
Dynamic Regulation of GH–IGF1 Signaling in Injury and Recovery in Hyperoxia-Induced Neonatal Lung Injury

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Studies
2.1.1. Experimental Setting for Hyperoxia-Induced Lung Injury
2.1.2. Physiological Data of the Offspring
2.1.3. Tissue Preparation and Postmortem Processing of the Lungs
2.1.4. Quantitative Histomorphometric Analysis of Lung Sections
2.2. Cell Culture Experiments
2.2.1. Murine Alveolar Epithelial Cells Type II (MLE-12)
2.2.2. Murine Primary Lung Fibroblasts (pLFs)
2.2.3. Rat Primary Neonatal Lung Myofibroblasts (pnFs)
2.2.4. Exposure of MLE-12 and pLFs to HYX
2.2.5. Treatment with GH, IGF1, and IGF1-R Inhibitor
2.2.6. siRNA Transfection Experiments
2.2.7. Viability Assay
2.2.8. Proliferation Assays (Bromodeoxyuridine (BrdU) Incorporation Assay)
2.3. Enzyme-Linked Immunosorbent Assay (ELISA)
2.3.1. Immunoblotting
2.3.2. Immunofluorescent Staining
2.3.3. RNA Extraction, Real-Time RT-PCR
2.4. Statistics
3. Results
3.1. HYX Induces an Arrest of Alveolarization That Persists in Adulthood
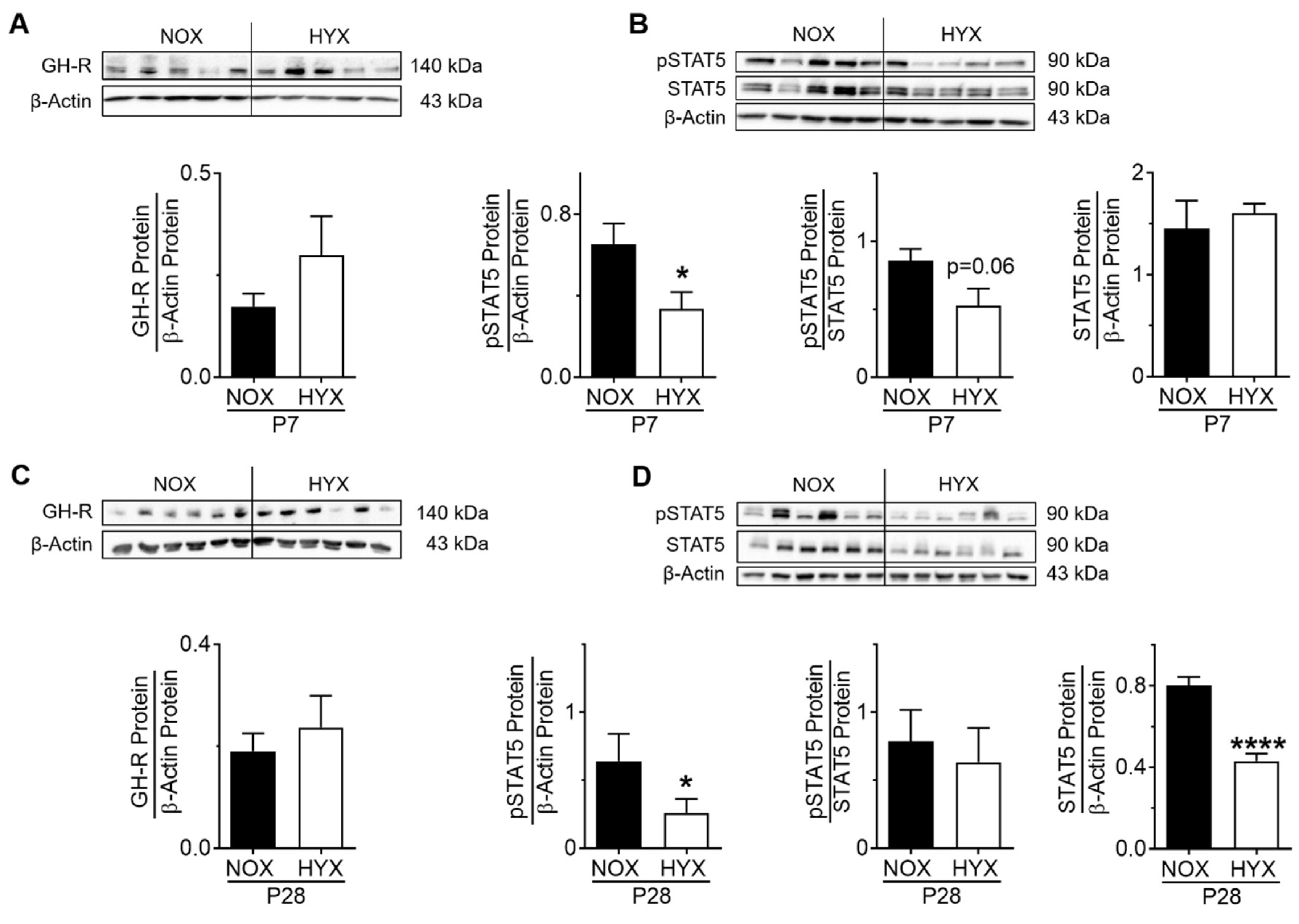
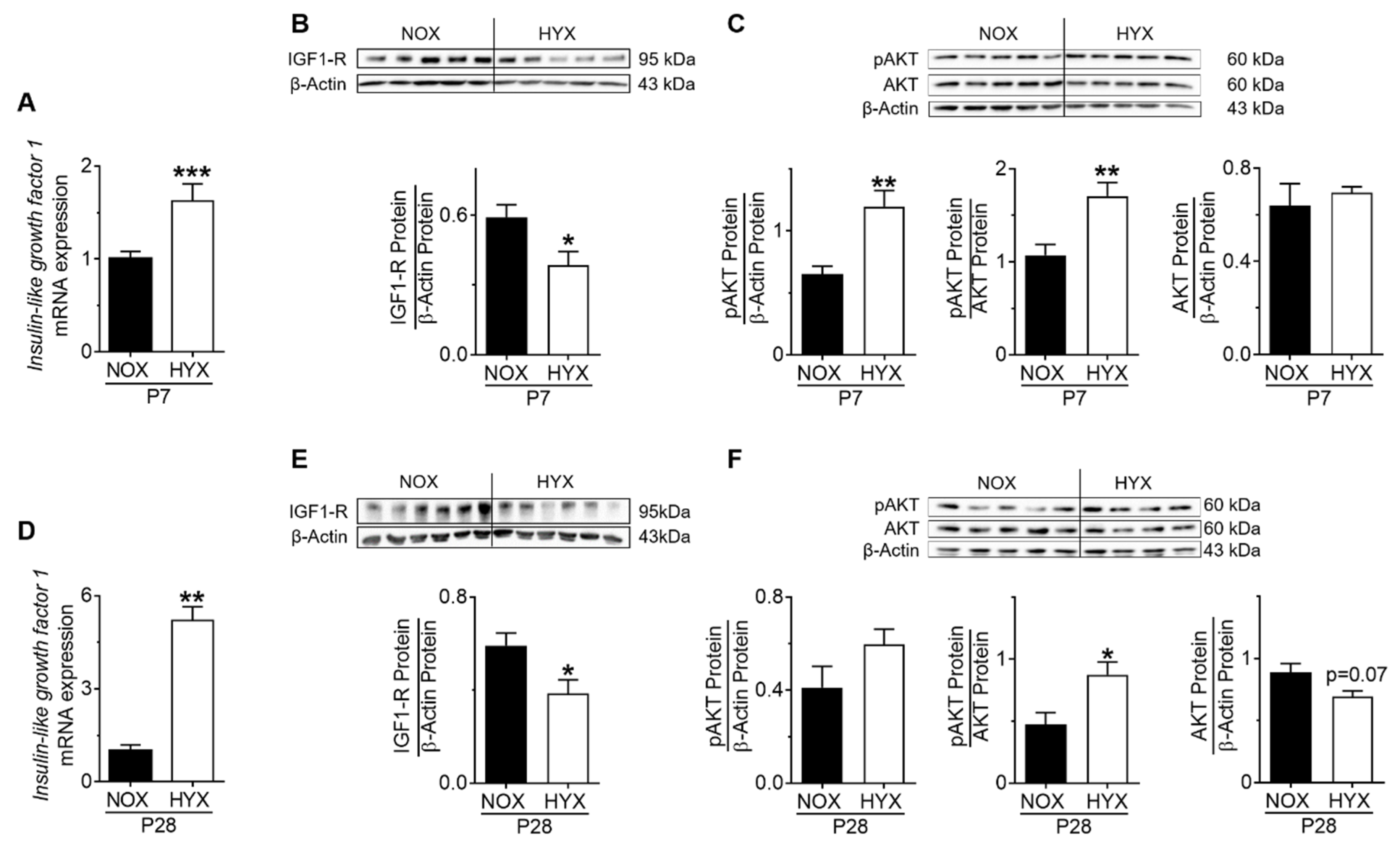
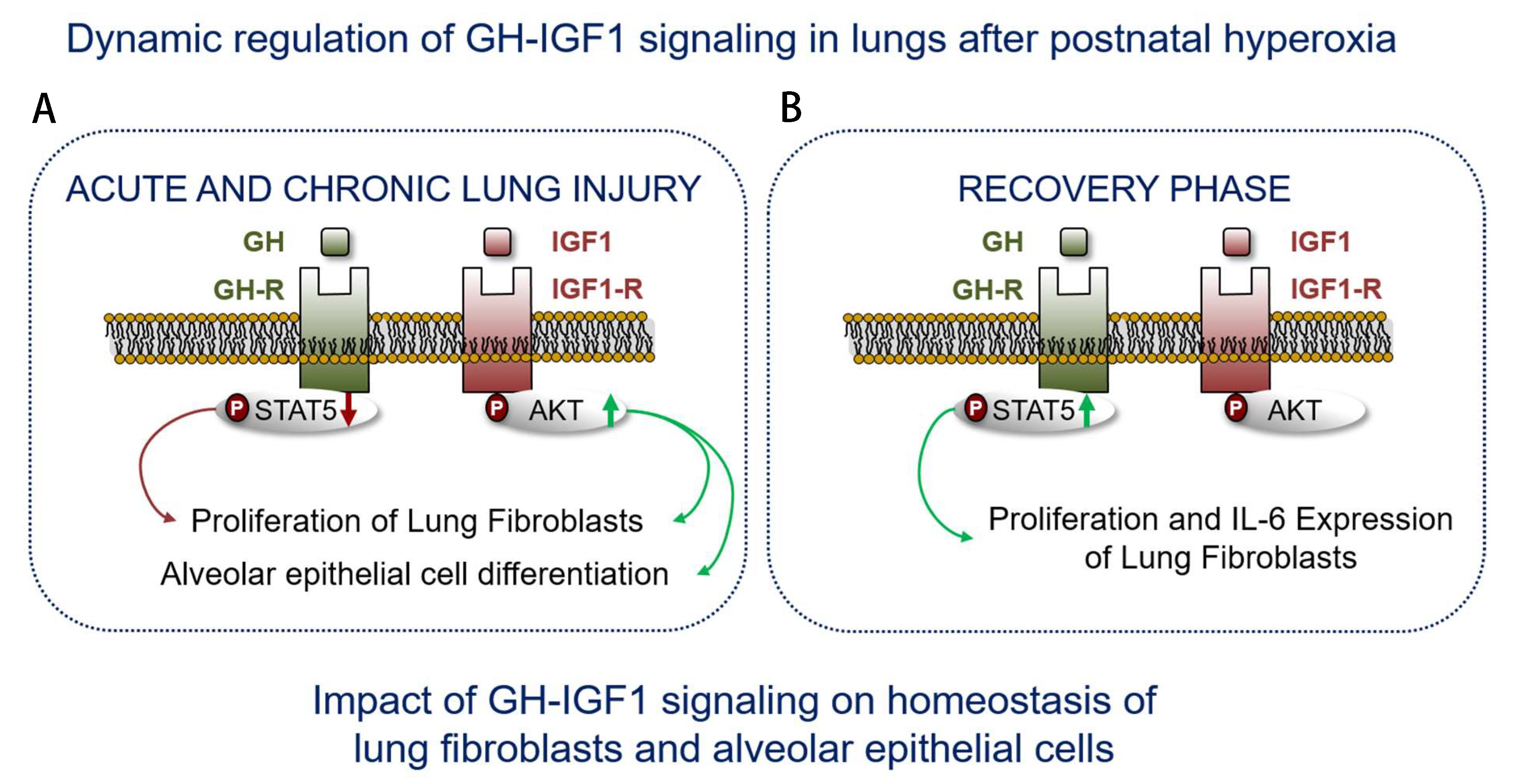
3.2. HYX Dynamically Regulates the GH–IGF1 Axis during Acute and Chronic Lung Injury
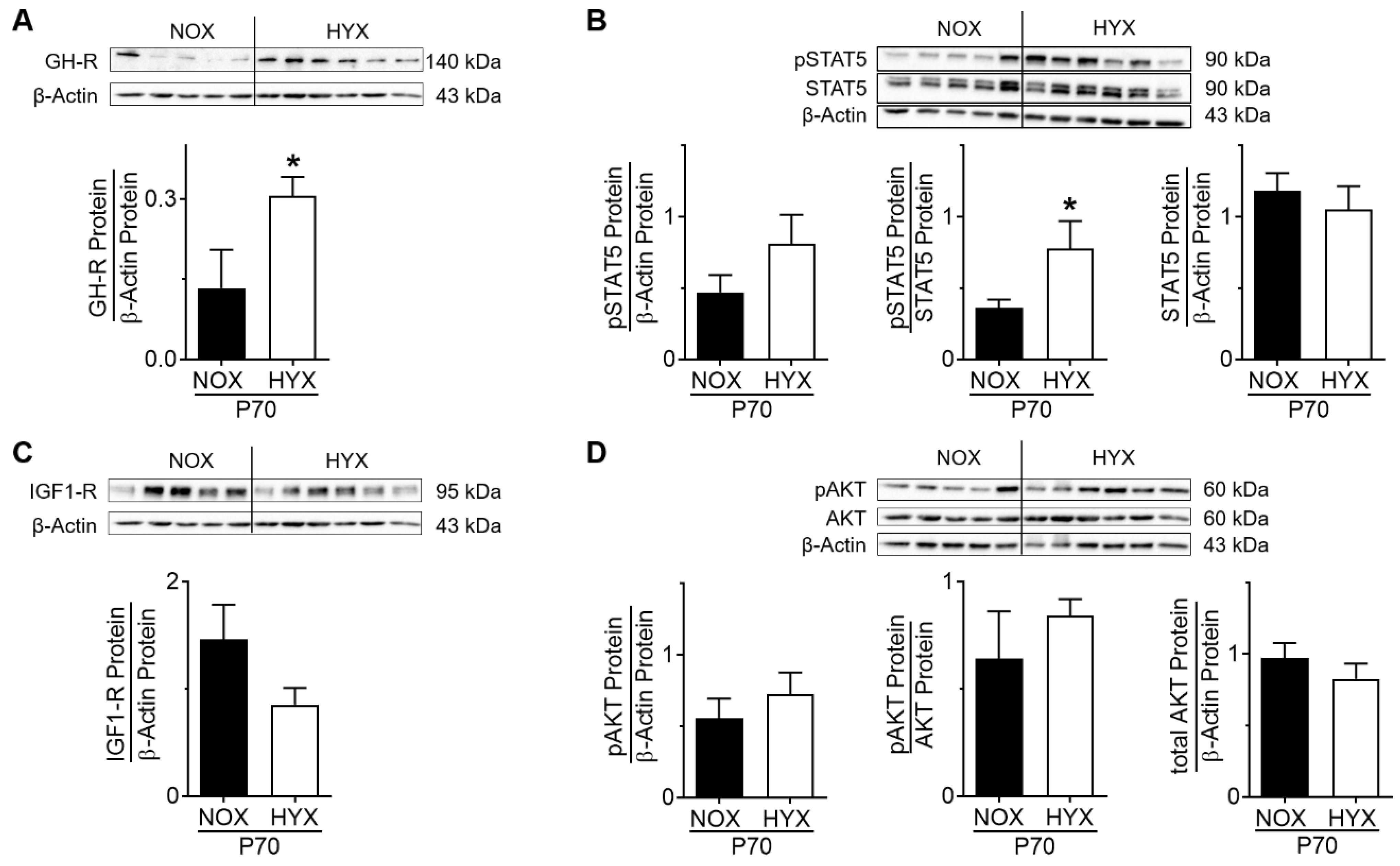
3.3. HYX Regulates the GH–IGF1 Axis during the Regenerative Phase of Lung Injury
3.4. Dysregulation of Lung-Intrinsic GH–IGF1 Signaling Is Associated with Proliferation of αSMA+ Cells, Indicative of Myofibroblasts
3.5. Alveolar Epithelial Cell Markers Are Dysregulated after HYX in a Time-Dependent Manner
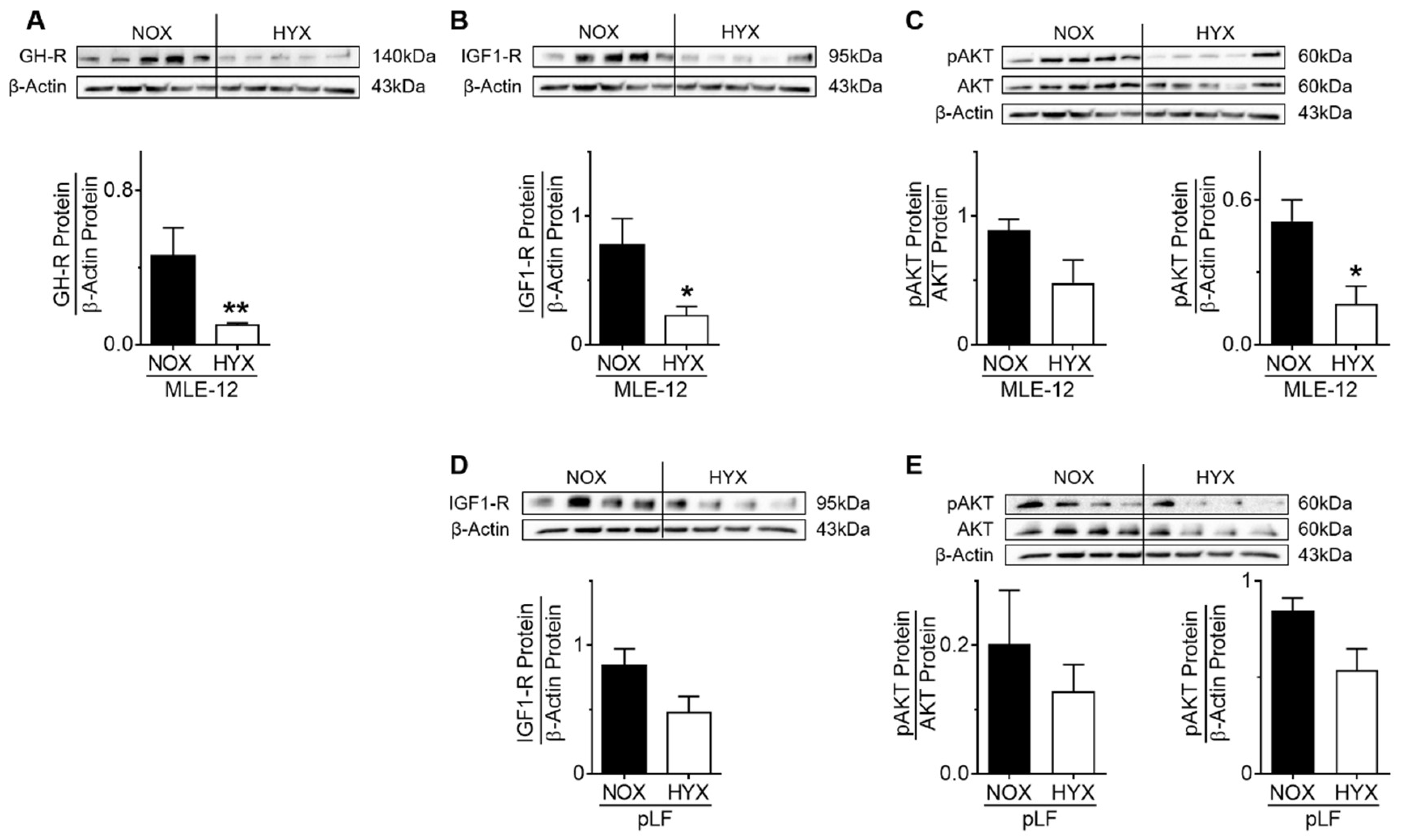
3.6. HYX Regulates GH–IGF1 Signaling in MLE-12
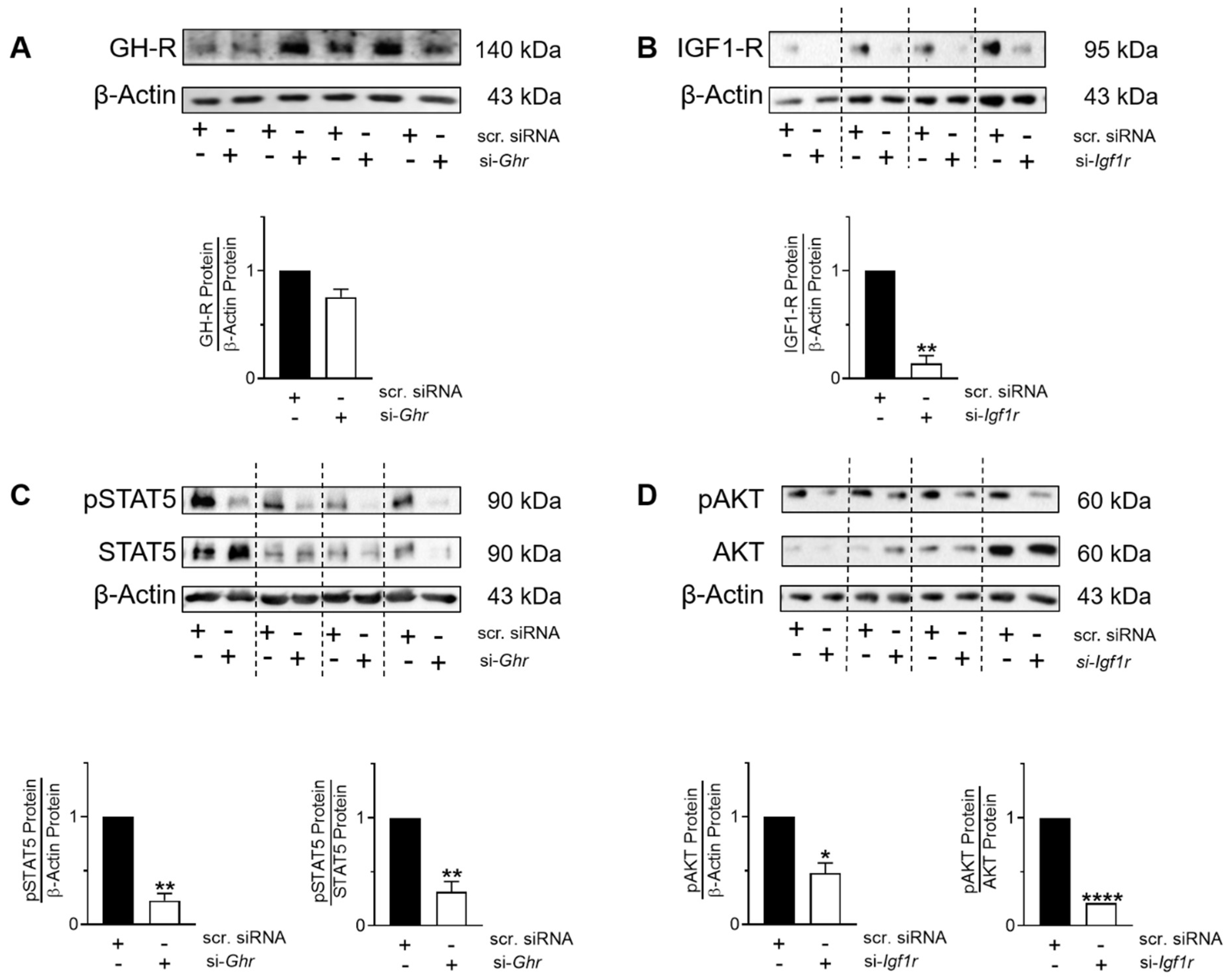
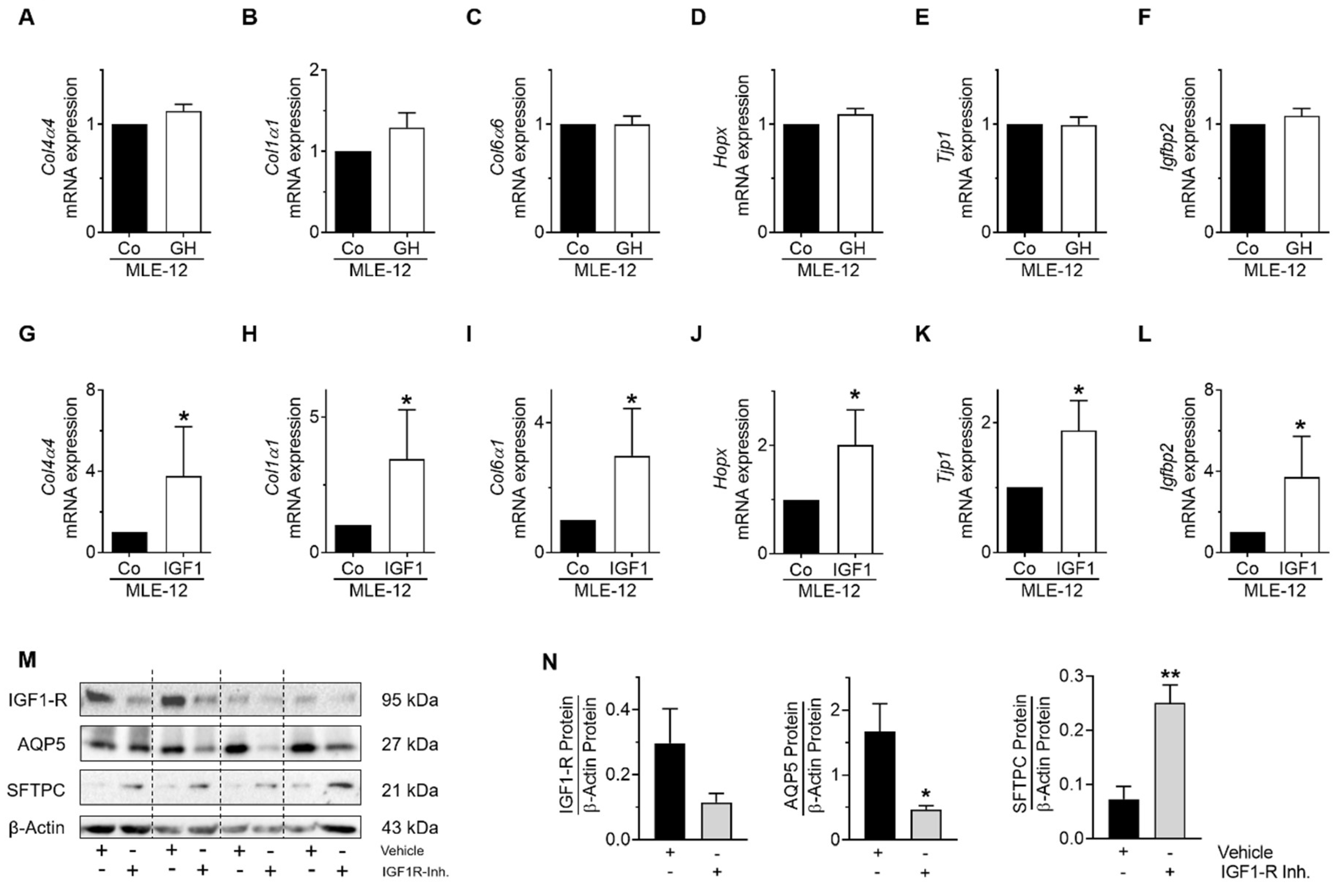
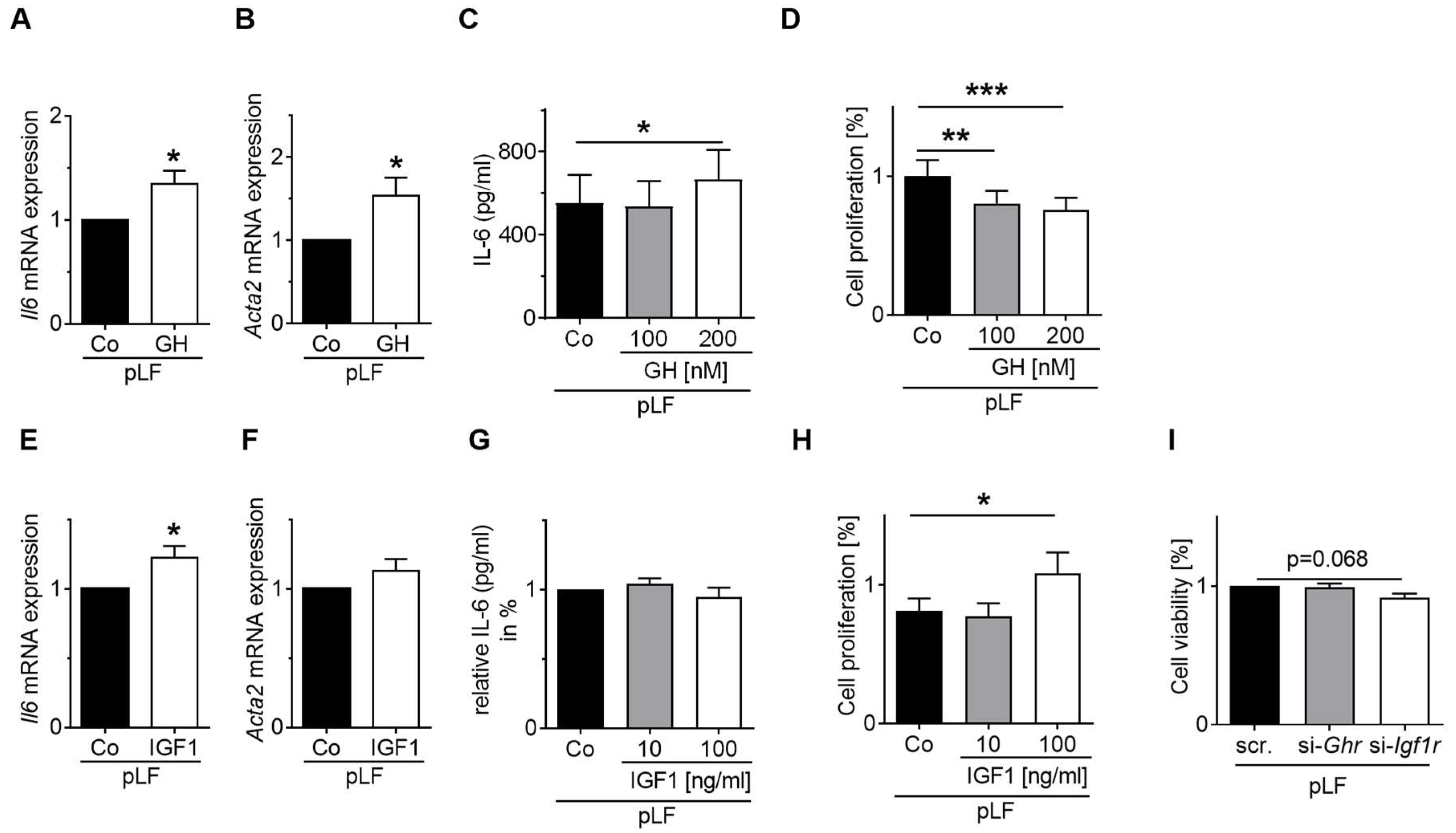
3.7. GH-R and IGF1-R Regulate STAT5 and AKT Signaling as Well as Gene Expression and Homeostasis of MLE-12 and pLFs
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Thebaud, B.; Goss, K.N.; Laughon, M.; Whitsett, J.A.; Abman, S.H.; Steinhorn, R.H.; Aschner, J.L.; Davis, P.G.; McGrath-Morrow, S.A.; Soll, R.F.; et al. Bronchopulmonary dysplasia. Nat. Rev. Dis. Primers 2019, 5, 78. [Google Scholar] [CrossRef] [PubMed]
- Jobe, A.H.; Abman, S.H. Bronchopulmonary Dysplasia: A Continuum of Lung Disease from the Fetus to the Adult. Am. J. Respir. Crit. Care Med. 2019, 200, 659–660. [Google Scholar] [CrossRef] [PubMed]
- Lignelli, E.; Palumbo, F.; Myti, D.; Morty, R.E. Recent advances in our understanding of the mechanisms of lung alveolarization and bronchopulmonary dysplasia. Am. J. Physiol. Cell. Mol. Physiol. 2019, 317, L832–L887. [Google Scholar] [CrossRef]
- Nawabi, J.; Vohlen, C.; Dinger, K.; Thangaratnarajah, C.; Klaudt, C.; Garcia, E.L.; Hirani, D.V.; Haznedar-Karakaya, P.H.; Macheleidt, I.; Odenthal, M.; et al. Novel functional role of GH/IGF-I in neonatal lung myofibroblasts and in rat lung growth after intrauterine growth restriction. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 315, L623–L637. [Google Scholar] [CrossRef] [PubMed]
- Iioka, Y.; Tatsumi, K.; Sugito, K.; Moriya, T.; Kuriyama, T. Effects of Insulin-like Growth Factor on Weight Gain in Chronic Hypoxic Rats. J. Cardiovasc. Pharmacol. 2002, 39, 636–642. [Google Scholar] [CrossRef]
- Wang, Z.; Li, W.; Guo, Q.; Wang, Y.; Ma, L.; Zhang, X. Insulin-Like Growth Factor-1 Signaling in Lung Development and Inflammatory Lung Diseases. BioMed Res. Int. 2018, 2018, 6057589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, H.; Snowball, J.; Sun, F.; Na, C.-L.; Whitsett, J.A. IGF1R controls mechanosignaling in myofibroblasts required for pulmonary alveologenesis. JCI Insight 2021, 6, e144863. [Google Scholar] [CrossRef]
- Epaud, R.; Aubey, F.; Xu, J.; Chaker, Z.; Clemessy, M.; Dautin, A.; Ahmed, K.; Bonora, M.; Hoyeau, N.; Flejou, J.-F.; et al. Knockout of insulin-like growth factor-1 receptor impairs distal lung morphogenesis. PLoS ONE 2012, 7, e48071. [Google Scholar] [CrossRef] [Green Version]
- Parimon, T.; Yao, C.; Stripp, B.R.; Noble, P.W.; Chen, P. Alveolar Epithelial Type II Cells as Drivers of Lung Fibrosis in Idiopathic Pulmonary Fibrosis. Int. J. Mol. Sci. 2020, 21, 2269. [Google Scholar] [CrossRef] [Green Version]
- Goldstein, R.H.; Poliks, C.F.; Pilch, P.F.; Smith, B.D.; Fine, A. Stimulation of collagen formation by insulin and insulin-like growth factor I in cultures of human lung fibroblasts. Endocrinology 1989, 124, 964–970. [Google Scholar] [CrossRef]
- Warnken, M.; Reitzenstein, U.; Sommer, A.; Fuhrmann, M.; Mayer, P.; Enzmann, H.; Juergens, U.R.; Racke, K. Characterization of proliferative effects of insulin, insulin analogues and insulin-like growth factor-1 (IGF-1) in human lung fibroblasts. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2010, 382, 511–524. [Google Scholar] [CrossRef] [PubMed]
- Chetty, A.; Cao, G.-J.; Nielsen, H.C. Insulin-like Growth Factor-I signaling mechanisms, type I collagen and alpha smooth muscle actin in human fetal lung fibroblasts. Pediatr. Res. 2006, 60, 389–394. [Google Scholar] [CrossRef] [Green Version]
- Seedorf, G.; Kim, C.; Wallace, B.; Mandell, E.W.; Nowlin, T.; Shepherd, D.; Abman, S.H. rhIGF-1/BP3 Preserves Lung Growth and Prevents Pulmonary Hypertension in Experimental Bronchopulmonary Dysplasia. Am. J. Respir Crit. Care Med. 2020, 201, 1120–1134. [Google Scholar] [CrossRef]
- Alejandre Alcazar, M.A.; Kwapiszewska, G.; Reiss, I.; Amarie, O.V.; Marsh, L.M.; Sevilla-Perez, J.; Wygrecka, M.; Eul, B.; Kobrich, S.; Hesse, M.; et al. Hyperoxia modulates TGF-beta/BMP signaling in a mouse model of bronchopulmonary dysplasia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 292, L537–L549. [Google Scholar] [CrossRef]
- Will, J.P.; Hirani, D.; Thielen, F.; Klein, F.; Vohlen, C.; Dinger, K.; Dotsch, J.; Alejandre Alcazar, M.A. Strain-dependent effects on lung structure, matrix remodeling, and Stat3/Smad2 signaling in C57BL/6N and C57BL/6J mice after neonatal hyperoxia. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2019, 317, R169–R181. [Google Scholar] [CrossRef]
- Thangaratnarajah, C.; Dinger, K.; Vohlen, C.; Klaudt, C.; Nawabi, J.; Lopez Garcia, E.; Kwapiszewska, G.; Dobner, J.; Nuesken, K.D.; van Koningsbruggen-Rietschel, S.; et al. Novel role of NPY in neuro-immune interaction and lung growth after intrauterine growth restriction. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 313, L491–L506. [Google Scholar] [CrossRef] [Green Version]
- Mohr, J.; Voggel, J.; Vohlen, C.; Dinger, K.; Dafinger, C.; Fink, G.; Göbel, H.; Liebau, M.C.; Doötsch, J.; Alejandre Alcazar, M.A. IL-6/Smad2 signaling mediates acute kidney injury and regeneration in a murine model of neonatal hyperoxia. FASEB J. 2019, 33, 5887–5902. [Google Scholar] [CrossRef] [PubMed]
- Hirani, D.; Alvira, C.M.; Danopoulos, S.; Milla, C.; Donato, M.; Tian, L.; Mohr, J.; Dinger, K.; Vohlen, C.; Selle, J.; et al. Macrophage-derived IL-6 trans-signaling as a novel target in the pathogenesis of bronchopulmonary dysplasia. Eur. Respir. J. 2021, 58, 2101657. [Google Scholar]
- Day, C.L.; Ryan, R.M. Bronchopulmonary dysplasia: New becomes old again! Pediatr. Res. 2017, 81, 210–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nardiello, C.; Mižíková, I.; Silva, D.M.; Ruiz-Camp, J.; Mayer, K.; Vadász, I.; Herold, S.; Seeger, W.; Morty, R.E. Standardisation of oxygen exposure in the development of mouse models for bronchopulmonary dysplasia. Dis. Models Mech. 2017, 10, 185–196. [Google Scholar] [CrossRef] [Green Version]
- Martinez, C.S.; Piazza, V.G.; Ratner, L.D.; Matos, M.N.; Gonzaález, L.; Rulli, S.B.; Miquet, J.G.; Sotelo, A.I. Growth hormone STAT5-mediated signaling and its modulation in mice liver during the growth period. Growth Horm. IGF Res. 2013, 23, 19–28. [Google Scholar] [CrossRef]
- Sanders, E.J.; Harvey, S. Growth hormone as an early embryonic growth and differentiation factor. Anat. Embryiol. 2004, 209, 1–9. [Google Scholar] [CrossRef]
- Perrini, S.; Laviola, L.; Carreira, M.C.; Cignarelli, A.; Natalicchio, A.; Giorgino, F. The GH/IGF1 axis and signaling pathways in the muscle and bone: Mechanisms underlying age-related skeletal muscle wasting and osteoporosis. J. Endocrinol. 2010, 205, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Beyea, J.A.; Olson, D.M.; Harvey, S. Growth hormone (GH) action in the developing lung: Changes in lung proteins after adenoviral GH overexpression. Dev. Dyn. 2005, 234, 404–412. [Google Scholar] [CrossRef]
- Beyea, J.A.; Olson, D.M.; Harvey, S. Growth hormone-dependent changes in the rat lung proteome during alveorization. Mol. Cell. Biochem. 2009, 321, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Zvonic, S.; Story, D.J.; Stephens, J.M.; Mynatt, R.L. Growth hormone, but not insulin, activates STAT5 proteins in adipocytes in vitro and in vivo. Biochem. Biophys. Res. Commun. 2003, 302, 359–362. [Google Scholar] [CrossRef]
- Barclay, J.L.; Nelson, C.N.; Ishikawa, M.; Murray, L.A.; Kerr, L.M.; McPhee, T.R.; Powell, E.E.; Waters, M.J. GH-Dependent STAT5 Signaling Plays an Important Role in Hepatic Lipid Metabolism. Endocrinology 2011, 152, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Masood, A.; Yi, M.; Lau, M.; Belcastro, R.; Ivanovska, J.; Jankov, R.P.; Tanswell, A.K. The IGF-I/IGF-R1 pathway regulates postnatal lung growth and is a nonspecific regulator of alveologenesis in the neonatal rat. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 304, L626–L637. [Google Scholar] [CrossRef] [Green Version]
- Chetty, A.; Andersson, S.; Lassus, P.; Nielsen, H.C. Insulin-like growth factor-1 (IGF-1) and IGF-1 receptor (IGF-1R) expression in human lung in RDS and BPD. Pediatr. Pulmonol. 2004, 37, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.-P.; Baker, J.; Perkins, A.S.; Robertson, E.J.; Efstratiadis, A. Mice carrying null mutations of the genes encoding insulin-like growth factor I (Igf-1) and type 1 IGF receptor (Igf1r). Cell 1993, 75, 59–72. [Google Scholar] [CrossRef]
- Beyea, J.A.; Sawicki, G.; Olson, D.M.; List, E.; Kopchick, J.J.; Harvey, S. Growth hormone (GH) receptor knockout mice reveal actions of GH in lung development. Proteomics 2006, 6, 341–348. [Google Scholar] [CrossRef]
- Greenbaum, D.; Colangelo, C.; Williams, K.; Gerstein, M. Comparing protein abundance and mRNA expression levels on a genomic scale. Genome Biol. 2003, 4, 117. [Google Scholar] [CrossRef] [Green Version]
- Pajares, M.; Jimeénez-Moreno, N.; Dias, I.H.; Debelec, B.; Vucetic, M.; Fladmark, K.E.; Basaga, H.; Ribaric, S.; Milisav, I.; Cuadrado, A. Redox control of protein degradation. Redox Biol. 2015, 6, 409–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, V.; Quintanilha, A.; Moradas-Ferreira, P. Protein oxidation, repair mechanisms and proteolysis in Saccharomyces cerevisiae. IUBMB Life 2007, 59, 293–298. [Google Scholar] [CrossRef]
- Reichmann, D.; Voth, W.; Jakob, U. Maintaining a Healthy Proteome during Oxidative Stress. Mol. Cell 2018, 69, 203–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stiles, A.D.; D’Ercole, A.J. The insulin-like growth factors and the lung. Am. J. Respir. Cell Mol. Biol. 1990, 3, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Krein, P.M.; Sabatini, P.J.B.; Tinmouth, W.; Green, F.H.Y.; Winston, B.W. Localization of insulin-like growth factor-I in lung tissues of patients with fibroproliferative acute respiratory distress syndrome. Am. J. Respir. Crit. 2003, 167, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Batth, I.S.; Qu, X.; Xu, L.; Song, N.; Wang, R.; Liu, Y. IGF-IR signaling in epithelial to mesenchymal transition and targeting IGF-IR therapy: Overview and new insights. Mol. Cancer 2017, 16, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hellstrom, A.; Ley, D.; Hansen-Pupp, I.; Hallberg, B.; Lofqvist, C.; van Marter, L.; van Weissenbruch, M.; Ramenghi, L.A.; Beardsall, K.; Dunger, D.; et al. Insulin-like growth factor 1 has multisystem effects on foetal and preterm infant development. Acta Paediatr. 2016, 105, 576–586. [Google Scholar] [CrossRef] [Green Version]
- Capoluongo, E.; Ameglio, F.; Zuppi, C. Insulin-like growth factor-I and complications of prematurity: A focus on bronchopulmonary dysplasia. Clin. Chem. Lab. Med. 2008, 46, 1061–1066. [Google Scholar] [CrossRef]
- Perkett, E.A.; Badesch, D.B.; Roessler, M.K.; Stenmark, K.R.; Meyrick, B. Insulin-like growth factor I and pulmonary hypertension induced by continuous air embolization in sheep. Am. J. Respir. Cell Mol. Biol. 1992, 6, 82–87. [Google Scholar] [CrossRef]
- Sun, M.; Ramchandran, R.; Chen, J.; Qiwei, Y.; Raj, J.U. Smooth Muscle Insulin-Like Growth Factor-1 Mediates Hypoxia-Induced Pulmonary Hypertension in Neonatal Mice. Am. J. Respir. Cell Mol. Biol. 2016, 55, 779–791. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.-E.; Sung-soon, L.; Sunde, D.A.; Huizar, I.; Haugk, K.L.; Thannickal, V.J.; Vittal, R.; Plymate, S.R.; Schnapp, L.M. Insulin-like growth factor-I receptor blockade improves outcome in mouse model of lung injury. Am. J. Respir. Crit. 2009, 179, 212–219. [Google Scholar] [CrossRef] [Green Version]
- Ionescu, L.; Byrne, R.N.; van Haaften, T.; Vadivel, A.; Alphonse, R.S.; Rey-Parra, G.J.; Weissmann, G.; Hall, A.; Eaton, F.; Thebaud, B. Stem cell conditioned medium improves acute lung injury in mice: In vivo evidence for stem cell paracrine action. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 303, L967–L977. [Google Scholar] [CrossRef] [Green Version]
- Lofqvist, C.; Hellgren, G.; Niklasson, A.; Engstrom, E.; Ley, D.; Hansen-Pupp, I. Low postnatal serum IGF-I levels are associated with bronchopulmonary dysplasia (BPD). Acta Paediatr. 2012, 101, 1211–1216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ley, D.; Hallberg, B.; Hansen-Pupp, I.; Dani, C.; Ramenghi, L.A.; Marlow, N.; Beardsall, K.; Bhatti, F.; Dunger, D.; Higginson, J.D.; et al. rhIGF-1/rhIGFBP-3 in Preterm Infants: A Phase 2 Randomized Controlled Trial. J. Pediatr. 2019, 206, 56–65.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunscmann, C.; Stachelschied, H.; Akyuz, M.D.; Schmitz, A.; Missero, C.; Bruning, J.C.; Niessen, C.N. Insulin/IGF-1 controls epidermal morphogenesis via regulation of FoxO-mediated p63 inhibition. Dev. Cell 2013, 26, 176–187. [Google Scholar] [CrossRef] [Green Version]
- Galvis, L.A.; Holik, A.; Short, K.M.; Pasquet, J.; Lun, A.T.L.; Blewitt, M.E.; Smyth, I.M.; Ritchie, M.E.; Asselin-Labat, M.-L. Repression of Igf1 expression by Ezh2 prevents basal cell differentiation in the developing lung. Development 2015, 142, 1458–1469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomi, K.; Tang, Y.; Arbelaez, V.; Crystal, R.G.; Walters, M.S. Endothelial Cell Mediated Promotion of Ciliated Cell Differentiation of Human Airway Basal Cells via Insulin and Insulin-Like Growth Factor 1 Receptor Mediated Signaling. Stem Cell Rev. Rep. 2017, 13, 309–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, M.C.; Goranthla, V.; Makena, P.S.; Luellen, C.; Sinclair, S.E.; Schwingshackl, A.; Waters, C.M. Insulin-like growth factor-I stimulates differentiation of ATII cells to ATI-like cells through activation of Wnt5a. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 305, L222–L228. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| P7 | P28 | P70 | ||||
|---|---|---|---|---|---|---|
| NOX | HYX | NOX | HYX | NOX | HYX | |
| Body weight (g) | 3.576 ± 0.113 | 3.455 ± 0.137 | 13. 90 ± 0.328 | 9.469 ± 0.154 **** | 25.220 ± 0.944 | 23.780 ± 1.494 |
| Lung weight (g) | 0.076 ± 0.004 | 0.062 ± 0.003 * | 0.072 ± 0.002 | 0.077 ± 0.002 * | 0.081 ± 0.003 | 0.199 ± 0.103 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vohlen, C.; Mohr, J.; Fomenko, A.; Kuiper-Makris, C.; Grzembke, T.; Aydogmus, R.; Wilke, R.; Hirani, D.; Dötsch, J.; Alejandre Alcazar, M.A. Dynamic Regulation of GH–IGF1 Signaling in Injury and Recovery in Hyperoxia-Induced Neonatal Lung Injury. Cells 2021, 10, 2947. https://doi.org/10.3390/cells10112947
Vohlen C, Mohr J, Fomenko A, Kuiper-Makris C, Grzembke T, Aydogmus R, Wilke R, Hirani D, Dötsch J, Alejandre Alcazar MA. Dynamic Regulation of GH–IGF1 Signaling in Injury and Recovery in Hyperoxia-Induced Neonatal Lung Injury. Cells. 2021; 10(11):2947. https://doi.org/10.3390/cells10112947
Chicago/Turabian StyleVohlen, Christina, Jasmine Mohr, Alexey Fomenko, Celien Kuiper-Makris, Tiffany Grzembke, Rabia Aydogmus, Rebecca Wilke, Dharmesh Hirani, Jörg Dötsch, and Miguel A. Alejandre Alcazar. 2021. "Dynamic Regulation of GH–IGF1 Signaling in Injury and Recovery in Hyperoxia-Induced Neonatal Lung Injury" Cells 10, no. 11: 2947. https://doi.org/10.3390/cells10112947
APA StyleVohlen, C., Mohr, J., Fomenko, A., Kuiper-Makris, C., Grzembke, T., Aydogmus, R., Wilke, R., Hirani, D., Dötsch, J., & Alejandre Alcazar, M. A. (2021). Dynamic Regulation of GH–IGF1 Signaling in Injury and Recovery in Hyperoxia-Induced Neonatal Lung Injury. Cells, 10(11), 2947. https://doi.org/10.3390/cells10112947