
Mitochondria in Diabetic Kidney Disease



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Diabetic Kidney Disease: Generalities
2. Diabetic Kidney Disease: Pathogenesis
3. Crosstalk between Glomeruli and Tubules in the Diabetic Kidney. Relationship with Kidney Bioenergetics
4. Mitochondrial Energy Metabolism in the Normal Kidney Cells
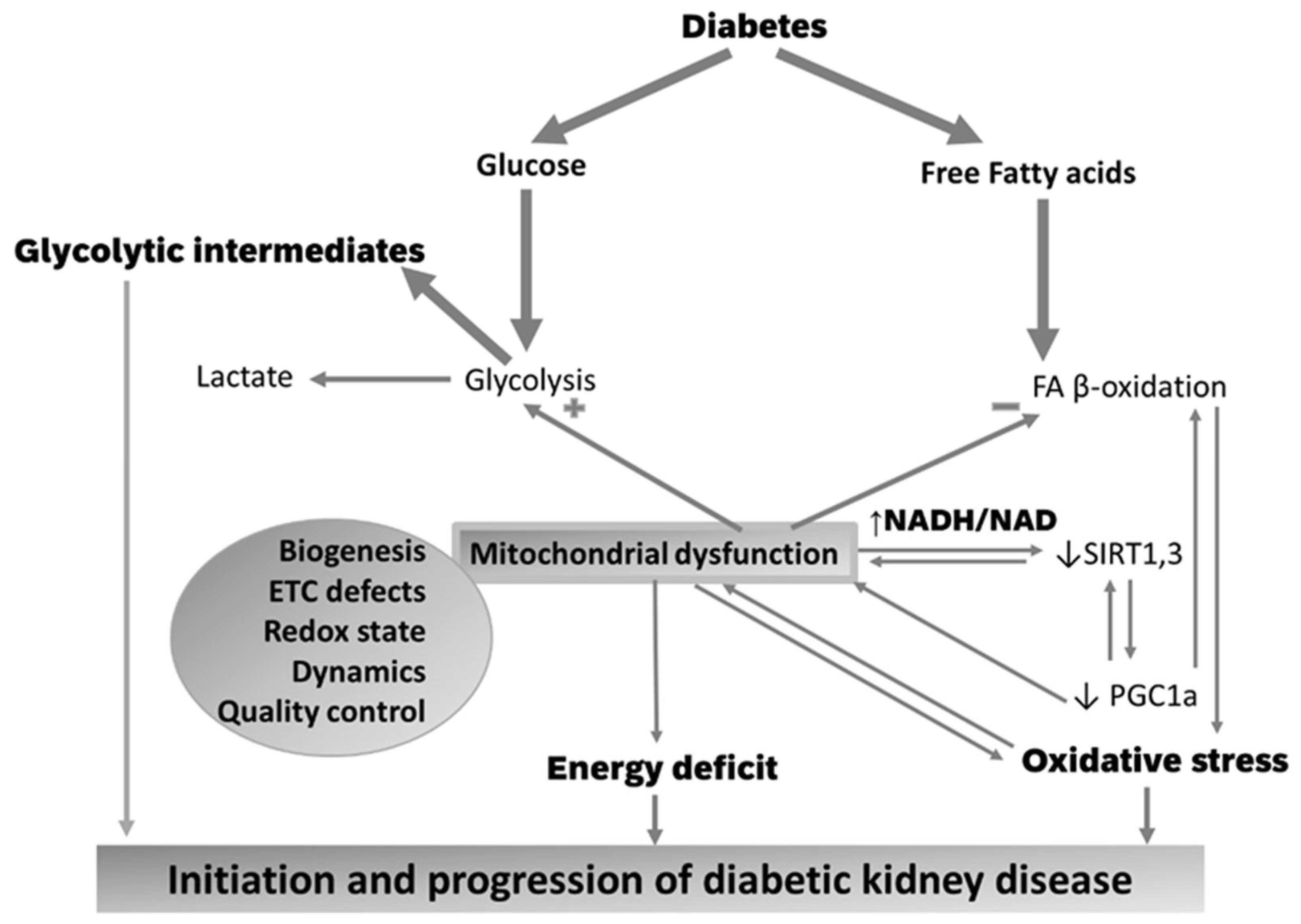
5. Mitochondrial Abnormalities in the Diabetic Kidney
5.1. Alterations in Substrate Selection
5.1.1. Glomerular Cells
5.1.2. Tubular Cells
5.2. Mitochondrial Biogenesis
5.3. Mitochondrial ETC Defects
5.4. Mitochondrial Oxidative Stress
5.5. NADH/NAD Redox State
5.5.1. NAD Pool
5.5.2. NADH/NAD+ Redox Ratio
5.6. Mitochondrial Dynamics and Quality Control
6. Therapeutic Approaches to Improve Kidney Bioenergetics and DKD
7. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Han, Q.; Zhu, H.; Chen, X.; Liu, Z. Non-genetic mechanisms of diabetic nephropathy. Front. Med. 2017, 11, 319–332. [Google Scholar] [CrossRef] [PubMed]
- Alicic, R.Z.; Rooney, M.T.; Tuttle, K.R. Diabetic Kidney Disease: Challenges, Progress, and Possibilities. Clin. J. Am. Soc. Nephrol. 2017, 12, 2032–2045. [Google Scholar] [CrossRef] [PubMed]
- Pugliese, G.; Penno, G.; Natali, A.; Barutta, F.; Di Paolo, S.; Reboldi, G.; Gesualdo, L.; De Nicola, L.; Italian Diabetes Society; the Italian Society of Nephrology. Diabetic kidney disease: New clinical and therapeutic issues. Joint position statement of the Italian Diabetes Society and the Italian Society of Nephrology on “The natural history of diabetic kidney disease and treatment of hyperglycemia in patients with type 2 diabetes and impaired renal function”. Nutr. Metab. Cardiovasc. Dis. 2019, 29, 1127–1150. [Google Scholar] [CrossRef]
- Afkarian, M.; Zelnick, L.R.; Hall, Y.N.; Heagerty, P.J.; Tuttle, K.; Weiss, N.S.; de Boer, I.H. Clinical Manifestations of Kidney Disease Among US Adults with Diabetes, 1988–2014. JAMA 2016, 316, 602–610. [Google Scholar] [CrossRef] [PubMed]
- Minutolo, R.; Gabbai, F.B.; Provenzano, M.; Chiodini, P.; Borrelli, S.; Garofalo, C.; Sasso, F.C.; Santoro, D.; Bellizzi, V.; Conte, G.; et al. Cardiorenal prognosis by residual proteinuria level in diabetic chronic kidney disease: Pooled analysis of four cohort studies. Nephrol. Dial. Transplant. 2018, 33, 1942–1949. [Google Scholar] [CrossRef]
- Heerspink, H.J.L.; Coresh, J.; Gansevoort, R.T.; Inker, L.A. Change in albuminuria as a surrogate endpoint in chronic kidney disease—Authors’ reply. Lancet Diabetes Endocrinol. 2019, 7, 336–337. [Google Scholar] [CrossRef]
- Introduction: Standards of Medical Care in Diabetes-2018. Diabetes Care 2018, 41, S1–S2. [CrossRef]
- Qi, C.; Mao, X.; Zhang, Z.; Wu, H. Classification and Differential Diagnosis of Diabetic Nephropathy. J. Diabetes Res. 2017, 2017, 8637138. [Google Scholar] [CrossRef]
- Fu, J.; Lee, K.; Chuang, P.Y.; Liu, Z.; He, J.C. Glomerular endothelial cell injury and cross talk in diabetic kidney disease. Am. J. Physiol. Renal. Physiol. 2015, 308, F287–F297. [Google Scholar] [CrossRef]
- Nieuwdorp, M.; van Haeften, T.W.; Gouverneur, M.C.; Mooij, H.L.; van Lieshout, M.H.; Levi, M.; Meijers, J.C.; Holleman, F.; Hoekstra, J.B.; Vink, H.; et al. Loss of endothelial glycocalyx during acute hyperglycemia coincides with endothelial dysfunction and coagulation activation in vivo. Diabetes 2006, 55, 480–486. [Google Scholar] [CrossRef]
- Umanath, K.; Lewis, J.B. Update on Diabetic Nephropathy: Core Curriculum 2018. Am. J. Kidney Dis 2018, 71, 884–895. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.C.; Brownlee, M.; Susztak, K.; Sharma, K.; Jandeleit-Dahm, K.A.; Zoungas, S.; Rossing, P.; Groop, P.H.; Cooper, M.E. Diabetic kidney disease. Nat. Rev. Dis. Primers 2015, 1, 15018. [Google Scholar] [CrossRef] [PubMed]
- Papademetriou, V.; Lovato, L.; Doumas, M.; Nylen, E.; Mottl, A.; Cohen, R.M.; Applegate, W.B.; Puntakee, Z.; Yale, J.F.; Cushman, W.C.; et al. Chronic kidney disease and intensive glycemic control increase cardiovascular risk in patients with type 2 diabetes. Kidney Int. 2015, 87, 649–659. [Google Scholar] [CrossRef]
- Vallon, V.; Komers, R. Pathophysiology of the diabetic kidney. Compr. Physiol. 2011, 1, 1175–1232. [Google Scholar] [CrossRef]
- Vallon, V. The proximal tubule in the pathophysiology of the diabetic kidney. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 300, R1009–R1022. [Google Scholar] [CrossRef] [PubMed]
- Bak, M.; Thomsen, K.; Christiansen, T.; Flyvbjerg, A. Renal enlargement precedes renal hyperfiltration in early experimental diabetes in rats. J. Am. Soc. Nephrol. 2000, 11, 1287–1292. [Google Scholar] [CrossRef] [PubMed]
- Helal, I.; Fick-Brosnahan, G.M.; Reed-Gitomer, B.; Schrier, R.W. Glomerular hyperfiltration: Definitions, mechanisms and clinical implications. Nat. Rev. Nephrol. 2012, 8, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Vallon, V.; Thomson, S.C. The tubular hypothesis of nephron filtration and diabetic kidney disease. Nat. Rev. Nephrol. 2020, 16, 317–336. [Google Scholar] [CrossRef] [PubMed]
- Persson, P.; Hansell, P.; Palm, F. Tubular reabsorption and diabetes-induced glomerular hyperfiltration. Acta Physiol. 2010, 200, 3–10. [Google Scholar] [CrossRef]
- Ziyadeh, F.N.; Hoffman, B.B.; Han, D.C.; Iglesias-De La Cruz, M.C.; Hong, S.W.; Isono, M.; Chen, S.; McGowan, T.A.; Sharma, K. Long-term prevention of renal insufficiency, excess matrix gene expression, and glomerular mesangial matrix expansion by treatment with monoclonal antitransforming growth factor-beta antibody in db/db diabetic mice. Proc. Natl. Acad. Sci. USA 2000, 97, 8015–8020. [Google Scholar] [CrossRef]
- Bai, Y.; Wang, L.; Li, Y.; Liu, S.; Li, J.; Wang, H.; Huang, H. High ambient glucose levels modulates the production of MMP-9 and alpha5(IV) collagen by cultured podocytes. Cell Physiol. Biochem. 2006, 17, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Stieger, N.; Worthmann, K.; Teng, B.; Engeli, S.; Das, A.M.; Haller, H.; Schiffer, M. Impact of high glucose and transforming growth factor-beta on bioenergetic profiles in podocytes. Metabolism 2012, 61, 1073–1086. [Google Scholar] [CrossRef] [PubMed]
- Susztak, K.; Raff, A.C.; Schiffer, M.; Bottinger, E.P. Glucose-induced reactive oxygen species cause apoptosis of podocytes and podocyte depletion at the onset of diabetic nephropathy. Diabetes 2006, 55, 225–233. [Google Scholar] [CrossRef]
- Li, X.; Chuang, P.Y.; D’Agati, V.D.; Dai, Y.; Yacoub, R.; Fu, J.; Xu, J.; Taku, O.; Premsrirut, P.K.; Holzman, L.B.; et al. Nephrin Preserves Podocyte Viability and Glomerular Structure and Function in Adult Kidneys. J. Am. Soc. Nephrol. 2015, 26, 2361–2377. [Google Scholar] [CrossRef]
- Doublier, S.; Salvidio, G.; Lupia, E.; Ruotsalainen, V.; Verzola, D.; Deferrari, G.; Camussi, G. Nephrin expression is reduced in human diabetic nephropathy: Evidence for a distinct role for glycated albumin and angiotensin II. Diabetes 2003, 52, 1023–1030. [Google Scholar] [CrossRef]
- Wharram, B.L.; Goyal, M.; Wiggins, J.E.; Sanden, S.K.; Hussain, S.; Filipiak, W.E.; Saunders, T.L.; Dysko, R.C.; Kohno, K.; Holzman, L.B.; et al. Podocyte depletion causes glomerulosclerosis: Diphtheria toxin-induced podocyte depletion in rats expressing human diphtheria toxin receptor transgene. J. Am. Soc. Nephrol. 2005, 16, 2941–2952. [Google Scholar] [CrossRef]
- Coward, R.J.; Welsh, G.I.; Yang, J.; Tasman, C.; Lennon, R.; Koziell, A.; Satchell, S.; Holman, G.D.; Kerjaschki, D.; Tavare, J.M.; et al. The human glomerular podocyte is a novel target for insulin action. Diabetes 2005, 54, 3095–3102. [Google Scholar] [CrossRef]
- Meyer, T.W.; Bennett, P.H.; Nelson, R.G. Podocyte number predicts long-term urinary albumin excretion in Pima Indians with Type II diabetes and microalbuminuria. Diabetologia 1999, 42, 1341–1344. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Liu, S.; Bastacky, S.I.; Wang, X.; Tian, X.J.; Zhou, D. Diabetic kidney diseases revisited: A new perspective for a new era. Mol. Metab. 2019, 30, 250–263. [Google Scholar] [CrossRef]
- Hashimoto, Y.; Yamagishi, S.; Mizukami, H.; Yabe-Nishimura, C.; Lim, S.W.; Kwon, H.M.; Yagihashi, S. Polyol pathway and diabetic nephropathy revisited: Early tubular cell changes and glomerulopathy in diabetic mice overexpressing human aldose reductase. J. Diabetes Investig. 2011, 2, 111–122. [Google Scholar] [CrossRef]
- White, K.E.; Marshall, S.M.; Bilous, R.W. Prevalence of atubular glomeruli in type 2 diabetic patients with nephropathy. Nephrol. Dial. Transplant. 2008, 23, 3539–3545. [Google Scholar] [CrossRef][Green Version]
- Sayyed, S.G.; Ryu, M.; Kulkarni, O.P.; Schmid, H.; Lichtnekert, J.; Gruner, S.; Green, L.; Mattei, P.; Hartmann, G.; Anders, H.J. An orally active chemokine receptor CCR2 antagonist prevents glomerulosclerosis and renal failure in type 2 diabetes. Kidney Int. 2011, 80, 68–78. [Google Scholar] [CrossRef]
- Shahzad, K.; Bock, F.; Dong, W.; Wang, H.; Kopf, S.; Kohli, S.; Al-Dabet, M.M.; Ranjan, S.; Wolter, J.; Wacker, C.; et al. Nlrp3-inflammasome activation in non-myeloid-derived cells aggravates diabetic nephropathy. Kidney Int. 2015, 87, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Santoro, D.; Torreggiani, M.; Pellicano, V.; Cernaro, V.; Messina, R.M.; Longhitano, E.; Siligato, R.; Gembillo, G.; Esposito, C.; Piccoli, G.B. Kidney Biopsy in Type 2 Diabetic Patients: Critical Reflections on Present Indications and Diagnostic Alternatives. Int. J. Mol. Sci. 2021, 22, 5425. [Google Scholar] [CrossRef]
- Porrini, E.; Ruggenenti, P.; Mogensen, C.E.; Barlovic, D.P.; Praga, M.; Cruzado, J.M.; Hojs, R.; Abbate, M.; de Vries, A.P.; the ERA-EDTA Diabesity Working Group. Non-proteinuric pathways in loss of renal function in patients with type 2 diabetes. Lancet Diabetes Endocrinol. 2015, 3, 382–391. [Google Scholar] [CrossRef]
- Verzola, D.; Gandolfo, M.T.; Gaetani, G.; Ferraris, A.; Mangerini, R.; Ferrario, F.; Villaggio, B.; Gianiorio, F.; Tosetti, F.; Weiss, U.; et al. Accelerated senescence in the kidneys of patients with type 2 diabetic nephropathy. Am. J. Physiol. Ren. Physiol. 2008, 295, F1563–F1573. [Google Scholar] [CrossRef] [PubMed]
- Mise, K.; Hoshino, J.; Ueno, T.; Hazue, R.; Hasegawa, J.; Sekine, A.; Sumida, K.; Hiramatsu, R.; Hasegawa, E.; Yamanouchi, M.; et al. Prognostic Value of Tubulointerstitial Lesions, Urinary N-Acetyl-beta-d-Glucosaminidase, and Urinary beta2-Microglobulin in Patients with Type 2 Diabetes and Biopsy-Proven Diabetic Nephropathy. Clin. J. Am. Soc. Nephrol. 2016, 11, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.; Park, J.; Kim, J.; Jang, H.R.; Kwon, G.Y.; Huh, W.; Kim, Y.G.; Kim, D.J.; Oh, H.Y.; Lee, J.E. Tissue expression of tubular injury markers is associated with renal function decline in diabetic nephropathy. J. Diabetes Complicat. 2017, 31, 1704–1709. [Google Scholar] [CrossRef]
- Vallon, V.; Osswald, H. Adenosine receptors and the kidney. Handb. Exp. Pharmacol. 2009, 443–470. [Google Scholar] [CrossRef]
- Vallon, V.; Schroth, J.; Satriano, J.; Blantz, R.C.; Thomson, S.C.; Rieg, T. Adenosine A(1) receptors determine glomerular hyperfiltration and the salt paradox in early streptozotocin diabetes mellitus. Nephron Physiol. 2009, 111, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Vallon, V.; Platt, K.A.; Cunard, R.; Schroth, J.; Whaley, J.; Thomson, S.C.; Koepsell, H.; Rieg, T. SGLT2 mediates glucose reabsorption in the early proximal tubule. J. Am. Soc. Nephrol. 2011, 22, 104–112. [Google Scholar] [CrossRef]
- Rieg, T.; Masuda, T.; Gerasimova, M.; Mayoux, E.; Platt, K.; Powell, D.R.; Thomson, S.C.; Koepsell, H.; Vallon, V. Increase in SGLT1-mediated transport explains renal glucose reabsorption during genetic and pharmacological SGLT2 inhibition in euglycemia. Am. J. Physiol. Ren. Physiol. 2014, 306, F188–F193. [Google Scholar] [CrossRef] [PubMed]
- Hallow, K.M.; Gebremichael, Y.; Helmlinger, G.; Vallon, V. Primary proximal tubule hyperreabsorption and impaired tubular transport counterregulation determine glomerular hyperfiltration in diabetes: A modeling analysis. Am. J. Physiol. Ren. Physiol. 2017, 312, F819–F835. [Google Scholar] [CrossRef]
- Vallon, V.; Gerasimova, M.; Rose, M.A.; Masuda, T.; Satriano, J.; Mayoux, E.; Koepsell, H.; Thomson, S.C.; Rieg, T. SGLT2 inhibitor empagliflozin reduces renal growth and albuminuria in proportion to hyperglycemia and prevents glomerular hyperfiltration in diabetic Akita mice. Am. J. Physiol. Ren. Physiol. 2014, 306, F194–F204. [Google Scholar] [CrossRef] [PubMed]
- Vallon, V.; Rose, M.; Gerasimova, M.; Satriano, J.; Platt, K.A.; Koepsell, H.; Cunard, R.; Sharma, K.; Thomson, S.C.; Rieg, T. Knockout of Na-glucose transporter SGLT2 attenuates hyperglycemia and glomerular hyperfiltration but not kidney growth or injury in diabetes mellitus. Am. J. Physiol. Ren. Physiol. 2013, 304, F156–F167. [Google Scholar] [CrossRef] [PubMed]
- Marks, J.; Carvou, N.J.; Debnam, E.S.; Srai, S.K.; Unwin, R.J. Diabetes increases facilitative glucose uptake and GLUT2 expression at the rat proximal tubule brush border membrane. J. Physiol. 2003, 553, 137–145. [Google Scholar] [CrossRef]
- Hinden, L.; Udi, S.; Drori, A.; Gammal, A.; Nemirovski, A.; Hadar, R.; Baraghithy, S.; Permyakova, A.; Geron, M.; Cohen, M.; et al. Modulation of Renal GLUT2 by the Cannabinoid-1 Receptor: Implications for the Treatment of Diabetic Nephropathy. J. Am. Soc. Nephrol. 2018, 29, 434–448. [Google Scholar] [CrossRef]
- Rahmoune, H.; Thompson, P.W.; Ward, J.M.; Smith, C.D.; Hong, G.; Brown, J. Glucose transporters in human renal proximal tubular cells isolated from the urine of patients with non-insulin-dependent diabetes. Diabetes 2005, 54, 3427–3434. [Google Scholar] [CrossRef]
- Wang, X.X.; Levi, J.; Luo, Y.; Myakala, K.; Herman-Edelstein, M.; Qiu, L.; Wang, D.; Peng, Y.; Grenz, A.; Lucia, S.; et al. SGLT2 Protein Expression Is Increased in Human Diabetic Nephropathy: SGLT2 Protein Inhibition Decreases Renal Lipid Accumulation, Inflammation, and the Development of Nephropathy in Diabetic Mice. J. Biol. Chem. 2017, 292, 5335–5348. [Google Scholar] [CrossRef]
- Weinstein, A.M.; Weinbaum, S.; Duan, Y.; Du, Z.; Yan, Q.; Wang, T. Flow-dependent transport in a mathematical model of rat proximal tubule. Am. J. Physiol. Ren. Physiol. 2007, 292, F1164–F1181. [Google Scholar] [CrossRef]
- Fattah, H.; Layton, A.; Vallon, V. How Do Kidneys Adapt to a Deficit or Loss in Nephron Number? Physiology 2019, 34, 189–197. [Google Scholar] [CrossRef]
- Ghezzi, C.; Wright, E.M. Regulation of the human Na+-dependent glucose cotransporter hSGLT2. Am. J. Physiol. Cell Physiol. 2012, 303, C348–C354. [Google Scholar] [CrossRef] [PubMed]
- Phillips, A.O.; Steadman, R.; Morrisey, K.; Williams, J.D. Polarity of stimulation and secretion of transforming growth factor-beta 1 by cultured proximal tubular cells. Am. J. Pathol. 1997, 150, 1101–1111. [Google Scholar]
- Phillips, A.O.; Steadman, R. Diabetic nephropathy: The central role of renal proximal tubular cells in tubulointerstitial injury. Histol. Histopathol. 2002, 17, 247–252. [Google Scholar] [CrossRef] [PubMed]
- Onishi, A.; Fu, Y.; Darshi, M.; Crespo-Masip, M.; Huang, W.; Song, P.; Patel, R.; Kim, Y.C.; Nespoux, J.; Freeman, B.; et al. Effect of renal tubule-specific knockdown of the Na(+)/H(+) exchanger NHE3 in Akita diabetic mice. Am. J. Physiol. Ren. Physiol. 2019, 317, F419–F434. [Google Scholar] [CrossRef] [PubMed]
- Noh, H.; King, G.L. The role of protein kinase C activation in diabetic nephropathy. Kidney Int. Suppl. 2007, S49–S53. [Google Scholar] [CrossRef]
- Meier, M.; Park, J.K.; Overheu, D.; Kirsch, T.; Lindschau, C.; Gueler, F.; Leitges, M.; Menne, J.; Haller, H. Deletion of protein kinase C-beta isoform in vivo reduces renal hypertrophy but not albuminuria in the streptozotocin-induced diabetic mouse model. Diabetes 2007, 56, 346–354. [Google Scholar] [CrossRef]
- Layton, A.T.; Vallon, V.; Edwards, A. Predicted consequences of diabetes and SGLT inhibition on transport and oxygen consumption along a rat nephron. Am. J. Physiol. Ren. Physiol. 2016, 310, F1269–F1283. [Google Scholar] [CrossRef] [PubMed]
- Layton, A.T.; Vallon, V.; Edwards, A. Modeling oxygen consumption in the proximal tubule: Effects of NHE and SGLT2 inhibition. Am. J. Physiol. Ren. Physiol. 2015, 308, F1343–F1357. [Google Scholar] [CrossRef]
- O’Neill, J.; Fasching, A.; Pihl, L.; Patinha, D.; Franzen, S.; Palm, F. Acute SGLT inhibition normalizes O2 tension in the renal cortex but causes hypoxia in the renal medulla in anaesthetized control and diabetic rats. Am. J. Physiol. Ren. Physiol. 2015, 309, F227–F234. [Google Scholar] [CrossRef]
- Pruijm, M.; Milani, B.; Pivin, E.; Podhajska, A.; Vogt, B.; Stuber, M.; Burnier, M. Reduced cortical oxygenation predicts a progressive decline of renal function in patients with chronic kidney disease. Kidney Int. 2018, 93, 932–940. [Google Scholar] [CrossRef] [PubMed]
- Bhargava, P.; Schnellmann, R.G. Mitochondrial energetics in the kidney. Nat. Rev. Nephrol. 2017, 13, 629–646. [Google Scholar] [CrossRef]
- Cargill, K.; Sims-Lucas, S. Metabolic requirements of the nephron. Pediatr. Nephrol. 2020, 35, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, R.E. Proximal Tubulopathy: Prime Mover and Key Therapeutic Target in Diabetic Kidney Disease. Diabetes 2017, 66, 791–800. [Google Scholar] [CrossRef]
- Pfaller, W.; Rittinger, M. Quantitative morphology of the rat kidney. Int J. Biochem 1980, 12, 17–22. [Google Scholar] [CrossRef]
- Brinkkoetter, P.T.; Bork, T.; Salou, S.; Liang, W.; Mizi, A.; Ozel, C.; Koehler, S.; Hagmann, H.H.; Ising, C.; Kuczkowski, A.; et al. Anaerobic Glycolysis Maintains the Glomerular Filtration Barrier Independent of Mitochondrial Metabolism and Dynamics. Cell Rep. 2019, 27, 1551–1566. [Google Scholar] [CrossRef] [PubMed]
- Abe, Y.; Sakairi, T.; Kajiyama, H.; Shrivastav, S.; Beeson, C.; Kopp, J.B. Bioenergetic characterization of mouse podocytes. Am. J. Physiol. Cell Physiol. 2010, 299, C464–C476. [Google Scholar] [CrossRef]
- Muller-Deile, J.; Schiffer, M. The podocyte power-plant disaster and its contribution to glomerulopathy. Front. Endocrinol. 2014, 5, 209. [Google Scholar] [CrossRef]
- Schaffer, J.E.; Lodish, H.F. Expression cloning and characterization of a novel adipocyte long chain fatty acid transport protein. Cell 1994, 79, 427–436. [Google Scholar] [CrossRef]
- Watanabe, S.; Ichikawa, D.; Sugaya, T.; Ohata, K.; Inoue, K.; Hoshino, S.; Kimura, K.; Shibagaki, Y.; Kamijo-Ikemori, A. Urinary Level of Liver-Type Fatty Acid Binding Protein Reflects the Degree of Tubulointerstitial Damage in Polycystic Kidney Disease. Kidney Blood Press Res. 2018, 43, 1716–1729. [Google Scholar] [CrossRef]
- Morita, M.; Kanasaki, K. Sodium-glucose cotransporter-2 inhibitors for diabetic kidney disease: Targeting Warburg effects in proximal tubular cells. Diabetes Metab. 2020, 46, 353–361. [Google Scholar] [CrossRef]
- Bolon, C.; Gauthier, C.; Simonnet, H. Glycolysis inhibition by palmitate in renal cells cultured in a two-chamber system. Am. J. Physiol. 1997, 273, C1732–C1738. [Google Scholar] [CrossRef]
- Coughlan, M.T.; Nguyen, T.V.; Penfold, S.A.; Higgins, G.C.; Thallas-Bonke, V.; Tan, S.M.; Van Bergen, N.J.; Sourris, K.C.; Harcourt, B.E.; Thorburn, D.R.; et al. Mapping time-course mitochondrial adaptations in the kidney in experimental diabetes. Clin. Sci. 2016, 130, 711–720. [Google Scholar] [CrossRef] [PubMed]
- Mima, A.; Ohshiro, Y.; Kitada, M.; Matsumoto, M.; Geraldes, P.; Li, C.; Li, Q.; White, G.S.; Cahill, C.; Rask-Madsen, C.; et al. Glomerular-specific protein kinase C-beta-induced insulin receptor substrate-1 dysfunction and insulin resistance in rat models of diabetes and obesity. Kidney Int. 2011, 79, 883–896. [Google Scholar] [CrossRef] [PubMed]
- Tejada, T.; Catanuto, P.; Ijaz, A.; Santos, J.V.; Xia, X.; Sanchez, P.; Sanabria, N.; Lenz, O.; Elliot, S.J.; Fornoni, A. Failure to phosphorylate AKT in podocytes from mice with early diabetic nephropathy promotes cell death. Kidney Int. 2008, 73, 1385–1393. [Google Scholar] [CrossRef]
- Weigert, C.; Brodbeck, K.; Brosius, F.C., 3rd; Huber, M.; Lehmann, R.; Friess, U.; Facchin, S.; Aulwurm, S.; Haring, H.U.; Schleicher, E.D.; et al. Evidence for a novel TGF-beta1-independent mechanism of fibronectin production in mesangial cells overexpressing glucose transporters. Diabetes 2003, 52, 527–535. [Google Scholar] [CrossRef]
- Gnudi, L.; Thomas, S.M.; Viberti, G. Mechanical forces in diabetic kidney disease: A trigger for impaired glucose metabolism. J. Am. Soc. Nephrol. 2007, 18, 2226–2232. [Google Scholar] [CrossRef]
- Lewko, B.; Bryl, E.; Witkowski, J.M.; Latawiec, E.; Angielski, S.; Stepinski, J. Mechanical stress and glucose concentration modulate glucose transport in cultured rat podocytes. Nephrol. Dial. Transplant. 2005, 20, 306–311. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Guzman, J.; Jauregui, A.N.; Merscher-Gomez, S.; Maiguel, D.; Muresan, C.; Mitrofanova, A.; Diez-Sampedro, A.; Szust, J.; Yoo, T.H.; Villarreal, R.; et al. Podocyte-specific GLUT4-deficient mice have fewer and larger podocytes and are protected from diabetic nephropathy. Diabetes 2014, 63, 701–714. [Google Scholar] [CrossRef]
- Coward, R.; Fornoni, A. Insulin signaling: Implications for podocyte biology in diabetic kidney disease. Curr. Opin. Nephrol. Hypertens. 2015, 24, 104–110. [Google Scholar] [CrossRef]
- Nishikawa, T.; Edelstein, D.; Du, X.L.; Yamagishi, S.; Matsumura, T.; Kaneda, Y.; Yorek, M.A.; Beebe, D.; Oates, P.J.; Hammes, H.P.; et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 2000, 404, 787–790. [Google Scholar] [CrossRef]
- Imasawa, T.; Obre, E.; Bellance, N.; Lavie, J.; Imasawa, T.; Rigothier, C.; Delmas, Y.; Combe, C.; Lacombe, D.; Benard, G.; et al. High glucose repatterns human podocyte energy metabolism during differentiation and diabetic nephropathy. FASEB J. 2017, 31, 294–307. [Google Scholar] [CrossRef]
- Sharma, K.; Karl, B.; Mathew, A.V.; Gangoiti, J.A.; Wassel, C.L.; Saito, R.; Pu, M.; Sharma, S.; You, Y.H.; Wang, L.; et al. Metabolomics reveals signature of mitochondrial dysfunction in diabetic kidney disease. J. Am. Soc. Nephrol. 2013, 24, 1901–1912. [Google Scholar] [CrossRef]
- Sas, K.M.; Kayampilly, P.; Byun, J.; Nair, V.; Hinder, L.M.; Hur, J.; Zhang, H.; Lin, C.; Qi, N.R.; Michailidis, G.; et al. Tissue-specific metabolic reprogramming drives nutrient flux in diabetic complications. JCI Insight 2016, 1, e86976. [Google Scholar] [CrossRef] [PubMed]
- Mima, A.; Qi, W.; King, G.L. Implications of treatment that target protective mechanisms against diabetic nephropathy. Semin. Nephrol. 2012, 32, 471–478. [Google Scholar] [CrossRef]
- Mima, A.; Hiraoka-Yamomoto, J.; Li, Q.; Kitada, M.; Li, C.; Geraldes, P.; Matsumoto, M.; Mizutani, K.; Park, K.; Cahill, C.; et al. Protective effects of GLP-1 on glomerular endothelium and its inhibition by PKCbeta activation in diabetes. Diabetes 2012, 61, 2967–2979. [Google Scholar] [CrossRef] [PubMed]
- Rosca, M.G.; Mustata, T.G.; Kinter, M.T.; Ozdemir, A.M.; Kern, T.S.; Szweda, L.I.; Brownlee, M.; Monnier, V.M.; Weiss, M.F. Glycation of mitochondrial proteins from diabetic rat kidney is associated with excess superoxide formation. Am. J. Physiol. 2005, 289, F420–F430. [Google Scholar] [CrossRef]
- Nam, D.H.; Han, J.H.; Lee, T.J.; Shishido, T.; Lim, J.H.; Kim, G.Y.; Woo, C.H. CHOP deficiency prevents methylglyoxal-induced myocyte apoptosis and cardiac dysfunction. J. Mol. Cell. Cardiol. 2015, 85, 168–177. [Google Scholar] [CrossRef]
- Szablewski, L. Distribution of glucose transporters in renal diseases. J. Biomed. Sci. 2017, 24, 64. [Google Scholar] [CrossRef]
- Jiang, Y.K.; Xin, K.Y.; Ge, H.W.; Kong, F.J.; Zhao, G. Upregulation of Renal GLUT2 and SGLT2 is Involved in High-Fat Diet-Induced Gestational Diabetes in Mice. Diabetes Metab. Syndr. Obes. 2019, 12, 2095–2105. [Google Scholar] [CrossRef] [PubMed]
- Lan, R.; Geng, H.; Singha, P.K.; Saikumar, P.; Bottinger, E.P.; Weinberg, J.M.; Venkatachalam, M.A. Mitochondrial Pathology and Glycolytic Shift during Proximal Tubule Atrophy after Ischemic AKI. J. Am. Soc. Nephrol. 2016, 27, 3356–3367. [Google Scholar] [CrossRef]
- Burch, H.B.; Choi, S.; Dence, C.N.; Alvey, T.R.; Cole, B.R.; Lowry, O.H. Metabolic effects of large fructose loads in different parts of the rat nephron. J. Biol. Chem. 1980, 255, 8239–8244. [Google Scholar] [CrossRef]
- Diggle, C.P.; Shires, M.; Leitch, D.; Brooke, D.; Carr, I.M.; Markham, A.F.; Hayward, B.E.; Asipu, A.; Bonthron, D.T. Ketohexokinase: Expression and localization of the principal fructose-metabolizing enzyme. J. Histochem. Cytochem. 2009, 57, 763–774. [Google Scholar] [CrossRef]
- Lanaspa, M.A.; Ishimoto, T.; Cicerchi, C.; Tamura, Y.; Roncal-Jimenez, C.A.; Chen, W.; Tanabe, K.; Andres-Hernando, A.; Orlicky, D.J.; Finol, E.; et al. Endogenous fructose production and fructokinase activation mediate renal injury in diabetic nephropathy. J. Am. Soc. Nephrol. 2014, 25, 2526–2538. [Google Scholar] [CrossRef]
- Berthiaume, J.M.; Kurdys, J.G.; Muntean, D.M.; Rosca, M.G. Mitochondrial NAD(+)/NADH Redox State and Diabetic Cardiomyopathy. Antioxid. Redox Signal. 2019, 30, 375–398. [Google Scholar] [CrossRef]
- Kang, H.M.; Ahn, S.H.; Choi, P.; Ko, Y.A.; Han, S.H.; Chinga, F.; Park, A.S.; Tao, J.; Sharma, K.; Pullman, J.; et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat. Med. 2015, 21, 37–46. [Google Scholar] [CrossRef]
- Meyer, C.; Nadkarni, V.; Stumvoll, M.; Gerich, J. Human kidney free fatty acid and glucose uptake: Evidence for a renal glucose-fatty acid cycle. Am. J. Physiol. 1997, 273, E650–E654. [Google Scholar] [CrossRef] [PubMed]
- Kovesdy, C.P.; Anderson, J.E.; Kalantar-Zadeh, K. Inverse association between lipid levels and mortality in men with chronic kidney disease who are not yet on dialysis: Effects of case mix and the malnutrition-inflammation-cachexia syndrome. J. Am. Soc. Nephrol. 2007, 18, 304–311. [Google Scholar] [CrossRef]
- Gai, Z.; Wang, T.; Visentin, M.; Kullak-Ublick, G.A.; Fu, X.; Wang, Z. Lipid Accumulation and Chronic Kidney Disease. Nutrients 2019, 11, 722. [Google Scholar] [CrossRef] [PubMed]
- Tontonoz, P.; Nagy, L.; Alvarez, J.G.; Thomazy, V.A.; Evans, R.M. PPARgamma promotes monocyte/macrophage differentiation and uptake of oxidized LDL. Cell 1998, 93, 241–252. [Google Scholar] [CrossRef]
- Hua, W.; Huang, H.Z.; Tan, L.T.; Wan, J.M.; Gui, H.B.; Zhao, L.; Ruan, X.Z.; Chen, X.M.; Du, X.G. CD36 Mediated Fatty Acid-Induced Podocyte Apoptosis via Oxidative Stress. PLoS ONE 2015, 10, e0127507. [Google Scholar] [CrossRef]
- Cui, W.; Maimaitiyiming, H.; Zhou, Q.; Norman, H.; Zhou, C.; Wang, S. Interaction of thrombospondin1 and CD36 contributes to obesity-associated podocytopathy. Biochim. Biophys. Acta 2015, 1852, 1323–1333. [Google Scholar] [CrossRef]
- Hou, Y.; Wu, M.; Wei, J.; Ren, Y.; Du, C.; Wu, H.; Li, Y.; Shi, Y. CD36 is involved in high glucose-induced epithelial to mesenchymal transition in renal tubular epithelial cells. Biochem. Biophys. Res. Commun. 2015, 468, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Gu, C.; Li, Y.; Huang, J. High Glucose Promotes CD36 Expression by Upregulating Peroxisome Proliferator-Activated Receptor gamma Levels to Exacerbate Lipid Deposition in Renal Tubular Cells. Biomed. Res. Int. 2017, 2017, 1414070. [Google Scholar] [CrossRef] [PubMed]
- Rosca, M.G.; Vazquez, E.J.; Chen, Q.; Kerner, J.; Kern, T.S.; Hoppel, C.L. Oxidation of fatty acids is the source of increased mitochondrial reactive oxygen species production in kidney cortical tubules in early diabetes. Diabetes 2012, 61, 2074–2083. [Google Scholar] [CrossRef]
- Herman-Edelstein, M.; Scherzer, P.; Tobar, A.; Levi, M.; Gafter, U. Altered renal lipid metabolism and renal lipid accumulation in human diabetic nephropathy. J. Lipid Res. 2014, 55, 561–572. [Google Scholar] [CrossRef] [PubMed]
- Proctor, G.; Jiang, T.; Iwahashi, M.; Wang, Z.; Li, J.; Levi, M. Regulation of renal fatty acid and cholesterol metabolism, inflammation, and fibrosis in Akita and OVE26 mice with type 1 diabetes. Diabetes 2006, 55, 2502–2509. [Google Scholar] [CrossRef]
- Ducasa, G.M.; Mitrofanova, A.; Mallela, S.K.; Liu, X.; Molina, J.; Sloan, A.; Pedigo, C.E.; Ge, M.; Santos, J.V.; Hernandez, Y.; et al. ATP-binding cassette A1 deficiency causes cardiolipin-driven mitochondrial dysfunction in podocytes. J. Clin. Investig. 2019, 129, 3387–3400. [Google Scholar] [CrossRef] [PubMed]
- Bottinger, E.P.; Bitzer, M. TGF-beta signaling in renal disease. J. Am. Soc. Nephrol. 2002, 13, 2600–2610. [Google Scholar] [CrossRef]
- Yadav, H.; Quijano, C.; Kamaraju, A.K.; Gavrilova, O.; Malek, R.; Chen, W.; Zerfas, P.; Zhigang, D.; Wright, E.C.; Stuelten, C.; et al. Protection from obesity and diabetes by blockade of TGF-beta/Smad3 signaling. Cell Metab. 2011, 14, 67–79. [Google Scholar] [CrossRef]
- Tran, M.; Tam, D.; Bardia, A.; Bhasin, M.; Rowe, G.C.; Kher, A.; Zsengeller, Z.K.; Akhavan-Sharif, M.R.; Khankin, E.V.; Saintgeniez, M.; et al. PGC-1alpha promotes recovery after acute kidney injury during systemic inflammation in mice. J. Clin. Invest. 2011, 121, 4003–4014. [Google Scholar] [CrossRef]
- Scarpulla, R.C.; Vega, R.B.; Kelly, D.P. Transcriptional integration of mitochondrial biogenesis. Trends Endocrinol. Metab. 2012, 23, 459–466. [Google Scholar] [CrossRef]
- Wang, X.X.; Wang, D.; Luo, Y.; Myakala, K.; Dobrinskikh, E.; Rosenberg, A.Z.; Levi, J.; Kopp, J.B.; Field, A.; Hill, A.; et al. FXR/TGR5 Dual Agonist Prevents Progression of Nephropathy in Diabetes and Obesity. J. Am. Soc. Nephrol. 2018, 29, 118–137. [Google Scholar] [CrossRef]
- Xiao, L.; Zhu, X.; Yang, S.; Liu, F.; Zhou, Z.; Zhan, M.; Xie, P.; Zhang, D.; Li, J.; Song, P.; et al. Rap1 ameliorates renal tubular injury in diabetic nephropathy. Diabetes 2014, 63, 1366–1380. [Google Scholar] [CrossRef] [PubMed]
- Dugan, L.L.; You, Y.H.; Ali, S.S.; Diamond-Stanic, M.; Miyamoto, S.; DeCleves, A.E.; Andreyev, A.; Quach, T.; Ly, S.; Shekhtman, G.; et al. AMPK dysregulation promotes diabetes-related reduction of superoxide and mitochondrial function. J. Clin. Investig. 2013, 123, 4888–4899. [Google Scholar] [CrossRef]
- Long, J.; Badal, S.S.; Ye, Z.; Wang, Y.; Ayanga, B.A.; Galvan, D.L.; Green, N.H.; Chang, B.H.; Overbeek, P.A.; Danesh, F.R. Long noncoding RNA Tug1 regulates mitochondrial bioenergetics in diabetic nephropathy. J. Clin. Invest. 2016, 126, 4205–4218. [Google Scholar] [CrossRef] [PubMed]
- Platt, C.; Coward, R.J. Peroxisome proliferator activating receptor-gamma and the podocyte. Nephrol. Dial. Transplant. 2017, 32, 423–433. [Google Scholar] [CrossRef]
- Yuan, Y.; Huang, S.; Wang, W.; Wang, Y.; Zhang, P.; Zhu, C.; Ding, G.; Liu, B.; Yang, T.; Zhang, A. Activation of peroxisome proliferator-activated receptor-gamma coactivator 1alpha ameliorates mitochondrial dysfunction and protects podocytes from aldosterone-induced injury. Kidney Int. 2012, 82, 771–789. [Google Scholar] [CrossRef] [PubMed]
- Hershberger, K.A.; Martin, A.S.; Hirschey, M.D. Role of NAD(+) and mitochondrial sirtuins in cardiac and renal diseases. Nat. Rev. Nephrol. 2017, 13, 213–225. [Google Scholar] [CrossRef] [PubMed]
- Perico, L.; Morigi, M.; Benigni, A. Mitochondrial Sirtuin 3 and Renal Diseases. Nephron 2016, 134, 14–19. [Google Scholar] [CrossRef]
- Holthofer, H.; Kretzler, M.; Haltia, A.; Solin, M.L.; Taanman, J.W.; Schagger, H.; Kriz, W.; Kerjaschki, D.; Schlondorff, D. Altered gene expression and functions of mitochondria in human nephrotic syndrome. FASEB J. 1999, 13, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Qi, H.; Casalena, G.; Shi, S.; Yu, L.; Ebefors, K.; Sun, Y.; Zhang, W.; D’Agati, V.; Schlondorff, D.; Haraldsson, B.; et al. Glomerular Endothelial Mitochondrial Dysfunction Is Essential and Characteristic of Diabetic Kidney Disease Susceptibility. Diabetes 2017, 66, 763–778. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Shao, X.; Jia, S.; Qu, L.; Weng, C.; Shen, X.; Wang, Y.; Huang, H.; Wang, Y.; Wang, C.; et al. The Mitochondria-Targeted Metabolic Tubular Injury in Diabetic Kidney Disease. Cell Physiol. Biochem. 2019, 52, 156–171. [Google Scholar] [CrossRef] [PubMed]
- Czajka, A.; Ajaz, S.; Gnudi, L.; Parsade, C.K.; Jones, P.; Reid, F.; Malik, A.N. Altered Mitochondrial Function, Mitochondrial DNA and Reduced Metabolic Flexibility in Patients with Diabetic Nephropathy. EBioMedicine 2015, 2, 499–512. [Google Scholar] [CrossRef]
- Wu, M.; Li, S.; Yu, X.; Chen, W.; Ma, H.; Shao, C.; Zhang, Y.; Zhang, A.; Huang, S.; Jia, Z. Mitochondrial activity contributes to impaired renal metabolic homeostasis and renal pathology in STZ-induced diabetic mice. Am. J. Physiol. Ren. Physiol. 2019, 317, F593–F605. [Google Scholar] [CrossRef]
- Liu, H.W.; Kao, H.H.; Wu, C.H. Exercise training upregulates SIRT1 to attenuate inflammation and metabolic dysfunction in kidney and liver of diabetic db/db mice. Nutr. Metab. 2019, 16, 22. [Google Scholar] [CrossRef]
- Hunter, C.A.; Kartal, F.; Koc, Z.C.; Murphy, T.; Kim, J.H.; Denvir, J.; Koc, E.C. Mitochondrial oxidative phosphorylation is impaired in TALLYHO mice, a new obesity and type 2 diabetes animal model. Int. J. Biochem. Cell Biol. 2019, 116, 105616. [Google Scholar] [CrossRef]
- Mustata, G.T.; Rosca, M.; Biemel, K.M.; Reihl, O.; Smith, M.A.; Viswanathan, A.; Strauch, C.; Du, Y.; Tang, J.; Kern, T.S.; et al. Paradoxical effects of green tea (Camellia sinensis) and antioxidant vitamins in diabetic rats: Improved retinopathy and renal mitochondrial defects but deterioration of collagen matrix glycoxidation and cross-linking. Diabetes 2005, 54, 517–526. [Google Scholar] [CrossRef]
- Wu, J.; Luo, X.; Yan, L.J. Two dimensional blue native/SDS-PAGE to identify mitochondrial complex I subunits modified by 4-hydroxynonenal (HNE). Front. Physiol. 2015, 6, 98. [Google Scholar] [CrossRef]
- Coughlan, M.T.; Thorburn, D.R.; Penfold, S.A.; Laskowski, A.; Harcourt, B.E.; Sourris, K.C.; Tan, A.L.; Fukami, K.; Thallas-Bonke, V.; Nawroth, P.P.; et al. RAGE-induced cytosolic ROS promote mitochondrial superoxide generation in diabetes. J. Am. Soc. Nephrol. 2009, 20, 742–752. [Google Scholar] [CrossRef]
- Galvan, D.L.; Badal, S.S.; Long, J.; Chang, B.H.; Schumacker, P.T.; Overbeek, P.A.; Danesh, F.R. Real-time in vivo mitochondrial redox assessment confirms enhanced mitochondrial reactive oxygen species in diabetic nephropathy. Kidney Int. 2017, 92, 1282–1287. [Google Scholar] [CrossRef]
- Gordin, D.; Shah, H.; Shinjo, T.; St-Louis, R.; Qi, W.; Park, K.; Paniagua, S.M.; Pober, D.M.; Wu, I.H.; Bahnam, V.; et al. Characterization of Glycolytic Enzymes and Pyruvate Kinase M2 in Type 1 and 2 Diabetic Nephropathy. Diabetes Care 2019, 42, 1263–1273. [Google Scholar] [CrossRef]
- Friederich, M.; Fasching, A.; Hansell, P.; Nordquist, L.; Palm, F. Diabetes-induced up-regulation of uncoupling protein-2 results in increased mitochondrial uncoupling in kidney proximal tubular cells. Biochim. Biophys. Acta 2008, 1777, 935–940. [Google Scholar] [CrossRef] [PubMed]
- Palm, F.; Cederberg, J.; Hansell, P.; Liss, P.; Carlsson, P.O. Reactive oxygen species cause diabetes-induced decrease in renal oxygen tension. Diabetologia 2003, 46, 1153–1160. [Google Scholar] [CrossRef] [PubMed]
- Christensen, M.; Schiffer, T.A.; Gustafsson, H.; Krag, S.P.; Norregaard, R.; Palm, F. Metformin attenuates renal medullary hypoxia in diabetic nephropathy through inhibition uncoupling protein-2. Diabetes Metab. Res. Rev. 2019, 35, e3091. [Google Scholar] [CrossRef] [PubMed]
- Friederich-Persson, M.; Persson, P. Mitochondrial angiotensin II receptors regulate oxygen consumption in kidney mitochondria from healthy and type 1 diabetic rats. Am. J. Physiol. Ren. Physiol. 2020, 318, F683–F688. [Google Scholar] [CrossRef]
- Forbes, J.M.; Thorburn, D.R. Mitochondrial dysfunction in diabetic kidney disease. Nat. Rev. Nephrol. 2018, 14, 291–312. [Google Scholar] [CrossRef] [PubMed]
- Wirthensohn, G.; Guder, W.G. Renal substrate metabolism. Physiol. Rev. 1986, 66, 469–497. [Google Scholar] [CrossRef] [PubMed]
- Sourris, K.C.; Harcourt, B.E.; Tang, P.H.; Morley, A.L.; Huynh, K.; Penfold, S.A.; Coughlan, M.T.; Cooper, M.E.; Nguyen, T.V.; Ritchie, R.H.; et al. Ubiquinone (coenzyme Q10) prevents renal mitochondrial dysfunction in an experimental model of type 2 diabetes. Free Radic. Biol. Med. 2012, 52, 716–723. [Google Scholar] [CrossRef] [PubMed]
- Stanton, R.C. Oxidative stress and diabetic kidney disease. Curr. Diabetes Rep. 2011, 11, 330–336. [Google Scholar] [CrossRef] [PubMed]
- Wong, H.S.; Dighe, P.A.; Mezera, V.; Monternier, P.A.; Brand, M.D. Production of superoxide and hydrogen peroxide from specific mitochondrial sites under different bioenergetic conditions. J. Biol. Chem. 2017, 292, 16804–16809. [Google Scholar] [CrossRef]
- Fisher-Wellman, K.H.; Lin, C.T.; Ryan, T.E.; Reese, L.R.; Gilliam, L.A.; Cathey, B.L.; Lark, D.S.; Smith, C.D.; Muoio, D.M.; Neufer, P.D. Pyruvate dehydrogenase complex and nicotinamide nucleotide transhydrogenase constitute an energy-consuming redox circuit. Biochem. J. 2015, 467, 271–280. [Google Scholar] [CrossRef]
- Walsh, M.E.; Shi, Y.; Van Remmen, H. The effects of dietary restriction on oxidative stress in rodents. Free Radic. Biol. Med. 2014, 66, 88–99. [Google Scholar] [CrossRef]
- Sinclair, D.A. Toward a unified theory of caloric restriction and longevity regulation. Mech. Ageing Dev. 2005, 126, 987–1002. [Google Scholar] [CrossRef] [PubMed]
- Fulco, M.; Cen, Y.; Zhao, P.; Hoffman, E.P.; McBurney, M.W.; Sauve, A.A.; Sartorelli, V. Glucose restriction inhibits skeletal myoblast differentiation by activating SIRT1 through AMPK-mediated regulation of Nampt. Dev. Cell 2008, 14, 661–673. [Google Scholar] [CrossRef]
- Treberg, J.R.; Quinlan, C.L.; Brand, M.D. Evidence for two sites of superoxide production by mitochondrial NADH-ubiquinone oxidoreductase (complex I). J. Biol. Chem. 2011, 286, 27103–27110. [Google Scholar] [CrossRef] [PubMed]
- Lambert, A.J.; Brand, M.D. Superoxide production by NADH:ubiquinone oxidoreductase (complex I) depends on the pH gradient across the mitochondrial inner membrane. Biochem. J. 2004, 382, 511–517. [Google Scholar] [CrossRef]
- Quinlan, C.L.; Goncalves, R.L.; Hey-Mogensen, M.; Yadava, N.; Bunik, V.I.; Brand, M.D. The 2-oxoacid dehydrogenase complexes in mitochondria can produce superoxide/hydrogen peroxide at much higher rates than complex I. J. Biol. Chem. 2014, 289, 8312–8325. [Google Scholar] [CrossRef]
- Teodoro, J.S.; Gomes, A.P.; Varela, A.T.; Duarte, F.V.; Rolo, A.P.; Palmeira, C.M. Uncovering the beginning of diabetes: The cellular redox status and oxidative stress as starting players in hyperglycemic damage. Mol. Cell Biochem. 2013, 376, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Nigro, M.; De Sanctis, C.; Formisano, P.; Stanzione, R.; Forte, M.; Capasso, G.; Gigliotti, G.; Rubattu, S.; Viggiano, D. Cellular and subcellular localization of uncoupling protein 2 in the human kidney. J. Mol. Histol. 2018, 49, 437–445. [Google Scholar] [CrossRef]
- Persson, M.F.; Franzen, S.; Catrina, S.B.; Dallner, G.; Hansell, P.; Brismar, K.; Palm, F. Coenzyme Q10 prevents GDP-sensitive mitochondrial uncoupling, glomerular hyperfiltration and proteinuria in kidneys from db/db mice as a model of type 2 diabetes. Diabetologia 2012, 55, 1535–1543. [Google Scholar] [CrossRef]
- Li, J.; Jiang, R.; Cong, X.; Zhao, Y. UCP2 gene polymorphisms in obesity and diabetes, and the role of UCP2 in cancer. FEBS Lett. 2019, 593, 2525–2534. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.S.; Brachmann, C.B.; Celic, I.; Kenna, M.A.; Muhammad, S.; Starai, V.J.; Avalos, J.L.; Escalante-Semerena, J.C.; Grubmeyer, C.; Wolberger, C.; et al. A phylogenetically conserved NAD+-dependent protein deacetylase activity in the Sir2 protein family. Proc. Natl. Acad. Sci. USA 2000, 97, 6658–6663. [Google Scholar] [CrossRef] [PubMed]
- Canto, C.; Sauve, A.A.; Bai, P. Crosstalk between poly(ADP-ribose) polymerase and sirtuin enzymes. Mol. Aspects Med. 2013, 34, 1168–1201. [Google Scholar] [CrossRef]
- Katsyuba, E.; Mottis, A.; Zietak, M.; De Franco, F.; van der Velpen, V.; Gariani, K.; Ryu, D.; Cialabrini, L.; Matilainen, O.; Liscio, P.; et al. De novo NAD(+) synthesis enhances mitochondrial function and improves health. Nature 2018, 563, 354–359. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, M.T.; Soares, S.M.; Novak, C.M.; Sinclair, D.; Levine, J.A.; Aksoy, P.; Chini, E.N. The enzyme CD38 (a NAD glycohydrolase, EC 3.2.2.5) is necessary for the development of diet-induced obesity. FASEB J. 2007, 21, 3629–3639. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liang, Y.; Hu, T.; Wei, R.; Cai, C.; Wang, P.; Wang, L.; Qiao, W.; Feng, L. Endogenous Nampt upregulation is associated with diabetic nephropathy inflammatory-fibrosis through the NF-kappaB p65 and Sirt1 pathway; NMN alleviates diabetic nephropathy inflammatory-fibrosis by inhibiting endogenous Nampt. Exp. Ther. Med. 2017, 14, 4181–4193. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Jones, D.P.; Sies, H. The Redox Code. Antioxid. Redox Signal. 2015, 23, 734–746. [Google Scholar] [CrossRef] [PubMed]
- Ido, Y.; Nyengaard, J.R.; Chang, K.; Tilton, R.G.; Kilo, C.; Mylari, B.L.; Oates, P.J.; Williamson, J.R. Early neural and vascular dysfunctions in diabetic rats are largely sequelae of increased sorbitol oxidation. Antioxid. Redox Signal. 2010, 12, 39–51. [Google Scholar] [CrossRef]
- Williamson, J.R.; Chang, K.; Frangos, M.; Hasan, K.S.; Ido, Y.; Kawamura, T.; Nyengaard, J.R.; van den Enden, M.; Kilo, C.; Tilton, R.G. Hyperglycemic pseudohypoxia and diabetic complications. Diabetes 1993, 42, 801–813. [Google Scholar] [CrossRef] [PubMed]
- Lorenzi, M. The polyol pathway as a mechanism for diabetic retinopathy: Attractive, elusive, and resilient. Exp. Diabetes Res. 2007, 2007, 61038. [Google Scholar] [CrossRef]
- Masutani, M.; Suzuki, H.; Kamada, N.; Watanabe, M.; Ueda, O.; Nozaki, T.; Jishage, K.; Watanabe, T.; Sugimoto, T.; Nakagama, H.; et al. Poly(ADP-ribose) polymerase gene disruption conferred mice resistant to streptozotocin-induced diabetes. Proc. Natl. Acad. Sci. USA 1999, 96, 2301–2304. [Google Scholar] [CrossRef] [PubMed]
- Bai, P.; Canto, C.; Oudart, H.; Brunyanszki, A.; Cen, Y.; Thomas, C.; Yamamoto, H.; Huber, A.; Kiss, B.; Houtkooper, R.H.; et al. PARP-1 inhibition increases mitochondrial metabolism through SIRT1 activation. Cell Metab. 2011, 13, 461–468. [Google Scholar] [CrossRef]
- Mekala, N.K.; Kurdys, J.; Depuydt, M.M.; Vazquez, E.J.; Rosca, M.G. Apoptosis inducing factor deficiency causes retinal photoreceptor degeneration. The protective role of the redox compound methylene blue. Redox Biol. 2019, 20, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Karamanlidis, G.; Lee, C.F.; Garcia-Menendez, L.; Kolwicz, S.C., Jr.; Suthammarak, W.; Gong, G.; Sedensky, M.M.; Morgan, P.G.; Wang, W.; Tian, R. Mitochondrial complex I deficiency increases protein acetylation and accelerates heart failure. Cell Metab. 2013, 18, 239–250. [Google Scholar] [CrossRef]
- Sung, H.J.; Ma, W.; Wang, P.Y.; Hynes, J.; O’Riordan, T.C.; Combs, C.A.; McCoy, J.P., Jr.; Bunz, F.; Kang, J.G.; Hwang, P.M. Mitochondrial respiration protects against oxygen-associated DNA damage. Nat. Commun. 2010, 1, 5. [Google Scholar] [CrossRef]
- Akie, T.E.; Liu, L.; Nam, M.; Lei, S.; Cooper, M.P. OXPHOS-Mediated Induction of NAD+ Promotes Complete Oxidation of Fatty Acids and Interdicts Non-Alcoholic Fatty Liver Disease. PLoS ONE 2015, 10, e0125617. [Google Scholar] [CrossRef]
- Ogura, Y.; Kitada, M.; Xu, J.; Monno, I.; Koya, D. CD38 inhibition by apigenin ameliorates mitochondrial oxidative stress through restoration of the intracellular NAD(+)/NADH ratio and Sirt3 activity in renal tubular cells in diabetic rats. Aging Albany NY 2020, 12, 11325–11336. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Li, S.; Cruz, P.; Kone, B.C. Sirtuin 1 functionally and physically interacts with disruptor of telomeric silencing-1 to regulate alpha-ENaC transcription in collecting duct. J. Biol. Chem. 2009, 284, 20917–20926. [Google Scholar] [CrossRef]
- Zhao, Y.; Wei, J.; Hou, X.; Liu, H.; Guo, F.; Zhou, Y.; Zhang, Y.; Qu, Y.; Gu, J.; Zhou, Y.; et al. SIRT1 rs10823108 and FOXO1 rs17446614 responsible for genetic susceptibility to diabetic nephropathy. Sci. Rep. 2017, 7, 10285. [Google Scholar] [CrossRef]
- Hasegawa, K.; Wakino, S.; Simic, P.; Sakamaki, Y.; Minakuchi, H.; Fujimura, K.; Hosoya, K.; Komatsu, M.; Kaneko, Y.; Kanda, T.; et al. Renal tubular Sirt1 attenuates diabetic albuminuria by epigenetically suppressing Claudin-1 overexpression in podocytes. Nat. Med. 2013, 19, 1496–1504. [Google Scholar] [CrossRef] [PubMed]
- Hong, Q.; Zhang, L.; Das, B.; Li, Z.; Liu, B.; Cai, G.; Chen, X.; Chuang, P.Y.; He, J.C.; Lee, K. Increased podocyte Sirtuin-1 function attenuates diabetic kidney injury. Kidney Int. 2018, 93, 1330–1343. [Google Scholar] [CrossRef]
- Perico, L.; Morigi, M.; Rota, C.; Breno, M.; Mele, C.; Noris, M.; Introna, M.; Capelli, C.; Longaretti, L.; Rottoli, D.; et al. Human mesenchymal stromal cells transplanted into mice stimulate renal tubular cells and enhance mitochondrial function. Nat. Commun. 2017, 8, 983. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.X.; Edelstein, M.H.; Gafter, U.; Qiu, L.; Luo, Y.; Dobrinskikh, E.; Lucia, S.; Adorini, L.; D’Agati, V.D.; Levi, J.; et al. G Protein-Coupled Bile Acid Receptor TGR5 Activation Inhibits Kidney Disease in Obesity and Diabetes. J. Am. Soc. Nephrol. 2016, 27, 1362–1378. [Google Scholar] [CrossRef]
- Yi, W.; Xie, X.; Du, M.; Bu, Y.; Wu, N.; Yang, H.; Tian, C.; Xu, F.; Xiang, S.; Zhang, P.; et al. Green Tea Polyphenols Ameliorate the Early Renal Damage Induced by a High-Fat Diet via Ketogenesis/SIRT3 Pathway. Oxid. Med. Cell Longev. 2017, 2017, 9032792. [Google Scholar] [CrossRef]
- Zhuo, L.; Fu, B.; Bai, X.; Zhang, B.; Wu, L.; Cui, J.; Cui, S.; Wei, R.; Chen, X.; Cai, G. NAD blocks high glucose induced mesangial hypertrophy via activation of the sirtuins-AMPK-mTOR pathway. Cell Physiol. Biochem. 2011, 27, 681–690. [Google Scholar] [CrossRef] [PubMed]
- Ogura, Y.; Kitada, M.; Monno, I.; Kanasaki, K.; Watanabe, A.; Koya, D. Renal mitochondrial oxidative stress is enhanced by the reduction of Sirt3 activity, in Zucker diabetic fatty rats. Redox Rep. 2018, 23, 153–159. [Google Scholar] [CrossRef]
- Jiao, X.; Li, Y.; Zhang, T.; Liu, M.; Chi, Y. Role of Sirtuin3 in high glucose-induced apoptosis in renal tubular epithelial cells. Biochem. Biophys. Res. Commun. 2016, 480, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Tran, M.T.; Zsengeller, Z.K.; Berg, A.H.; Khankin, E.V.; Bhasin, M.K.; Kim, W.; Clish, C.B.; Stillman, I.E.; Karumanchi, S.A.; Rhee, E.P.; et al. PGC1alpha drives NAD biosynthesis linking oxidative metabolism to renal protection. Nature 2016, 531, 528–532. [Google Scholar] [CrossRef]
- Kong, X.; Wang, R.; Xue, Y.; Liu, X.; Zhang, H.; Chen, Y.; Fang, F.; Chang, Y. Sirtuin 3, a new target of PGC-1alpha, plays an important role in the suppression of ROS and mitochondrial biogenesis. PLoS ONE 2010, 5, e11707. [Google Scholar] [CrossRef]
- Huang, W.; Liu, H.; Zhu, S.; Woodson, M.; Liu, R.; Tilton, R.G.; Miller, J.D.; Zhang, W. Sirt6 deficiency results in progression of glomerular injury in the kidney. Aging Albany NY 2017, 9, 1069–1083. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.J. Pathogenesis of chronic hyperglycemia: From reductive stress to oxidative stress. J. Diabetes Res. 2014, 2014, 137919. [Google Scholar] [CrossRef]
- Rydstrom, J. Mitochondrial NADPH, transhydrogenase and disease. Biochim. Biophys. Acta 2006, 1757, 721–726. [Google Scholar] [CrossRef]
- Nickel, A.G.; von Hardenberg, A.; Hohl, M.; Loffler, J.R.; Kohlhaas, M.; Becker, J.; Reil, J.C.; Kazakov, A.; Bonnekoh, J.; Stadelmaier, M.; et al. Reversal of Mitochondrial Transhydrogenase Causes Oxidative Stress in Heart Failure. Cell Metab. 2015, 22, 472–484. [Google Scholar] [CrossRef]
- Arkblad, E.L.; Egorov, M.; Shakhparonov, M.; Romanova, L.; Polzikov, M.; Rydstrom, J. Expression of proton-pumping nicotinamide nucleotide transhydrogenase in mouse, human brain and C elegans. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2002, 133, 13–21. [Google Scholar] [CrossRef]
- Meimaridou, E.; Goldsworthy, M.; Chortis, V.; Fragouli, E.; Foster, P.A.; Arlt, W.; Cox, R.; Metherell, L.A. NNT is a key regulator of adrenal redox homeostasis and steroidogenesis in male mice. J. Endocrinol. 2018, 236, 13–28. [Google Scholar] [CrossRef]
- Ronchi, J.A.; Figueira, T.R.; Ravagnani, F.G.; Oliveira, H.C.; Vercesi, A.E.; Castilho, R.F. A spontaneous mutation in the nicotinamide nucleotide transhydrogenase gene of C57BL/6J mice results in mitochondrial redox abnormalities. Free Radical Biol. Med. 2013, 63, 446–456. [Google Scholar] [CrossRef] [PubMed]
- Galvan, D.L.; Long, J.; Green, N.; Chang, B.H.; Lin, J.S.; Schumacker, P.; Truong, L.D.; Overbeek, P.; Danesh, F.R. Drp1S600 phosphorylation regulates mitochondrial fission and progression of nephropathy in diabetic mice. J. Clin. Investig. 2019, 129, 2807–2823. [Google Scholar] [CrossRef]
- Huang, C.; Lin, M.Z.; Cheng, D.; Braet, F.; Pollock, C.A.; Chen, X.M. KCa3.1 mediates dysfunction of tubular autophagy in diabetic kidneys via PI3k/Akt/mTOR signaling pathways. Sci. Rep. 2016, 6, 23884. [Google Scholar] [CrossRef]
- Mourier, A.; Motori, E.; Brandt, T.; Lagouge, M.; Atanassov, I.; Galinier, A.; Rappl, G.; Brodesser, S.; Hultenby, K.; Dieterich, C.; et al. Mitofusin 2 is required to maintain mitochondrial coenzyme Q levels. J. Cell Biol. 2015, 208, 429–442. [Google Scholar] [CrossRef]
- Ayanga, B.A.; Badal, S.S.; Wang, Y.; Galvan, D.L.; Chang, B.H.; Schumacker, P.T.; Danesh, F.R. Dynamin-Related Protein 1 Deficiency Improves Mitochondrial Fitness and Protects against Progression of Diabetic Nephropathy. J. Am. Soc. Nephrol. 2016, 27, 2733–2747. [Google Scholar] [CrossRef]
- Qin, X.; Zhao, Y.; Gong, J.; Huang, W.; Su, H.; Yuan, F.; Fang, K.; Wang, D.; Li, J.; Zou, X.; et al. Berberine Protects Glomerular Podocytes via Inhibiting Drp1-Mediated Mitochondrial Fission and Dysfunction. Theranostics 2019, 9, 1698–1713. [Google Scholar] [CrossRef]
- Lee, W.C.; Chiu, C.H.; Chen, J.B.; Chen, C.H.; Chang, H.W. Mitochondrial Fission Increases Apoptosis and Decreases Autophagy in Renal Proximal Tubular Epithelial Cells Treated with High Glucose. DNA Cell Biol. 2016, 35, 657–665. [Google Scholar] [CrossRef]
- Sheng, J.; Li, H.; Dai, Q.; Lu, C.; Xu, M.; Zhang, J.; Feng, J. NR4A1 Promotes Diabetic Nephropathy by Activating Mff-Mediated Mitochondrial Fission and Suppressing Parkin-Mediated Mitophagy. Cell Physiol. Biochem. 2018, 48, 1675–1693. [Google Scholar] [CrossRef]
- Higgins, G.C.; Coughlan, M.T. Mitochondrial dysfunction and mitophagy: The beginning and end to diabetic nephropathy? Br. J. Pharmacol. 2014, 171, 1917–1942. [Google Scholar] [CrossRef]
- Lazarou, M.; Jin, S.M.; Kane, L.A.; Youle, R.J. Role of PINK1 binding to the TOM complex and alternate intracellular membranes in recruitment and activation of the E3 ligase Parkin. Dev. Cell 2012, 22, 320–333. [Google Scholar] [CrossRef]
- McWilliams, T.G.; Prescott, A.R.; Allen, G.F.; Tamjar, J.; Munson, M.J.; Thomson, C.; Muqit, M.M.; Ganley, I.G. mito-QC illuminates mitophagy and mitochondrial architecture in vivo. J. Cell Biol. 2016, 214, 333–345. [Google Scholar] [CrossRef]
- Hartleben, B.; Godel, M.; Meyer-Schwesinger, C.; Liu, S.; Ulrich, T.; Kobler, S.; Wiech, T.; Grahammer, F.; Arnold, S.J.; Lindenmeyer, M.T.; et al. Autophagy influences glomerular disease susceptibility and maintains podocyte homeostasis in aging mice. J. Clin. Investig. 2010, 120, 1084–1096. [Google Scholar] [CrossRef]
- Huang, C.; Zhang, Y.; Kelly, D.J.; Tan, C.Y.; Gill, A.; Cheng, D.; Braet, F.; Park, J.S.; Sue, C.M.; Pollock, C.A.; et al. Thioredoxin interacting protein (TXNIP) regulates tubular autophagy and mitophagy in diabetic nephropathy through the mTOR signaling pathway. Sci. Rep. 2016, 6, 29196. [Google Scholar] [CrossRef]
- Huang, C.; Lin, M.Z.; Cheng, D.; Braet, F.; Pollock, C.A.; Chen, X.M. Thioredoxin-interacting protein mediates dysfunction of tubular autophagy in diabetic kidneys through inhibiting autophagic flux. Lab. Investig. 2014, 94, 309–320. [Google Scholar] [CrossRef]
- Li, W.; Du, M.; Wang, Q.; Ma, X.; Wu, L.; Guo, F.; Ji, H.; Huang, F.; Qin, G. FoxO1 Promotes Mitophagy in the Podocytes of Diabetic Male Mice via the PINK1/Parkin Pathway. Endocrinology 2017, 158, 2155–2167. [Google Scholar] [CrossRef]
- Tagawa, A.; Yasuda, M.; Kume, S.; Yamahara, K.; Nakazawa, J.; Chin-Kanasaki, M.; Araki, H.; Araki, S.; Koya, D.; Asanuma, K.; et al. Impaired Podocyte Autophagy Exacerbates Proteinuria in Diabetic Nephropathy. Diabetes 2016, 65, 755–767. [Google Scholar] [CrossRef]
- Yamahara, K.; Kume, S.; Koya, D.; Tanaka, Y.; Morita, Y.; Chin-Kanasaki, M.; Araki, H.; Isshiki, K.; Araki, S.; Haneda, M.; et al. Obesity-mediated autophagy insufficiency exacerbates proteinuria-induced tubulointerstitial lesions. J. Am. Soc. Nephrol. 2013, 24, 1769–1781. [Google Scholar] [CrossRef]
- Neumiller, J.J.; Alicic, R.Z.; Tuttle, K.R. Therapeutic Considerations for Antihyperglycemic Agents in Diabetic Kidney Disease. J. Am. Soc. Nephrol. 2017, 28, 2263–2274. [Google Scholar] [CrossRef]
- Lonn, E.; Yusuf, S.; Hoogwerf, B.; Pogue, J.; Yi, Q.; Zinman, B.; Bosch, J.; Dagenais, G.; Mann, J.F.; Gerstein, H.C.; et al. Effects of vitamin E on cardiovascular and microvascular outcomes in high-risk patients with diabetes: Results of the HOPE study and MICRO-HOPE substudy. Diabetes Care 2002, 25, 1919–1927. [Google Scholar] [CrossRef]
- de Zeeuw, D.; Akizawa, T.; Audhya, P.; Bakris, G.L.; Chin, M.; Christ-Schmidt, H.; Goldsberry, A.; Houser, M.; Krauth, M.; Lambers Heerspink, H.J.; et al. Bardoxolone methyl in type 2 diabetes and stage 4 chronic kidney disease. N. Engl. J. Med. 2013, 369, 2492–2503. [Google Scholar] [CrossRef]
- Birk, A.V.; Liu, S.; Soong, Y.; Mills, W.; Singh, P.; Warren, J.D.; Seshan, S.V.; Pardee, J.D.; Szeto, H.H. The mitochondrial-targeted compound SS-31 re-energizes ischemic mitochondria by interacting with cardiolipin. J. Am. Soc. Nephrol. 2013, 24, 1250–1261. [Google Scholar] [CrossRef]
- Hou, Y.; Li, S.; Wu, M.; Wei, J.; Ren, Y.; Du, C.; Wu, H.; Han, C.; Duan, H.; Shi, Y. Mitochondria-targeted peptide SS-31 attenuates renal injury via an antioxidant effect in diabetic nephropathy. Am. J. Physiol. Ren. Physiol. 2016, 310, F547–F559. [Google Scholar] [CrossRef]
- Escribano-Lopez, I.; Diaz-Morales, N.; Iannantuoni, F.; Lopez-Domenech, S.; de Maranon, A.M.; Abad-Jimenez, Z.; Banuls, C.; Rovira-Llopis, S.; Herance, J.R.; Rocha, M.; et al. The mitochondrial antioxidant SS-31 increases SIRT1 levels and ameliorates inflammation, oxidative stress and leukocyte-endothelium interactions in type 2 diabetes. Sci. Rep. 2018, 8, 15862. [Google Scholar] [CrossRef]
- Chacko, B.K.; Reily, C.; Srivastava, A.; Johnson, M.S.; Ye, Y.; Ulasova, E.; Agarwal, A.; Zinn, K.R.; Murphy, M.P.; Kalyanaraman, B.; et al. Prevention of diabetic nephropathy in Ins2(+/)(-)(AkitaJ) mice by the mitochondria-targeted therapy MitoQ. Biochem. J. 2010, 432, 9–19. [Google Scholar] [CrossRef]
- Ward, M.S.; Flemming, N.B.; Gallo, L.A.; Fotheringham, A.K.; McCarthy, D.A.; Zhuang, A.; Tang, P.H.; Borg, D.J.; Shaw, H.; Harvie, B.; et al. Targeted mitochondrial therapy using MitoQ shows equivalent renoprotection to angiotensin converting enzyme inhibition but no combined synergy in diabetes. Sci. Rep. 2017, 7, 15190. [Google Scholar] [CrossRef]
- Gao, P.; Yang, M.; Chen, X.; Xiong, S.; Liu, J.; Sun, L. DsbA-L deficiency exacerbates mitochondrial dysfunction of tubular cells in diabetic kidney disease. Clin. Sci. 2020, 134, 677–694. [Google Scholar] [CrossRef]
- Han, Y.; Xu, X.; Tang, C.; Gao, P.; Chen, X.; Xiong, X.; Yang, M.; Yang, S.; Zhu, X.; Yuan, S.; et al. Reactive oxygen species promote tubular injury in diabetic nephropathy: The role of the mitochondrial ros-txnip-nlrp3 biological axis. Redox Biol. 2018, 16, 32–46. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Chen, J.K.; Harris, R.C. EGF receptor deletion in podocytes attenuates diabetic nephropathy. J. Am. Soc. Nephrol. 2015, 26, 1115–1125. [Google Scholar] [CrossRef]
- Sims, C.R.; MacMillan-Crow, L.A.; Mayeux, P.R. Targeting mitochondrial oxidants may facilitate recovery of renal function during infant sepsis. Clin. Pharmacol. Ther. 2014, 96, 662–664. [Google Scholar] [CrossRef]
- Rogacka, D.; Audzeyenka, I.; Rychlowski, M.; Rachubik, P.; Szrejder, M.; Angielski, S.; Piwkowska, A. Metformin overcomes high glucose-induced insulin resistance of podocytes by pleiotropic effects on SIRT1 and AMPK. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 115–125. [Google Scholar] [CrossRef]
- Eid, A.A.; Ford, B.M.; Block, K.; Kasinath, B.S.; Gorin, Y.; Ghosh-Choudhury, G.; Barnes, J.L.; Abboud, H.E. AMP-activated protein kinase (AMPK) negatively regulates Nox4-dependent activation of p53 and epithelial cell apoptosis in diabetes. J. Biol. Chem. 2010, 285, 37503–37512. [Google Scholar] [CrossRef]
- Lee, M.J.; Feliers, D.; Mariappan, M.M.; Sataranatarajan, K.; Mahimainathan, L.; Musi, N.; Foretz, M.; Viollet, B.; Weinberg, J.M.; Choudhury, G.G.; et al. A role for AMP-activated protein kinase in diabetes-induced renal hypertrophy. Am. J. Physiol. Ren. Physiol. 2007, 292, F617–F627. [Google Scholar] [CrossRef]
- Habib, S.L.; Yadav, A.; Kidane, D.; Weiss, R.H.; Liang, S. Novel protective mechanism of reducing renal cell damage in diabetes: Activation AMPK by AICAR increased NRF2/OGG1 proteins and reduced oxidative DNA damage. Cell Cycle 2016, 15, 3048–3059. [Google Scholar] [CrossRef]
- Szeto, H.H.; Liu, S.; Soong, Y.; Alam, N.; Prusky, G.T.; Seshan, S.V. Protection of mitochondria prevents high-fat diet-induced glomerulopathy and proximal tubular injury. Kidney Int. 2016, 90, 997–1011. [Google Scholar] [CrossRef]
- Kim, Y.; Park, C.W. Adenosine monophosphate-activated protein kinase in diabetic nephropathy. Kidney Res. Clin. Pract. 2016, 35, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.Q.; Wang, Y.; Senitko, M.; Meyer, C.; Wigley, W.C.; Ferguson, D.A.; Grossman, E.; Chen, J.; Zhou, X.J.; Hartono, J.; et al. Bardoxolone methyl (BARD) ameliorates ischemic AKI and increases expression of protective genes Nrf2, PPARgamma, and HO-1. Am. J. Physiol. Ren. Physiol. 2011, 300, F1180–F1192. [Google Scholar] [CrossRef]
- Al-Rasheed, N.M.; Al-Rasheed, N.M.; Al-Amin, M.A.; Hasan, I.H.; Al-Ajmi, H.N.; Mohammad, R.A.; Attia, H.A. Fenofibrate attenuates diabetic nephropathy in experimental diabetic rat’s model via suppression of augmented TGF-beta1/Smad3 signaling pathway. Arch. Physiol. Biochem. 2016, 122, 186–194. [Google Scholar] [CrossRef]
- Hong, Y.A.; Lim, J.H.; Kim, M.Y.; Kim, T.W.; Kim, Y.; Yang, K.S.; Park, H.S.; Choi, S.R.; Chung, S.; Kim, H.W.; et al. Fenofibrate improves renal lipotoxicity through activation of AMPK-PGC-1alpha in db/db mice. PLoS ONE 2014, 9, e96147. [Google Scholar] [CrossRef]
- Kawanami, D.; Utsunomiya, K. Epidemiology of diabetic nephropathy. Nihon Rinsho 2016, 74 (Suppl. 2), 153–157. [Google Scholar] [PubMed]
- Vallorz, E.L.; Blohm-Mangone, K.; Schnellmann, R.G.; Mansour, H.M. Formoterol PLGA-PEG Nanoparticles Induce Mitochondrial Biogenesis in Renal Proximal Tubules. AAPS J. 2021, 23, 88. [Google Scholar] [CrossRef]
- Arif, E.; Solanki, A.K.; Srivastava, P.; Rahman, B.; Fitzgibbon, W.R.; Deng, P.; Budisavljevic, M.N.; Baicu, C.F.; Zile, M.R.; Megyesi, J.; et al. Mitochondrial biogenesis induced by the beta2-adrenergic receptor agonist formoterol accelerates podocyte recovery from glomerular injury. Kidney Int. 2019, 96, 656–673. [Google Scholar] [CrossRef]
- Cameron, R.B.; Peterson, Y.K.; Beeson, C.C.; Schnellmann, R.G. Structural and pharmacological basis for the induction of mitochondrial biogenesis by formoterol but not clenbuterol. Sci. Rep. 2017, 7, 10578. [Google Scholar] [CrossRef]
- Akhtar, S.; Siragy, H.M. Pro-renin receptor suppresses mitochondrial biogenesis and function via AMPK/SIRT-1/ PGC-1alpha pathway in diabetic kidney. PLoS ONE 2019, 14, e0225728. [Google Scholar] [CrossRef]
- Yasuda, I.; Hasegawa, K.; Sakamaki, Y.; Muraoka, H.; Kawaguchi, T.; Kusahana, E.; Ono, T.; Kanda, T.; Tokuyama, H.; Wakino, S.; et al. Pre-emptive Short-term Nicotinamide Mononucleotide Treatment in a Mouse Model of Diabetic Nephropathy. J. Am. Soc. Nephrol. 2021, 32, 1355–1370. [Google Scholar] [CrossRef]
- Benito-Martin, A.; Ucero, A.C.; Izquierdo, M.C.; Santamaria, B.; Picatoste, B.; Carrasco, S.; Lorenzo, O.; Ruiz-Ortega, M.; Egido, J.; Ortiz, A. Endogenous NAMPT dampens chemokine expression and apoptotic responses in stressed tubular cells. Biochim. Biophys. Acta 2014, 1842, 293–303. [Google Scholar] [CrossRef]
- Kourtzidis, I.A.; Stoupas, A.T.; Gioris, I.S.; Veskoukis, A.S.; Margaritelis, N.V.; Tsantarliotou, M.; Taitzoglou, I.; Vrabas, I.S.; Paschalis, V.; Kyparos, A.; et al. The NAD(+) precursor nicotinamide riboside decreases exercise performance in rats. J. Int. Soc. Sports Nutr. 2016, 13, 32. [Google Scholar] [CrossRef] [PubMed]
- Kourtzidis, I.A.; Dolopikou, C.F.; Tsiftsis, A.N.; Margaritelis, N.V.; Theodorou, A.A.; Zervos, I.A.; Tsantarliotou, M.P.; Veskoukis, A.S.; Vrabas, I.S.; Paschalis, V.; et al. Nicotinamide riboside supplementation dysregulates redox and energy metabolism in rats: Implications for exercise performance. Exp. Physiol. 2018, 103, 1357–1366. [Google Scholar] [CrossRef] [PubMed]
- Simic, P.; Vela Parada, X.F.; Parikh, S.M.; Dellinger, R.; Guarente, L.P.; Rhee, E.P. Nicotinamide riboside with pterostilbene (NRPT) increases NAD(+) in patients with acute kidney injury (AKI): A randomized, double-blind, placebo-controlled, stepwise safety study of escalating doses of NRPT in patients with AKI. BMC Nephrol. 2020, 21, 342. [Google Scholar] [CrossRef]
- Estrela, G.R.; Wasinski, F.; Batista, R.O.; Hiyane, M.I.; Felizardo, R.J.; Cunha, F.; de Almeida, D.C.; Malheiros, D.M.; Camara, N.O.; Barros, C.C.; et al. Caloric Restriction Is More Efficient than Physical Exercise to Protect from Cisplatin Nephrotoxicity via PPAR-Alpha Activation. Front. Physiol. 2017, 8, 116. [Google Scholar] [CrossRef]
- Ning, Y.C.; Cai, G.Y.; Zhuo, L.; Gao, J.J.; Dong, D.; Cui, S.Y.; Shi, S.Z.; Feng, Z.; Zhang, L.; Sun, X.F.; et al. Beneficial effects of short-term calorie restriction against cisplatin-induced acute renal injury in aged rats. Nephron Exp. Nephrol. 2013, 124, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Tikoo, K.; Lodea, S.; Karpe, P.A.; Kumar, S. Calorie restriction mimicking effects of roflumilast prevents diabetic nephropathy. Biochem. Biophys. Res. Commun. 2014, 450, 1581–1586. [Google Scholar] [CrossRef] [PubMed]
- Kitada, M.; Takeda, A.; Nagai, T.; Ito, H.; Kanasaki, K.; Koya, D. Dietary restriction ameliorates diabetic nephropathy through anti-inflammatory effects and regulation of the autophagy via restoration of Sirt1 in diabetic Wistar fatty (fa/fa) rats: A model of type 2 diabetes. Exp. Diabetes Res. 2011, 2011, 908185. [Google Scholar] [CrossRef]
- Wang, F.; Nguyen, M.; Qin, F.X.; Tong, Q. SIRT2 deacetylates FOXO3a in response to oxidative stress and caloric restriction. Aging Cell 2007, 6, 505–514. [Google Scholar] [CrossRef]
- Qiu, X.; Brown, K.; Hirschey, M.D.; Verdin, E.; Chen, D. Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell Metab. 2010, 12, 662–667. [Google Scholar] [CrossRef] [PubMed]
- Koyama, T.; Kume, S.; Koya, D.; Araki, S.; Isshiki, K.; Chin-Kanasaki, M.; Sugimoto, T.; Haneda, M.; Sugaya, T.; Kashiwagi, A.; et al. SIRT3 attenuates palmitate-induced ROS production and inflammation in proximal tubular cells. Free Radic. Biol. Med. 2011, 51, 1258–1267. [Google Scholar] [CrossRef]
- Zhang, T.; Chi, Y.; Kang, Y.; Lu, H.; Niu, H.; Liu, W.; Li, Y. Resveratrol ameliorates podocyte damage in diabetic mice via SIRT1/PGC-1alpha mediated attenuation of mitochondrial oxidative stress. J. Cell Physiol. 2019, 234, 5033–5043. [Google Scholar] [CrossRef]
- Zhang, T.; Chi, Y.; Ren, Y.; Du, C.; Shi, Y.; Li, Y. Resveratrol Reduces Oxidative Stress and Apoptosis in Podocytes via Sir2-Related Enzymes, Sirtuins1 (SIRT1)/Peroxisome Proliferator-Activated Receptor gamma Co-Activator 1alpha (PGC-1alpha) Axis. Med. Sci. Monit. 2019, 25, 1220–1231. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Deng, Q.; Zhang, J.; Ke, J.; Zhu, Y.; Wen, R.W.; Ye, Z.; Peng, H.; Su, Z.Z.; Wang, C.; et al. Activation of the Nrf2-ARE Pathway Ameliorates Hyperglycemia-Mediated Mitochondrial Dysfunction in Podocytes Partly Through Sirt1. Cell Physiol. Biochem. 2018, 48, 1–15. [Google Scholar] [CrossRef]
- Qi, Y.Y.; Zhou, X.J.; Cheng, F.J.; Hou, P.; Ren, Y.L.; Wang, S.X.; Zhao, M.H.; Yang, L.; Martinez, J.; Zhang, H. Increased autophagy is cytoprotective against podocyte injury induced by antibody and interferon-alpha in lupus nephritis. Ann. Rheum. Dis. 2018, 77, 1799–1809. [Google Scholar] [CrossRef] [PubMed]
- Ren, H.; Shao, Y.; Wu, C.; Ma, X.; Lv, C.; Wang, Q. Metformin alleviates oxidative stress and enhances autophagy in diabetic kidney disease via AMPK/SIRT1-FoxO1 pathway. Mol. Cell Endocrinol. 2020, 500, 110628. [Google Scholar] [CrossRef]
- Umino, H.; Hasegawa, K.; Minakuchi, H.; Muraoka, H.; Kawaguchi, T.; Kanda, T.; Tokuyama, H.; Wakino, S.; Itoh, H. High Basolateral Glucose Increases Sodium-Glucose Cotransporter 2 and Reduces Sirtuin-1 in Renal Tubules through Glucose Transporter-2 Detection. Sci. Rep. 2018, 8, 6791. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Liu, H.; Takagi, S.; Nitta, K.; Kitada, M.; Srivastava, S.P.; Takagaki, Y.; Kanasaki, K.; Koya, D. Renal protective effects of empagliflozin via inhibition of EMT and aberrant glycolysis in proximal tubules. JCI Insight 2020, 5. [Google Scholar] [CrossRef]
- Peixoto, E.B.; Papadimitriou, A.; Lopes de Faria, J.M.; Lopes de Faria, J.B. Tempol reduces podocyte apoptosis via PARP signaling pathway in experimental diabetes mellitus. Nephron Exp. Nephrol. 2012, 120, e81–e90. [Google Scholar] [CrossRef]
- Szabo, C.; Biser, A.; Benko, R.; Bottinger, E.; Susztak, K. Poly(ADP-ribose) polymerase inhibitors ameliorate nephropathy of type 2 diabetic Leprdb/db mice. Diabetes 2006, 55, 3004–3012. [Google Scholar] [CrossRef]
- Chiu, J.; Xu, B.Y.; Chen, S.; Feng, B.; Chakrabarti, S. Oxidative stress-induced, poly(ADP-ribose) polymerase-dependent upregulation of ET-1 expression in chronic diabetic complications. Can. J. Physiol. Pharmacol. 2008, 86, 365–372. [Google Scholar] [CrossRef]
- Xu, B.; Chiu, J.; Feng, B.; Chen, S.; Chakrabarti, S. PARP activation and the alteration of vasoactive factors and extracellular matrix protein in retina and kidney in diabetes. Diabetes Metab. Res. Rev. 2008, 24, 404–412. [Google Scholar] [CrossRef] [PubMed]
- Tarrago, M.G.; Chini, C.C.S.; Kanamori, K.S.; Warner, G.M.; Caride, A.; de Oliveira, G.C.; Rud, M.; Samani, A.; Hein, K.Z.; Huang, R.; et al. A Potent and Specific CD38 Inhibitor Ameliorates Age-Related Metabolic Dysfunction by Reversing Tissue NAD(+) Decline. Cell Metab. 2018, 27, 1081–1095. [Google Scholar] [CrossRef]
- Camacho-Pereira, J.; Tarrago, M.G.; Chini, C.C.S.; Nin, V.; Escande, C.; Warner, G.M.; Puranik, A.S.; Schoon, R.A.; Reid, J.M.; Galina, A.; et al. CD38 Dictates Age-Related NAD Decline and Mitochondrial Dysfunction through an SIRT3-Dependent Mechanism. Cell Metab. 2016, 23, 1127–1139. [Google Scholar] [CrossRef]
- Malik, S.; Suchal, K.; Khan, S.I.; Bhatia, J.; Kishore, K.; Dinda, A.K.; Arya, D.S. Apigenin ameliorates streptozotocin-induced diabetic nephropathy in rats via MAPK-NF-kappaB-TNF-alpha and TGF-beta1-MAPK-fibronectin pathways. Am. J. Physiol. Ren. Physiol. 2017, 313, F414–F422. [Google Scholar] [CrossRef]
- Zhang, J.; Zhao, X.; Zhu, H.; Wang, J.; Ma, J.; Gu, M. Apigenin Protects Against Renal Tubular Epithelial Cell Injury and Oxidative Stress by High Glucose via Regulation of NF-E2-Related Factor 2 (Nrf2) Pathway. Med. Sci. Monit. 2019, 25, 5280–5288. [Google Scholar] [CrossRef]
- Vallon, V.; Thomson, S.C. Targeting renal glucose reabsorption to treat hyperglycaemia: The pleiotropic effects of SGLT2 inhibition. Diabetologia 2017, 60, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Wanner, C.; Inzucchi, S.E.; Lachin, J.M.; Fitchett, D.; von Eynatten, M.; Mattheus, M.; Johansen, O.E.; Woerle, H.J.; Broedl, U.C.; Zinman, B.; et al. Empagliflozin and Progression of Kidney Disease in Type 2 Diabetes. N. Engl. J. Med. 2016, 375, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Zelniker, T.A.; Wiviott, S.D.; Raz, I.; Sabatine, M.S. SGLT-2 inhibitors for people with type 2 diabetes–Authors’ reply. Lancet 2019, 394, 560–561. [Google Scholar] [CrossRef]
- Heerspink, H.J.; Desai, M.; Jardine, M.; Balis, D.; Meininger, G.; Perkovic, V. Canagliflozin Slows Progression of Renal Function Decline Independently of Glycemic Effects. J. Am. Soc. Nephrol. 2017, 28, 368–375. [Google Scholar] [CrossRef]
- Sa-Nguanmoo, P.; Tanajak, P.; Kerdphoo, S.; Jaiwongkam, T.; Pratchayasakul, W.; Chattipakorn, N.; Chattipakorn, S.C. SGLT2-inhibitor and DPP-4 inhibitor improve brain function via attenuating mitochondrial dysfunction, insulin resistance, inflammation, and apoptosis in HFD-induced obese rats. Toxicol. Appl. Pharmacol. 2017, 333, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Durak, A.; Olgar, Y.; Degirmenci, S.; Akkus, E.; Tuncay, E.; Turan, B. A SGLT2 inhibitor dapagliflozin suppresses prolonged ventricular-repolarization through augmentation of mitochondrial function in insulin-resistant metabolic syndrome rats. Cardiovasc. Diabetol. 2018, 17, 144. [Google Scholar] [CrossRef]
- Yang, X.; Liu, Q.; Li, Y.; Tang, Q.; Wu, T.; Chen, L.; Pu, S.; Zhao, Y.; Zhang, G.; Huang, C.; et al. The diabetes medication canagliflozin promotes mitochondrial remodelling of adipocyte via the AMPK-Sirt1-Pgc-1alpha signalling pathway. Adipocyte 2020, 9, 484–494. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Kim, S.H.; Kang, J.M.; Heo, J.H.; Kim, D.J.; Park, S.H.; Sung, M.; Kim, J.; Oh, J.; Yang, D.H.; et al. Empagliflozin attenuates diabetic tubulopathy by improving mitochondrial fragmentation and autophagy. Am. J. Physiol. Ren. Physiol. 2019, 317, F767–F780. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmad, A.A.; Draves, S.O.; Rosca, M. Mitochondria in Diabetic Kidney Disease. Cells 2021, 10, 2945. https://doi.org/10.3390/cells10112945
Ahmad AA, Draves SO, Rosca M. Mitochondria in Diabetic Kidney Disease. Cells. 2021; 10(11):2945. https://doi.org/10.3390/cells10112945
Chicago/Turabian StyleAhmad, Amna Ayesha, Shayna Odeal Draves, and Mariana Rosca. 2021. "Mitochondria in Diabetic Kidney Disease" Cells 10, no. 11: 2945. https://doi.org/10.3390/cells10112945
APA StyleAhmad, A. A., Draves, S. O., & Rosca, M. (2021). Mitochondria in Diabetic Kidney Disease. Cells, 10(11), 2945. https://doi.org/10.3390/cells10112945