
Mechanosensitive Regulation of Fibrosis


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
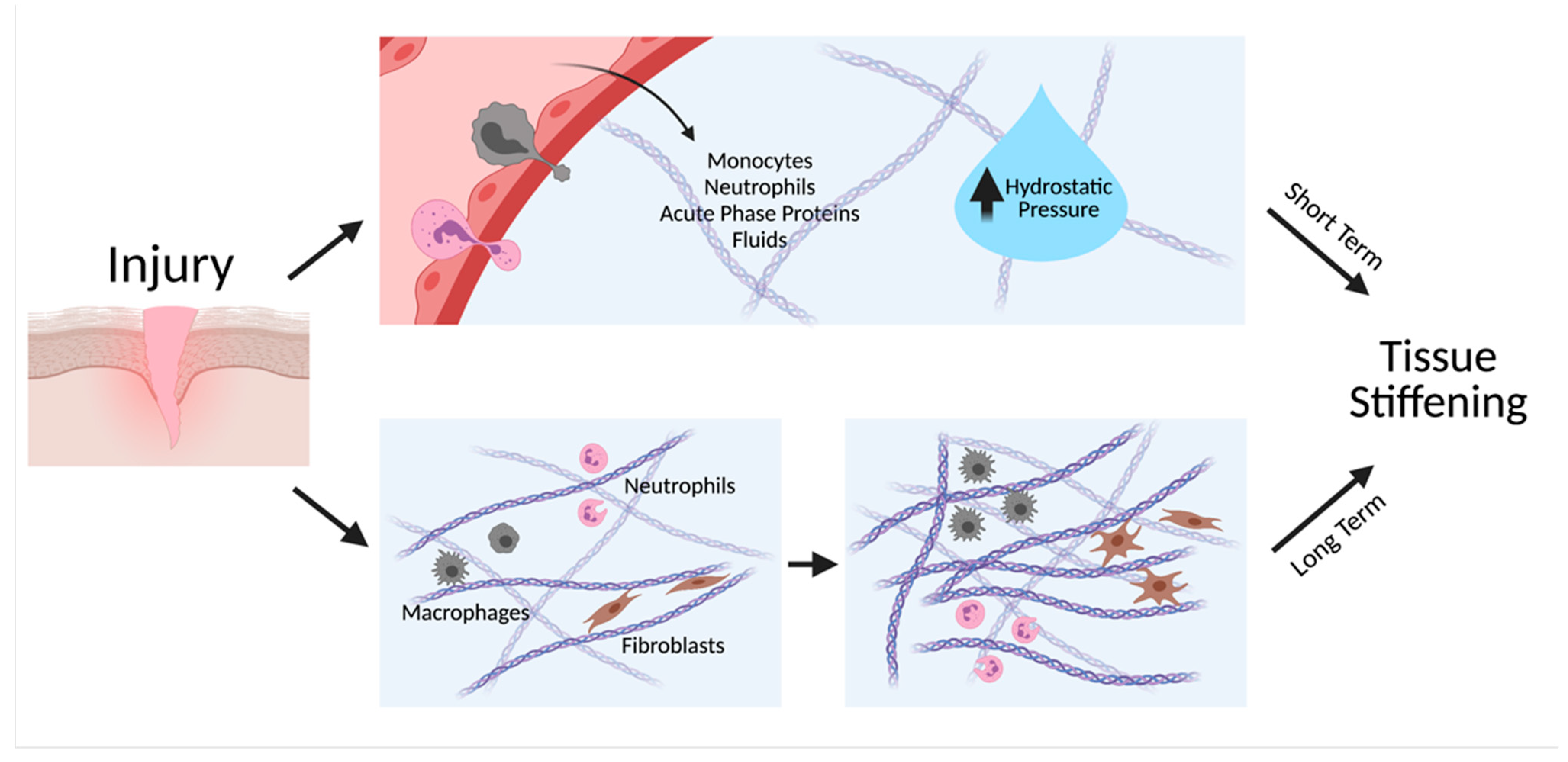
2. Tissue Stiffness—A Hallmark of Inflammation
3. Tissue Stiffness—A Driver of Inflammation
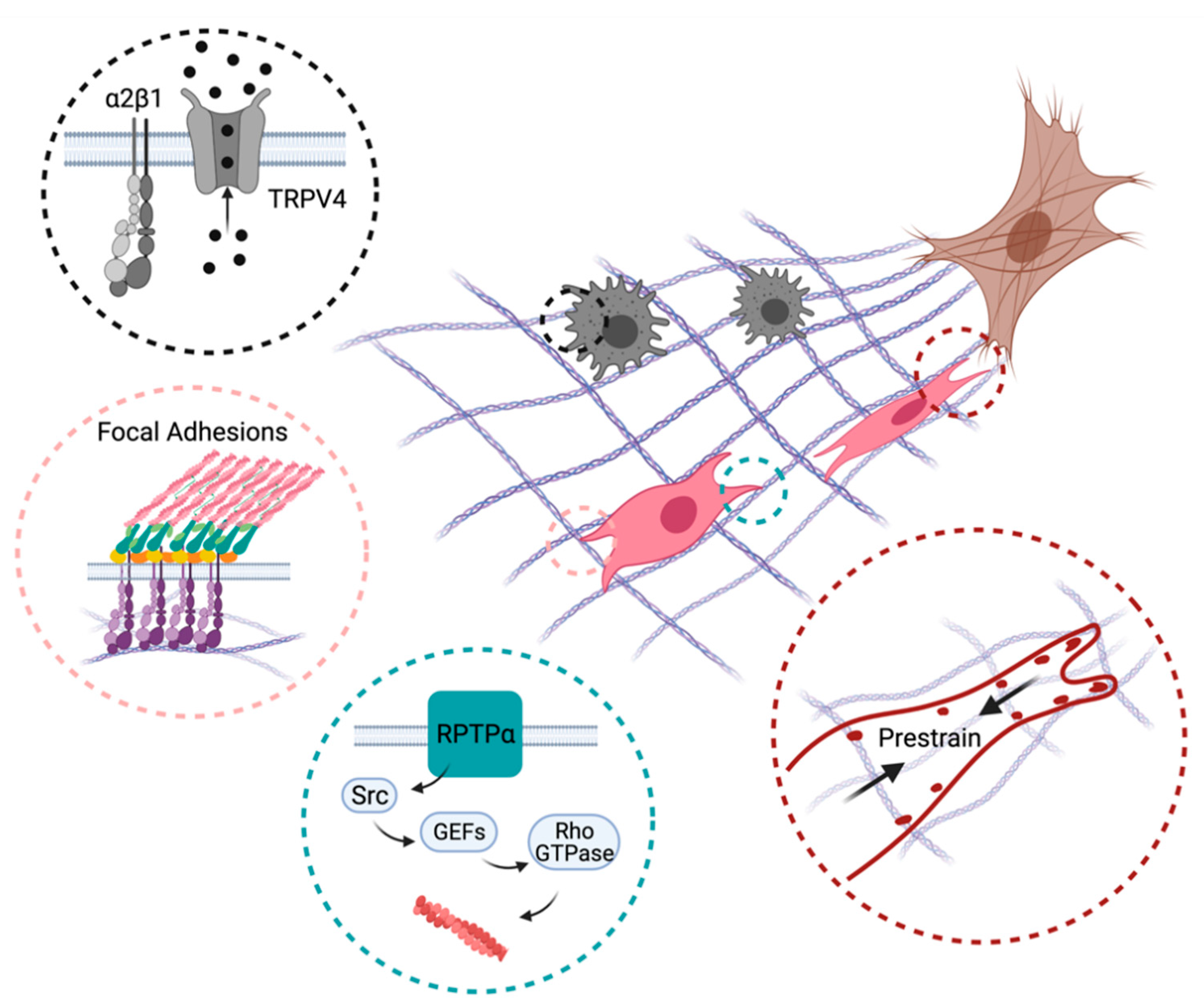
3.1. Molecular Sensors of Stiffness
3.1.1. Focal Adhesions/Focal Contacts
3.1.2. Actin Cytoskeleton
3.1.3. Nucleus
3.1.4. Mechanosensitive Ion Channels
3.1.5. G-Protein-Coupled Receptors
3.2. Inflammatory and Mechanosensitive Pathways Intertwined
4. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Dixit, U.S.; Hazarika, M.; Davim, J.P.; Dixit, U.S.; Hazarika, M.; Davim, J.P. History of Mechanics; Springer International Publishing: Berlin/Heidelberg, Germany, 2017; pp. 47–72. ISBN 9783319429144. [Google Scholar]
- Engler, A.J.; Sen, S.; Sweeney, H.L.; Discher, D.E. Matrix Elasticity Directs Stem Cell Lineage Specification. Cell 2006, 126, 677–689. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, B.D.; Grashoff, C.; Schwartz, M.A. Dynamic Molecular Processes Mediate Cellular Mechanotransduction. Nature 2011, 475, 316–323. [Google Scholar] [CrossRef]
- Salvi, A.M.; DeMali, K.A. Mechanisms Linking Mechanotransduction and Cell Metabolism. Curr. Opin. Cell Biol. 2018, 54, 114–120. [Google Scholar] [CrossRef]
- Kai, F.; Drain, A.P.; Weaver, V.M. The Extracellular Matrix Modulates the Metastatic Journey. Dev. Cell. 2019, 49, 332–346. [Google Scholar] [CrossRef] [PubMed]
- Oudin, M.J.; Weaver, V.M. Physical and Chemical Gradients in the Tumor Microenvironment Regulate Tumor Cell Invasion, Migration, and Metastasis. Cold Spring Harb. Symp. Quant. Biol. 2016, 81, 189–205. [Google Scholar] [CrossRef]
- Sundararaghavan, H.G.; Monteiro, G.A.; Firestein, B.L.; Shreiber, D.I. Neurite growth in 3D collagen gels with gradients of mechanical properties. Biotechnol. Bioeng. 2009, 102, 632–643. [Google Scholar] [CrossRef] [PubMed]
- Budday, S.; Sommer, G.; Birkl, C.; Langkammer, C.; Haybaeck, J.; Kohnert, J.; Bauer, M.; Paulsen, F.; Steinmann, P.; Kuhl, E.; et al. Mechanical characterization of human brain tissue. Acta Biomater. 2017, 48, 319–340. [Google Scholar] [CrossRef]
- McIlvain, G.; Schwarb, H.; Cohen, N.J.; Telzer, E.H.; Johnson, C.L. Mechanical properties of the in vivo adolescent human brain. Dev. Cogn. Neurosci. 2018, 34, 27–33. [Google Scholar] [CrossRef]
- Zysset, P.K.; Guo, X.E.; Hoffler, C.E.; Moore, K.E.; Goldstein, S.A. Elastic modulus and hardness of cortical and trabecular bone lamellae measured by nanoindentation in the human femur. J. Biomech. 1999, 32, 1005–1012. [Google Scholar] [CrossRef]
- Guimarães, C.F.; Gasperini, L.; Marques, A.P.; Reis, R.L. The stiffness of living tissues and its implications for tissue engineering. Nat. Rev. Mater. 2020, 5, 351–370. [Google Scholar] [CrossRef]
- Cox, T.R.; Erler, J.T. Remodeling and homeostasis of the extracellular matrix: Implications for fibrotic diseases and cancer. Dis. Model. Mech. 2011, 4, 165–178. [Google Scholar] [CrossRef]
- Huang, A.L.; Vita, J.A. Effects of Systemic Inflammation on Endothelium-Dependent Vasodilation. Trends Cardiovasc. Med. 2006, 16, 15–20. [Google Scholar] [CrossRef]
- Wiig, H.; Swartz, M.A. Interstitial Fluid and Lymph Formation and Transport: Physiological Regulation and Roles in Inflammation and Cancer. Physiol. Rev. 2012, 92, 1005–1060. [Google Scholar] [CrossRef]
- Lawrence, T.; Gilroy, D.W. Chronic inflammation: A failure of resolution? Int. J. Exp. Pathol. 2006, 88, 85–94. [Google Scholar] [CrossRef]
- Georges, P.C.; Hui, J.-J.; Gombos, Z.; McCormick, M.E.; Wang, A.Y.; Uemura, M.; Mick, R.; Janmey, P.A.; Furth, E.E.; Wells, R.G. Increased stiffness of the rat liver precedes matrix deposition: Implications for fibrosis. Am. J. Physiol. Liver Physiol. 2007, 293, G1147–G1154. [Google Scholar] [CrossRef]
- Rich, A.; Crick, F. The molecular structure of collagen. J. Mol. Biol. 1961, 3, 483-IN4. [Google Scholar] [CrossRef]
- Yamauchi, M.; Sricholpech, M. Lysine post-translational modifications of collagen. Essays Biochem. 2012, 52, 113–133. [Google Scholar] [CrossRef] [PubMed]
- Panwar, P.; Lamour, G.; Mackenzie, N.C.W.; Yang, H.; Ko, F.; Li, H.; Brömme, D. Changes in Structural-Mechanical Properties and Degradability of Collagen during Aging-associated Modifications. J. Biol. Chem. 2015, 290, 23291–23306. [Google Scholar] [CrossRef]
- Kadler, K.E.; Baldock, C.; Bella, J.; Boot-Handford, R.P. Collagens at a glance. J. Cell Sci. 2007, 120, 1955–1958. [Google Scholar] [CrossRef] [PubMed]
- Loke, P.; Gallagher, I.; Nair, M.G.; Zang, X.; Brombacher, F.; Mohrs, M.; Allison, J.P.; Allen, J.E. Alternative Activation Is an Innate Response to Injury That Requires CD4+T Cells to be Sustained during Chronic Infection. J. Immunol. 2007, 179, 3926–3936. [Google Scholar] [CrossRef]
- Duque, G.A.; Descoteaux, A. Macrophage Cytokines: Involvement in Immunity and Infectious Diseases. Front. Immunol. 2014, 5, 491. [Google Scholar] [CrossRef]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A.; Vannella, K.M. Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity 2016, 44, 450–462. [Google Scholar] [CrossRef] [PubMed]
- Knipper, J.A.; Willenborg, S.; Brinckmann, J.; Bloch, W.; Maaß, T.; Wagener, R.; Krieg, T.; Sutherland, T.; Munitz, A.; Rothenberg, M.E.; et al. Interleukin-4 Receptor α Signaling in Myeloid Cells Controls Collagen Fibril Assembly in Skin Repair. Immunity 2015, 43, 803–816. [Google Scholar] [CrossRef] [PubMed]
- Wurster, A.L.; Tanaka, T.; Grusby, M.J. The biology of Stat4 and Stat6. Oncogene 2000, 19, 2577–2584. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, S.; Ciechomska, M.; Fullard, N.; Przyborski, S.; Van Laar, J.M. IL-13 mediates collagen deposition via STAT6 and microRNA-135b: A role for epigenetics. Sci. Rep. 2016, 6, 25066. [Google Scholar] [CrossRef] [PubMed]
- Aoudjehane, L.; Pissaia, A.; Scatton, O.; Podevin, P.; Massault, P.-P.; Chouzenoux, S.; Soubrane, O.; Calmus, Y.; Conti, F. Interleukin-4 induces the activation and collagen production of cultured human intrahepatic fibroblasts via the STAT-6 pathway. Lab. Investig. 2008, 88, 973–985. [Google Scholar] [CrossRef] [PubMed]
- Sheng, J.; Yang, Y.; Cui, Y.; He, S.; Wang, L.; Liu, L.; He, Q.; Lv, T.; Han, W.; Yu, W.; et al. M2 macrophage-mediated interleukin-4 signalling induces myofibroblast phenotype during the progression of benign prostatic hyperplasia. Cell Death Dis. 2018, 9, 1–13. [Google Scholar] [CrossRef]
- Hashimoto, S.; Gon, Y.; Takeshita, I.; Maruoka, S.; Horie, T. IL-4 and IL-13 induce myofibroblastic phenotype of human lung fibroblasts through c-Jun NH2-terminal kinase–dependent pathway. J. Allergy Clin. Immunol. 2001, 107, 1001–1008. [Google Scholar] [CrossRef]
- Yue, T.; Wang, X.; Olson, B.A.; Feuerstein, G.Z. Interleukin-1β (IL-1β) Induces Transforming Growth Factor-β1 (TGF-β1) Production by Rat Aortic Smooth Muscle Cells. Biochem. Biophys. Res. Commun. 1994, 204, 1186–1192. [Google Scholar] [CrossRef]
- Sullivan, D.E.; Ferris, M.; Nguyen, H.; Abboud, E.; Brody, A.R. TNF-α induces TGF-β1expression in lung fibroblasts at the transcriptional levelviaAP-1 activation. J. Cell. Mol. Med. 2009, 13, 1866–1876. [Google Scholar] [CrossRef]
- Au, P.Y.B.; Martin, N.; Chau, H.; Moemeni, B.; Chia, M.; Liu, F.-F.; Minden, M.; Yeh, W.-C. The oncogene PDGF-B provides a key switch from cell death to survival induced by TNF. Oncogene 2005, 24, 3196–3205. [Google Scholar] [CrossRef][Green Version]
- Reitamo, S.; Remitz, A.; Tamai, K.; Uitto, J. Interleukin-10 modulates type I collagen and matrix metalloprotease gene expression in cultured human skin fibroblasts. J. Clin. Investig. 1994, 94, 2489–2492. [Google Scholar] [CrossRef]
- Ma, Y.; Thornton, S.; Duwel, L.E.; Boivin, G.P.; Giannini, E.H.; Leiden, J.M.; Bluestone, J.A.; Hirsch, R. Inhibition of Collagen-Induced Arthritis in Mice by Viral IL-10 Gene Transfer. J. Immunol. 1998, 161, 1516–1524. [Google Scholar]
- Simões, F.C.; Cahill, T.J.; Kenyon, A.; Gavriouchkina, D.; Vieira, J.M.; Sun, X.; Pezzolla, D.; Ravaud, C.; Masmanian, E.; Weinberger, M.; et al. Macrophages directly contribute collagen to scar formation during zebrafish heart regeneration and mouse heart repair. Nat. Commun. 2020, 11, 1–17. [Google Scholar] [CrossRef]
- Massagué, J. How cells read TGF-β signals. Nat. Rev. Mol. Cell Biol. 2000, 1, 169–178. [Google Scholar] [CrossRef]
- David, C.J.; Massagué, J. Contextual determinants of TGFβ action in development, immunity and cancer. Nat. Rev. Mol. Cell Biol. 2018, 19, 419–435. [Google Scholar] [CrossRef] [PubMed]
- Karsdal, M.A.; Larsen, L.; Engsig, M.T.; Lou, H.; Ferreras, M.; Lochter, A.; Delaissé, J.-M.; Foged, N.T. Matrix Metalloproteinase-dependent Activation of Latent Transforming Growth Factor-β Controls the Conversion of Osteoblasts into Osteocytes by Blocking Osteoblast Apoptosis. J. Biol. Chem. 2002, 277, 44061–44067. [Google Scholar] [CrossRef]
- Hinz, B. Formation and Function of the Myofibroblast during Tissue Repair. J. Investig. Dermatol. 2007, 127, 526–537. [Google Scholar] [CrossRef] [PubMed]
- Cirka, H.; Monterosso, M.; Diamantides, N.; Favreau, J.; Wen, Q.; Billiar, K. Active Traction Force Response to Long-Term Cyclic Stretch Is Dependent on Cell Pre-stress. Biophys. J. 2016, 110, 1845–1857. [Google Scholar] [CrossRef]
- Hinz, B.; Phan, S.H.; Thannickal, V.J.; Galli, A.; Bochaton-Piallat, M.-L.; Gabbiani, G. The Myofibroblast: One Function, Multiple Origins. Am. J. Pathol. 2007, 170, 1807–1816. [Google Scholar] [CrossRef]
- Theiss, A.L.; Simmons, J.G.; Jobin, C.; Lund, P.K. Tumor Necrosis Factor (TNF) α Increases Collagen Accumulation and Proliferation in Intestinal Myofibroblasts via TNF Receptor 2. J. Biol. Chem. 2005, 280, 36099–36109. [Google Scholar] [CrossRef]
- Harris, A.K.; Stopak, D.; Wild, P. Fibroblast traction as a mechanism for collagen morphogenesis. Nat. Cell Biol. 1981, 290, 249–251. [Google Scholar] [CrossRef]
- Miron-Mendoza, M.; Seemann, J.; Grinnell, F. Collagen Fibril Flow and Tissue Translocation Coupled to Fibroblast Migration in 3D Collagen Matrices. Mol. Biol. Cell 2008, 19, 2051–2058. [Google Scholar] [CrossRef]
- Pakshir, P.; Alizadehgiashi, M.; Wong, B.; Coelho, N.M.; Chen, X.; Gong, Z.; Shenoy, V.B.; McCulloch, C.A.; Hinz, B. Dynamic fibroblast contractions attract remote macrophages in fibrillar collagen matrix. Nat. Commun. 2019, 10, 1–17. [Google Scholar] [CrossRef]
- Taufalele, P.V.; Vanderburgh, J.A.; Muñoz, A.; Zanotelli, M.R.; Reinhart-King, C.A. Fiber alignment drives changes in architectural and mechanical features in collagen matrices. PLoS ONE 2019, 14, e0216537. [Google Scholar] [CrossRef]
- Huang, W.-C.; Sala-Newby, G.B.; Susana, A.; Johnson, J.L.; Newby, A.C. Classical Macrophage Activation Up-Regulates Several Matrix Metalloproteinases through Mitogen Activated Protein Kinases and Nuclear Factor-κB. PLoS ONE 2012, 7, e42507. [Google Scholar] [CrossRef]
- Craig, V.J.; Zhang, L.; Hagood, J.S.; Owen, C.A. Matrix Metalloproteinases as Therapeutic Targets for Idiopathic Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2015, 53, 585–600. [Google Scholar] [CrossRef]
- Giannandrea, M.; Parks, W.C. Diverse functions of matrix metalloproteinases during fibrosis. Dis. Model. Mech. 2014, 7, 193–203. [Google Scholar] [CrossRef]
- Pilcher, B.K.; Dumin, J.A.; Sudbeck, B.D.; Krane, S.M.; Welgus, H.G.; Parks, W.C. The Activity of Collagenase-1 Is Required for Keratinocyte Migration on a Type I Collagen Matrix. J. Cell Biol. 1997, 137, 1445–1457. [Google Scholar] [CrossRef]
- Mazor, R.; Alsaigh, T.; Shaked, H.; Altshuler, A.E.; Pocock, E.S.; Kistler, E.B.; Karin, M.; Schmid-Schönbein, G.W. Matrix Metalloproteinase-1-mediated Up-regulation of Vascular Endothelial Growth Factor-2 in Endothelial Cells. J. Biol. Chem. 2013, 288, 598–607. [Google Scholar] [CrossRef]
- Page-McCaw, A.; Ewald, A.J.; Werb, Z. Matrix metalloproteinases and the regulation of tissue remodelling. Nat. Rev. Mol. Cell Biol. 2007, 8, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Egeblad, M.; Werb, Z. New functions for the matrix metalloproteinases in cancer progression. Nat. Rev. Cancer 2002, 2, 161–174. [Google Scholar] [CrossRef]
- Fingleton, B. MMP inhibitor clinical trials—The past, present, and future. In The Cancer Degradome: Proteases and Cancer Biology; Springer: New York, NY, USA, 2008; pp. 759–785. [Google Scholar]
- Cai, L.; Xiong, X.; Kong, X.; Xie, J. The Role of the Lysyl Oxidases in Tissue Repair and Remodeling: A Concise Review. Tissue Eng. Regen. Med. 2017, 14, 15–30. [Google Scholar] [CrossRef]
- Trackman, P.C.; Graham, R.J.; Bittner, H.K.; Carnes, D.L.; Gilles, J.A.; Graves, D.T. Inflammation-associated lysyl oxidase protein expression in vivo, and modulation by FGF-2 plus IGF-1. Histochem. Cell Biol. 1998, 110, 9–14. [Google Scholar] [CrossRef]
- Wirtz, D.; Konstantopoulos, K.; Searson, P.C. The physics of cancer: The role of physical interactions and mechanical forces in metastasis. Nat. Rev. Cancer 2011, 11, 512–522. [Google Scholar] [CrossRef]
- Kumari, S.; Mak, M.; Poh, Y.; Tohme, M.; Watson, N.; Melo, M.; Janssen, E.; Dustin, M.; Geha, R.; Irvine, D.J. Cytoskeletal tension actively sustains the migratory T-cell synaptic contact. EMBO J. 2020, 39, e102783. [Google Scholar] [CrossRef] [PubMed]
- Saitakis, M.; Dogniaux, S.; Goudot, C.; Bufi, N.; Asnacios, S.; Maurin, M.; Randriamampita, C.; Asnacios, A.; Hivroz, C. Different TCR-induced T lymphocyte responses are potentiated by stiffness with variable sensitivity. eLife 2017, 6, e23190. [Google Scholar] [CrossRef]
- Kuo, J.-C.; Han, X.; Hsiao, C.-T.; Iii, J.R.Y.; Waterman, C.M. Analysis of the myosin-II-responsive focal adhesion proteome reveals a role for β-Pix in negative regulation of focal adhesion maturation. Nat. Cell Biol. 2011, 13, 383–393. [Google Scholar] [CrossRef]
- Kanchanawong, P.; Shtengel, G.; Pasapera, A.M.; Ramko, E.B.; Davidson, M.W.; Hess, H.F.; Waterman, C.M. Nanoscale architecture of integrin-based cell adhesions. Nat. Cell Biol. 2010, 468, 580–584. [Google Scholar] [CrossRef]
- Pasapera, A.M.; Schneider, I.C.; Rericha, E.; Schlaepfer, D.D.; Waterman, C.M. Myosin II activity regulates vinculin recruitment to focal adhesions through FAK-mediated paxillin phosphorylation. J. Cell Biol. 2010, 188, 877–890. [Google Scholar] [CrossRef]
- Zaidel-Bar, R.; Milo, R.; Kam, Z.; Geiger, B. A paxillin tyrosine phosphorylation switch regulates the assembly and form of cell-matrix adhesions. J. Cell Sci. 2006, 120, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Zaidel-Bar, R.; Ballestrem, C.; Kam, Z.; Geiger, B. Early molecular events in the assembly of matrix adhesions at the leading edge of migrating cells. J. Cell Sci. 2003, 116, 4605–4613. [Google Scholar] [CrossRef]
- Del Rio, A.; Perez-Jimenez, R.; Liu, R.; Roca-Cusachs, P.; Fernandez, J.M.; Sheetz, M.P. Stretching Single Talin Rod Molecules Activates Vinculin Binding. Scence 2009, 323, 638–641. [Google Scholar] [CrossRef]
- Margadant, F.; Chew, L.L.; Hu, X.; Yu, H.; Bate, N.; Zhang, X.; Sheetz, M. Mechanotransduction In Vivo by Repeated Talin Stretch-Relaxation Events Depends upon Vinculin. PLoS Biol. 2011, 9, e1001223. [Google Scholar] [CrossRef]
- Thievessen, I.; Thompson, P.M.; Berlemont, S.; Plevock, K.M.; Plotnikov, S.V.; Zemljic-Harpf, A.; Ross, R.S.; Davidson, M.W.; Danuser, G.; Campbell, S.L.; et al. Vinculin–actin interaction couples actin retrograde flow to focal adhesions, but is dispensable for focal adhesion growth. J. Cell Biol. 2013, 202, 163–177. [Google Scholar] [CrossRef]
- Lessey, E.C.; Guilluy, C.; Burridge, K. From Mechanical Force to RhoA Activation. Biochemistry 2012, 51, 7420–7432. [Google Scholar] [CrossRef]
- Lo, C.-M.; Wang, H.-B.; Dembo, M.; Wang, Y.-L. Cell Movement Is Guided by the Rigidity of the Substrate. Biophys. J. 2000, 79, 144–152. [Google Scholar] [CrossRef]
- Chan, C.E.; Odde, D.J. Traction Dynamics of Filopodia on Compliant Substrates. Science 2008, 322, 1687–1691. [Google Scholar] [CrossRef]
- Plotnikov, S.V.; Pasapera, A.M.; Sabass, B.; Waterman, C.M. Force Fluctuations within Focal Adhesions Mediate ECM-Rigidity Sensing to Guide Directed Cell Migration. Cell 2012, 151, 1513–1527. [Google Scholar] [CrossRef] [PubMed]
- Baneyx, G.; Baugh, L.; Vogel, V. Fibronectin extension and unfolding within cell matrix fibrils controlled by cytoskeletal tension. Proc. Natl. Acad. Sci. USA 2002, 99, 5139–5143. [Google Scholar] [CrossRef]
- Pankov, R.; Cukierman, E.; Katz, B.-Z.; Matsumoto, K.; Lin, D.C.; Lin, S.; Hahn, C.; Yamada, K.M. Integrin Dynamics and Matrix Assembly. J. Cell Biol. 2000, 148, 1075–1090. [Google Scholar] [CrossRef]
- Chrzanowska-Wodnicka, M.; Burridge, K. Rho-stimulated contractility drives the formation of stress fibers and focal adhesions. J. Cell Biol. 1996, 133, 1403–1415. [Google Scholar] [CrossRef]
- Oakes, P.W.; Beckham, Y.; Stricker, J.; Gardel, M.L. Tension is required but not sufficient for focal adhesion maturation without a stress fiber template. J. Cell Biol. 2012, 196, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Plotnikov, S.V.; Moalim, A.Y.; Waterman, C.M.; Liu, J. Two Distinct Actin Networks Mediate Traction Oscillations to Confer Focal Adhesion Mechanosensing. Biophys. J. 2017, 112, 780–794. [Google Scholar] [CrossRef]
- Puleo, J.I.; Parker, S.S.; Roman, M.R.; Watson, A.W.; Eliato, K.R.; Peng, L.; Saboda, K.; Roe, D.J.; Ros, R.; Gertler, F.B.; et al. Mechanosensing during directed cell migration requires dynamic actin polymerization at focal adhesions. J. Cell Biol. 2019, 218, 4215–4235. [Google Scholar] [CrossRef]
- Kirby, T.J.; Lammerding, J. Emerging views of the nucleus as a cellular mechanosensor. Nat. Cell Biol. 2018, 20, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Graham, D.M.; Andersen, T.; Sharek, L.; Uzer, G.; Rothenberg, K.; Hoffman, B.D.; Rubin, J.; Balland, M.; Bear, J.E.; Burridge, K. Enucleated Cells Reveal Differential Roles of the Nucleus in Cell Migration, Polarity, and Mechanotransduction. J. Cell Biol. 2018, 217, 895–914. [Google Scholar] [CrossRef] [PubMed]
- Swift, J.; Ivanovska, I.L.; Buxboim, A.; Harada, T.; Dingal, P.C.D.P.; Pinter, J.; Pajerowski, J.D.; Spinler, K.R.; Shin, J.W.; Tewari, M.; et al. Nuclear Lamin-A Scales with Tissue Stiffness and Enhances Matrix-Directed Differentiation. Science 2013, 341, 1240104. [Google Scholar] [CrossRef] [PubMed]
- Dupont, S.; Morsut, L.; Aragona, M.; Enzo, E.; Giulitti, S.; Cordenonsi, M.; Zanconato, F.; Digabel, J.L.; Forcato, M.; Bicciato, S.; et al. Role of YAP/TAZ in Mechanotransduction. Nature 2011, 474, 179–183. [Google Scholar] [CrossRef]
- Miralles, F.; Posern, G.; Zaromytidou, A.-I.; Treisman, R. Actin Dynamics Control SRF Activity by Regulation of Its Coactivator MAL. Cell 2003, 113, 329–342. [Google Scholar] [CrossRef]
- Zhao, B.; Wei, X.; Li, W.; Udan, R.S.; Yang, Q.; Kim, J.; Xie, J.; Ikenoue, T.; Yu, J.; Li, L.; et al. Inactivation of YAP Oncoprotein by the Hippo Pathway Is Involved in Cell Contact Inhibition and Tissue Growth Control. Gene Dev. 2007, 21, 2747–2761. [Google Scholar] [CrossRef] [PubMed]
- Wagh, K.; Ishikawa, M.; Garcia, D.A.; Stavreva, D.A.; Upadhyaya, A.; Hager, G.L. Mechanical Regulation of Transcription: Recent Advances. Trends Cell Biol 2021. [Google Scholar] [CrossRef] [PubMed]
- Lomakin, A.J.; Cattin, C.J.; Cuvelier, D.; Alraies, Z.; Molina, M.; Nader, G.P.F.; Srivastava, N.; Sáez, P.J.; Garcia-Arcos, J.M.; Zhitnyak, I.Y.; et al. The Nucleus Acts as a Ruler Tailoring Cell Responses to Spatial Constraints. Science 2020, 370, eaba2894. [Google Scholar] [CrossRef]
- Kung, C. A Possible Unifying Principle for Mechanosensation. Nature 2005, 436, 647–654. [Google Scholar] [CrossRef]
- Lundbæk, J.A.; Maer, A.M.; Andersen, O.S. Lipid Bilayer Electrostatic Energy, Curvature Stress, and Assembly of Gramicidin Channels. Biochemistry 1997, 36, 5695–5701. [Google Scholar] [CrossRef] [PubMed]
- Roux, A.-L.; Quiroga, X.; Walani, N.; Arroyo, M.; Roca-Cusachs, P. The Plasma Membrane as a Mechanochemical Transducer. Philos. Trans. R. Soc. B 2019, 374, 20180221. [Google Scholar] [CrossRef]
- Sugio, S.; Nagasawa, M.; Kojima, I.; Ishizaki, Y.; Shibasaki, K. Transient Receptor Potential Vanilloid 2 Activation by Focal Mechanical Stimulation Requires Interaction with the Actin Cytoskeleton and Enhances Growth Cone Motility. FASEB J. 2016, 31, 1368–1381. [Google Scholar] [CrossRef] [PubMed]
- Goswami, C.; Kuhn, J.; Heppenstall, P.A.; Hucho, T. Importance of Non-Selective Cation Channel TRPV4 Interaction with Cytoskeleton and Their Reciprocal Regulations in Cultured Cells. PLoS ONE 2010, 5, e11654. [Google Scholar] [CrossRef]
- Tang, Y.; Tang, J.; Chen, Z.; Trost, C.; Flockerzi, V.; Li, M.; Ramesh, V.; Zhu, M.X. Association of Mammalian Trp4 and Phospholipase C Isozymes with a PDZ Domain-Containing Protein, NHERF. J. Biol. Chem. 2000, 275, 37559–37564. [Google Scholar] [CrossRef]
- Hudspeth, A.J.; Choe, Y.; Mehta, A.D.; Martin, P. Putting Ion Channels to Work: Mechanoelectrical Transduction, Adaptation, and Amplification by Hair Cells. Proc. Natl. Acad. Sci. USA 2000, 97, 11765–11772. [Google Scholar] [CrossRef]
- Zou, Y.; Akazawa, H.; Qin, Y.; Sano, M.; Takano, H.; Minamino, T.; Makita, N.; Iwanaga, K.; Zhu, W.; Kudoh, S.; et al. Mechanical Stress Activates Angiotensin II Type 1 Receptor without the Involvement of Angiotensin II. Nat. Cell Biol. 2004, 6, 499–506. [Google Scholar] [CrossRef]
- Zhang, Y.-L.; Frangos, J.A.; Chachisvilis, M. Mechanical Stimulus Alters Conformation of Type 1 Parathyroid Hormone Receptor in Bone Cells. Am. J. Physiol. Cell Physiol. 2009, 296, C1391–C1399. [Google Scholar] [CrossRef]
- Schnitzler, M.M.; Storch, U.; Meibers, S.; Nurwakagari, P.; Breit, A.; Essin, K.; Gollasch, M.; Gudermann, T. Gq-coupled Receptors as Mechanosensors Mediating Myogenic Vasoconstriction. EMBO J. 2008, 27, 3092–3103. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, M.G.; Vanek, M.; Guerini, D.; Gasser, J.A.; Jones, C.E.; Junker, U.; Hofstetter, H.; Wolf, R.M.; Seuwen, K. Proton-Sensing G-Protein-Coupled Receptors. Nature 2003, 425, 93–98. [Google Scholar] [CrossRef]
- Xu, J.; Mathur, J.; Vessières, E.; Hammack, S.; Nonomura, K.; Favre, J.; Grimaud, L.; Petrus, M.; Francisco, A.; Li, J.; et al. GPR68 Senses Flow and Is Essential for Vascular Physiology. Cell 2018, 173, 762–775.e16. [Google Scholar] [CrossRef] [PubMed]
- Wiley, S.Z.; Sriram, K.; Liang, W.; Chang, S.E.; French, R.; McCann, T.; Sicklick, J.; Nishihara, H.; Lowy, A.M.; Insel, P.A. GPR68, a Proton-sensing GPCR, Mediates Interaction of Cancer-associated Fibroblasts and Cancer Cells. FASEB J. 2018, 32, 1170–1183. [Google Scholar] [CrossRef] [PubMed]
- Maeyashiki, C.; Melhem, H.; Hering, L.; Baebler, K.; Cosin-Roger, J.; Schefer, F.; Weder, B.; Hausmann, M.; Scharl, M.; Rogler, G.; et al. Activation of pH-Sensing Receptor OGR1 (GPR68) Induces ER Stress Via the IRE1α/JNK Pathway in an Intestinal Epithelial Cell Model. Sci. Rep. 2020, 10, 1–12. [Google Scholar] [CrossRef]
- Chachisvilis, M.; Zhang, Y.-L.; Frangos, J.A. G protein-coupled receptors sense fluid shear stress in endothelial cells. Proc. Natl. Acad. Sci. USA 2006, 103, 15463–15468. [Google Scholar] [CrossRef] [PubMed]
- Erdogmus, S.; Storch, U.; Danner, L.; Becker, J.; Winter, M.; Ziegler, N.; Wirth, A.; Offermanns, S.; Hoffmann, C.; Gudermann, T.; et al. Helix 8 is the essential structural motif of mechanosensitive GPCRs. Nat. Commun. 2019, 10, 1–15. [Google Scholar] [CrossRef]
- Tschumperlin, D.J.; Ligresti, G.; Hilscher, M.B.; Shah, V.H. Mechanosensing and fibrosis. J. Clin. Investig. 2018, 128, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Herrera, J.; Henke, C.A.; Bitterman, P.B. Extracellular matrix as a driver of progressive fibrosis. J. Clin. Investig. 2018, 128, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Hartman, C.D.; Isenberg, B.C.; Chua, S.G.; Wong, J.Y. Vascular smooth muscle cell durotaxis depends on extracellular matrix composition. Proc. Natl. Acad. Sci. USA 2016, 113, 11190–11195. [Google Scholar] [CrossRef]
- Harland, B.; Walcott, S.; Sun, S.X. Adhesion dynamics and durotaxis in migrating cells. Phys. Biol. 2011, 8, 015011. [Google Scholar] [CrossRef]
- Vincent, L.G.; Choi, Y.S.; Alonso-Latorre, B.; Del Álamo, J.C.; Engler, A.J. Mesenchymal stem cell durotaxis depends on substrate stiffness gradient strength. Biotechnol. J. 2013, 8, 472–484. [Google Scholar] [CrossRef]
- Raab, M.; Swift, J.; Dingal, P.D.P.; Shah, P.; Shin, J.-W.; Discher, D.E. Crawling from soft to stiff matrix polarizes the cytoskeleton and phosphoregulates myosin-II heavy chain. J. Cell Biol. 2012, 199, 669–683. [Google Scholar] [CrossRef]
- Wang, H.-B.; Dembo, M.; Hanks, S.K.; Wang, Y.-L. Focal adhesion kinase is involved in mechanosensing during fibroblast migration. Proc. Natl. Acad. Sci. USA 2001, 98, 11295–11300. [Google Scholar] [CrossRef]
- Von Wichert, G.; Jiang, G.; Kostic, A.D.; De Vos, K.; Sap, J.; Sheetz, M.P. RPTP-α acts as a transducer of mechanical force on αv/β3-integrin–cytoskeleton linkages. J. Cell Biol. 2003, 161, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Jiang, G.; Huang, A.H.; Cai, Y.; Tanase, M.; Sheetz, M.P. Rigidity Sensing at the Leading Edge through αvβ3 Integrins and RPTPα. Biophys. J. 2006, 90, 1804–1809. [Google Scholar] [CrossRef]
- Wormer, D.B.; Davis, K.A.; Henderson, J.H.; Turner, C.E. The Focal Adhesion-Localized CdGAP Regulates Matrix Rigidity Sensing and Durotaxis. PLoS ONE 2014, 9, e91815. [Google Scholar] [CrossRef]
- Lachowski, D.; Cortes, E.; Robinson, B.; Rice, A.; Rombouts, K.; Hernández, A.E.D.R. FAK controls the mechanical activation of YAP, a transcriptional regulator required for durotaxis. FASEB J. 2018, 32, 1099–1107. [Google Scholar] [CrossRef] [PubMed]
- Lachowski, D.; Cortes, E.; Pink, D.; Chronopoulos, A.; Karim, S.A.; Morton, J.P.; Hernández, A.E.D.R. Substrate Rigidity Controls Activation and Durotaxis in Pancreatic Stellate Cells. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef]
- Yang, H.-W.; Liu, X.-Y.; Shen, Z.-F.; Yao, W.; Gong, X.-B.; Huang, H.-X.; Ding, G.-H. An investigation of the distribution and location of mast cells affected by the stiffness of substrates as a mechanical niche. Int. J. Biol. Sci. 2018, 14, 1142–1152. [Google Scholar] [CrossRef]
- Tisler, M.; Alkmin, S.; Chang, H.-Y.; Leet, J.; Bernau, K.; Sandbo, N.; Campagnola, P.J. Analysis of fibroblast migration dynamics in idiopathic pulmonary fibrosis using image-based scaffolds of the lung extracellular matrix. Am. J. Physiol. Cell. Mol. Physiol. 2020, 318, L276–L286. [Google Scholar] [CrossRef]
- Sunyer, R.; Conte, V.; Escribano, J.; Elosegui-Artola, A.; Labernadie, A.; Valon, L.; Navajas, D.; García-Aznar, J.M.; Muñoz, J.J.; Roca-Cusachs, P.; et al. Collective cell durotaxis emerges from long-range intercellular force transmission. Science 2016, 353, 1157–1161. [Google Scholar] [CrossRef]
- DuChez, B.J.; Doyle, A.D.; Dimitriadis, E.K.; Yamada, K.M. Durotaxis by Human Cancer Cells. Biophys. J. 2019, 116, 670–683. [Google Scholar] [CrossRef]
- Doyle, A.D.; Sykora, D.J.; Pacheco, G.G.; Kutys, M.L.; Yamada, K.M. 3D mesenchymal cell migration is driven by anterior cellular contraction that generates an extracellular matrix prestrain. Dev. Cell 2021, 56, 826–841.e4. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Wu, P.-F.; Ma, J.-X.; Liao, M.-J.; Xu, L.-S.; Yi, L. TRPV4 activates the Cdc42/N-wasp pathway to promote glioblastoma invasion by altering cellular protrusions. Sci. Rep. 2020, 10, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Potla, R.; Hirano-Kobayashi, M.; Wu, H.; Chen, H.; Mammoto, A.; Matthews, B.D.; Ingber, D.E. Molecular mapping of transmembrane mechanotransduction through the β1 integrin–CD98hc–TRPV4 axis. J. Cell Sci. 2020, 133, jcs248823. [Google Scholar] [CrossRef] [PubMed]
- Arya, R.K.; Goswami, R.; Rahaman, S.O. Mechanotransduction via a TRPV4-Rac1 signaling axis plays a role in multinucleated giant cell formation. J. Biol. Chem. 2021, 296, 100129. [Google Scholar] [CrossRef]
- Ridley, A.J. Rho GTPase signalling in cell migration. Curr. Opin. Cell Biol. 2015, 36, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Pasapera, A.M.; Plotnikov, S.V.; Fischer, R.S.; Case, L.B.; Egelhoff, T.T.; Waterman, C.M. Rac1-dependent phosphorylation and focal adhesion recruitment of myosin IIA regulates migration and mechanosensing. Curr. Biol. 2015, 25, 175–186. [Google Scholar] [CrossRef]
- Zambruno, G.; Marchisio, P.C.; Marconi, A.; Vaschieri, C.; Melchiori, A.; Giannetti, A.; De Luca, M. Transforming growth factor-beta 1 modulates beta 1 and beta 5 integrin receptors and induces the de novo expression of the alpha v beta 6 heterodimer in normal human keratinocytes: Implications for wound healing. J. Cell Biol. 1995, 129, 853–865. [Google Scholar] [CrossRef]
- Brown, N.F.; Marshall, J.F. Integrin-Mediated TGFβ Activation Modulates the Tumour Microenvironment. Cancers 2019, 11, 1221. [Google Scholar] [CrossRef]
- Kinoshita, K.; Aono, Y.; Azuma, M.; Kishi, J.; Takezaki, A.; Kishi, M.; Makino, H.; Okazaki, H.; Uehara, H.; Izumi, K.; et al. Antifibrotic Effects of Focal Adhesion Kinase Inhibitor in Bleomycin-Induced Pulmonary Fibrosis in Mice. Am. J. Respir. Cell Mol. Biol. 2013, 49, 536–543. [Google Scholar] [CrossRef]
- Andrikopoulos, P.; Kieswich, J.; Pacheco, S.; Nadarajah, L.; Harwood, S.M.; O’Riordan, C.E.; Thiemermann, C.; Yaqoob, M.M. The MEK Inhibitor Trametinib Ameliorates Kidney Fibrosis by Suppressing ERK1/2 and mTORC1 Signaling. J. Am. Soc. Nephrol. 2019, 30, 33–49. [Google Scholar] [CrossRef]
- Chen, X.; Wang, H.; Liao, H.-J.; Hu, W.; Gewin, L.; Mernaugh, G.; Zhang, S.; Zhang, Z.-Y.; Vega-Montoto, L.; Vanacore, R.M.; et al. Integrin-mediated type II TGF-β receptor tyrosine dephosphorylation controls SMAD-dependent profibrotic signaling. J. Clin. Investig. 2014, 124, 3295–3310. [Google Scholar] [CrossRef]
- Chaudhary, N.I.; Roth, G.J.; Hilberg, F.; Muller-Quernheim, J.; Prasse, A.; Zissel, G.; Schnapp, A.; Park, J.E. Inhibition of PDGF, VEGF and FGF signalling attenuates fibrosis. Eur. Respir. J. 2007, 29, 976–985. [Google Scholar] [CrossRef]
- Shochet, G.E.; Brook, E.; Eyal, O.; Edelstein, E.; Shitrit, D. Epidermal growth factor receptor paracrine upregulation in idiopathic pulmonary fibrosis fibroblasts is blocked by nintedanib. Am. J. Physiol. Cell. Mol. Physiol. 2019, 316, L1025–L1034. [Google Scholar] [CrossRef]
- Yang, L.; Kwon, J.; Popov, Y.; Gajdos, G.B.; Ordog, T.; Brekken, R.A.; Mukhopadhyay, D.; Schuppan, D.; Bi, Y.; Simonetto, D.; et al. Vascular Endothelial Growth Factor Promotes Fibrosis Resolution and Repair in Mice. Gastroenterology 2014, 146, 1339–1350.e1. [Google Scholar] [CrossRef]
- Moro, L.; Venturino, M.; Bozzo, C.; Silengo, L.; Altruda, F.; Beguinot, L.; Tarone, G.; Defilippi, P. Integrins induce activation of EGF receptor: Role in MAP kinase induction and adhesion-dependent cell survival. EMBO J. 1998, 17, 6622–6632. [Google Scholar] [CrossRef] [PubMed]
- Morello, V.; Cabodi, S.; Sigismund, S.; Camacho-Leal, M.P.; Repetto, D.; Volante, M.; Papotti, M.; Turco, E.; Defilippi, P. β1 integrin controls EGFR signaling and tumorigenic properties of lung cancer cells. Oncogene 2011, 30, 4087–4096. [Google Scholar] [CrossRef]
- Vlahakis, N.E.; Young, B.A.; Atakilit, A.; Hawkridge, A.E.; Issaka, R.B.; Boudreau, N.; Sheppard, D. Integrin α9β1 Directly Binds to Vascular Endothelial Growth Factor (VEGF)-A and Contributes to VEGF-A-induced Angiogenesis. J. Biol. Chem. 2007, 282, 15187–15196. [Google Scholar] [CrossRef] [PubMed]
- Somanath, P.R.; Malinin, N.L.; Byzova, T.V. Cooperation between integrin ανβ3 and VEGFR2 in angiogenesis. Angiogenesis 2009, 12, 177–185. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, S.; Plotnikov, S.V. Mechanosensitive Regulation of Fibrosis. Cells 2021, 10, 994. https://doi.org/10.3390/cells10050994
Yang S, Plotnikov SV. Mechanosensitive Regulation of Fibrosis. Cells. 2021; 10(5):994. https://doi.org/10.3390/cells10050994
Chicago/Turabian StyleYang, Shuying, and Sergey V. Plotnikov. 2021. "Mechanosensitive Regulation of Fibrosis" Cells 10, no. 5: 994. https://doi.org/10.3390/cells10050994
APA StyleYang, S., & Plotnikov, S. V. (2021). Mechanosensitive Regulation of Fibrosis. Cells, 10(5), 994. https://doi.org/10.3390/cells10050994