
Fast Self-Healing at Room Temperature in Diels–Alder Elastomers

 ,
,  ,
,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
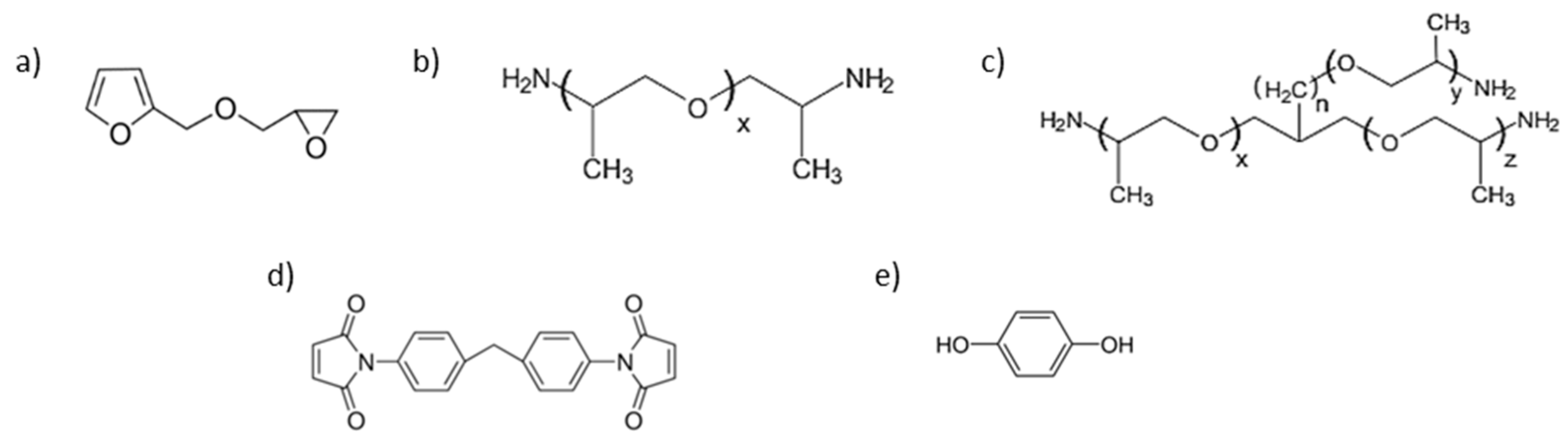
2.1.1. Reagents
2.1.2. Synthesis
2.1.3. Polymer Network Composition
2.2. Instruments
2.2.1. Uniaxial Tensile Testing
2.2.2. Dynamic Mechanical Analysis
2.2.3. Dynamic Rheometry
2.2.4. Differential Scanning Calorimetry
2.2.5. Thermogravimetric Analysis
2.3. Methods
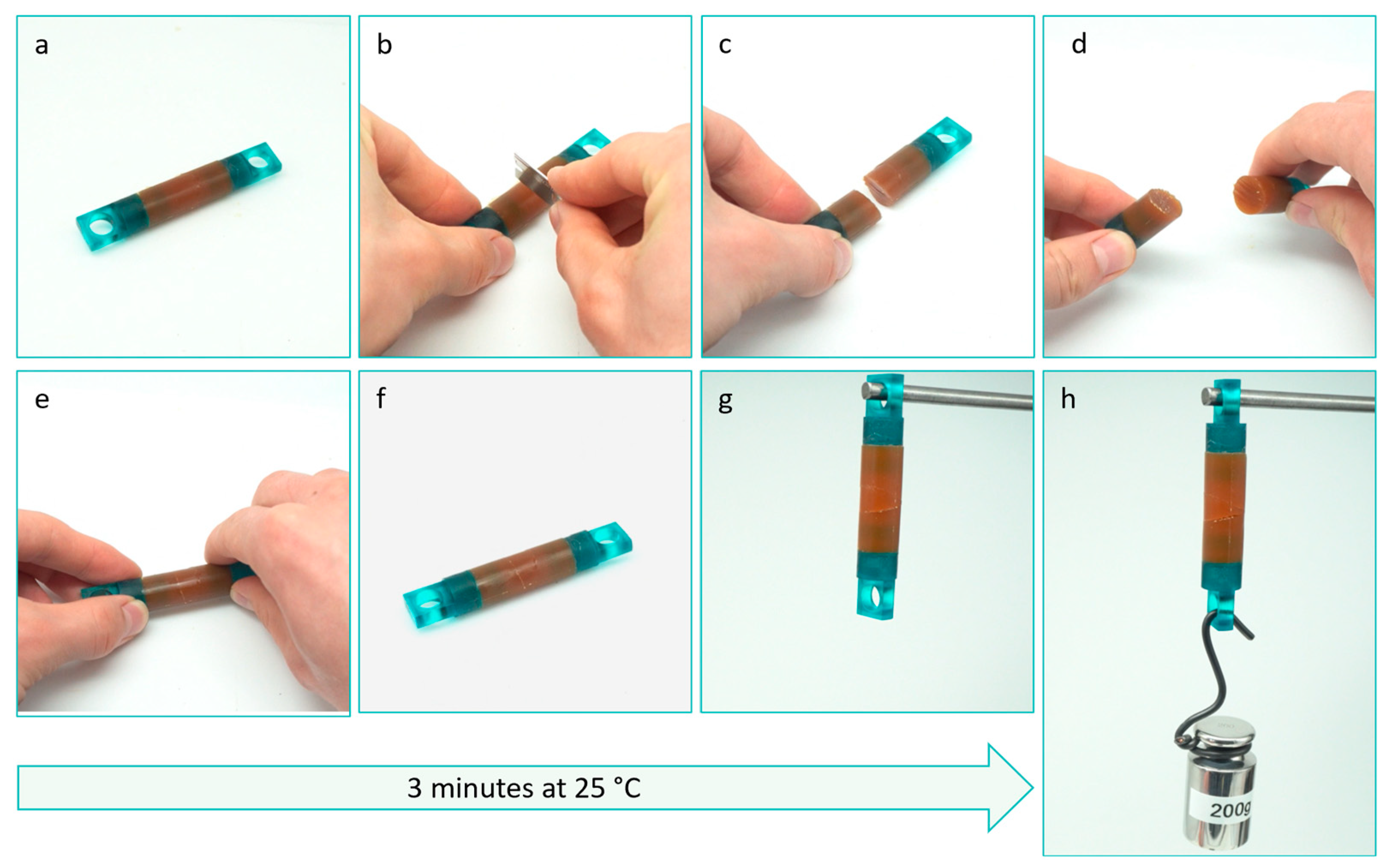
2.3.1. Self-Healing and Healing Efficiency
2.3.2. Kinetics Simulation
3. Results
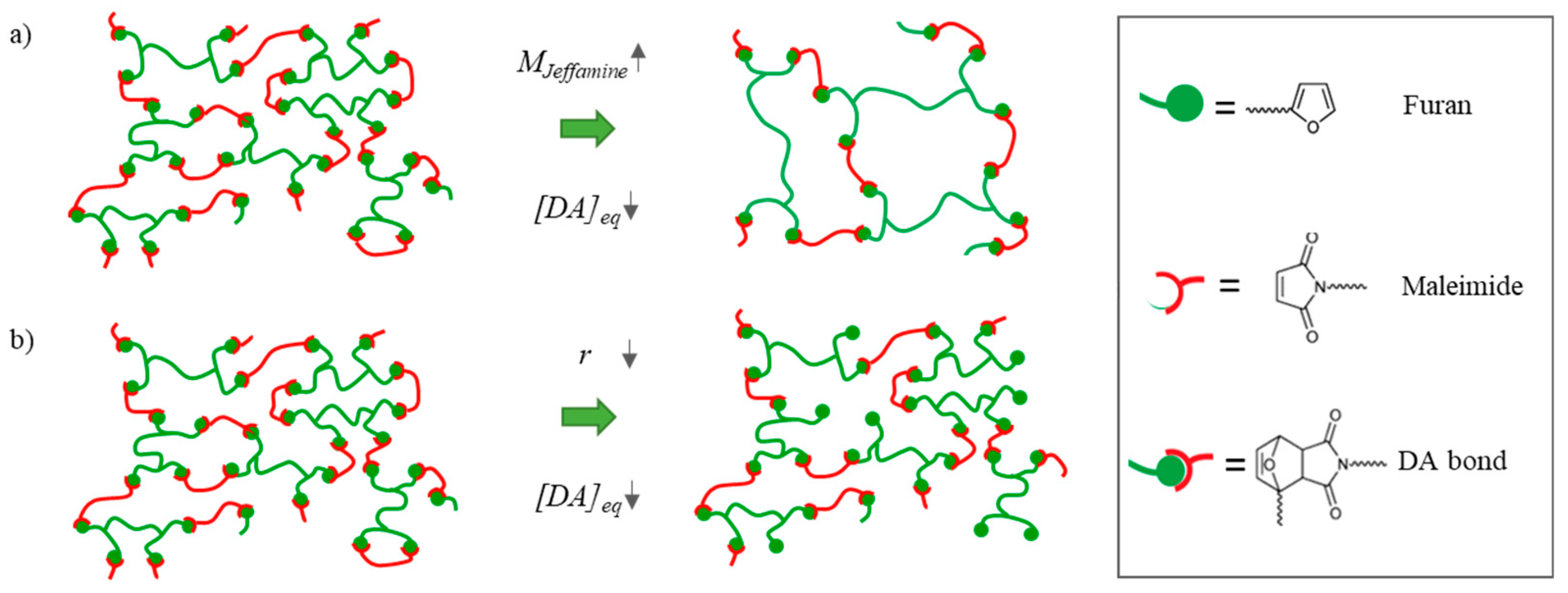
3.1. Varying the Diels–Alder Concentration through the Molecular Weight of the Monomers
3.1.1. Quasistatic Mechanical Performance at Room Temperature
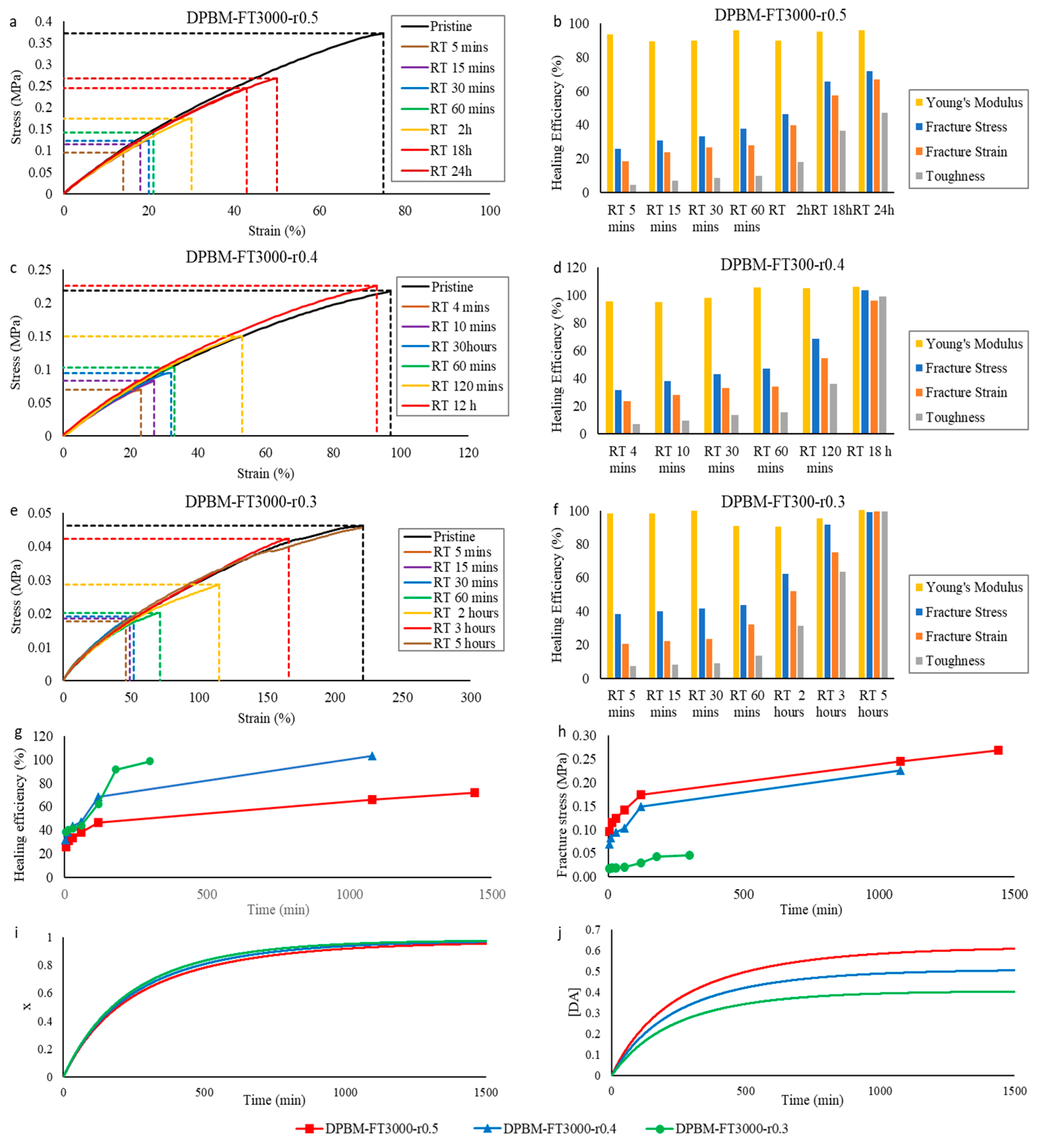
3.1.2. Self-Healing Performance at Room Temperature
3.1.3. Visco-Elastic Behaviour as Function of Temperature
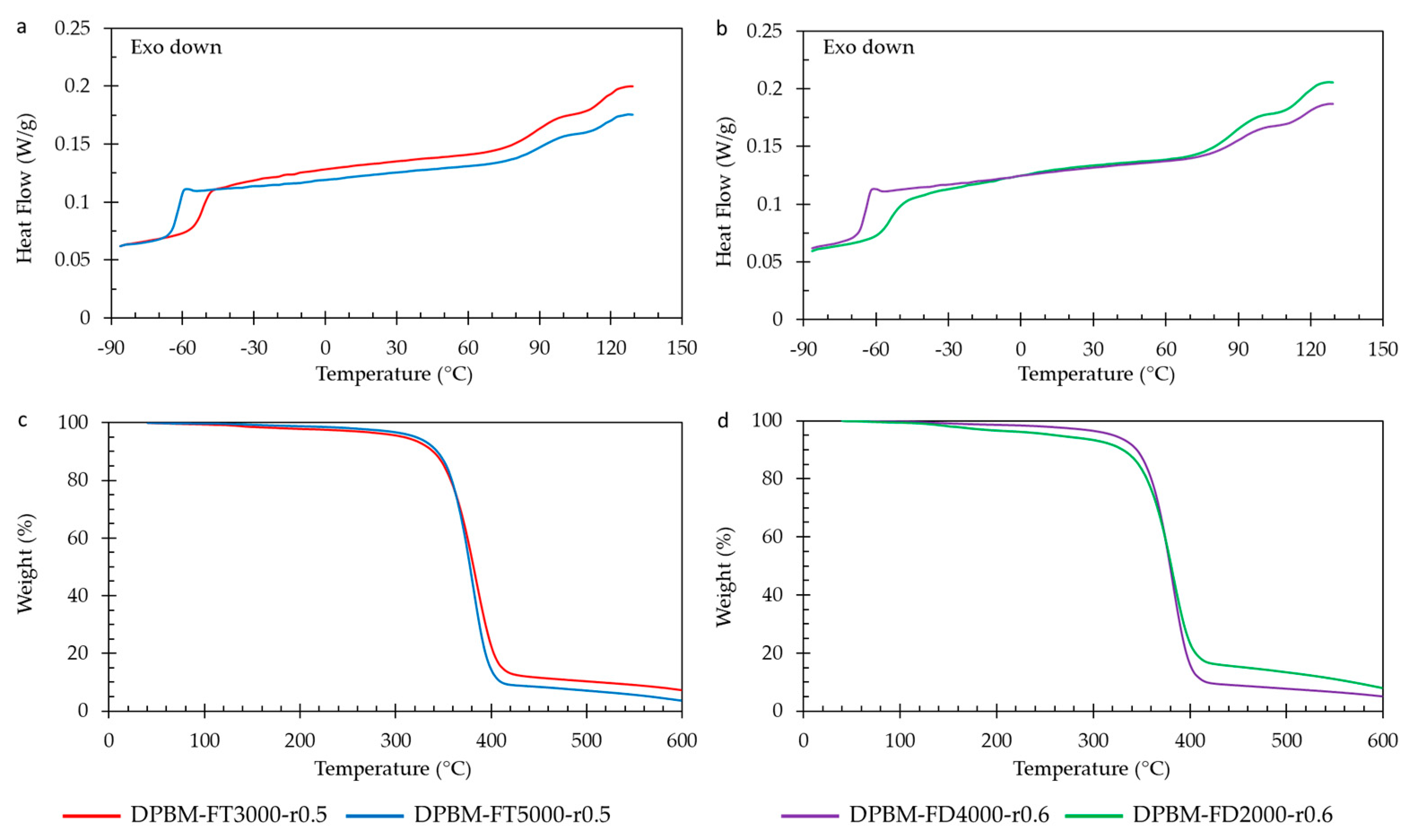
3.1.4. Thermal Behaviour as Function of Temperature
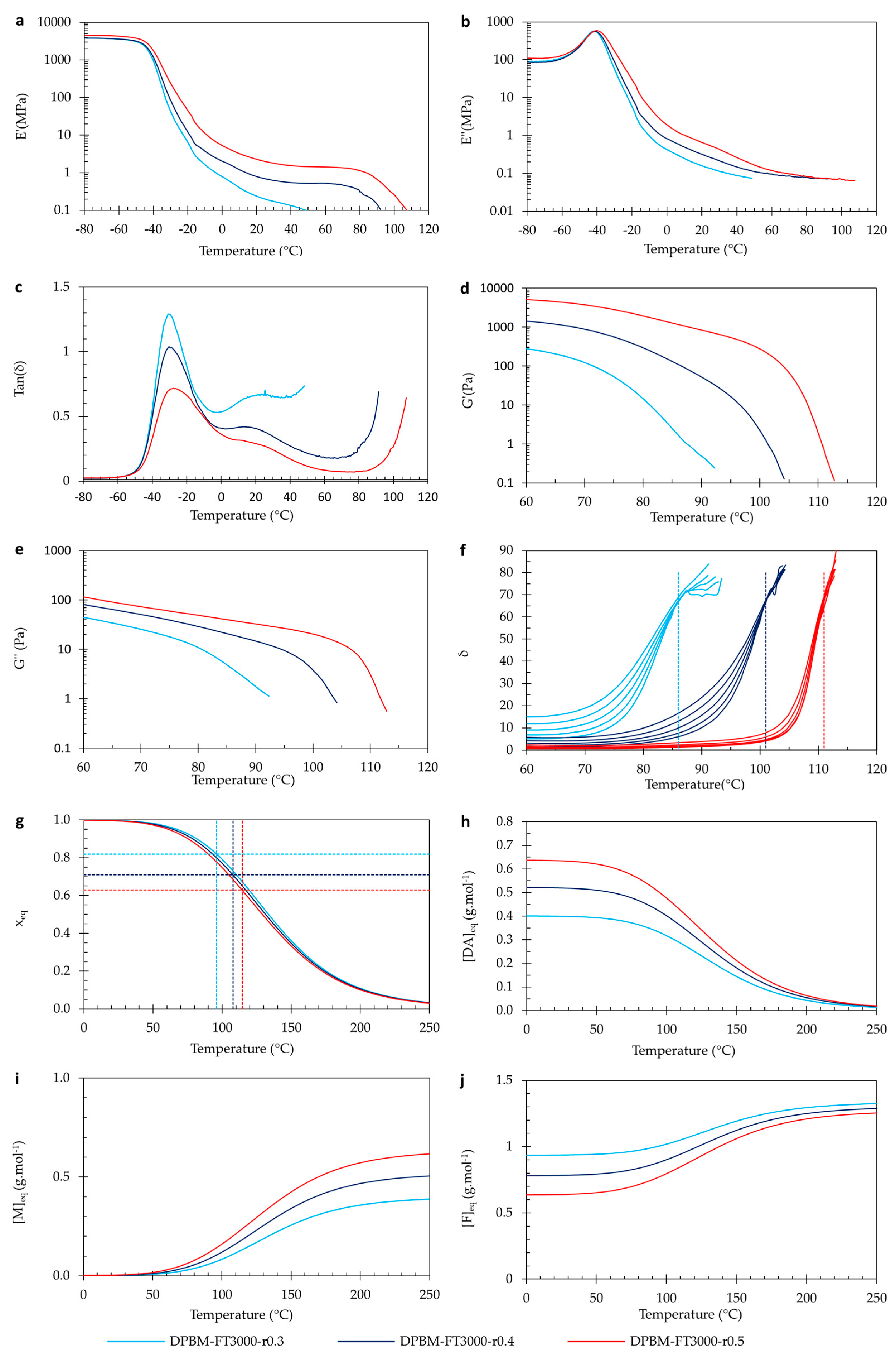
3.2. Varying the Diels–Alder Concentration through the Stoichiometric Ratio
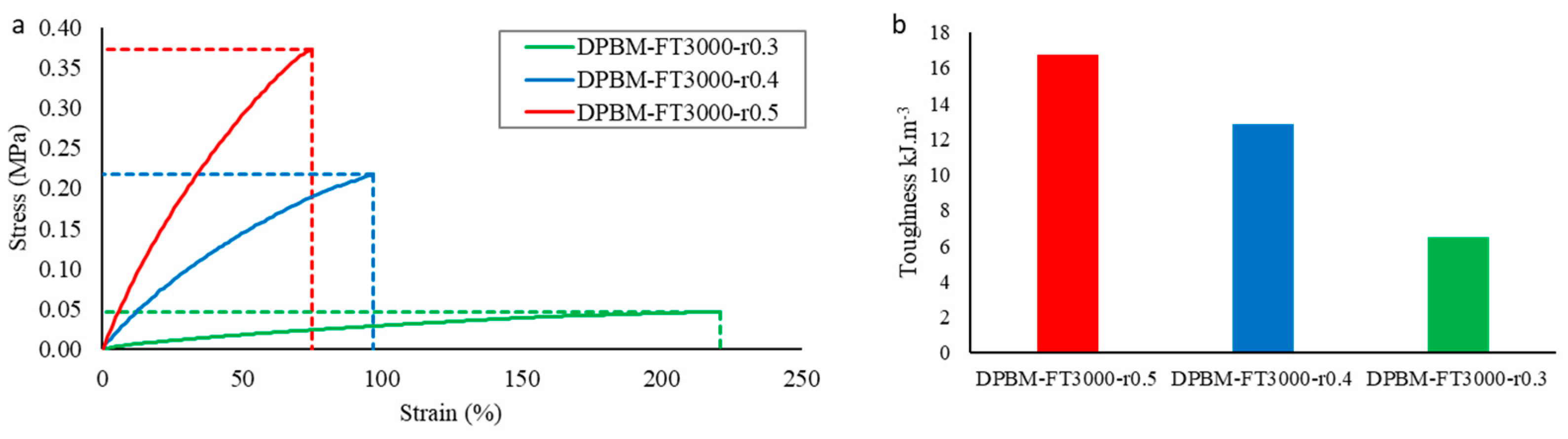
3.2.1. Quasistatic Mechanical Performance at Room Temperature
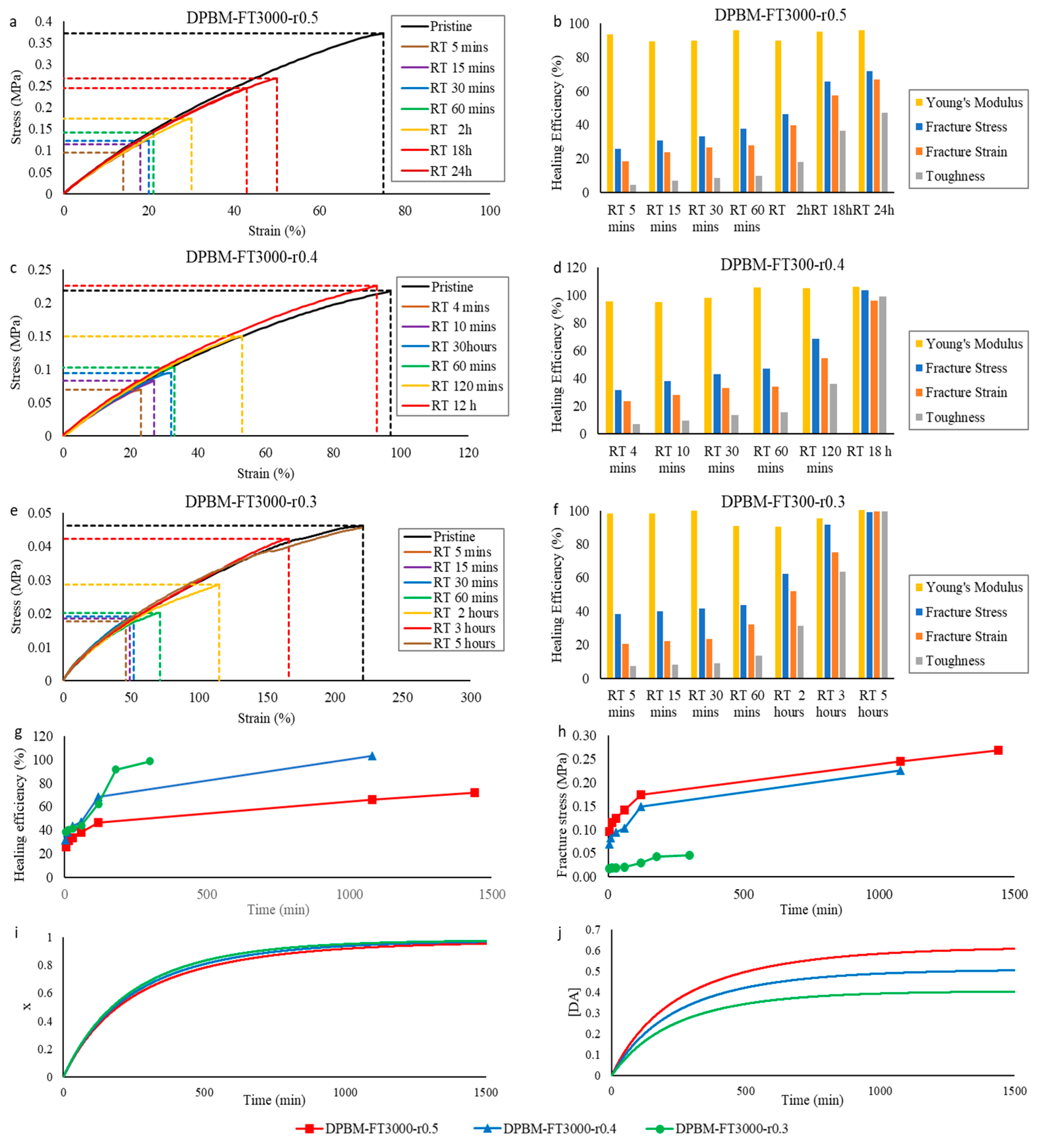
3.2.2. Self-Healing Performance at Room Temperature
3.2.3. Visco-Elastic Behaviour as Function of Temperature
3.2.4. Thermal Behaviour as Function of Temperature
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zhu, D.Y.; Rong, M.Z.; Zhang, M.Q. Self-Healing Polymeric Materials Based on Microencapsulated Healing Agents: From Design to Preparation. Prog. Polym. Sci. 2015, 49–50, 175–220. [Google Scholar] [CrossRef]
- Li, B.; Cao, P.; Saito, T.; Sokolov, A.P. Intrinsically Self-Healing Polymers: From Mechanistic Insight to Current Challenges. Chem. Rev. 2023, 123, 701–735. [Google Scholar] [CrossRef] [PubMed]
- Terryn, S.; Langenbach, J.; Roels, E.; Brancart, J.; Thuruthel, T.G.; Safaei, A.; Ferrentino, P.; Sebastian, T.; Bakkali, C.; Norvez, S.; et al. A Review on Self-Healing Polymers for Soft Robotics. Mater. Today 2021, 47, 187–205. [Google Scholar] [CrossRef]
- Hoogenboom, R. Hard Autonomous Self-Healing Supramolecular Materials—A Contradiction in Terms? Angew. Chem. Int. Ed. 2012, 51, 11942–11944. [Google Scholar] [CrossRef] [PubMed]
- Dahlke, J.; Zechel, S.; Hager, M.D.; Schubert, U.S. How to Design a Self-Healing Polymer: General Concepts of Dynamic Covalent Bonds and Their Application for Intrinsic Healable Materials. Adv. Mater. Interfaces 2018, 5, 1800051. [Google Scholar] [CrossRef]
- Denissen, W.; Winne, J.M.; Du Prez, F.E. Vitrimers: Permanent Organic Networks with Glass-like Fluidity. Chem. Sci. 2016, 7, 30–38. [Google Scholar] [CrossRef]
- Ling, L.; Li, J.; Zhang, G.; Sun, R.; Wong, C.P. Self-Healing and Shape Memory Linear Polyurethane Based on Disulfide Linkages with Excellent Mechanical Property. Macromol. Res. 2018, 26, 365–373. [Google Scholar] [CrossRef]
- Bakkali-Hassani, C.; Poutrel, Q.A.; Langenbach, J.; Chappuis, S.; Blaker, J.J.; Gresil, M.; Tournilhac, F. Lipase-Catalyzed Epoxy-Acid Addition and Transesterification: From Model Molecule Studies to Network Build-Up. Biomacromolecules 2021, 22, 4544–4551. [Google Scholar] [CrossRef]
- Herbst, F.; Döhler, D.; Michael, P.; Binder, W.H. Self-healing Polymers via Supramolecular Forces. Macromol. Rapid Commun. 2013, 34, 203–22018. [Google Scholar] [CrossRef]
- Xie, Z.; Hu, B.; Li, R.; Zhang, Q. Hydrogen Bonding in Self-Healing Elastomers. ACS Omega 2021, 6, 9319–9333. [Google Scholar] [CrossRef]
- Shi, Y.; Wang, M.; Wang, Y.; Li, X.; Yu, G. A Conductive Self-Healing Hybrid Gel Enabled by Metal–Ligand Supramolecule and Nanostructured Conductive Polymer. Nano Lett. 2015, 15, 6276–6281. [Google Scholar] [CrossRef]
- Deriabin, K.V.; Ignatova, N.A.; Kirichenko, S.O.; Novikov, A.S.; Islamova, M. Nickel(II)-Pyridinedicarboxamide-co-Polydimethylsiloxane Complexes as Elastic Self-Healing Silicone Materials with Reversible Coordination. Polymer 2021, 212, 123119. [Google Scholar] [CrossRef]
- Deriabin, K.V.; Ignatova, N.A.; Kirichenko, S.O.; Novikov, A.S.; Kryukova, M.A.; Kukushkin, V.Y.; Islamova, R.M. Structural Features of Polymer Ligand Environments DramaticallyAffect the Mechanical and Room-Temperature Self-HealingProperties of Cobalt(II)-Incorporating Polysiloxanes. Organometallics 2021, 40, 2750–2760. [Google Scholar] [CrossRef]
- Roels, E.; Terryn, S.; Iida, F.; Bosman, A.W.; Norvez, S.; Clemens, F.J.; Van Assche, G.; Vanderborght, B.; Brancart, J. Processing of Self-Healing Polymers for Soft Robotics. Adv. Mater. 2022, 34, 2104798. [Google Scholar] [CrossRef]
- Terryn, S.; Brancart, J.; Lefeber, D.; Van Assche, G.; Vanderborght, B. Self-Healing Soft Pneumatic Robots. Sci. Robot. 2017, 2, eaan4268. [Google Scholar] [CrossRef]
- Orozco, F.; Kaveh, M.; Santosa, D.S.; Lima, G.M.R.; Gomes, D.R.; Pei, Y.; Araya-hermosilla, R.; Moreno-villoslada, I.; Picchioni, F.; Bose, R.K. Electroactive Self-Healing Shape Memory Polymer Composites Based on Diels-Alder Chemistry. Appl. Polym. Mater. 2021, 3, 6147–6156. [Google Scholar] [CrossRef]
- Tiwari, N.; Ho, F.; Mathews, N. A Rapid Low Temperature Self-Healable Polymeric Composite for Fl Exible Electronic Devices. J. Mater. Chem. A 2018, 6, 21428–21434. [Google Scholar] [CrossRef]
- Turkenburg, D.H.; Durant, Y.; Fischer, H.R. Bio-Based Self-Healing Coatings Based on Thermo-Reversible Diels-Alder Reaction. Prog. Org. Coat. 2017, 111, 38–46. [Google Scholar] [CrossRef]
- Terryn, S.; Brancart, J.; Roels, E.; Van Assche, G.; Vanderborght, B. Room Temperature Self-Healing in Soft Pneumatic Robotics: Autonomous Self-Healing in a Diels-Alder Polymer Network. IEEE Robot. Autom. Mag. 2020, 27, 44–55. [Google Scholar] [CrossRef]
- Pratama, P.A.; Shari, M.; Peterson, A.M.; Palmese, G.R. Room Temperature Self-Healing Thermoset Based on the Diels-Alder Reaction. ACS Appl. Mater. Interfaces 2013, 5, 12425–12431. [Google Scholar] [CrossRef]
- Terryn, S.; Brancart, J.; Roels, E.; Verhelle, R.; Cuvellier, A.; Safaei, A.; Bram, V.; Guy, V.A. Structure-Property Relationships of Self-Healing Polymer Networks Based on Reversible Diels-Alder Chemistry. Macromolecules 2022, 55, 5497–5513. [Google Scholar] [CrossRef]
- Terryn, S.; Mathijssen, G.; Brancart, J.; Van Assche, G.; Vanderborght, B. Towards Self-Healing Compliant Actuators: A Preliminary Design. Submitt. IEEE Trans. Robot. 2016, 32, 736–743. [Google Scholar] [CrossRef]
- Safaei, A.; Terryn, S.; Vanderborght, B.; Van Assche, G.; Brancar, J. The Influence of the Furan and Maleimide Stoichiometry on the Thermoreversible Diels-Alder Network Polymerization. Polym. Spec. Issue Dyn. Covalent Polym. Netw. 2021, 13, 2522. [Google Scholar] [CrossRef]
- Orozco, F.; Li, J.; Ezekiel, U.; Niyazov, Z.; Floyd, L.; Lima, G.M.R.; Winkelman, J.G.M.; Moreno-Villoslada, I.; Picchioni, F.; Bose, R.K. Diels-Alder-Based Thermo-Reversibly Crosslinked Polymers: Interplay of Crosslinking Density, Network Mobility, Kinetics and Stereoisomerism. Eur. Polym. J. 2020, 135, 109882. [Google Scholar] [CrossRef]
- Zeng, C.; Seino, H.; Ren, J.; Hatanaka, K.; Yoshie, N. Self-Healing Bio-Based Furan Polymers Cross-Linked with Various. Polymer 2013, 54, 5351–5357. [Google Scholar] [CrossRef]
- Roels, E.; Terryn, S.; Ferrentino, P.; Brancart, J.; Van Assche, G.; Vanderborght, B. An Interdisciplinary Tutorial: A Self-Healing Soft Finger with Embedded Sensor. Sensors 2023, 23, 811. [Google Scholar] [CrossRef]
- Yang, L.; Lu, X.; Wang, Z. Diels–Alder Dynamic Crosslinked Polyurethane/Polydopamine Composites with NIR Triggered Self-Healing Function. Polym. Chem. 2018, 9, 2166–2172. [Google Scholar] [CrossRef]
- Wang, Z.; Terryn, S.; Legrand, J.; Ferrentino, P.; Kashef Tabrizian, S.; Brancart, J.; Roels, E.; Van Assche, G.; Vanderborght, B. Topology Optimized Multi-Material Self-Healing actuator with Reduced out of Plane Deformation. In Proceedings of the 2022 IEEE/RSJ International Conference on Intelligent Robots and Systems (IROS), Kyoto, Japan, 23–27 October 2022. [Google Scholar]
- Roels, E.; Terryn, S.; Brancart, J.; Verhelle, R.; Van Assche, G.; Vanderborght, B. Additive Manufacturing for Self-Healing Soft Robots. Soft Robot. 2020, 7, 711–723. [Google Scholar] [CrossRef]
- Yang, K.; Grant, J.C.; Lamey, P.; Joshi-imre, A.; Lund, B.R.; Smaldone, R.A.; Voit, W. Diels–Alder Reversible Thermoset 3D Printing Isotropic Thermoset Polymers via Fused Filament Fabrication. Adv. Funct. Mater. 2017, 27, 1700318. [Google Scholar] [CrossRef]
- Yuan, T.; Zhang, L.; Li, T.; Tu, R.; Sodano, H.A. 3D Printing of a Self-Healing, High Strength, and Reprocessable Thermoset. Polym. Chem. 2020, 11, 6441–6452. [Google Scholar] [CrossRef]
- Cerdan, K.; Brancart, J.; De Coninck, H.; Van Hooreweder, B.; Van, G.; Van Puyvelde, P. Laser Sintering of Self-Healable and Recyclable Thermoset Networks. Eur. Polym. J. 2022, 175, 111383. [Google Scholar] [CrossRef]
- Cuvellier, A.; Verhelle, R.; Brancart, J.; Vanderborght, B.; Van Assche, G.; Rahier, H. The Influence of Stereochemistry on the Reactivity of the Diels-Alder Cycloaddition and the Implications for Reversible Network Polymerization. Polym. Chem. 2019, 10, 473–485. [Google Scholar] [CrossRef]
- Stockmayer, W.H. Theory of Molecular Size Distribution and Gel Formation in Branched Polymers II. General Cross Linking. J. Chem. Phys. 1944, 12, 125–131. [Google Scholar] [CrossRef]
- Winter, H.H. Can the Gel Point of a Cross-Linking Polymer Be Detected. Polym. Eng. Sci. 1987, 27, 1698–1702. [Google Scholar] [CrossRef]
- Wang, H.; Terryn, S.; Wang, Z.; Van Assche, G.; Iida, F.; Vanderborght, B. Self-Regulated Self-Healing Robotic Gripper for Resilient and Adaptive Grasping. Adv. Intell. Syst. 2023, in press.
- Hopewell, J.L.; Hill, D.J.T.; Pomery, P.J. Electron Spin Resonance Study of the Homopolumerization of Aromatic Bismaleimides. Polymer 1998, 39, 5601–5607. [Google Scholar] [CrossRef]
- Brown, I.M.; Sandreczki, T.C. Cross-Linking Reactions in Maleimide and Bis (Maleimide) Polymers. An ESR Study. Macromolecules 1990, 23, 94–100. [Google Scholar] [CrossRef]
- Garcia, S.J. Effect of Polymer Architecture on the Intrinsic Self-Healing Character of Polymers. Eur. Polym. J. 2014, 53, 118–125. [Google Scholar] [CrossRef]
- Ferrentino, P.; Kashef Tabrizian, S.; Brancart, J.; Van Assche, G.; Vanderborght, B.; Terryn, S. FEA Based Inverse Kinematic Control on Hyperelasticmaterial Characterization of Self Healing Soft Robots. IEEE Robot. Autom. Mag. 2021, 29, 78–88. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Monomers | f | M/f (g.mol−1) | Elastomers | r | [M]0 (mol.kg−1) | [F]0 (mol.kg−1) | [DA]eq,25 °C (mol.kg−1) | [M]eq,25 °C (mol.kg−1) | [F]eq,25 °C (mol.kg−1) |
|---|---|---|---|---|---|---|---|---|---|
| DPBM | 2 | 179 | |||||||
| FT5000 | 6 | 1112 | DPBM-FT5000 | 0.5 | 0.41 | 0.83 | 0.61 | 0.01 | 0.42 |
| FT3000 | 6 | 691 | DPBM-FT3000 | 0.5 | 0.63 | 1.27 | 0.63 | 0.01 | 0.64 |
| FD4000 | 4 | 1297 | DPBM-FD4000 | 0.6 | 0.43 | 0.71 | 0.43 | 0.01 | 0.28 |
| FD2000 | 4 | 609 | DPBM-FD2000 | 0.6 | 0.83 | 1.38 | 0.83 | 0.01 | 0.55 |
| Monomers | f | M/f (g.mol−1) | Elastomers | r | [M]0 (mol.kg−1) | [F]0 (mol.kg−1) | [DA]eq,25 °C (mol.kg−1) | [M]eq,25 °C (mol.kg−1) | [F]eq,25 °C (mol.kg−1) |
|---|---|---|---|---|---|---|---|---|---|
| DPBM | 2 | 179 | |||||||
| FT3000 | 6 | 691 | DPBM-FT3000 | 0.3 | 0.40 | 1.34 | 0.40 | 0.01 | 0.94 |
| FT3000 | 6 | 691 | DPBM-FT3000 | 0.4 | 0.52 | 1.30 | 0.52 | 0.01 | 0.78 |
| FT3000 | 6 | 691 | DPBM-FT3000 | 0.5 | 0.64 | 1.27 | 0.64 | 0.01 | 0.63 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Safaei, A.; Brancart, J.; Wang, Z.; Yazdani, S.; Vanderborght, B.; Van Assche, G.; Terryn, S. Fast Self-Healing at Room Temperature in Diels–Alder Elastomers. Polymers 2023, 15, 3527. https://doi.org/10.3390/polym15173527
Safaei A, Brancart J, Wang Z, Yazdani S, Vanderborght B, Van Assche G, Terryn S. Fast Self-Healing at Room Temperature in Diels–Alder Elastomers. Polymers. 2023; 15(17):3527. https://doi.org/10.3390/polym15173527
Chicago/Turabian StyleSafaei, Ali, Joost Brancart, Zhanwei Wang, Sogol Yazdani, Bram Vanderborght, Guy Van Assche, and Seppe Terryn. 2023. "Fast Self-Healing at Room Temperature in Diels–Alder Elastomers" Polymers 15, no. 17: 3527. https://doi.org/10.3390/polym15173527
APA StyleSafaei, A., Brancart, J., Wang, Z., Yazdani, S., Vanderborght, B., Van Assche, G., & Terryn, S. (2023). Fast Self-Healing at Room Temperature in Diels–Alder Elastomers. Polymers, 15(17), 3527. https://doi.org/10.3390/polym15173527