; Standen, C.L.; Morel, C.; Jung, D.Y.; Kim, J.K.; Swat, W.; Flavell, R.A.; Davis, R.J. A Protein Scaffold Coordinates SRC-Mediated JNK Activation in Response to Metabolic Stress. Cell Rep. 2017, 20, 2775–2783. [Google Scholar] [CrossRef] [Green Version]
- Natalicchio, A.; Tortosa, F.; Labarbuta, R.; Biondi, G.; Marrano, N.; Carchia, E.; Leonardini, A.; Cignarelli, A.; Bugliani, M.; Marchetti, P.; et al. The p66(Shc) redox adaptor protein is induced by saturated fatty acids and mediates lipotoxicity-induced apoptosis in pancreatic beta cells. Diabetologia 2015, 58, 1260–1271. [Google Scholar] [CrossRef] [PubMed]
- Bonfini, L.; Migliaccio, E.; Pelicci, G.; Lanfrancone, L.; Pelicci, P.G. Not all Shc’s roads lead to Ras. Trends Biochem. Sci. 1996, 21, 257–261. [Google Scholar] [CrossRef]
- Ravichandran, K.S. Signaling via Shc family adapter proteins. Oncogene 2001, 20, 6322–6330. [Google Scholar] [CrossRef] [Green Version]
- van der Geer, P.; Wiley, S.; Gish, G.D.; Pawson, T. The Shc adaptor protein is highly phosphorylated at conserved, twin tyrosine residues (Y239/240) that mediate protein-protein interactions. Curr. Biol. 1996, 6, 1435–1444. [Google Scholar] [CrossRef] [Green Version]
- Khalid, S.; Drasche, A.; Thurner, M.; Hermann, M.; Ashraf, M.I.; Fresser, F.; Baier, G.; Kremser, L.; Lindner, H.; Troppmair, J. cJun N-terminal kinase (JNK) phosphorylation of serine 36 is critical for p66Shc activation. Sci. Rep. 2016, 6, 20930. [Google Scholar] [CrossRef] [Green Version]
- Vilas-Boas, E.A.; Nalbach, L.; Ampofo, E.; Lucena, C.F.; Naudet, L.; Ortis, F.; Carpinelli, A.R.; Morgan, B.; Roma, L.P. Transient NADPH oxidase 2-dependent H2O2 production drives early palmitate-induced lipotoxicity in pancreatic islets. Free Radic. Biol. Med. 2021, 162, 1–13. [Google Scholar] [CrossRef]
- Li, F.; Munsey, T.S.; Sivaprasadarao, A. TRPM2-mediated rise in mitochondrial Zn(2+) promotes palmitate-induced mitochondrial fission and pancreatic beta-cell death in rodents. Cell Death Differ. 2017, 24, 1999–2012. [Google Scholar] [CrossRef] [Green Version]
- Cnop, M.; Hannaert, J.C.; Grupping, A.Y.; Pipeleers, D.G. Low density lipoprotein can cause death of islet beta-cells by its cellular uptake and oxidative modification. Endocrinology 2002, 143, 3449–3453. [Google Scholar] [CrossRef] [Green Version]
- Grupping, A.Y.; Cnop, M.; Van Schravendijk, C.F.; Hannaert, J.C.; Van Berkel, T.J.; Pipeleers, D.G. Low density lipoprotein binding and uptake by human and rat islet beta cells. Endocrinology 1997, 138, 4064–4068. [Google Scholar] [CrossRef] [PubMed]
- Awasthi, D.; Nagarkoti, S.; Kumar, A.; Dubey, M.; Singh, A.K.; Pathak, P.; Chandra, T.; Barthwal, M.K.; Dikshit, M. Oxidized LDL induced extracellular trap formation in human neutrophils via TLR-PKC-IRAK-MAPK and NADPH-oxidase activation. Free Radic. Biol. Med. 2016, 93, 190–203. [Google Scholar] [CrossRef] [PubMed]
- Plaisance, V.; Brajkovic, S.; Tenenbaum, M.; Favre, D.; Ezanno, H.; Bonnefond, A.; Bonner, C.; Gmyr, V.; Kerr-Conte, J.; Gauthier, B.R.; et al. Endoplasmic Reticulum Stress Links Oxidative Stress to Impaired Pancreatic Beta-Cell Function Caused by Human Oxidized LDL. PLoS ONE 2016, 11, e0163046. [Google Scholar] [CrossRef] [Green Version]
- Honjo, T.; Otsui, K.; Shiraki, R.; Kawashima, S.; Sawamura, T.; Yokoyama, M.; Inoue, N. Essential role of NOXA1 in generation of reactive oxygen species induced by oxidized low-density lipoprotein in human vascular endothelial cells. Endothelium 2008, 15, 137–141. [Google Scholar] [CrossRef]
- Li, M.; Dou, L.; Jiao, J.; Lu, Y.; Guo, H.B.; Man, Y.; Wang, S.; Li, J. NADPH oxidase 2-derived reactive oxygen species are involved in dysfunction and apoptosis of pancreatic beta-cells induced by low density lipoprotein. Cell. Physiol. Biochem. 2012, 30, 439–449. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.; Bai, Y.P.; Gao, H.C.; Wang, X.; Li, L.F.; Zhang, G.G.; Hu, C.P. Ox-LDL induces endothelial cell apoptosis via the LOX-1-dependent endoplasmic reticulum stress pathway. Atherosclerosis 2014, 235, 310–317. [Google Scholar] [CrossRef]
- Riganti, C.; Gazzano, E.; Polimeni, M.; Costamagna, C.; Bosia, A.; Ghigo, D. Diphenyleneiodonium inhibits the cell redox metabolism and induces oxidative stress. J. Biol. Chem. 2004, 279, 47726–47731. [Google Scholar] [CrossRef] [Green Version]
- Vejrazka, M.; Micek, R.; Stipek, S. Apocynin inhibits NADPH oxidase in phagocytes but stimulates ROS production in non-phagocytic cells. Biochim. Biophys. Acta 2005, 1722, 143–147. [Google Scholar] [CrossRef]
- Schildknecht, S.; Weber, A.; Gerding, H.R.; Pape, R.; Robotta, M.; Drescher, M.; Marquardt, A.; Daiber, A.; Ferger, B.; Leist, M. The NOX1/4 inhibitor GKT136901 as selective and direct scavenger of peroxynitrite. Curr. Med. Chem. 2014, 21, 365–376. [Google Scholar] [CrossRef] [Green Version]
- Teixeira, G.; Szyndralewiez, C.; Molango, S.; Carnesecchi, S.; Heitz, F.; Wiesel, P.; Wood, J.M. Therapeutic potential of NADPH oxidase 1/4 inhibitors. Br. J. Pharmacol. 2017, 174, 1647–1669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Augsburger, F.; Filippova, A.; Rasti, D.; Seredenina, T.; Lam, M.; Maghzal, G.; Mahiout, Z.; Jansen-Durr, P.; Knaus, U.G.; Doroshow, J.; et al. Pharmacological characterization of the seven human NOX isoforms and their inhibitors. Redox Biol. 2019, 26, 101272. [Google Scholar] [CrossRef]
- Dao, V.T.; Elbatreek, M.H.; Altenhofer, S.; Casas, A.I.; Pachado, M.P.; Neullens, C.T.; Knaus, U.G.; Schmidt, H. Isoform-selective NADPH oxidase inhibitor panel for pharmacological target validation. Free Radic. Biol. Med. 2020, 148, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Reis, J.; Massari, M.; Marchese, S.; Ceccon, M.; Aalbers, F.S.; Corana, F.; Valente, S.; Mai, A.; Magnani, F.; Mattevi, A. A closer look into NADPH oxidase inhibitors: Validation and insight into their mechanism of action. Redox Biol. 2020, 32, 101466. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.I.; Lee, J.S.; Kwak, B.K.; Hwang, W.M.; Kim, M.J.; Kim, Y.B.; Chung, S.S.; Park, K.S. Metformin Ameliorates Lipotoxic beta-Cell Dysfunction through a Concentration-Dependent Dual Mechanism of Action. Diabetes Metab. J. 2019, 43, 854–866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Hua, N.; Fu, X.; Pan, Y.; Li, B.; Li, X. Metformin Regulates the Expression of SK2 and SK3 in the Atria of Rats With Type 2 Diabetes Mellitus Through the NOX4/p38MAPK Signaling Pathway. J. Cardiovasc. Pharmacol. 2018, 72, 205–213. [Google Scholar] [CrossRef]
- Takeno, A.; Kanazawa, I.; Tanaka, K.; Notsu, M.; Yokomoto, M.; Yamaguchi, T.; Sugimoto, T. Activation of AMP-activated protein kinase protects against homocysteine-induced apoptosis of osteocytic MLO-Y4 cells by regulating the expressions of NADPH oxidase 1 (Nox1) and Nox2. Bone 2015, 77, 135–141. [Google Scholar] [CrossRef]
- Palacios-Ramirez, R.; Hernanz, R.; Martin, A.; Perez-Giron, J.V.; Barrus, M.T.; Gonzalez-Carnicero, Z.; Aguado, A.; Jaisser, F.; Briones, A.M.; Salaices, M.; et al. Pioglitazone Modulates the Vascular Contractility in Hypertension by Interference with ET-1 Pathway. Sci. Rep. 2019, 9, 16461. [Google Scholar] [CrossRef] [Green Version]
- Salim, H.M.; Fukuda, D.; Higashikuni, Y.; Tanaka, K.; Hirata, Y.; Yagi, S.; Soeki, T.; Shimabukuro, M.; Sata, M. Teneligliptin, a dipeptidyl peptidase-4 inhibitor, attenuated pro-inflammatory phenotype of perivascular adipose tissue and inhibited atherogenesis in normoglycemic apolipoprotein-E-deficient mice. Vascul. Pharmacol. 2017, 96–98, 19–25. [Google Scholar] [CrossRef]
- Elumalai, S.; Karunakaran, U.; Moon, J.S.; Won, K.C. High glucose-induced PRDX3 acetylation contributes to glucotoxicity in pancreatic beta-cells: Prevention by Teneligliptin. Free Radic. Biol. Med. 2020, 160, 618–629. [Google Scholar] [CrossRef]
- Wosniak, J., Jr.; Santos, C.X.; Kowaltowski, A.J.; Laurindo, F.R. Cross-talk between mitochondria and NADPH oxidase: Effects of mild mitochondrial dysfunction on angiotensin II-mediated increase in Nox isoform expression and activity in vascular smooth muscle cells. Antioxid. Redox Signal. 2009, 11, 1265–1278. [Google Scholar] [CrossRef] [PubMed]
- Yahfoufi, N.; Alsadi, N.; Jambi, M.; Matar, C. The Immunomodulatory and Anti-Inflammatory Role of Polyphenols. Nutrients 2018, 10, 1618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yousefian, M.; Shakour, N.; Hosseinzadeh, H.; Hayes, A.W.; Hadizadeh, F.; Karimi, G. The natural phenolic compounds as modulators of NADPH oxidases in hypertension. Phytomedicine 2019, 55, 200–213. [Google Scholar] [CrossRef]
- Karunakaran, U.; Lee, J.E.; Elumalai, S.; Moon, J.S.; Won, K.C. Myricetin prevents thapsigargin-induced CDK5-P66Shc signalosome mediated pancreatic beta-cell dysfunction. Free Radic. Biol. Med. 2019, 141, 59–66. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elumalai, S.; Karunakaran, U.; Moon, J.-S.; Won, K.-C. NADPH Oxidase (NOX) Targeting in Diabetes: A Special Emphasis on Pancreatic β-Cell Dysfunction. Cells 2021, 10, 1573. https://doi.org/10.3390/cells10071573
Elumalai S, Karunakaran U, Moon J-S, Won K-C. NADPH Oxidase (NOX) Targeting in Diabetes: A Special Emphasis on Pancreatic β-Cell Dysfunction. Cells. 2021; 10(7):1573. https://doi.org/10.3390/cells10071573
Chicago/Turabian StyleElumalai, Suma, Udayakumar Karunakaran, Jun-Sung Moon, and Kyu-Chang Won. 2021. "NADPH Oxidase (NOX) Targeting in Diabetes: A Special Emphasis on Pancreatic β-Cell Dysfunction" Cells 10, no. 7: 1573. https://doi.org/10.3390/cells10071573
APA StyleElumalai, S., Karunakaran, U., Moon, J.-S., & Won, K.-C. (2021). NADPH Oxidase (NOX) Targeting in Diabetes: A Special Emphasis on Pancreatic β-Cell Dysfunction. Cells, 10(7), 1573. https://doi.org/10.3390/cells10071573