
RAFT-Based Polymers for Click Reactions



Abstract
:1. Introduction
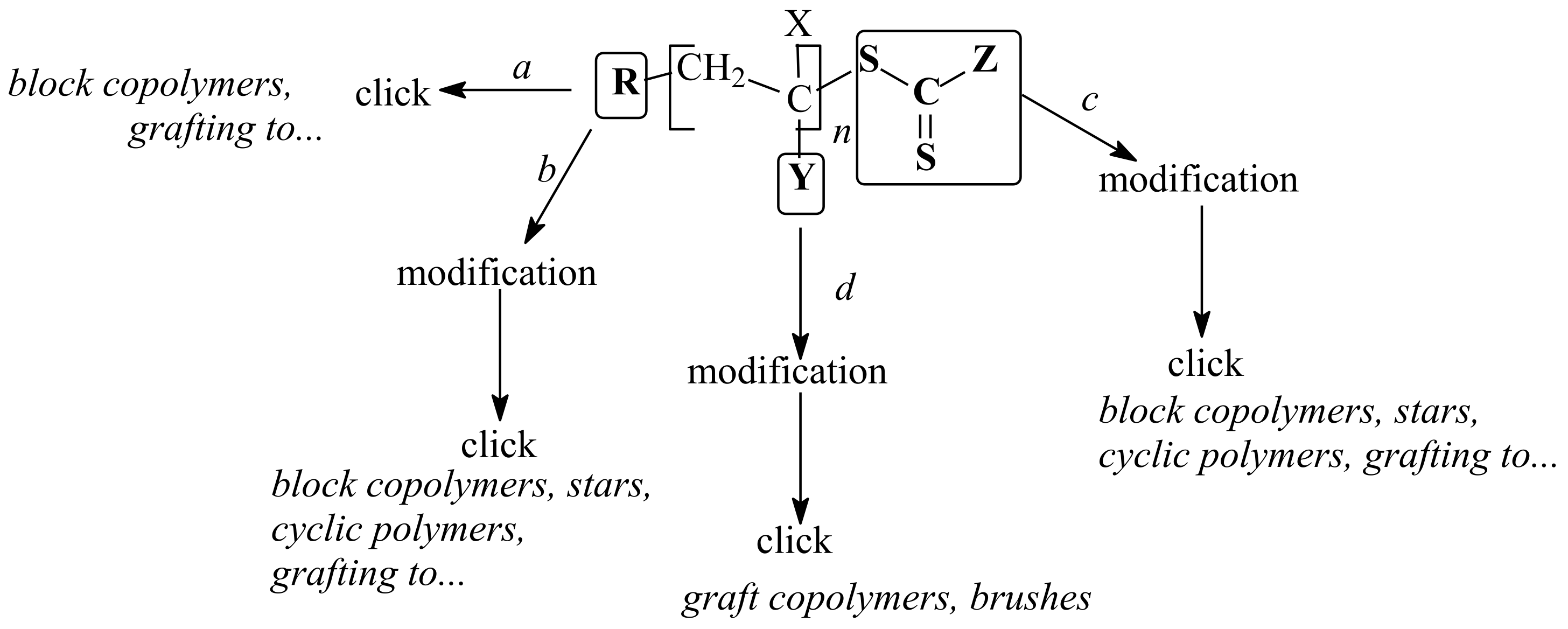
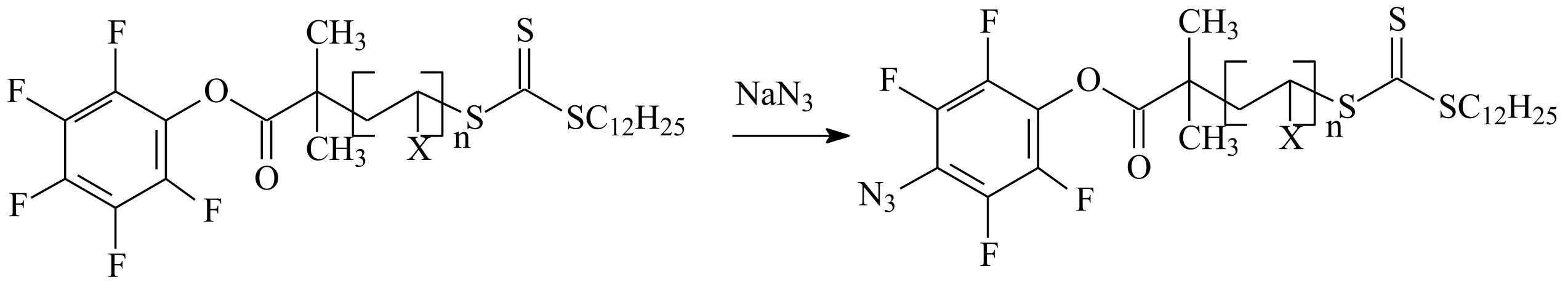
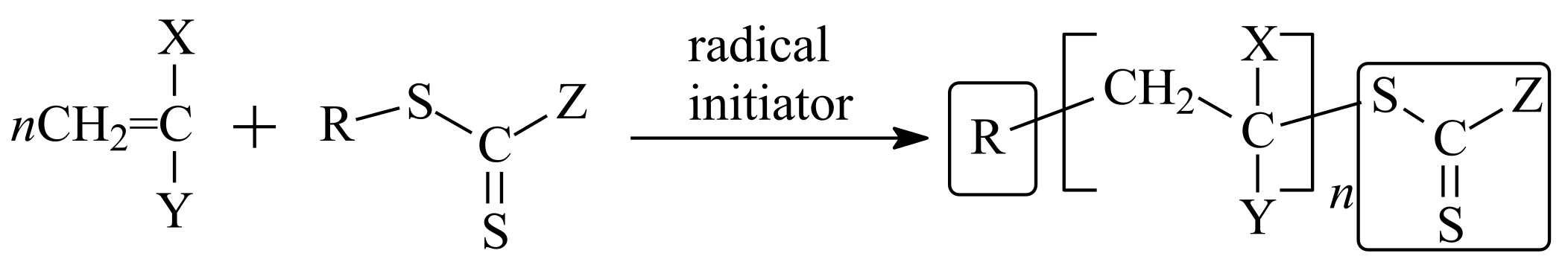
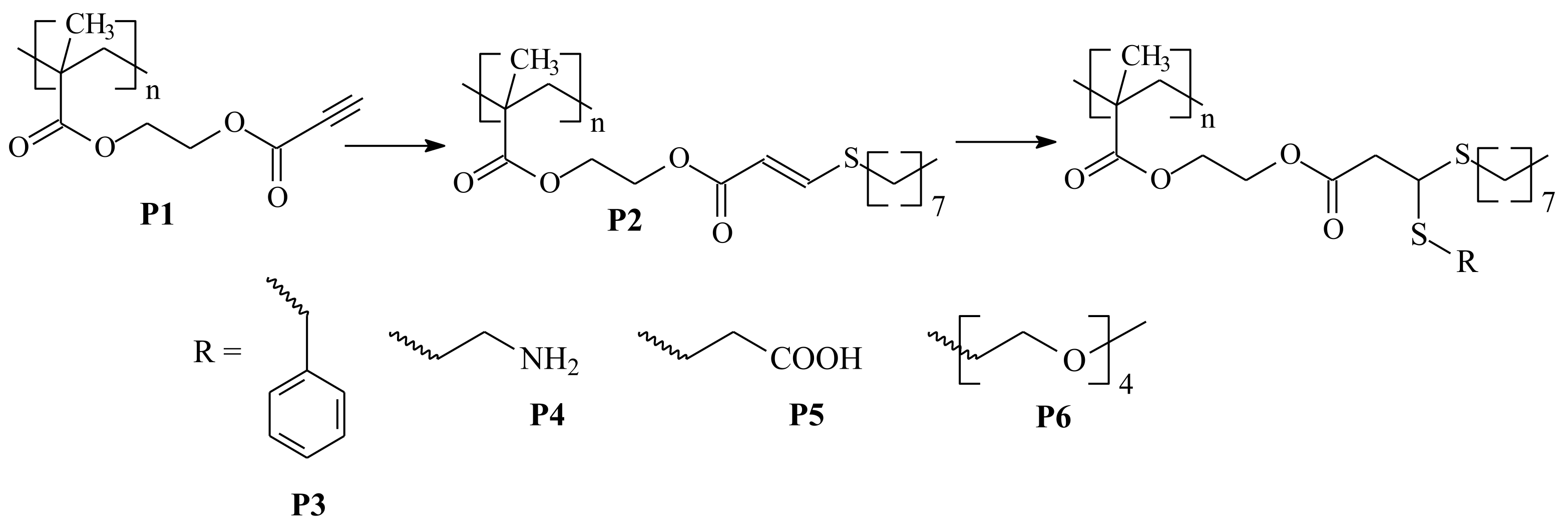
2. R/Z Group Approach

{kind=link}
{kind=link}
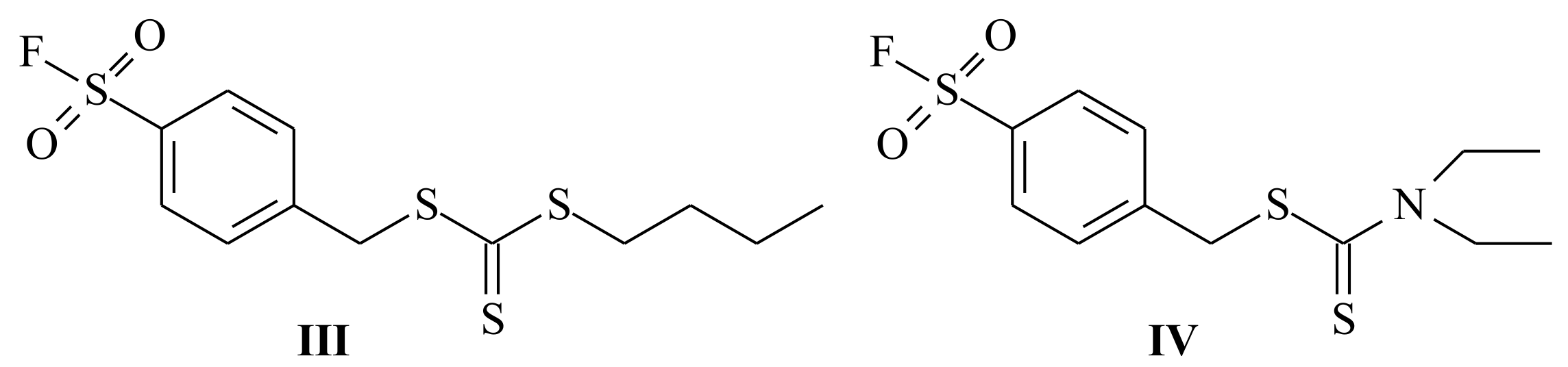{kind=link}
{kind=link}
{kind=link}
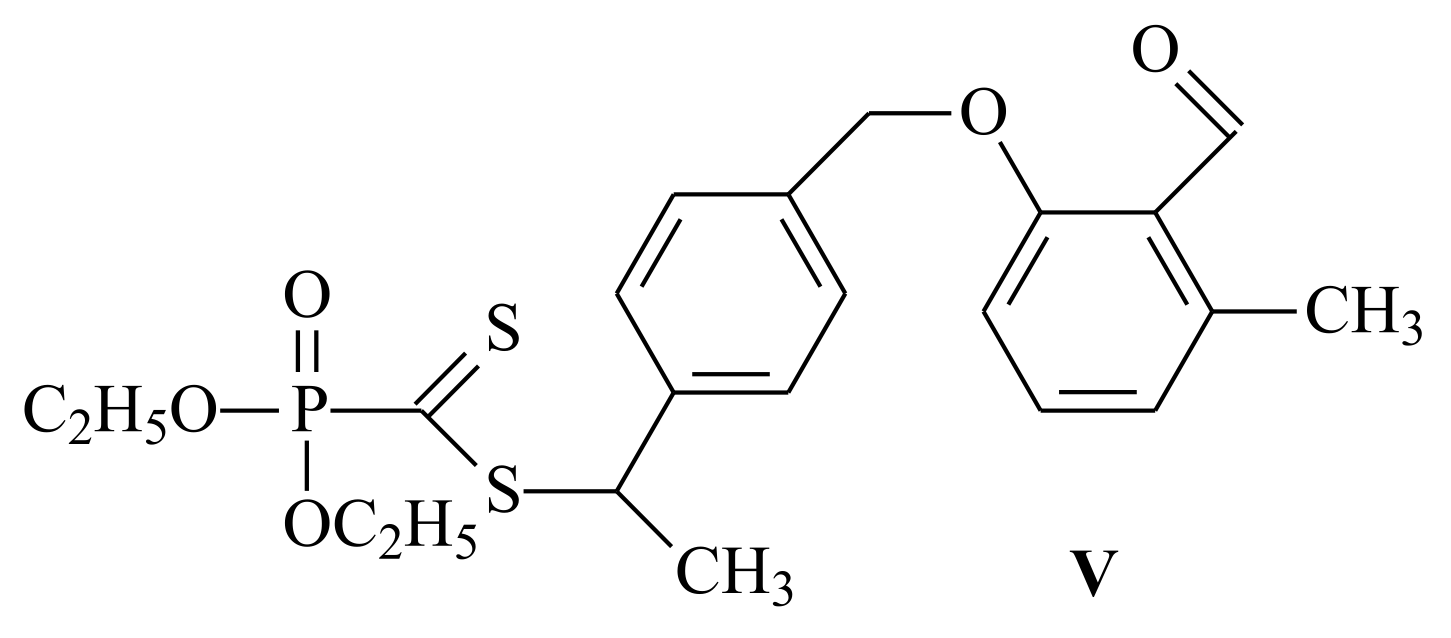
{kind=link}
{kind=link}
{kind=link}
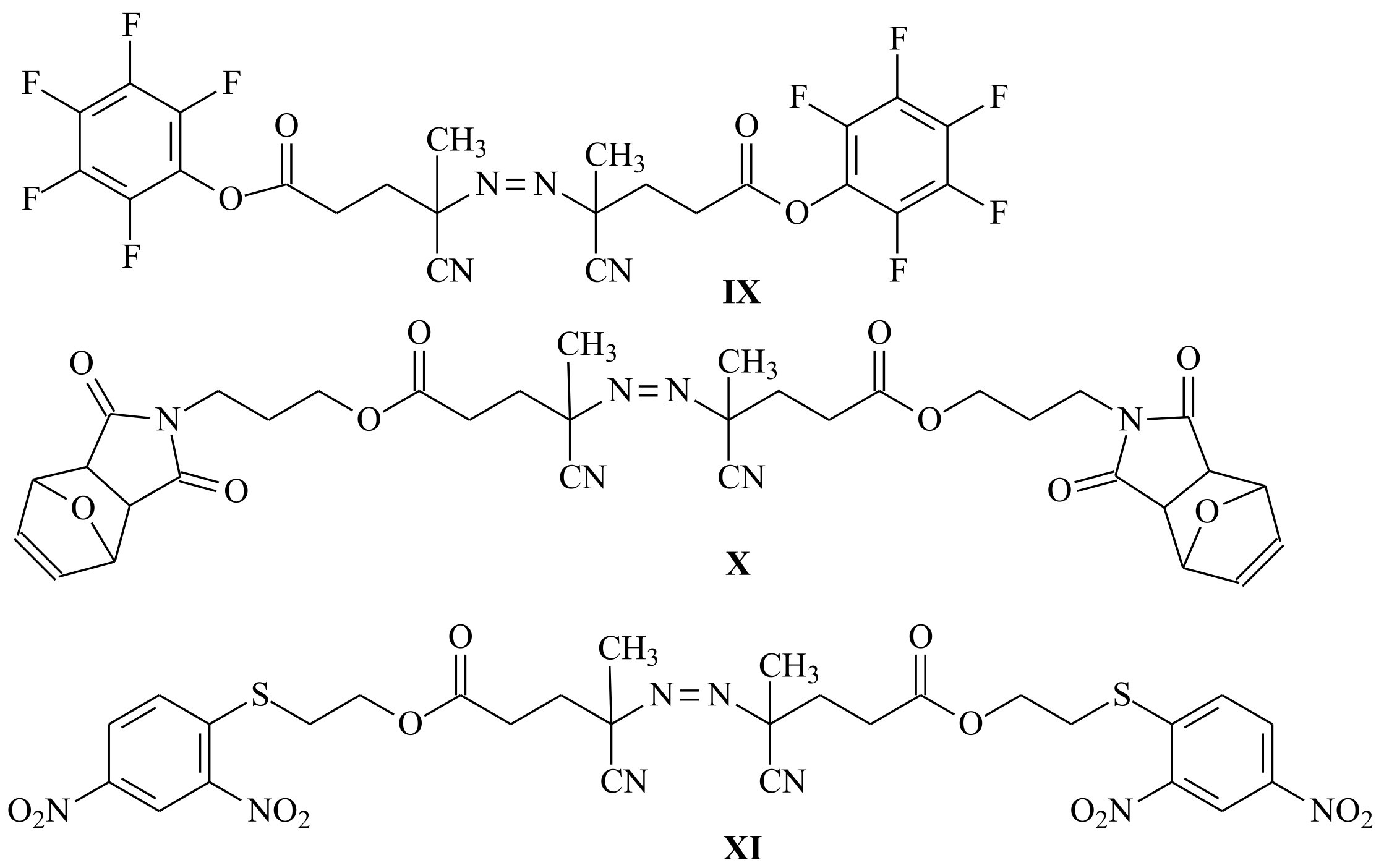
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
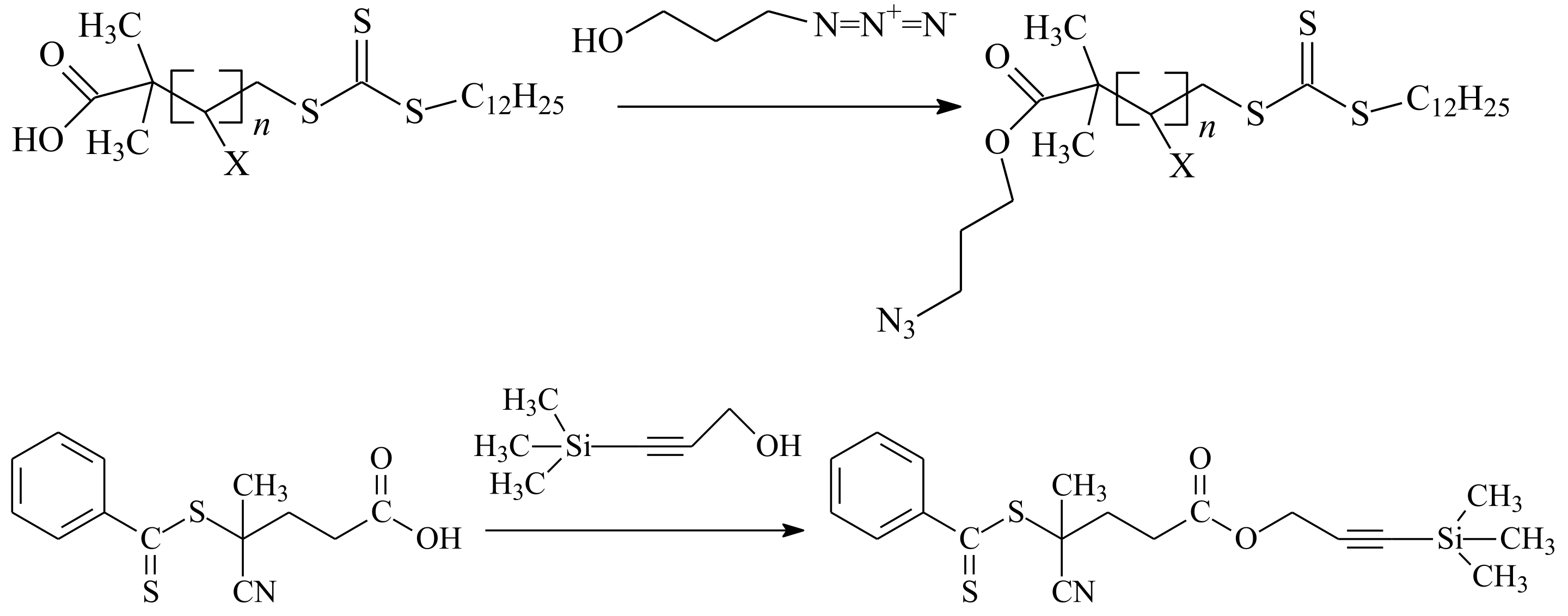{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type | Z Group | R Group | Ref. |
|---|---|---|---|
| Dithioester |  | 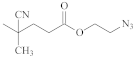 | [56] |
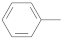 |  | [49,57,59] | |
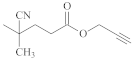 | [49,59] | ||
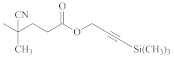 | [49,51,60] | ||
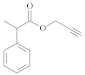 | [54] | ||
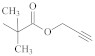 | [58,66,67] | ||
| Trithiocarbonate | C12H25S– | 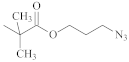 | [57,61,62,63] |
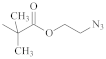 | [64,65] | ||
 | [55,68,69,70,71] | ||
| CH3S– |  | [52,53,74] | |
| C3H7S– |  | [72] | |
| iC4H9S– |  | [73] | |
| Xanthate | C2H5O– | 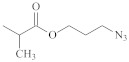 | [75,76] |
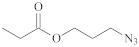 | [49,51,60] | ||
| N3CH2CH2O– | 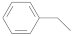 | [77] | |
| Dithiocarbamate |  | 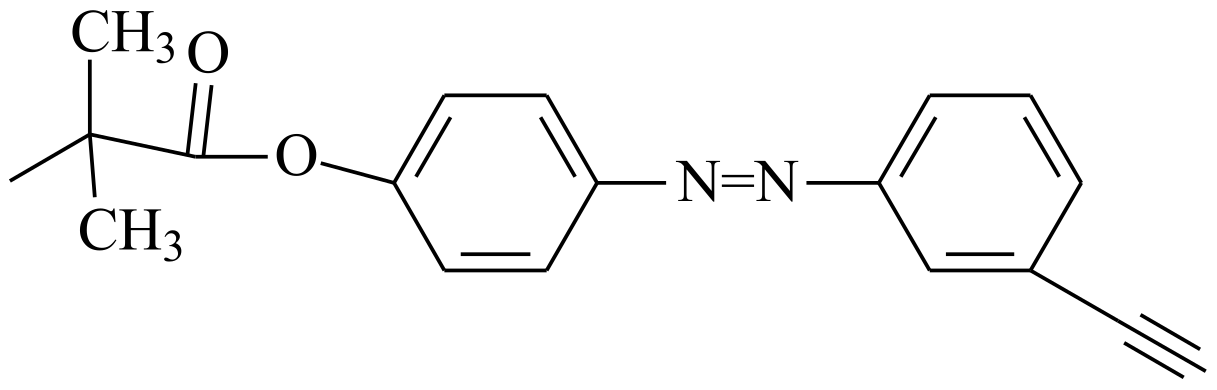 | [50] |
3. ZC(=S)S Group Modification Approach
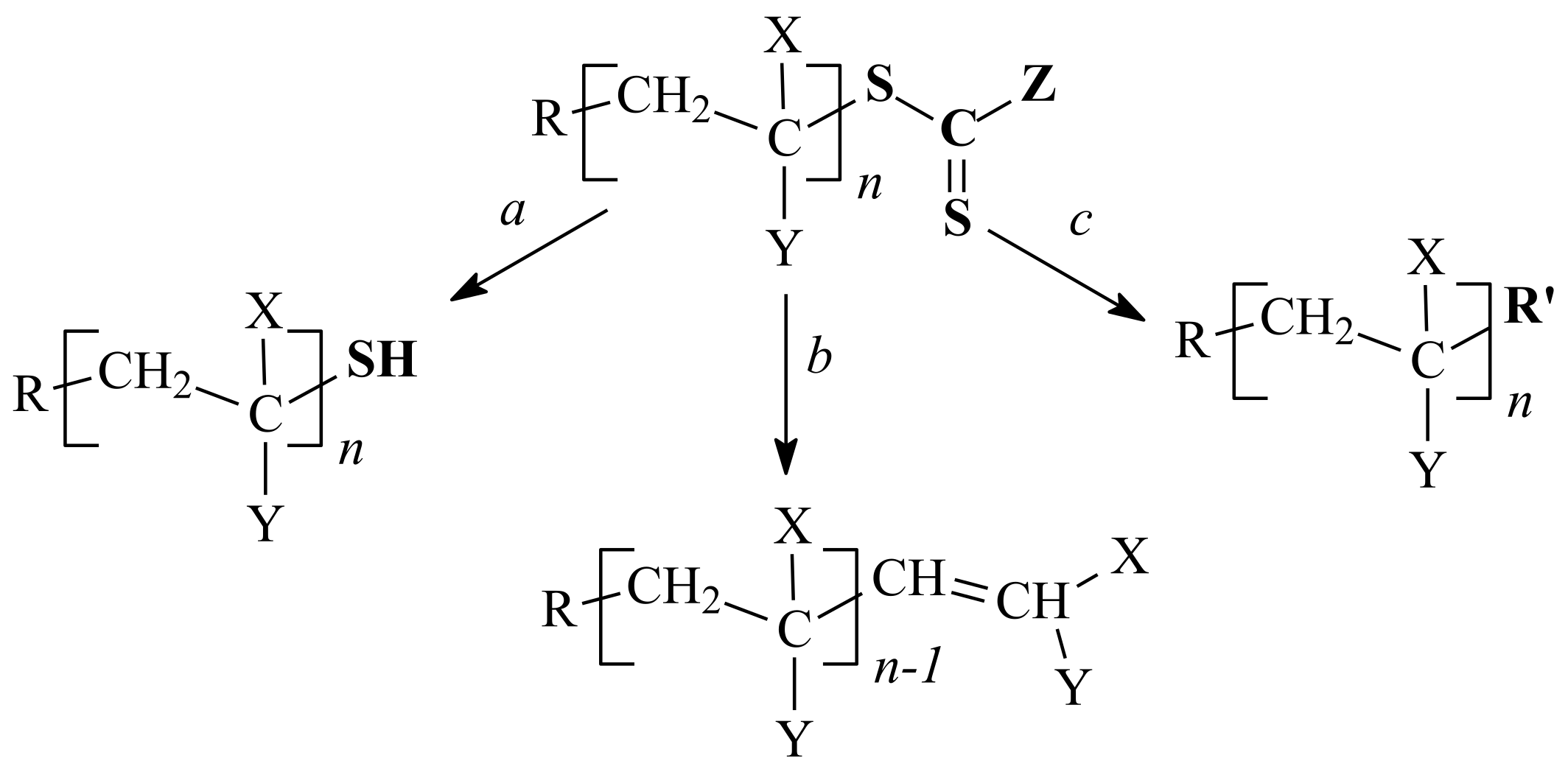
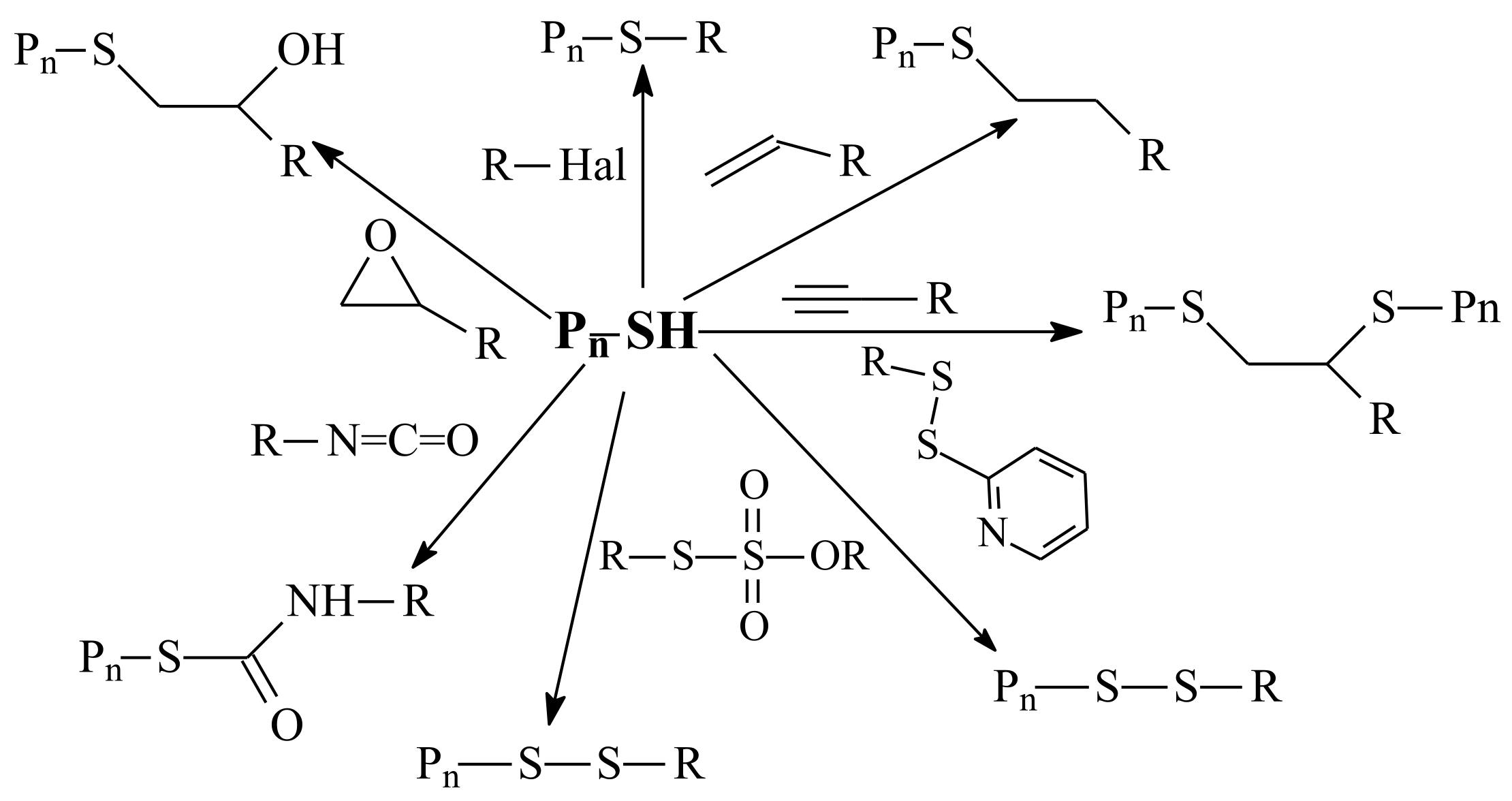
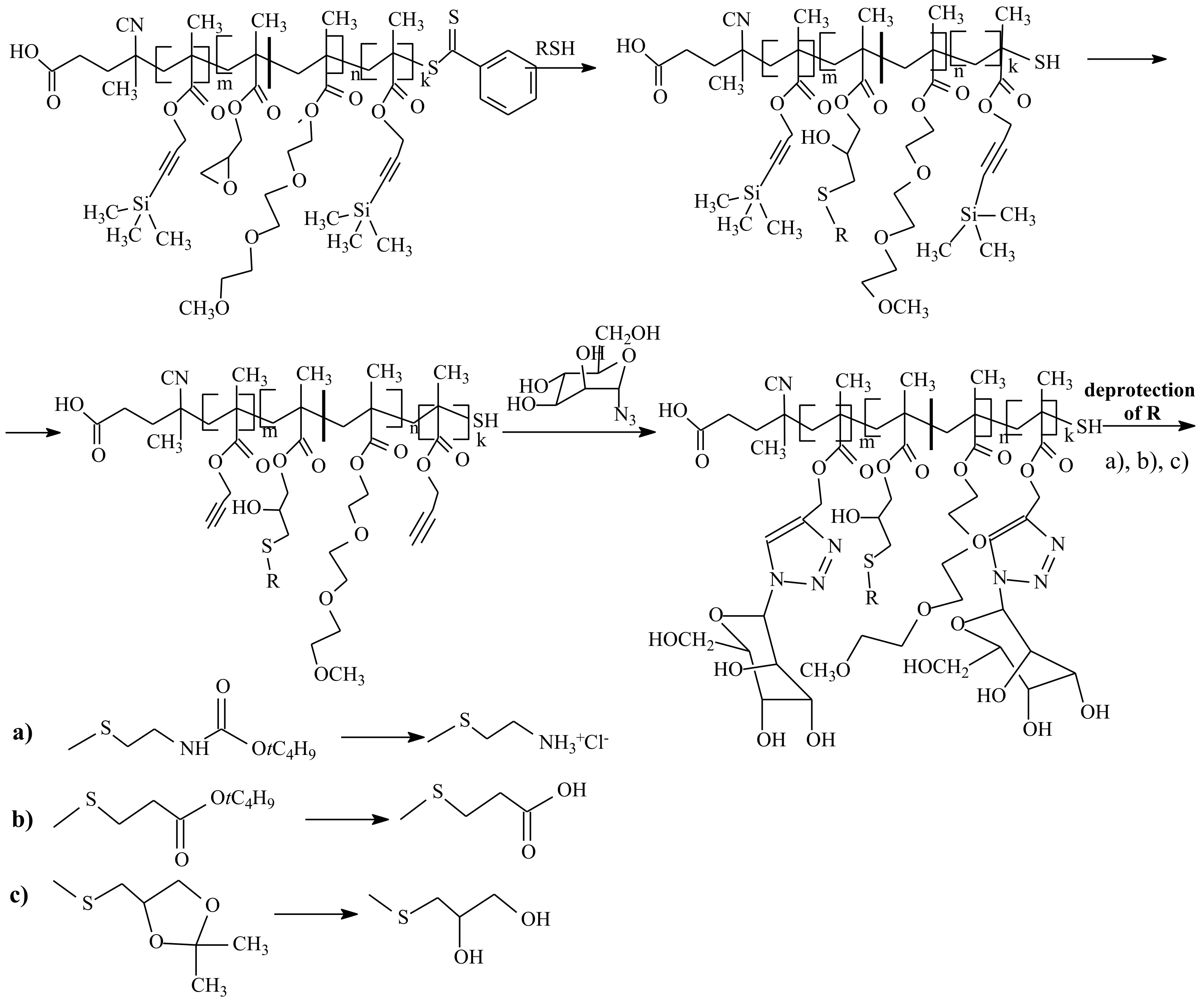
3.1. Conversion of the ZC(=S)S Group to the Thiol Group
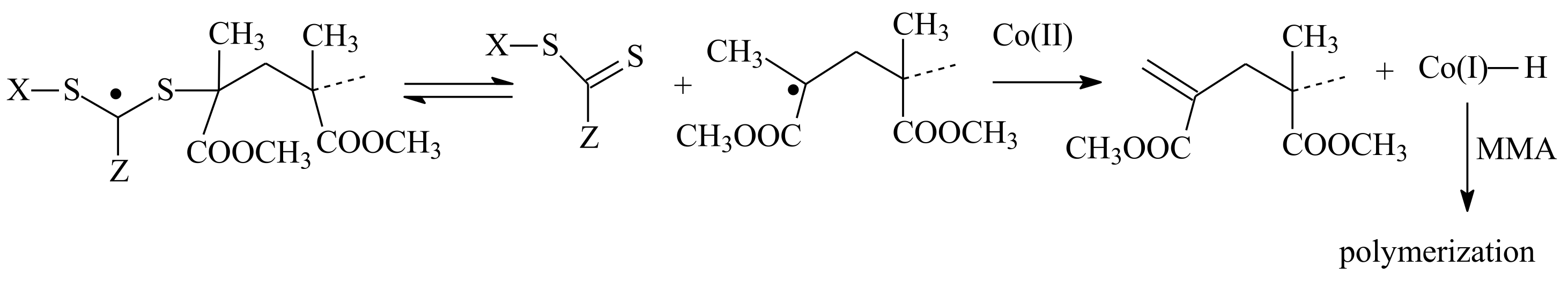
3.2. Conversion of the ZC(=S)S Group to the C=C Group
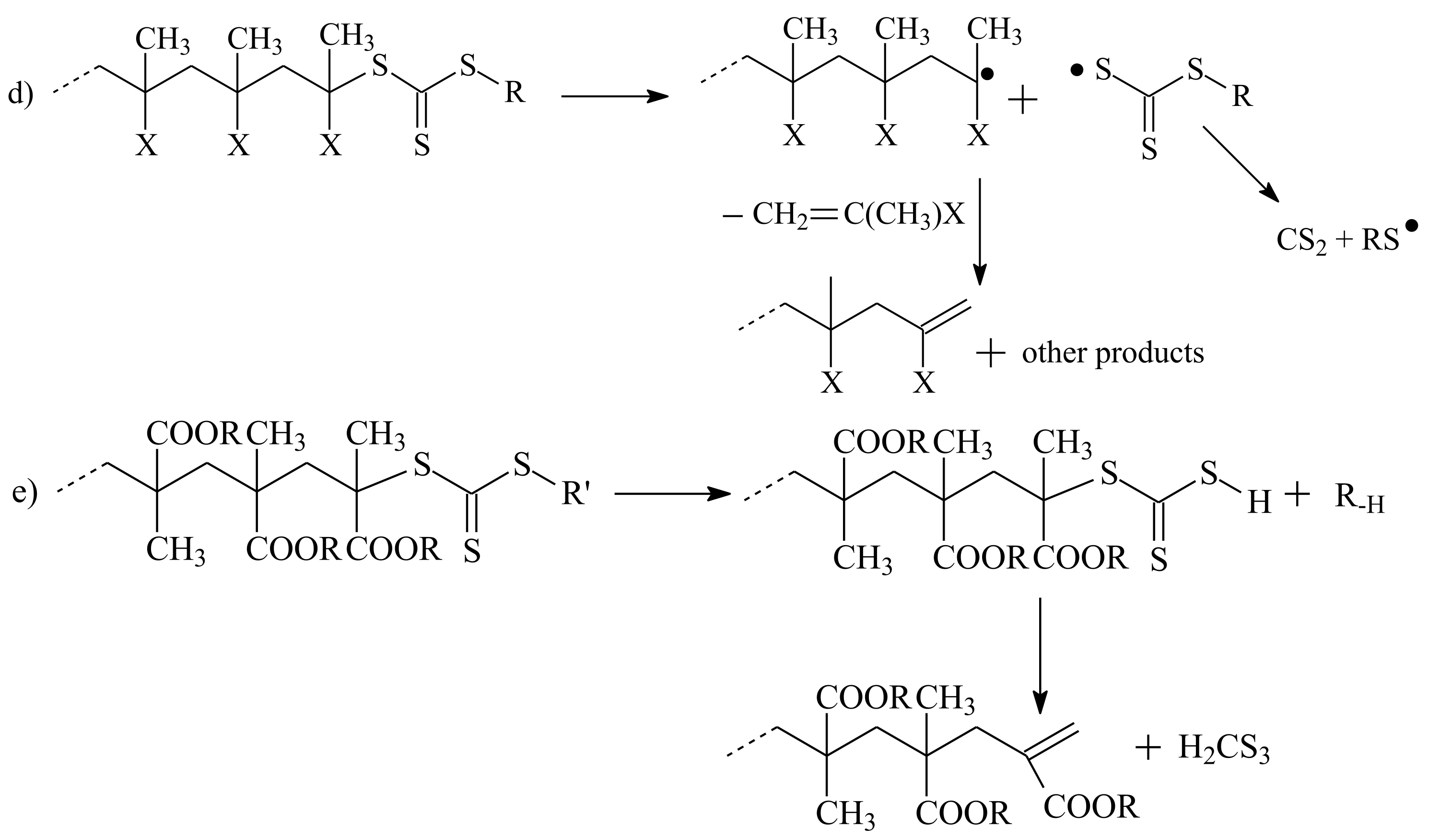
3.3. Reaction of Addition–Fragmentation Coupling of the ZC(=S)S Group with Radical Initiator
4. R Group Modification Approach
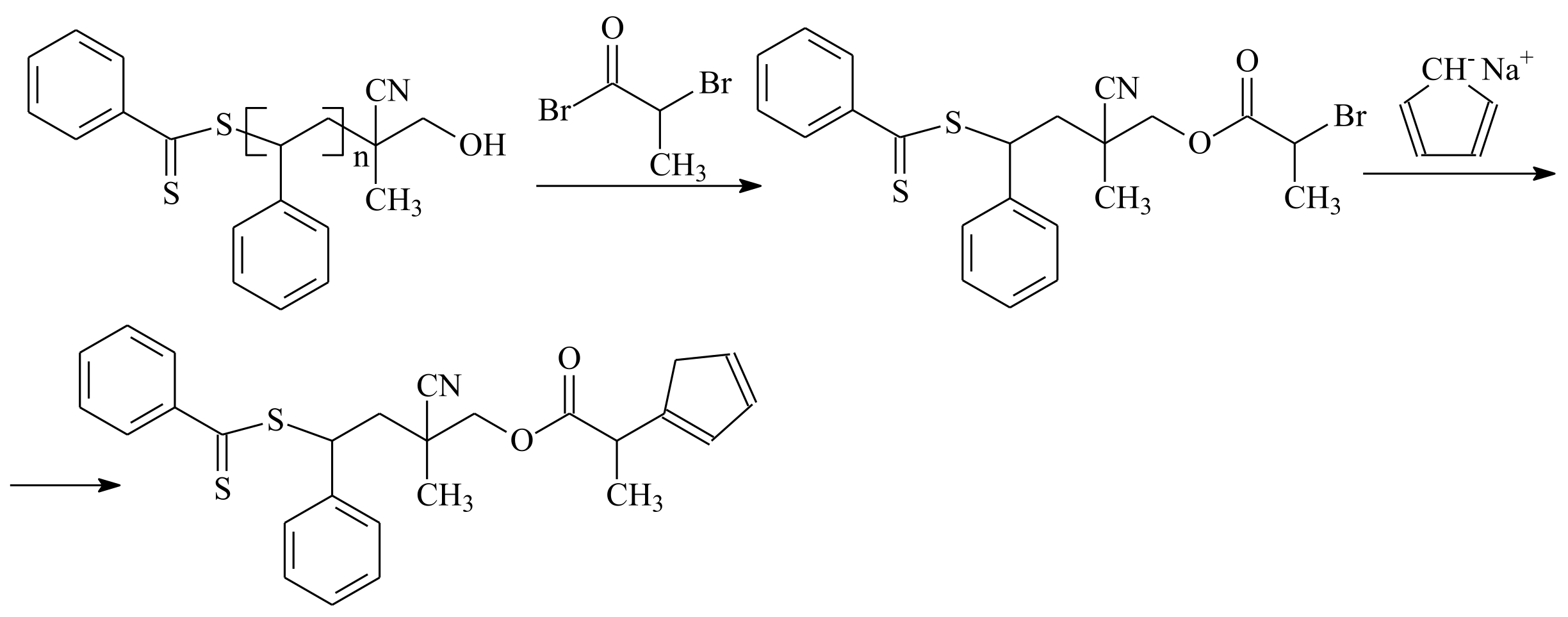
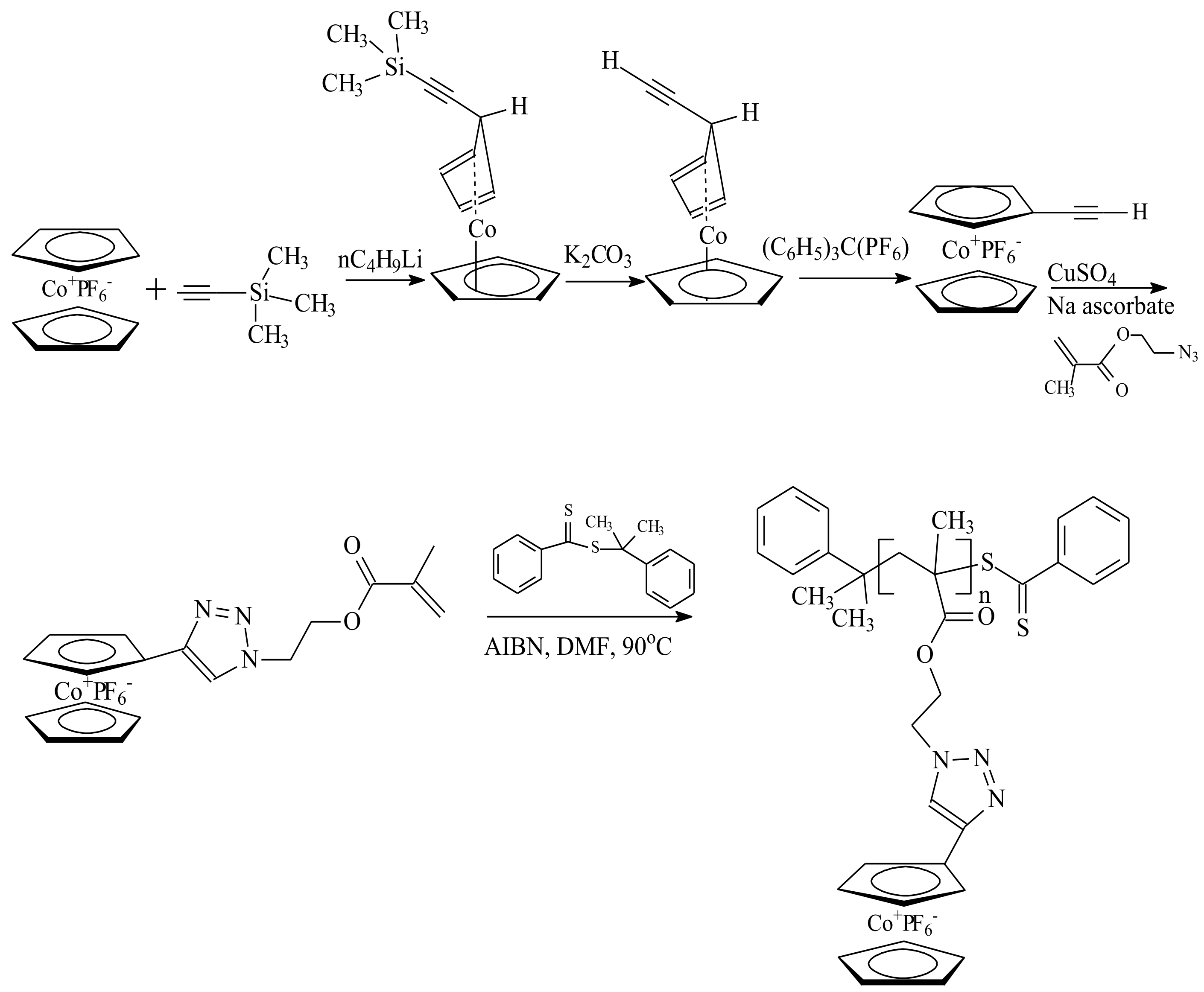
5. Post-Modification of Side-Group Approach
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Muller, A.H.E.; Matyjaszewski, K. (Eds.) Controlled and Living Polymerizations: From Mechanisms to Applicatons; Wiley: Weinheim, Germany, 2009. [Google Scholar] [CrossRef]
- Grubbs, R.B.; Grubbs, R.H. 50th Anniversary Perspective: Living Polymerization—Emphasizing the Molecule in Macromolecules. Macromolecules 2017, 50, 6979–6997. [Google Scholar] [CrossRef]
- Hadjichristidis, N.; Iatrou, H.; Pitsikalis, M.; Mays, J. Macromolecular architectures by living and controlled/living polymerizations. Prog. Polym. Sci. 2006, 31, 1068–1132. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, P.; Wang, U.; Zhang, W. Recent advances in organic/inorganic well-defined hybrid polymers using controlled living radical polymerization techniques. Polym. Chem. 2016, 7, 3950–3976. [Google Scholar] [CrossRef]
- Kolb, H.C.; Finn, M.G.; Sharpless, K.B. Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angew. Chem. Int. Ed. 2001, 40, 2004–2021. [Google Scholar] [CrossRef]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective Ligation of Azides and Terminal Alkynes. Angew. Chem. Int. Ed. 2002, 41, 2596–2599. [Google Scholar] [CrossRef]
- Himo, F.; Lovell, T.; Hilgraf, R.; Rostovtsev, V.V.; Noodleman, L.; Sharpless, K.B.; Fokin, V.V. Copper(I)-Catalyzed Synthesis of Azoles. DFT Study Predicts Unprecedented Reactivity and Intermediates. J. Am. Chem. Soc. 2005, 127, 210–216. [Google Scholar] [CrossRef]
- Worell, B.T.; Malik, J.A.; Fokin, V.V. Direct Evidence of a Dinuclear Copper Intermediate in Cu(I)-Catalyzed Azide-Alkyne Cycloadditions. Science 2013, 340, 457–460. [Google Scholar] [CrossRef] [Green Version]
- Golas, P.L.; Matyjaszewski, K. Click Chemistry and ATRP: A Beneficial Union for the Preparation of Functional Materials. QSAR Comb. Sci. 2007, 26, 1116–1134. [Google Scholar] [CrossRef]
- Binder, W.H.; Sachsenhofer, R. ‘Click’ Chemistry in Polymer and Materials Science. Macromol. Rapid Commun. 2007, 28, 15–54. [Google Scholar] [CrossRef]
- Binder, W.H.; Sachsenhofer, R. ‘Click’ Chemistry in Polymer and Material Science: An Update. Macromol. Rapid Commun. 2008, 29, 952–981. [Google Scholar] [CrossRef]
- Lundberg, P.; Hawker, C.J.; Hult, A.; Malkoch, M. Click Assisted One-Pot Multi-Step Reactions in Polymer Science: Accelerated Synthetic Protocols. Macromol. Rapid Commun. 2008, 29, 998–1015. [Google Scholar] [CrossRef]
- Meldal, M. Polymer “Clicking” by CuAAC Reactions. Macromol. Rapid Commun. 2008, 29, 1016–1051. [Google Scholar] [CrossRef]
- Johnson, J.A.; Finn, M.G.; Koberstein, J.T.; Turro, N.J. Construction of Linear Polymers, Dendrimers, Networks, and Other Polymeric Architectures by Copper-Catalyzed Azide-Alkyne Cycloaddition “Click” Chemistry. Macromol. Rapid Commun. 2008, 29, 1052–1072. [Google Scholar] [CrossRef]
- Le Droumaguet, B.; Velonia, K. Click Chemistry: A Powerful Tool to Create Polymer-Based Macromolecular Chimeras. Macromol. Rapid Commun. 2008, 29, 1073–1089. [Google Scholar] [CrossRef]
- Harvison, M.A.; Lowe, A.B. Combining RAFT Radical Polymerization and Click/Highly Efficient Coupling Chemistries: A Powerful Strategy for the Preparation of Novel Materials. Macromol. Rapid Commun. 2011, 32, 779–800. [Google Scholar] [CrossRef]
- Mansfeld, U.; Pietsch, C.; Hoogenboom, R.; Becer, R.; Schubert, U.S. Clickable initiators, monomers and polymers in controlled radical polymerizations–a prospective combination in polymer science. Polym. Chem. 2010, 1, 1560–1598. [Google Scholar] [CrossRef]
- Gauthier, M.A.; Gibson, M.I.; Klok, H.-A. Synthesis of Functional Polymers by Post-Polymerization Modification. Angew. Chem. Int. Ed. 2009, 48, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Golas, P.L.; Matyjaszewski, K. Marrying click chemistry with polymerization: Expanding the scope of polymeric materials. Chem. Soc. Rev. 2010, 39, 1338–1354. [Google Scholar] [CrossRef]
- Hoyle, C.E.; Lowe, A.B.; Bowman, C.N. Thiol-click chemistry: A multifaceted toolbox for small molecule and polymer synthesis. Chem. Soc. Rev. 2010, 39, 1355–1387. [Google Scholar] [CrossRef]
- Lowe, A.B. Thiol-ene ‘‘click’’ reactions and recent applications in polymer and materials synthesis. Polym. Chem. 2010, 1, 17–36. [Google Scholar] [CrossRef]
- Lowe, A.B. Thiol-ene “click” reactions and recent applications in polymer and materials synthesis: A first update. Polym. Chem. 2014, 5, 4820–4870. [Google Scholar] [CrossRef]
- Lallana, E.; Fernandez-Trillo, F.; Sousa-Herves, A.; Riguera, R.; Fernandez-Megia, E. Click Chemistry with Polymers, Dendrimers, and Hydrogels for Drug Delivery. Pharm. Res. 2012, 29, 902–921. [Google Scholar] [CrossRef] [PubMed]
- Sumerlin, B.S.; Vogt, A.P. Macromolecular Engineering through Click Chemistry and Other Efficient Transformations. Macromolecules 2010, 43, 1–13. [Google Scholar] [CrossRef]
- Liu, Q.; Zhang, Q.; Chen, S.; Zhou, J.; Lei, X. Progress in Thiol-Ene/Yne Click Chemistry. Chin. J. Org. Chem. 2012, 32, 1846–1863. [Google Scholar] [CrossRef] [Green Version]
- Delaittre, G.; Guimard, N.K.; Barner-Kowollik, C. Cycloadditions in Modern Polymer Chemistry. Acc. Chem. Res. 2015, 48, 1296–1307. [Google Scholar] [CrossRef]
- Bhavsar, C.; Momin, M.; Gharat, S.; Omri, A. Functionalized and graft copolymers of chitosan and its pharmaceutical applications. Expert Opin. Drug Deliv. 2017, 14, 1189–1204. [Google Scholar] [CrossRef]
- Goethals, F.; Frank, D.; Du Prez, F.E. Protected Thiol Strategies in Macromolecular Design. Prog. Polym. Sci. 2017, 64, 76–113. [Google Scholar] [CrossRef]
- Huang, Z.; Zhou, Y.; Wang, Z.; Li, Y.; Zhang, W.; Zhou, N.; Zhang, Z.; Zhu, X. Recent Advances of CuAAC Click Reaction in Building Cyclic Polymer. Chinese J. Polym. Sci. 2017, 35, 317–341. [Google Scholar] [CrossRef]
- Wang, Y.; Wu, C. Site-Specific Conjugation of Polymers to Proteins. Biomacromolecules 2018, 19, 1804–1825. [Google Scholar] [CrossRef]
- Arslan, M.; Tasdelen, M.A. Click Chemistry in Macromolecular Design: Complex Architectures from Functional Polymers. Chem. Afr. 2019, 2, 195–214. [Google Scholar] [CrossRef] [Green Version]
- Mohammad, S.A.; Shingdilwar, S.; Banerjee, S.; Ameduri, B. Macromolecular engineering approach for the preparation of new architectures from fluorinated olefins and their applications. Prog. Polym. Sci. 2020, 106, 101255. [Google Scholar] [CrossRef]
- Mondal, P.; Behera, P.K.; Singha, N.K. Macromolecular Engineering in Functional Polymers via ‘Click Chemistry’ Using Triazolinedione Derivatives. Prog. Polym. Sci. 2020, 113, 101343. [Google Scholar] [CrossRef]
- Martin, J.; Desfoux, A.; Martinez, J.; Amblard, M.; Mehdi, A.; Vezenkov, L.; Subra, G. Bottom-up strategies for the synthesis of peptide-based polymers. Prog. Polym. Sci. 2021, 115, 101377. [Google Scholar] [CrossRef]
- D’Acunzo, F.; Masci, G. Playing construction with the monomer toy box for the synthesis of multi-stimuli responsive copolymers by reversible deactivation radical polymerization protocols. J. Polym. Sci. 2021, 59, 3059–3083. [Google Scholar] [CrossRef]
- Worch, J.C.; Stubbs, C.J.; Price, M.J.; Dove, A.P. Click Nucleophilic Conjugate Additions to Activated Alkynes: Exploring Thiol-yne, Amino-yne, and Hydroxyl-yne Reactions from (Bio)Organic to Polymer Chemistry. Chem. Rev. 2021, 121, 6744–6776. [Google Scholar] [CrossRef]
- Chen, C.; Ng, D.Y.W.; Weil, T. Polymer bioconjugates: Modern design concepts toward precision hybrid materials. Prog. Polym. Sci. 2020, 105, 101241. [Google Scholar] [CrossRef]
- Geng, Z.; Shin, J.J.; Xi, Y.; Hawker, C.J. Click chemistry strategies for the accelerated synthesis of functional macromolecules. J. Polym. Sci. 2021, 59, 963–1042. [Google Scholar] [CrossRef]
- Jenkins, A.D.; Jones, R.G.; Moad, G. Terminology for reversible-deactivation radicalpolymerization previously called “controlled”radical or “living” radical polymerization (IUPAC Recommendations 2010). Pure Appl. Chem. 2009, 82, 483–491. [Google Scholar] [CrossRef]
- RAFT Polymerization: Methods, Synthesis, Applications; Moad, G.; Rizzardo, E. (Eds.) Wiley: Weinheim, Germany, 2022. [Google Scholar]
- Moad, G. Dithioesters in RAFT polymerization. In RAFT polymerization. Methods, Synthesis, Applications; Moad, G., Rizzardo, E., Eds.; Wiley-VCH: Weinheim, Germany, 2022; Volume 2, pp. 223–358. [Google Scholar] [CrossRef]
- Moad, G. Trithiocarbonates in RAFT polymerization. In RAFT polymerization. Methods, Synthesis, Applications; Moad, G., Rizzardo, E., Eds.; Wiley-VCH: Weinheim, Germany, 2022; Volume 2, pp. 359–492. [Google Scholar] [CrossRef]
- Wang, M.; Marty, J.-D.; Destarac, M. Xanthates in RAFT polymerization. In RAFT Polymerization. Methods, Synthesis, Applications; Moad, G., Rizzardo, E., Eds.; Wiley-VCH: Weinheim, Germany, 2022; Volume 2, pp. 493–548. [Google Scholar] [CrossRef]
- Moad, G. Dithiocarbamates in RAFT polymerization. In RAFT Polymerization. Methods, Synthesis, Applications; Moad, G., Rizzardo, E., Eds.; Wiley-VCH: Weinheim, Germany, 2022; Volume 2, pp. 549–610. [Google Scholar] [CrossRef]
- Lowe, A.B.; Dallerba, E. RAFT-Functional End Groups: Installation and Transformation. In RAFT Polymerization. Methods, Synthesis, Applications; Moad, G., Rizzardo, E., Eds.; Wiley-VCH: Weinheim, Germany, 2022; Volume 2, pp. 753–804. [Google Scholar] [CrossRef]
- Moad, G.; Rizzardo, E.; Thang, S.H. End-functional polymers, thiocarbonylthio group removal/transformation and reversible addition–fragmentation–chain transfer (RAFT) polymerization. Polym. Int. 2011, 60, 9–25. [Google Scholar] [CrossRef]
- Willcock, H.; O’Reilly, R.K. End group removal and modification of RAFT polymers. Polym. Chem. 2010, 1, 149–157. [Google Scholar] [CrossRef]
- Quinn, J.F.; Moad, G.; Barner-Kowollik, C. RAFT polymerization: Mechanistic considerations. In RAFT Polymerization. Methods, Synthesis, Applications; Moad, G., Rizzardo, E., Eds.; Wiley-VCH: Weinheim, Germany, 2022; Volume 2, pp. 95–137. [Google Scholar] [CrossRef]
- Quémener, D.; Davis, T.P.; Barner-Kowollik, C.; Stenzel, M.H. RAFT and click chemistry: A versatile approach to well-defined block copolymers. Chem. Commun. 2006, 5051–5053. [Google Scholar] [CrossRef]
- Xue, X.; Zhu, J.; Zhang, Z.; Cheng, Z.; Tu, Y.; Zhu, X. Synthesis and characterization of azobenzene-functionalized poly(styrene)-b-poly(vinyl acetate) via the combination of RAFT and “click” chemistry. Polymer 2010, 51, 3083–3090. [Google Scholar] [CrossRef]
- Ting, S.R.S.; Granville, A.M.; Quémener, D.; Davis, T.P.; Stenzel, M.H.; Barner-Kowollik, C. RAFT chemistry and Huisgen 1,3-dipolar cycloaddition: A route to block copolymers of vinyl acetate and 6-O-methacryloyl mannose? Aust. J. Chem. 2007, 60, 405–409. [Google Scholar] [CrossRef]
- Huang, Y.; Hou, T.; Cao, X.; Perrier, S.; Zhao, Y. Synthesis of silica-polymer hybrids by combination of RAFT polymerization and azide-alkyne cycloaddition ‘click’ reactions. Polym. Chem. 2010, 1, 1615–1623. [Google Scholar] [CrossRef]
- Zhao, G.; Zhang, P.; Zhang, C.; Zhao, Y. Facile synthesis of highly pure block copolymers by combination of RAFT polymerization, click reaction and de-grafting process. Polym. Chem. 2012, 3, 1803–1812. [Google Scholar] [CrossRef]
- O’Reilly, R.K.; Joralemon, M.J.; Hawker, C.J.; Wooley, K.L. Facile syntheses of surface-functionalized micelles and shell cross-linked nanoparticles. J. Polym. Sci. Part A Polym. Chem. 2006, 44, 5203–5217. [Google Scholar] [CrossRef]
- Zhang, T.; Zheng, Z.; Ding, X.; Peng, Y. Smart surface of gold nanoparticles fabricated by combination of RAFT and click chemistry. Macromol. Rapid Commun. 2008, 29, 1716–1720. [Google Scholar] [CrossRef]
- Zhu, J.; Zhu, L.; Kang, E.T.; Neoh, K.G. Design and synthesis of star polymers with hetero-arms by the combination of controlled radical polymerizations and click chemistry. Polymer 2007, 48, 6992–6999. [Google Scholar] [CrossRef]
- Gondi, S.R.; Vogt, A.P.; Sumerlin, B.S. Versatile Pathway to Functional Telechelics via RAFT Polymerization and Click Chemistry. Macromolecules 2007, 40, 474–481. [Google Scholar] [CrossRef]
- Shi, G.Y.; Tang, X.Z.; Pan, C.Y. Tadpole-shaped amphiphilic copolymers prepared via RAFT polymerization and click reaction. J. Polym. Sci. Part A Polym. Chem. 2008, 46, 2390–2401. [Google Scholar] [CrossRef]
- Goldmann, A.S.; Quémener, D.; Millard, P.E.; Davis, T.P.; Stenzel, M.H.; Barner-Kowollik, C.; Mueller, A.H.E. Access to cyclic polystyrenes via a combination of reversible addition fragmentation chain transfer (RAFT) polymerization and click chemistry. Polymer 2008, 49, 2274–2281. [Google Scholar] [CrossRef]
- Barner, L.; Davis, T.P.; Stenzel, M.H.; Barner-Kowollik, C. Complex Macromolecular Architectures by Reversible Addition Fragmentation Chain Transfer Chemistry: Theory and Practice. Macromol. Rapid Commun. 2007, 28, 539–559. [Google Scholar] [CrossRef]
- Vogt, A.P.; Sumerlin, B.S. Tuning the Temperature Response of Branched Poly(N-isopropylacrylamide) Prepared by RAFT Polymerization. Macromolecules 2008, 41, 7368–7373. [Google Scholar] [CrossRef]
- De, P.; Gondi, S.R. Folate-Conjugated Thermoresponsive Block Copolymers: Highly Efficient Conjugation and Solution Self-Assembly. Biomacromolecules 2008, 9, 1064–1070. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; De, P.; Gondi, S.R.; Sumerlin, B.S. Responsive Polymer-Protein Bioconjugates Prepared by RAFT Polymerization and Copper-Catalyzed Azide-Alkyne Click Chemistry. Macromol. Rapid Commun. 2008, 29, 1172–1176. [Google Scholar] [CrossRef]
- Vora, A.; Singh, K.; Webster, D.C. A new approach to 3-miktoarm star polymers using a combination of reversible addition–fragmentation chain transfer (RAFT) and ring opening polymerization (ROP) via “Click” chemistry. Polymer 2009, 50, 2768–2774. [Google Scholar] [CrossRef]
- Vogt, A.P.; Gondi, S.R.; Sumerlin, B.S. Hyperbranched Polymers via RAFT Copolymerization of an Acryloyl Trithiocarbonate. Aust. J. Chem. 2007, 60, 396–399. [Google Scholar] [CrossRef]
- Ranjan, R.; Brittain, W.J. Synthesis of High Density Polymer Brushes on Nanoparticles by Combined RAFT Polymerization and Click Chemistry. Macromol. Rapid Commun. 2008, 29, 1104–1110. [Google Scholar] [CrossRef]
- Ranjan, R.; Brittain, W.J. Tandem RAFT Polymerization and Click Chemistry: An Efficient Approach to Surface Modification. Macromol. Rapid Commun. 2007, 28, 2084–2089. [Google Scholar] [CrossRef]
- Magenau, A.J.D.; Martinez-Castro, N.; Savin, D.A.; Storey, R.F. Polyisobutylene RAFT CTA by a Click Chemistry Site Transformation Approach: Synthesis of Poly(isobutylene-b-N-isopropylacrylamide). Macromolecules 2009, 42, 8044–8051. [Google Scholar] [CrossRef]
- Zhang, T.; Wu, V.P.; Pan, X.M.; Zheng, Z.H.; Ding, X.B.; Peng, Y.X. An approach for the surface functionalized gold nanoparticles with pH-responsive polymer by combination of RAFT and click chemistry. Europ. Polym. J. 2009, 45, 1625–1633. [Google Scholar] [CrossRef]
- Ranjan, R.; Brittain, W.J. Combination of Living Radical Polymerization and Click Chemistry for Surface Modification. Macromolecules 2007, 40, 6217–6223. [Google Scholar] [CrossRef]
- Puttick, S.; Irvin, D.J.; Licence, P.; Thurecht, K.J. RAFT-functional ionic liquids: Towards understanding controlled free radical polymerisation in ionic liquids. J. Mater. Chem. 2009, 19, 2679–2682. [Google Scholar] [CrossRef]
- An, Z.S.; Tang, W.; Wu, M.H.; Jiao, Z.; Stucky, G.D. Heterofunctional polymers and core–shell nanoparticlesvia cascade aminolysis/Michael addition and alkyne–azide click reaction of RAFT polymers. Chem. Commun. 2008, 6501–6503. [Google Scholar] [CrossRef]
- Qiu, X.P.; Tanaka, F.; Winnik, F.M. Temperature-Induced Phase Transition of Well-Defined Cyclic Poly(N-isopropylacrylamide)s in Aqueous Solution. Macromolecules 2007, 40, 7069–7071. [Google Scholar] [CrossRef]
- Ladmiral, V.; Legge, T.M.; Zhao, Y.L.; Perrier, S. “Click” Chemistry and Radical Polymerization: Potential Loss of Orthogonality. Macromolecules 2008, 41, 6728–6732. [Google Scholar] [CrossRef]
- Quémener, D.; Le Hellaye, M.; Bissett, C.; Davis, T.P.; Barner-Kowollik, C.; Stenzel, M.H. Graft block copolymers of propargyl methacrylate and vinyl acetate via a combination of RAFT/MADIX and click chemistry: Reaction analysis. J. Polym. Sci. Part. A Polym. Chem. 2008, 46, 155–173. [Google Scholar] [CrossRef]
- Bernard, J.; Save, M.; Arathoon, B.; Charleux, B. Preparation of a xanthate-terminated dextran by click chemistry: Application to the synthesis of polysaccharide-coated nanoparticles via surfactant-free ab initio emulsion polymerization of vinyl acetate. J. Polym. Sci. Part A Polym. Chem. 2008, 46, 2845–2857. [Google Scholar] [CrossRef]
- Chen, F.; Cheng, Z.P.; Zhu, J.; Zhang, W.; Zhu, X.L. Synthesis of poly(vinyl acetate) with fluorescence via a combination of RAFT/MADIX and “click” chemistry. Eur. Polym. J. 2008, 44, 1789–1795. [Google Scholar] [CrossRef]
- Akeroyd, N.; Klumperman, B. The combination of living radical polymerization and click chemistry for the synthesis of advanced macromolecular architectures. Eur. Polym. J. 2011, 47, 1207–1231. [Google Scholar] [CrossRef]
- Mendonca, P.V.; Serra, A.C.; Popov, A.V.; Guliashvili, T.; Coelho, J.F.J. Efficient RAFT polymerization of N-(3-aminopropyl)methacrylamide hydrochloride using unprotected ‘‘clickable’’ chain transfer agents. React. Funct. Polym. 2014, 81, 1–7. [Google Scholar] [CrossRef]
- Hansell, C.F.; Espeel, P.; Stamenovic, M.M.; Barker, I.A.; Dove, A.P.; Du Prez, F.E.; O’Reilly, R.K. Additive-free clicking for polymer functionalization and coupling by tetrazine–norbornene chemistry. J. Am. Chem. Soc. 2011, 133, 13828–13831. [Google Scholar] [CrossRef]
- Koo, S.P.S.; Stamenovic, M.M.; Prasath, R.A.; Inglis, A.J.; Du Prez, F.E.; Barner-Kowollik, C.; Van Camp, W.; Junkers, T. Limitations of radical thiol-ene reactions for polymer–polymer conjugation. J. Polym. Sci. Part A Polym. Chem. 2010, 48, 1699–1713. [Google Scholar] [CrossRef] [Green Version]
- Carboni, R.A.; Lindsey, R.V., Jr. Reactions of Tetrazines with Unsaturated Compounds. A New Synthesis of Pyridazines. J. Am. Chem. Soc. 1959, 81, 4342–4346. [Google Scholar] [CrossRef]
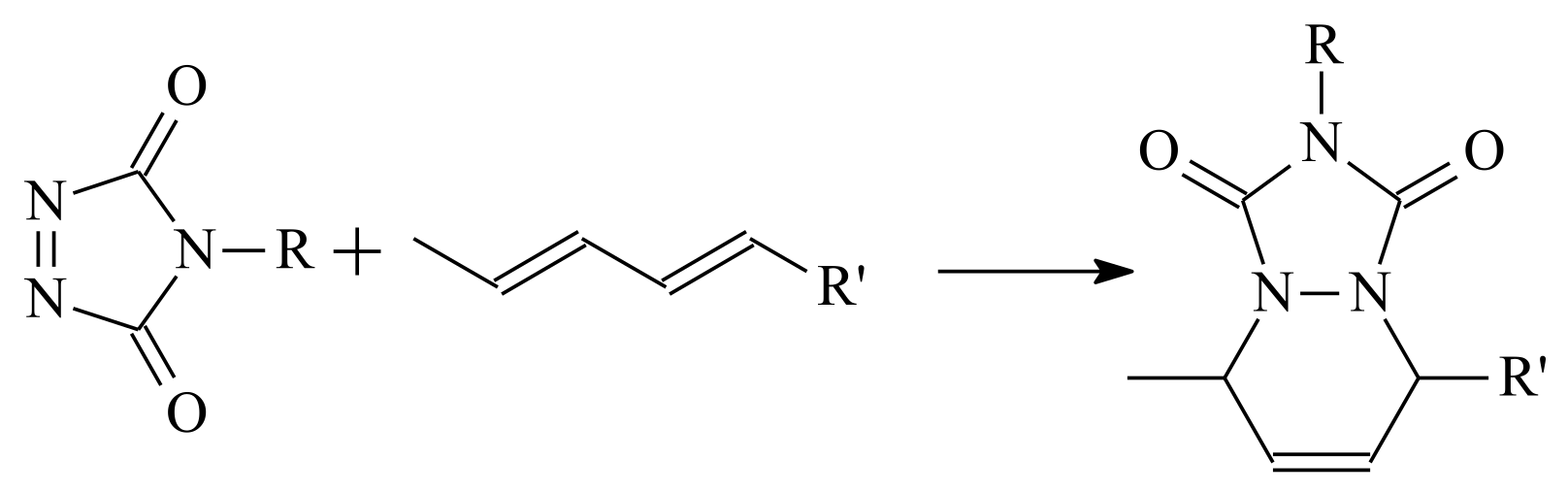
- Vandewalle, S.; Billiet, S.; Driessen, F.; Du Prez, F.E. Macromolecular coupling in seconds of triazolinedione end-functionalized polymers prepared by RAFT polymerization. ACS Macro Lett. 2016, 5, 766–771. [Google Scholar] [CrossRef]
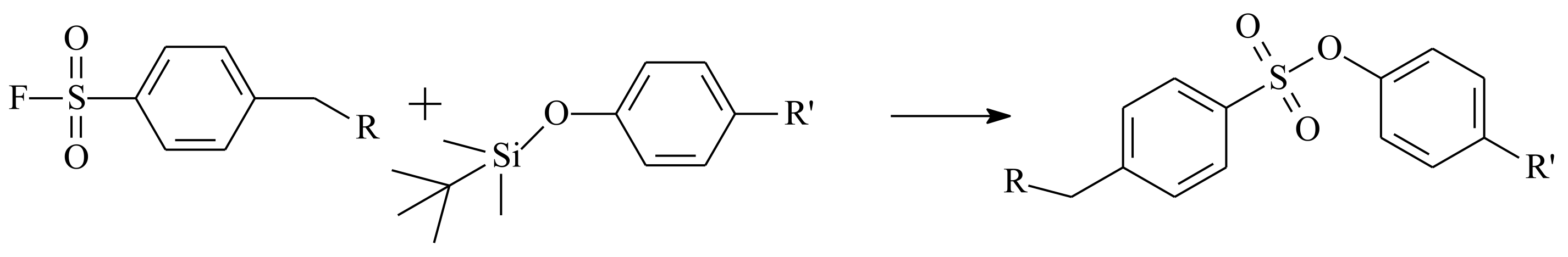
- Dong, J.; Krasnova, L.; Finn, M.G.; Sharpless, K.B. Sulfur(VI) Fluoride Exchange (SuFEx): Another Good Reaction for Click Chemistry. Angew. Chem. Int. Ed. 2014, 53, 9430–9448. [Google Scholar] [CrossRef]
- Dong, J.; Sharpless, K.B.; Kwisnek, L.; Oakdale, J.S.; Fokin, V.V. SuFEx-Based Synthesis of Polysulfates. Angew. Chem. Int. Ed. 2014, 53, 9466–9470. [Google Scholar] [CrossRef] [PubMed]
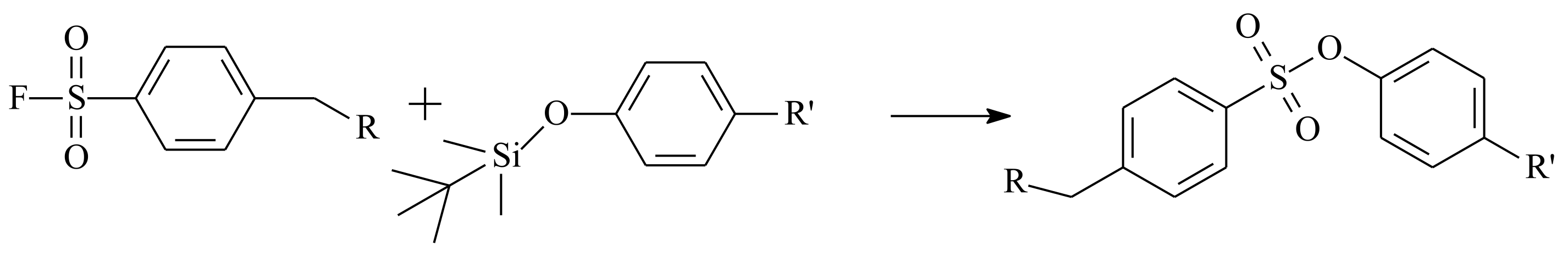
- Yatvin, J.; Brooks, K.; Locklin, J. SuFEx on the Surface: A Flexible Platform for Postpolymerization Modification of Polymer Brushes. Angew. Chem. Int. Ed. 2015, 54, 13370–13373. [Google Scholar] [CrossRef]
- Oakdale, J.S.; Kwisnek, L.; Fokin, V.V. Selective and Orthogonal Post-Polymerization Modification using Sulfur(VI) Fluoride Exchange (SuFEx) and Copper-Catalyzed Azide–Alkyne Cycloaddition (CuAAC) Reactions. Macromolecules 2016, 49, 4473–4479. [Google Scholar] [CrossRef]
- Chen, W.; Dong, J.; Plate, L.; Mortenson, D.E.; Brighty, G.J.; Li, S.; Liu, Y.; Galmozzi, A.; Lee, P.S.; Hulce, J.J.; et al. Arylfluorosulfates Inactivate Intracellular Lipid Binding Protein(s) through Chemoselective SuFEx Reaction with a Binding Site Tyr Residue. J. Am. Chem. Soc. 2016, 138, 7353–7364. [Google Scholar] [CrossRef] [Green Version]
- Brendel, J.C.; Martin, L.; Zhang, J.; Perrier, S. SuFEx–a selectively triggered chemistry for fast, efficient and equimolar polymer–polymer coupling reactions. Polym. Chem. 2017, 8, 7475–7485. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Dong, Y.; Lu, X.; Wu, Z.; Chen, H. Combining click sulfur (VI)-fluoride exchange with photoiniferters: A facile, fast, and efficient strategy for postpolymerization modification. Macromol. Rapid Commun. 2018, 39, 1700523. [Google Scholar] [CrossRef] [PubMed]
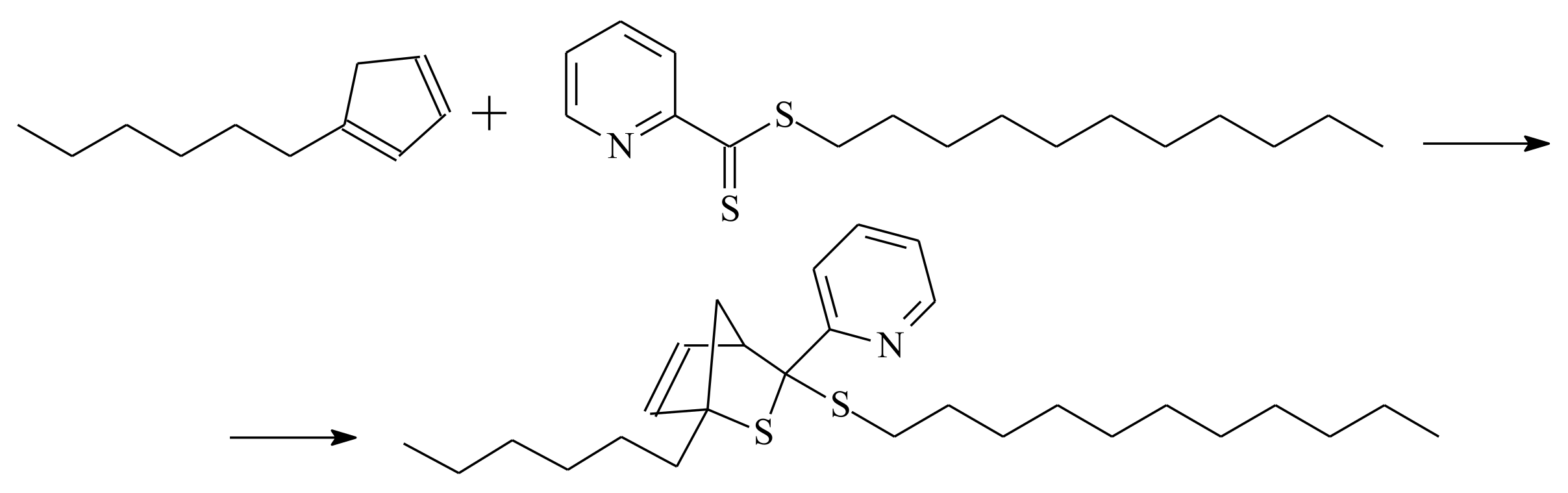
- Sinnwell, S.; Inglis, A.J.; Davis, T.P.; Stenzel, M.H.; Barner-Kowollik, C. An atom-efficient conjugation approach to well-defined block copolymers using RAFT chemistry and hetero Diels–Alder cycloaddition. Chem. Commun. 2008, 17, 2052–2054. [Google Scholar] [CrossRef] [PubMed]
- Inglis, A.J.; Stenzel, M.H.; Barner-Kowollik, C. Ultra-fast RAFT-HDA click conjugation: An efficient route to high molecular weight block copolymers. Macromol. Rapid Commun. 2009, 30, 1792–1798. [Google Scholar] [CrossRef]
- Langer, M.; Mueller, J.O.; Goldmann, A.S.; Schacher, F.H.; Barner-Kowollik, C. α,ω-Reactive building blocks based on a dual functional RAFT agent for thermal and light-induced ligation. ACS Macro Lett. 2016, 5, 597–601. [Google Scholar] [CrossRef]
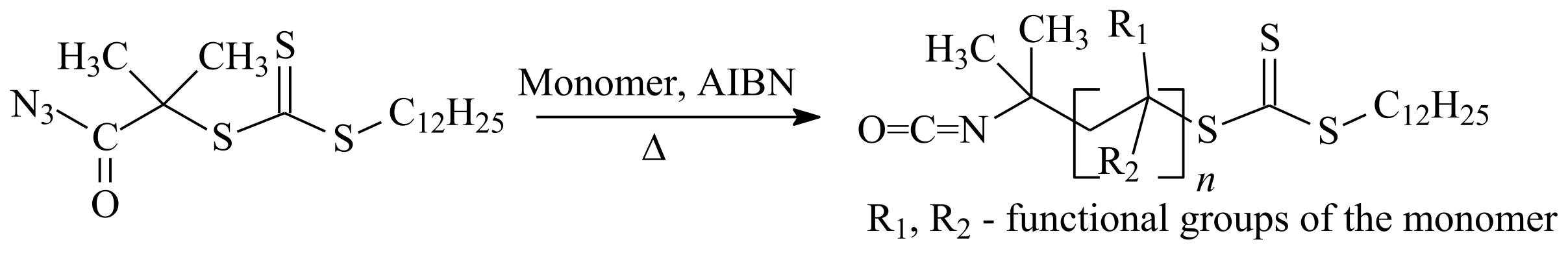
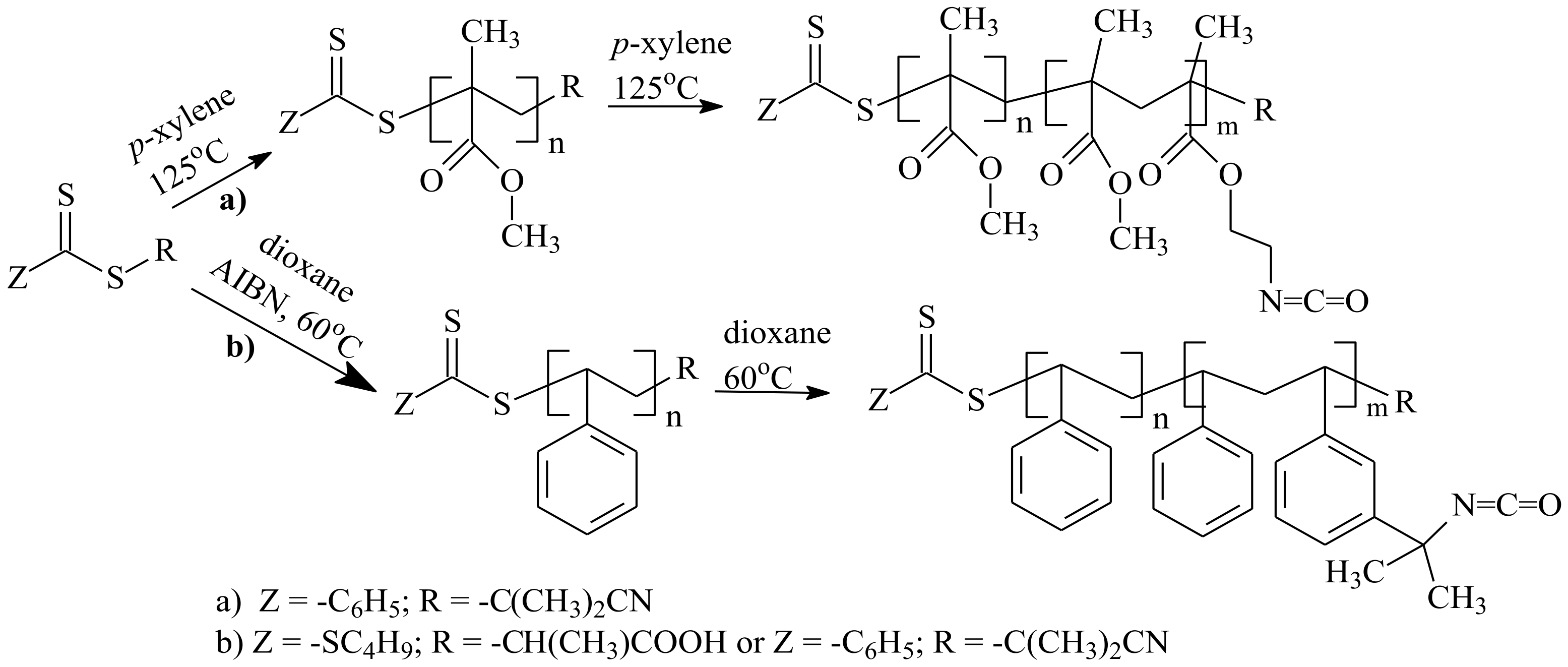
- Gody, G.; Rossner, C.; Moraes, J.; Vana, P.; Maschmeyer, T.; Perrier, S. One-Pot RAFT/“Click” Chemistry via Isocyanates: Efficient Synthesis of α-End-Functionalized Polymers. J. Am. Chem. Soc. 2012, 134, 12596–12603. [Google Scholar] [CrossRef] [PubMed]
- Boyer, C.; Liu, J.; Bulmus, V.; Davis, T.P.; Barner-Kowollik, C.; Stenzel, M.H. Direct Synthesis of Well-Defined Heterotelechelic Polymers for Bioconjugations. Macromolecules 2008, 41, 5641–5650. [Google Scholar] [CrossRef]
- Liu, J.; Bulmus, V.; Herlambang, D.L.; Barner-Kowollik, C.; Stenzel, M.H.; Davis, T.P. In Situ Formation of Protein–Polymer Conjugates through Reversible Addition Fragmentation Chain Transfer Polymerization. Angew. Chem. Int. Ed. 2007, 46, 3099–3103. [Google Scholar] [CrossRef] [PubMed]
- Heredia, K.L.; Nguyen, T.H.; Chang, C.-W.; Bulmus, V.; Davis, T.P.; Maynard, H.D. Reversible siRNA–polymer conjugates by RAFT polymerization. Chem. Commun. 2008, 3245–3247. [Google Scholar] [CrossRef] [Green Version]
- Boyer, C.; Bulmus, V.; Liu, J.; Davis, T.P.; Stenzel, M.H.; Barner-Kowollik, C. Well-Defined Protein−Polymer Conjugates via in Situ RAFT Polymerization. J. Am. Chem. Soc. 2007, 129, 7145–7154. [Google Scholar] [CrossRef]
- Boyer, C.; Liu, J.; Wong, M.; Tippett, M.; Bulmus, V.; Davis, T.P. Stability and utility of pyridyl disulfide functionality in RAFT and conventional radical polymerizations. J. Polym. Sci. Part A Polym. Chem. 2008, 46, 7207–7224. [Google Scholar] [CrossRef]
- Liu, J.; Liu, H.; Boyer, C.; Bulmus, V.; Davis, T.P. Approach to peptide decorated micelles via RAFT polymerization. J. Polym. Sci. Part A Polym. Chem. 2009, 47, 899–912. [Google Scholar] [CrossRef]
- Decker, C.G.; Maynard, H.D. Degradable PEGylated protein conjugates utilizing RAFT polymerization. Eur. Polym. J. 2015, 65, 305–312. [Google Scholar] [CrossRef] [Green Version]
- Martens, S.; Driessen, F.; Wallyn, S.; Türünç, O.; Du Prez, F.E.; Espeel, P. One-Pot Modular Synthesis of Functionalized RAFT Agents Derived from a Single Thiolactone Precursor. ACS Macro Lett. 2016, 5, 942–945. [Google Scholar] [CrossRef]
- Pereira, S.O.; Trindade, T.; Barros-Timmons, A. Biofunctional Polymer Coated Au Nanoparticles Prepared via RAFT-Assisted Encapsulating Emulsion Polymerization and Click Chemistry. Polymers 2020, 12, 1442. [Google Scholar] [CrossRef] [PubMed]
- Lowe, A.B. End-group functionalization of RAFT-prepared polymers using thiol-X chemistries. In Tiol-X Chemistries in Polymer and Materials Science; Lowe, A.B., Bowman, C.N., Eds.; RSC Publishing: London, UK, 2013; Chapter 2; pp. 28–58. [Google Scholar] [CrossRef]
- Duret, D.; Haftek-Terreau, Z.; Carretier, M.; Ladavière, C.; Charreyre, M.-T.; Favier, A. Fluorescent RAFT polymers bearing a nitrilotriacetic acid (NTA) ligand at the α-chainend for the site-specific labeling of histidinetagged proteins. Polym. Chem. 2017, 8, 1611–1615. [Google Scholar] [CrossRef]
- Lee, I.-H.; Discekici, E.H.; Shankel, S.L.; Anastasaki, A.; Read de Alaniz, J.; Hawker, C.J.; Lunn, D.J. Desulfurization–bromination: Direct chain-end modification of RAFT polymers. Polym. Chem. 2017, 8, 7188–7194. [Google Scholar] [CrossRef]
- Dao, V.H.; Cameron, N.R.; Saito, K. Synthesis of ultra-high molecular weight ABA triblock copolymers via aqueous RAFT-mediated gel polymerisation, end group modification and chain coupling. Polym. Chem. 2017, 8, 6834–6843. [Google Scholar] [CrossRef] [Green Version]
- Espeel, P.; Du Prez, F. One-pot multi-step reactions based on thiolactone chemistry: A powerful synthetic tool in polymer science. Eur. Polym. J. 2015, 62, 247–272. [Google Scholar] [CrossRef]
- Qui, X.; Winnik, F.M. Facile and Efficient One-Pot Transformation of RAFT Polymer End Groups via a Mild Aminolysis/Michael Addition Sequence. Macromol. Rapid Commun. 2006, 27, 1648–1653. [Google Scholar] [CrossRef]
- Zhang, Q.; Voorhaar, L.; Geest, B.G.D.; Hoogenboom, R. One-Pot Preparation of Inert Well-Defined Polymers by RAFT Polymerization and In Situ End Group Transformation. Macromol. Rapid. Commun. 2015, 36, 1177–1183. [Google Scholar] [CrossRef]
- Xu, L.; He, J.; Fan, D.; Wang, X.; Yang, Y. Aminolysis of Polymers with Thiocarbonylthio Termini Prepared by RAFT Polymerization: The Difference between Polystyrene and Polymethacrylates. Macromolecules 2006, 39, 8616–8624. [Google Scholar] [CrossRef]
- Langlais, M.; Coutelier, O.; Destarac, M. Thiolactone-Functional Reversible Deactivation Radical Polymerization Agents for Advanced Macromolecular Engineering. Macromolecules 2018, 51, 4315–4324. [Google Scholar] [CrossRef]
- Glaria, A.; Beija, M.; Bordes, R.; Desterac, M.; Marty, J. Understanding the Role of ω-End Groups and Molecular Weight in the Interaction of PNIPAM with Gold Surfaces. Chem. Mater. 2013, 25, 1868–1876. [Google Scholar] [CrossRef]
- Espeel, P.; Goethlas, F.; Du Prez, F.E. One-Pot Multistep Reactions Based on Thiolactones: Extending the Realm of Thiol−Ene Chemistry in Polymer Synthesis. J. Am. Chem. Soc. 2011, 133, 1678–1681. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zhou, Y.; Zhu, J.; Zhang, W.; Pan, X.; Zhang, Z.; Zhu, X. Fast conversion of terminal thiocarbonylthio groups of RAFT polymers to “clickable” thiol groups via versatile sodium azide. Polym. Chem. 2014, 5, 5546–5550. [Google Scholar] [CrossRef]
- Harvison, M.A.; Davis, T.P.; Lowe, A.B. Macromolecular thiolysis of oxiranes: End-group modification of RAFT prepared homopolymers. Polym. Chem. 2011, 2, 1347–1354. [Google Scholar] [CrossRef]
- Shen, W.; Qui, Q.; Wang, Y.; Miao, M.; Li, B.; Znang, T.; Cao, A.; An, Z. Hydrazine as a Nucleophile and Antioxidant for Fast Aminolysis of RAFT Polymers in Air. Macromol. Rapid Commun. 2010, 31, 1444–1448. [Google Scholar] [CrossRef]
- Shan, J.; Nuopponen, M.; Jiang, H.; Kauppinen, E.; Tenhu, H. Preparation of Poly(N-isopropylacrylamide)-Monolayer-Protected Gold Clusters: Synthesis Methods, Core Size, and Thickness of Monolayer. Macromolecules 2003, 36, 4526–4533. [Google Scholar] [CrossRef]
- Lee, C.-U.; Roy, D.; Sumerlin, B.S.; Dadmun, M.D. Facile synthesis of thiol-terminated poly(styrene-ran-vinyl phenol) (PSVPh) copolymers via reversible addition-fragmentation chain transfer (RAFT) polymerization and their use in the synthesis of gold nanoparticles with controllable hydrophilicity. Polymer 2010, 51, 1244–1251. [Google Scholar] [CrossRef]
- Yu, S.H.; Hu, J.; Ercole, F.; Truong, N.P.; Davis, T.P.; Whittaker, M.R.; Quinn, J.F. Transformation of RAFT Polymer End Groups into Nitric Oxide Donor Moieties: En Route to Biochemically Active Nanostructures. ACS Macro Lett. 2015, 4, 1278–1282. [Google Scholar] [CrossRef]
- Yang, K.; Huang, X.; Zhu, M.; Xie, L.; Tanaka, T.; Jiang, P. Combining RAFT Polymerization and Thiol−Ene Click Reaction for Core−Shell Structured Polymer@BaTiO3 Nanodielectrics with High Dielectric Constant, Low Dielectric Loss, and High Energy Storage Capability. ACS Appl. Mater. Interfaces 2014, 6, 1812–1822. [Google Scholar] [CrossRef] [PubMed]
- Llauro, M.-F.; Loiseau, J.; Boisson, F.; Delolme, F.; Ladavier, C. Unexpected end-groups of poly(acrylic acid) prepared by RAFT polymerization. J. Polym. Sci. Part A Polym. Chem. 2004, 42, 5439–5462. [Google Scholar] [CrossRef]
- Levit, M.; Zashikhina, N.; Dobrodumov, A.; Kashina, A.; Tarasenko, I.; Panarin, E.; Fiorucci, S.; Korzhikova-Vlakh, E.; Tennikova, T. Synthesis and characterization of well-defined poly(2-deoxy-2-methacrylamido-d-glucose) and its biopotential block copolymers via RAFT and ROP polymerization. Eur. Polym. J. 2018, 105, 26–37. [Google Scholar] [CrossRef]
- Litmanovich, E.A.; Bekanova, M.Z.; Shandryuk, G.A.; Chernikova, E.V.; Talroze, R.V. “Macromolecules–ghosts” in dynamic light scattering analysis: An approach to study interaction between CdSe quantum dots and RAFT-based poly(methyl methacrylate). Polymer 2018, 142, 1–10. [Google Scholar] [CrossRef]
- Lowe, A.B.; Sumerlin, B.S.; Donovan, M.S.; McCormick, C.L. Facile Preparation of Transition Metal Nanoparticles Stabilized by Well-Defined (Co)polymers Synthesized via Aqueous Reversible Addition-Fragmentation Chain Transfer Polymerization. J. Am. Chem. Soc. 2002, 124, 11562–11563. [Google Scholar] [CrossRef] [PubMed]
- Sumerlin, B.S.; Lowe, A.B.; Stroud, P.A.; Zhang, P.; Urban, M.W.; McCormick, C.L. Modification of Gold Surfaces with Water-Soluble (Co)polymers Prepared via Aqueous Reversible Addition-Fragmentation Chain Transfer (RAFT) Polymerization. Langmuir 2003, 19, 5559–5562. [Google Scholar] [CrossRef]
- Scales, C.W.; Convertine, A.J.; McCormick, C.L. Fluorescent Labeling of RAFT-Generated Poly(N-isopropylacrylamide) via a Facile Maleimide−Thiol Coupling Reaction. Biomacromolecules 2006, 7, 1389–1392. [Google Scholar] [CrossRef]
- Shan, J.; Zhao, Y.; Granqvist, N.; Tenhu, H. Thermoresponsive Properties of N-Isopropylacrylamide Oligomer Brushes Grafted to Gold Nanoparticles: Effects of Molar Mass and Gold Core Size. Macromolecules 2009, 42, 2696–2701. [Google Scholar] [CrossRef]
- Zhu, M.-Q.; Wang, L.-Q.; Exarhos, G.J.; Li, A.D.Q. Thermosensitive Gold Nanoparticles. J. Am. Chem. Soc. 2004, 126, 2656–2657. [Google Scholar] [CrossRef]
- Gibson, M.I.; Fröhlich, E.; Klok, H.-A. Postpolymerization modification of poly(pentafluorophenyl methacrylate): Synthesis of a diverse water-soluble polymer library. J. Polym. Sci. Part A Polym. Chem. 2009, 47, 4332–4345. [Google Scholar] [CrossRef]
- Nishi, H.; Kobatake, S. Reduction Reaction to Thiol Group of Dithiobenzoate End Group in Polystyrene Polymerized by Reversible Addition–Fragmentation Chain Transfer. Chem. Lett. 2008, 37, 630–631. [Google Scholar] [CrossRef]
- Spruell, J.M.; Levy, B.A.; Sutherland, A.; Dichtel, W.R.; Cheng, J.Y.; Stoddart, J.F. Facile postpolymerization end-modification of RAFT polymers. J. Polym. Sci. Part A Polym. Chem. 2009, 47, 346–356. [Google Scholar] [CrossRef]
- Yu, B.; Chan, J.W.; Hoyle, C.E.; Lowe, A.B. Sequential thiol-ene/thiol-ene and thiol-ene/thiol-yne reactions as a route to well-defined mono and bis end-functionalized poly(N-isopropylacrylamide). J. Polym. Sci. Part A Polym. Chem. 2009, 47, 3544–3557. [Google Scholar] [CrossRef]
- Lima, V.; Jiang, X.L.; Brokken-Zijp, J.; Schoenmakers, P.J.; Klumperman, B.; van der Linde, R. Synthesis and characterization of telechelic polymethacrylates via RAFT polymerization. J. Polym. Sci. Part A Polym. Chem. 2005, 43, 959–973. [Google Scholar] [CrossRef]
- Gemici, H.; Legge, T.M.; Whittaker, M.; Monteiro, M.J.; Perrier, S. Original approach to multiblock copolymers via reversible addition–fragmentation chain transfer polymerization. J. Polym. Sci. Part A Polym. Chem. 2007, 45, 2334–2340. [Google Scholar] [CrossRef]
- Wang, Z.M.; He, J.P.; Tao, Y.F.; Yang, L.; Jiang, H.J.; Yang, Y.L. Controlled Chain Branching by RAFT-Based Radical Polymerization. Macromolecules 2003, 20, 7446–7452. [Google Scholar] [CrossRef]
- Chang, C.-W.; Bays, E.; Tao, L.; Alconcel, S.N.S.; Maynard, H.D. Differences in cytotoxicity of poly(PEGA)s synthesized by reversible addition–fragmentation chain transfer polymerization. Chem. Commun. 2009, 3580–3582. [Google Scholar] [CrossRef] [Green Version]
- Carlson, J.S.; Hill, M.R.; Young, T.; Costanzo, P.J. Novel polymer coupling chemistry based upon latent cysteine-like residues and thiazolidine chemistry. Polym. Chem. 2010, 1, 1423–1426. [Google Scholar] [CrossRef] [Green Version]
- You, Y.-Z.; Manickam, D.S.; Zhou, Q.-H.; Oupicky, D. A Versatile Approach to Reducible Vinyl Polymers via Oxidation of Telechelic Polymers Prepared by Reversible Addition Fragmentation Chain Transfer Polymerization. Biomacromolecules 2007, 8, 2038–2044. [Google Scholar] [CrossRef]
- Patton, D.L.; Advincula, R.C. A Versatile Synthetic Route to Macromonomers via RAFT Polymerization. Macromolecules 2006, 39, 8674–8683. [Google Scholar] [CrossRef]
- Xu, J.; Tao, L.; Boyer, C.; Lowe, A.B.; Davis, T.P. Combining Thio−Bromo “Click” Chemistry and RAFT Polymerization: A Powerful Tool for Preparing Functionalized Multiblock and Hyperbranched Polymers. Macromolecules 2010, 43, 20–24. [Google Scholar] [CrossRef]
- Kim, B.J.; Bang, J.; Hawker, C.J.; Chiu, J.J.; Pine, D.; Jang, S.G.; Yang, S.-M.; Kramer, E.J. Creating Surfactant Nanoparticles for Block Copolymer Composites through Surface Chemistry. Langmuir 2007, 23, 12693–12703. [Google Scholar] [CrossRef]
- Goldmann, A.S.; Walther, A.; Nebhani, L.; Joso, R.; Ernst, D.; Loos, K. Surface Modification of Poly(divinylbenzene) Microspheres via Thiol−Ene Chemistry and Alkyne−Azide Click Reactions. Macromolecules 2009, 42, 3707–3714. [Google Scholar] [CrossRef] [Green Version]
- Segui, F.; Qui, X.-P.; Winnik, F.M. An efficient synthesis of telechelic poly (N-isopropylacrylamides) and its application to the preparation of α,ω-dicholesteryl and α,ω-dipyrenyl polymers. J. Polym. Sci. Part A Polym. Chem. 2008, 46, 314–326. [Google Scholar] [CrossRef]
- Qui, X.-P.; Winnik, F.M. Synthesis of α,ω-Dimercapto Poly(N-isopropylacrylamides) by RAFT Polymerization with a Hydrophilic Difunctional Chain Transfer Agent. Macromolecules 2007, 40, 872–878. [Google Scholar] [CrossRef]
- Ezhov, A.A.; Karpov, O.N.; Merekalov, A.S.; Abramchuk, S.S.; Bondarenko, G.N.; Talroze, R.V. Quantum dots-Polymer composites and the influence of gold nanoparticles on photoluminescence of polymer composite films. J. Luminesc. 2020, 220, 116992. [Google Scholar] [CrossRef]
- Malic, N.; Evans, R.A. Synthesis of Carboxylic Acid and Ester Mid-Functionalized Polymers using RAFT Polymerization and ATRP. Aust. J. Chem. 2006, 59, 763–771. [Google Scholar] [CrossRef]
- Kulkami, S.; Schili, C.; Grin, B.; Muller, A.H.E.; Hoffmann, A.S.; Stayton, P.S. Controlling the Aggregation of Conjugates of Streptavidin with Smart Block Copolymers Prepared via the RAFT Copolymerization Technique. Biomacromolecules 2006, 7, 2736–2741. [Google Scholar] [CrossRef]
- Zelikin, A.N.; Suck, G.K.; Postma, A.; Caruso, F. Poly(vinylpyrrolidone) for Bioconjugation and Surface Ligand Immobilization. Biomacromolecules 2007, 8, 2950–2953. [Google Scholar] [CrossRef]
- Holbein, B.E.; Ang, M.T.C.; Palaskar, D.V.; Satyanarayana, G.; Reddy, S.V.B.; Ali, S. Polymeric metal chelating compositions and methods of preparing same for controlling growth and activitiesof living cells and organisms. US Patent 20190169126A1, 2012. [Google Scholar]
- Shimoni, O.; Postma, A.; Yan, Y.; Scott, A.M.; Heath, J.K.; Nice, E.C.; Zelikin, A.N.; Caruso, F. Macromolecule functionalization of disulfide-bonded polymer hydrogel capsules and cancer cell targeting. ACS Nano 2012, 6, 1463–1472. [Google Scholar] [CrossRef] [Green Version]
- Kulkami, S.; Schili, C.; Muller, A.H.E.; Hoffmann, A.S.; Stayton, P.S. Reversible Meso-Scale Smart Polymer−Protein Particles of Controlled Sizes. Bioconj. Chem. 2004, 15, 747–753. [Google Scholar] [CrossRef]
- Le Neindre, M.; Magny, B.; Nicolay, R. Evaluation of thiocarbonyl and thioester moieties as thiol protecting groups for controlled radical polymerization. Polym. Chem. 2013, 4, 5577–5584. [Google Scholar] [CrossRef]
- Desmet, G.B.; D’hooge, D.R.; Sabbe, M.K.; Reyniers, M.-F.; Marin, G.B. Computational Investigation of the Aminolysis of RAFT Macromolecules. J. Org. Chem. 2016, 81, 11626–11634. [Google Scholar] [CrossRef] [PubMed]
- Lewis, R.W.; Evans, R.A.; Malic, N.; Saito, K.; Cameron, N.R. Cleavage of macromolecular RAFT chain transfer agents by sodium azide during characterization by aqueous GPC. Polym. Chem. 2017, 8, 3702–3711. [Google Scholar] [CrossRef] [Green Version]
- Leon, N.H.; Asquith, R.S. Preparation of dithioesters by ester interchange and the PMR spectral properties of these compounds. Tetrehedron 1970, 26, 1719–1725. [Google Scholar] [CrossRef]
- Chernikova, E.; Morozov, A.; Leonova, K.; Garina, E.; Golubev, V.; Bui, C.; Charleux, B. Controlled Free-Radical Polymerization of n-Butyl Acrylate by Reversible Addition−Fragmentation Chain Transfer in the Presence of tert-Butyl Dithiobenzoate. A Kinetic Study. Macromolecules 2004, 37, 6329–6339. [Google Scholar] [CrossRef]
- Chernikova, E.V.; Tarasenko, A.V.; Garina, E.S.; Golubev, V.B. Controlled radical polymerization of styrene mediated by dithiobenzoates as reversible addition-fragmentation chain-transfer agents. Polym. Sci. Ser. A 2006, 48, 1046–1057. [Google Scholar] [CrossRef]
- Cerda, M.M.; Newton, T.D.; Zhao, Y.; Collins, B.K.; Hendon, C.H.; Pluth, M.D. Dithioesters: Simple, tunable, cysteine-selective H2S donors. Chem. Sci. 2019, 10, 1773–1779. [Google Scholar] [CrossRef] [Green Version]
- Urquhart, M.C.; Dao, N.V.; Ercole, F.; Boyd, B.J.; Davis, T.P.; Whittaker, M.R.; Quinn, J.F. Polymers with Dithiobenzoate End Groups Constitutively Release Hydrogen Sulfide upon Exposure to Cysteine and Homocysteine. ACS Macro Lett. 2020, 9, 553–557. [Google Scholar] [CrossRef]
- Destarac, M.; Kalai, C.; Wilczewska, A.; Petit, L.; Gramberen, E.V.; Zard, S.Z. Various strategies for the chemical transformation of xanthate-functional chain termini in MADIX copolymers. Control./Living Radic. Polym. ACS Symp. Ser. 2006, 944, 564–577. [Google Scholar] [CrossRef]
- Postma, A.; Davis, T.P.; Li, G.; Moad, G.; O’Shea, M.S. RAFT Polymerization with Phthalimidomethyl Trithiocarbonates or Xanthates. On the Origin of Bimodal Molecular Weight Distributions in Living Radical Polymerization. Macromolecules 2006, 39, 5307–5318. [Google Scholar] [CrossRef]
- Coutelier, O.; Blidi, I.; Destarac, M. Xanthate-derived mercaptophosphonates for thiol-ene modification of styrene-butadiene rubber. Eur. Polym. J. 2021, 151, 110419. [Google Scholar] [CrossRef]
- Wu, J.; Huang, C.; Liang, W.; Wu, Y.; Yu, J.; Chen, H. Reactive Polymer Coatings: A General Route to Thiol-ene and Thiol-yne Click Reactions. Macromol. Rapid Commun. 2012, 33, 922–927. [Google Scholar] [CrossRef] [PubMed]
- Postma, A.; Davis, T.P.; Moad, G.; O’Shea, M.S. Thermolysis of RAFT-Synthesized Polymers. A Convenient Method for Trithiocarbonate Group Elimination. Macromolecules 2005, 38, 5371–5374. [Google Scholar] [CrossRef]
- Patton, D.L.; Mullings, M.; Fulghum, T.; Advincula, R.C. A Facile Synthesis Route to Thiol-Functionalized α,ω-Telechelic Polymers via Reversible Addition Fragmentation Chain Transfer Polymerization. Macromolecules 2005, 38, 8597–8602. [Google Scholar] [CrossRef]
- Chong, B.; Moad, G.; Rizzardo, E.; Skidmore, M.; Thang, S.H. Thermolysis of RAFT-Synthesized Poly(Methyl Methacrylate). Aust. J. Chem. 2006, 59, 755–762. [Google Scholar] [CrossRef]
- Altintas, O.; Riazi, K.; Lee, R.; Lin, C.Y.; Coote, M.L.; Wilhelm, M.; Barner-Kowollik, C. RAFT-based Polystyrene and Polyacrylate Melts under Thermal and Mechanical Stress. Macromolecules 2013, 46, 8079–8091. [Google Scholar] [CrossRef] [Green Version]
- Altintas, O.; Abbasi, M.; Riazi, K.; Goldmann, A.S.; Dingenouts, N.; Wilhelm, M.; Barner-Kowollik, C. Stability of star-shaped RAFT polystyrenes under mechanical and thermal stress. Polym. Chem. 2014, 5, 5009–5019. [Google Scholar] [CrossRef] [Green Version]
- Chernikova, E.V.; Plutalova, A.V.; Garina, E.S.; Vishnevetsky, D.V. Thermal stability of styrene/n-butyl acrylate RAFT-based copolymers. Polym. Chem. 2016, 7, 3622–3632. [Google Scholar] [CrossRef]
- Stace, S.J.; Fellows, C.M.; Moad, G.; Keddie, D.J. Effect of the Z- and Macro-R-Group on the Thermal Desulfurization of Polymers Synthesized with Acid/Base “Switchable” Dithiocarbamate RAFT Agents. Macromol. Rapid Commun. 2018, 39, 1800228. [Google Scholar] [CrossRef]
- Xu, J.; He, J.; Fan, D.; Tang, W.; Yang, Y. Thermal Decomposition of Dithioesters and Its Effect on RAFT Polymerization. Macromolecules 2006, 39, 3753–3759. [Google Scholar] [CrossRef]
- Zhou, Y.; He, J.; Li, C.; Hong, L.; Yang, Y. Dependence of Thermal Stability on Molecular Structure of RAFT/MADIX Agents: A Kinetic and Mechanistic Study. Macromolecules 2011, 44, 8446–8457. [Google Scholar] [CrossRef]
- Hornung, C.H.; Postma, A.; Saubern, S.; Chiefari, J. Sequential flow process for the controlled polymerisation and thermolysis of RAFT-synthesised polymers. Polymer 2014, 55, 1427–1435. [Google Scholar] [CrossRef]
- Bressy, C.; Ngo, V.G.; Margaillan, A. A first insight into the thermal degradation mechanism of silylated methacrylic homopolymers synthesized via the RAFT process. Polym. Degrad. Stab. 2013, 98, 115–121. [Google Scholar] [CrossRef]
- DePuy, C.H.; King, R.W. Pyrolytic Cis Eliminations. Chem. Rev. 1960, 60, 431–457. [Google Scholar] [CrossRef]
- Bekanova, M.Z.; Neumolotov, N.K.; Jablanovic, A.D.; Plutalova, A.V.; Chernikova, E.V.; Kudryavtsev, Y.V. Thermal stability of RAFT-based poly(methyl methacrylate): A kinetic study of the dithiobenzoate and trithiocarbonate end-group effect. Polym. Degrad. Stab. 2019, 164, 18–27. [Google Scholar] [CrossRef]
- Legge, T.M.; Slark, A.T.; Perrier, S. Thermal Stability of Reversible Addition–Fragmentation Chain Transfer/Macromolecular Architecture Design by Interchange of Xanthates Chain-Transfer Agents. J. Polym. Sci. Part A Polym. Chem. 2006, 44, 6980–6987. [Google Scholar] [CrossRef]
- Zhang, X.; Jiang, J.; Zhang, Y. Liquid-phase catalytic thermal cleavage of thiocarbonylthio end-groups for polymers synthesized by RAFT polymerization: Influences of different solvents. J. Appl. Polym. Sci. 2016, 133, 43992. [Google Scholar] [CrossRef]
- Roy, S.G.; Bauri, K.; Pal, S.; Goswami, A.; Madras, G.; De, P. Synthesis, characterization and thermal degradation of dual temperature- and pH-sensitive RAFT-made copolymers of N,N-(dimethylamino)ethyl methacrylate and methyl methacrylate. Polym. Int. 2013, 62, 463. [Google Scholar] [CrossRef]
- Chugaev, L. Ueber eine neue Methode zur Darstellung ungesättigter Kohlenwasserstoffe. Ber. Dtsch. Chem. Ges. 1899, 32, 3332–3335. [Google Scholar] [CrossRef] [Green Version]
- Soeriyadi, A.H.; Boyer, C.; Burns, J.; Becer, C.R.; Whittaker, M.R.; Haddleton, D.M.; Davis, T.P. High fidelity vinyl terminated polymers by combining RAFT and cobaltcatalytic chain transfer (CCT) polymerization methods. Chem. Commun. 2010, 46, 6338–6340. [Google Scholar] [CrossRef] [PubMed]
- Perrier, S.; Takolpuckdee, P.; Mars, C.A. Reversible Addition−Fragmentation Chain Transfer Polymerization: End Group Modification for Functionalized Polymers and Chain Transfer Agent Recovery. Macromolecules 2005, 38, 2033–2036. [Google Scholar] [CrossRef]
- Chong, Y.K.; Moad, G.; Rizzardo, E.; Thang, S.H. Thiocarbonylthio End Group Removal from RAFT-Synthesized Polymers by Radical-Induced Reduction. Macromolecules 2007, 40, 4446–4455. [Google Scholar] [CrossRef]
- Chen, M.; Moad, G.; Rizzardo, E. Thiocarbonylthio end group removal from RAFT-synthesized polymers by a radical-induced process. J. Polym. Sci. Part A Polym. Chem. 2009, 47, 6704–6714. [Google Scholar] [CrossRef]
- Vo, C.-D.; Rosselgong, J.; Armes, S.P.; Tirelli, N. Stimulus-responsive polymers based on 2-hydroxypropyl acrylate prepared by RAFT polymerization. J. Polym. Sci. Part A Polym. Chem. 2010, 48, 2032–2043. [Google Scholar] [CrossRef]
- Bekanova, M.Z.; Neumolotov, N.K.; Jablanovic, A.D.; Plutalova, A.V.; Chernikova, E.V. Radical Replacement of the Dithiocarbonyl Group of Poly(methyl methacrylate) Obtained by Reversible Addition–Fragmentation Chain Transfer Polymerization. Polym. Sci. Ser. C. 2019, 61, 188–199. [Google Scholar] [CrossRef]
- Scheibe, P.; Barz, M.; Hemmelmann, M.; Zentel, R. Langmuir−Blodgett Films of Biocompatible Poly(HPMA)-block-poly(lauryl methacrylate) and Poly(HPMA)-random-poly(lauryl methacrylate): Influence of Polymer Structure on Membrane Formation and Stability. Langmuir 2010, 26, 5661–5669. [Google Scholar] [CrossRef]
- Tanaka, S.; Nishida, H.; Endo, T. Miscibility of Polystyrene with One Hydroxystyrene Chain End into Poly(butyl methacrylate). Macromolecules 2009, 42, 293–298. [Google Scholar] [CrossRef]
- Zhou, Y.; Jiang, K.; Song, Q.; Liu, S. Thermo-Induced Formation of Unimolecular and Multimolecular Micelles from Novel Double Hydrophilic Multiblock Copolymers of N,N-Dimethylacrylamide and N-Isopropylacrylamide. Langmuir 2007, 23, 13076–13084. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, J.; Li, W.; Zhang, A. Synthesis and Characterization of Thermo- and pH-Responsive Double-Hydrophilic Diblock Copolypeptides. Biomacromolecules 2007, 8, 3557–3567. [Google Scholar] [CrossRef]
- Tong, Y.-Y.; Dong, Y.-Q.; Du, F.-S.; Li, Z.-C. Block Copolymers of Poly(ethylene oxide) and Poly(vinyl alcohol) Synthesized by the RAFT Methodology. J. Polym. Sci. Part A Polym. Chem. 2009, 47, 1901–1910. [Google Scholar] [CrossRef]
- Zhang, W.; Yan, Y.; Zhu, X.; Qui, Y.; Cheng, Z.; Zhu, J.; Zhang, Z. Chlorodithiocarbamate-Mediated RAFT Polymerization: A Novel Synthetic Method for ATRP Macroinitiators. Macromol. React. Eng. 2010, 4, 264–271. [Google Scholar] [CrossRef]
- Charmot, D.; Piotti, M. Removal of the Thiocarbonylthio or Thiophosphorylthio End Group of Polymers and Further Functionalization Thereof (Symyx Technologies, Inc.). US Patent 691,940,9B2, 2005. Available online: https://patents.google.com/patent/US6919409B2/en (accessed on 10 January 2022).
- Bohec, M.L.; Pioge, S.; Pascual, S.; Fontaine, L. Heterofunctional RAFT-derived PNIPAM via cascade trithiocarbonate removal and thiol-yne coupling click reaction. J. Polym. Sci. Part A Polym. Chem. 2017, 55, 3597–3606. [Google Scholar] [CrossRef]
- Kinoshita, K.; Takami, T.; Mori, Y.; Uchida, Y.; Murakami, Y. RAFT-based synthesis and the gelation property of telechelic polymers that can immobilize biomacromolecules. J. Polym. Sci. Part A Polym. Chem. 2017, 55, 1356–1365. [Google Scholar] [CrossRef]
- Roth, P.J.; Wiss, K.T.; Zentel, R.; Theato, P. Synthesis of Reactive Telechelic Polymers Based on Pentafluorophenyl Esters. Macromolecules 2008, 41, 8513–8519. [Google Scholar] [CrossRef] [Green Version]
- Heredia, K.L.; Grover, G.N.; Tao, L.; Maynard, H.D. Synthesis of Heterotelechelic Polymers for Conjugation of Two Different Proteins. Macromolecules 2009, 42, 2360–2367. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Zhou, Y.; Zhou, X.; Ma, L.; Wang, B.; Yu, C.-Y.; Wei, H. Synthesis of a triple-responsive double hydrophilic block copolymer prodrug using a reducible RAFT-ATRP double-head agent. ACS Appl. Polym. Mater. 2020, 2, 2126–2133. [Google Scholar] [CrossRef]
- Jin, Y.; Zhu, J.; Zhang, Z.; Cheng, Z.; Zhang, W.; Zhu, X. Synthesis and characterizations of 1,2,3-triazole containing polymers via reversible addition-fragmentation chain transfer (RAFT) polymerization. Eur. Polym. J. 2008, 44, 1743–1751. [Google Scholar] [CrossRef]
- Yan, Y.; Zhang, J.; Qiao, Y.; Tang, C. Facile preparation of cobaltocenium-containing polyelectrolyte via click chemistry and RAFT polymerization. Macromol. Rapid. Commun. 2014, 35, 254–259. [Google Scholar] [CrossRef]
- Wu, D.; Song, X.; Tang, T.; Zhao, H. Macromolecular brushes synthesized by ‘‘grafting from‘‘ approach based on ‘‘click chemistry’’ and RAFT polymerization. J. Polym. Sci. Part A Polym. Chem. 2010, 48, 443–453. [Google Scholar] [CrossRef]
- Li, Y.; Yang, J.; Benicewicz, B.C. Well-controlled polymerization of 2-azidoethyl methacrylate at near room temperature and click functionalization. J. Polym. Sci. Part A Polym. Chem. 2007, 45, 4300–4308. [Google Scholar] [CrossRef]
- Li, G.; Wang, H.; Zheng, H.; Bai, R. Room-temperature RAFT copolymerization of 2-chloroallyl azide with methyl acrylate and versatile applications of the azide copolymers. J. Polym. Sci. Part A Polym. Chem. 2010, 48, 1348–1356. [Google Scholar] [CrossRef]
- Ebbesen, M.F.; Schaffert, D.H.; Crowley, M.L.; Oupicky, D.; Howard, K.A. Synthesis of click-reactive HPMA copolymers using RAFT polymerization for drug delivery applications. J. Polym. Sci. Part A Polym. Chem. 2013, 51, 5091–5099. [Google Scholar] [CrossRef]
- Tian, J.; Xiao, C.; Huang, B.; Wang, C.; Zhang, W. Janus macromolecular brushes for synergistic cascade-amplified photodynamic therapy and enhanced chemotherapy. Acta Biomater. 2020, 101, 495–506. [Google Scholar] [CrossRef]
- Thankappan, H.; Semsarilar, M.; Li, S.; Chang, Y.; Bouyer, D.; Quémener, D. Synthesis of block copolymer brush by RAFT and click chemistry and its self-assembly as a thin film. Molecules 2020, 25, 4774. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.R.; Bach, L.G.; Park, J.M.; Hong, S.-S.; Lim, K.T. Synthesis and characterization of poly(HEMA-co-MMA)-g-POSS nanocomposites by combination of reversible addition fragmentation chain transfer polymerization and click chemistry. J. Appl. Polym. Sci. 2013, 127, 1569–1577. [Google Scholar] [CrossRef]
- Bach, L.G.; Cao, X.T.; Islam, M.R.; Jeong, Y.T.; Kim, J.S.; Lim, K.T. Synthesis and characterization of multiwalled carbon nanotubes/poly(HEMA-co-MMA) by utilizing click chemistry. J. Nanosci. Nanotechnol. 2016, 16, 2975–2978. [Google Scholar] [CrossRef]
- Li, Y.; Benicewicz, B.C. Functionalization of silica nanoparticles via the combination of surface-initiated RAFT polymerization and click reactions. Macromolecules 2008, 41, 7986–7992. [Google Scholar] [CrossRef]
- Zhang, W.; Zhang, W.; Zhang, Z.; Zhu, J.; Zhu, X. SET-RAFT Polymerization of propargyl methacrylate and a one-pot/one-step preparation of side-chain functionalized polymers via combination of SET-RAFT and click chemistry. Macromol. Rapid Commun. 2010, 31, 1354–1358. [Google Scholar] [CrossRef]
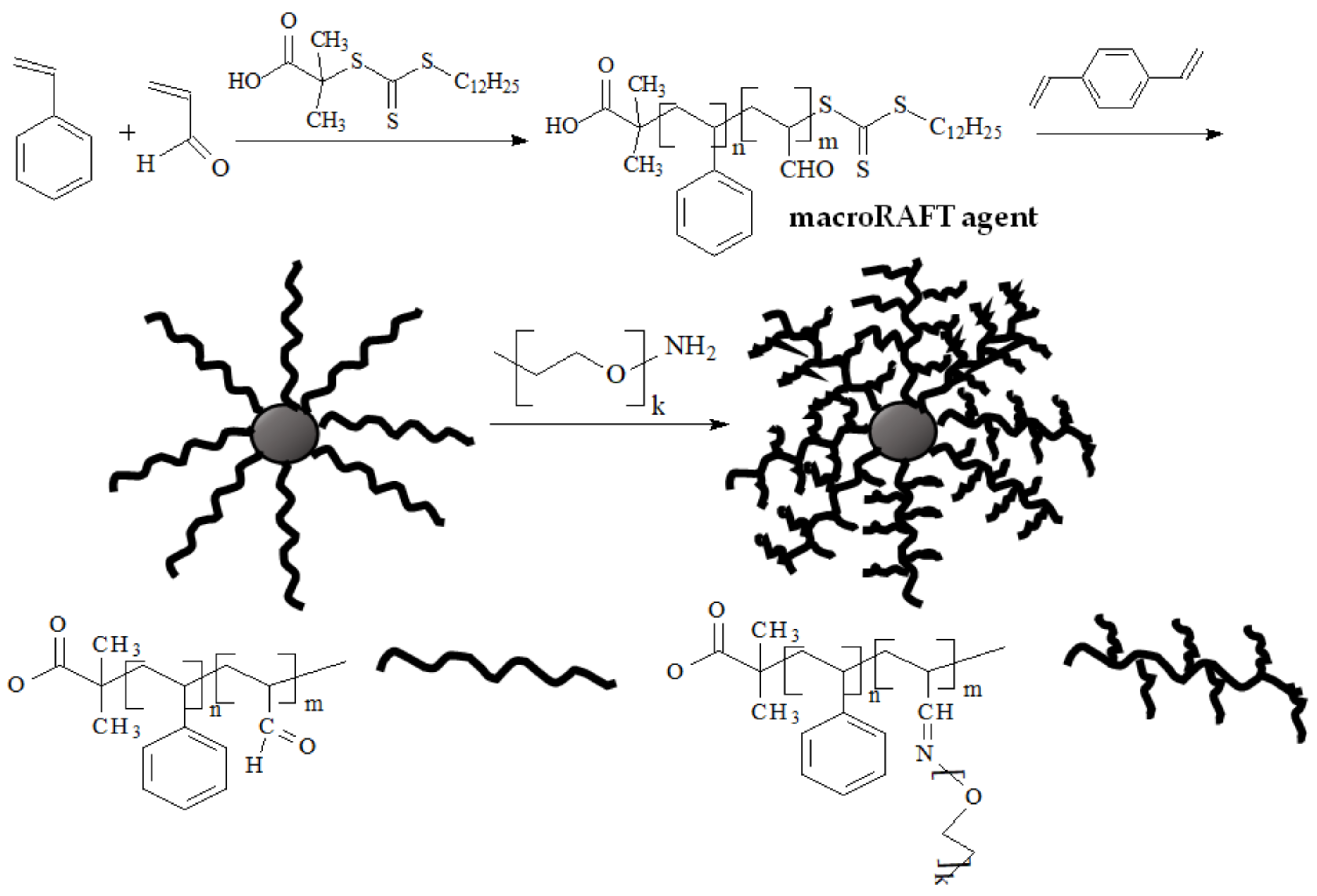
- Lu, J.; Zhang, W.; Richards, S.-J.; Gibson, M.I.; Chen, G. Glycopolymer-coated gold nanorods synthesised by a one pot copper(0) catalyzed tandem RAFT/click reaction. Polym. Chem. 2014, 5, 2326–2332. [Google Scholar] [CrossRef]
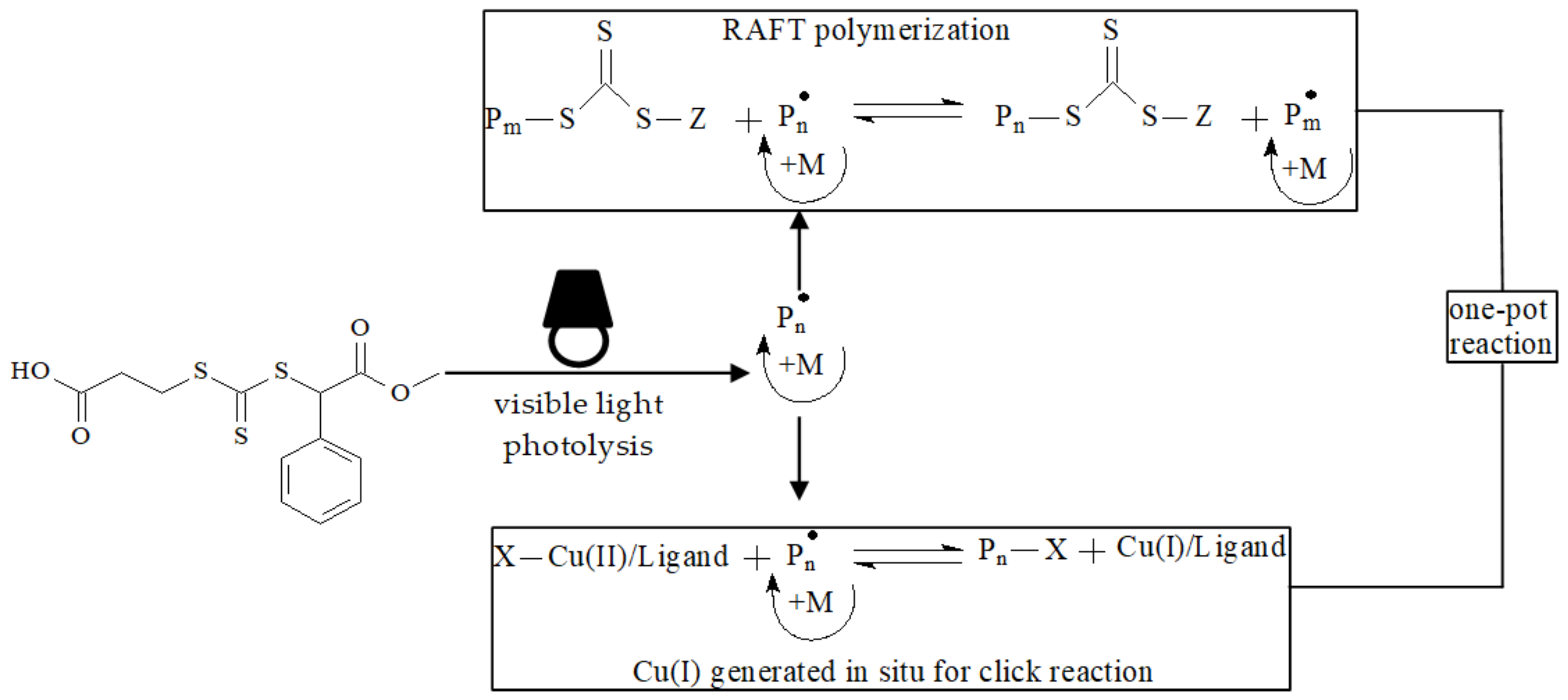
- Wang, J.; Wang, X.; Xue, W.; Chen, G.; Zhang, W.; Zhu, X. Initiator and photocatalyst-free visible light induced one-pot reaction: Concurrent RAFT polymerization and CuAAC click reaction. Macromol. Rapid. Commun. 2016, 37, 799–804. [Google Scholar] [CrossRef]
- Shen, Q.; Zhang, J.; Zhang, S.; Hao, Y.; Zhang, W.; Zhang, W.; Chen, G.; Zhang, Z.; Zhu, X. Facile One-Pot/One-Step Technique for Preparation of Side-Chain Functionalized Polymers: Combination of SET-RAFT Polymerization of Azide Vinyl Monomer and Click Chemistry. J. Polym. Sci. Part A Polym. Chem. 2012, 50, 1120–1126. [Google Scholar] [CrossRef]
- Li, Z.; Kosuri, S.; Foster, H.; Cohen, J.; Jumeaux, C.; Stevens, M.M.; Champan, R.; Gormley, A.J. A dual wavelength polymerization and bioconjugation strategy for high throughput synthesis of multivalent ligands. J. Am. Chem. Soc. 2019, 141, 19823–19830. [Google Scholar] [CrossRef] [PubMed]
- Bousquet, A.; Barner-Kowollik, C.; Davis, T.P.; Stenzel, M.H. Synthesis of comb polymers via grafting-onto macromolecules bearing pendant diene groups via the hetero-Diels-Alder-RAFT click concept. J. Polym. Sci. Part A Polym. Chem. 2010, 48, 1772–1781. [Google Scholar] [CrossRef]
- Bousquet, A.; Boyer, C.; Davis, T.P.; Stenzel, M.H. Electrostatic assembly of functional polymer combs onto gold nanoparticle surfaces: Combining RAFT, click and LbL to generate new hybrid nanomaterials. Polym. Chem. 2010, 1, 1186–1195. [Google Scholar] [CrossRef]
- Huang, J.; Xiao, Z.; Liang, H.; Lu, J. Star graft copolymer via grafting-onto strategy using a combination of reversible addition–fragmentation chain transfer arm-first technique and aldehyde–aminooxy click reaction. Polym. Int. 2014, 63, 1122–1128. [Google Scholar] [CrossRef]
- Moraes, J.; Maschmeyer, T.; Perrier, S. ‘‘Clickable’’ polymers via a combination of RAFT polymerization and isocyanate chemistry. J. Polym. Sci. Part A Polym. Chem. 2011, 49, 2771–2782. [Google Scholar] [CrossRef]
- Flores, J.D.; Shin, J.; Hoyle, C.E.; McCormick, C.L. Direct RAFT polymerization of an unprotected isocyanate-containing monomer and subsequent structopendant functionalization using ‘‘click’’-type reactions. Polym. Chem. 2010, 1, 213–220. [Google Scholar] [CrossRef]
- Flores, J.D.; Treat, N.J.; York, A.W.; McCormick, C.L. Facile, modular transformations of RAFT block copolymers via sequential isocyanate and thiol-ene reactions. Polym. Chem. 2011, 2, 1976–1985. [Google Scholar] [CrossRef]
- Wang, X.; Li, L.; Luo, Y.; Shi, H.; Li, J.; Zhao, X. Comb-shaped glycopolymer/peptide bioconjugates by combination of RAFT polymerization and thiol-ene ‘‘click’’ chemistry. Macromol. Biosci. 2012, 12, 1575–1582. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, Z.; Wang, H.; Feng, X.; He, C. Novel anti-biofouling soft contact lens: L-cysteine conjugated amphiphilic conetworks via RAFT and thiol–ene click chemistry. Macromol. Biosci. 2017, 17, 1600444. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Cai, Z.; Liu, H.; Cao, Z.; Xia, Y.; Ma, W.; Gong, F.; Tao, G.; Liu, C. Tailoring the surface of attapulgite by combining redox-initiated RAFT polymerization with alkynyl-thiol click reaction for polycarbonate nanocomposites:Effect of polymer brush chain length on mechanical, thermal and rheological properties. Mater. Chem. Phys. 2020, 241, 122334. [Google Scholar] [CrossRef]
- He, H.; Liu, B.; Wang, M.; Vachet, R.W.; Thayumanavan, S. Sequential nucleophilic “click” reactions for functional amphiphilic homopolymers. Polym. Chem. 2019, 10, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Oh, T.; Jono, K.; Kimoto, Y.; Hoshino, Y.; Miura, Y. Preparation of multifunctional glycopolymers using double orthogonal reactions and the effect of electrostatic groups on the glycopolymer–lectin interaction. Polym. J. 2019, 51, 1299–1308. [Google Scholar] [CrossRef]
- Hrsic, E.; Zografou, I.; Schulte, B.; Pich, A.; Keul, H.; Möller, M. Amphiphilic block copolymers with pendant thiol groups in side chains by RAFT polymerizationtion. Polymer 2013, 54, 495–504. [Google Scholar] [CrossRef]
- Platé, N.A.; Litmanovich, A.D.; Noah, O.V. Macromolecular Reactions: Peculiarities, Theory and Experimental Approaches, 1st ed.; Wiley: Chichester, UK, 1995; 450p, ISBN 978-0471943921. [Google Scholar]
- Forder, T.; Maschmeyer, P.; Zeng, H.; Roberts, D. Post-Synthetic ‘Click’ Synthesis of RAFT Polymers with Pendant Self-Immolative Triazoles. Chem. Asian J. 2021, 16, 287–291. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.; Cai, Y.; Fanslau, L.; Vana, P. Nanoengineering with RAFT polymers: From nanocomposite design to applications. Polym. Chem. 2021, 12, 6198–6229. [Google Scholar] [CrossRef]
- Truong, N.P.; Jones, G.R.; Bradford, K.G.E.; Konkolewicz, D.; Anastasaki, A.A. Comparison of RAFT and ATRP methods for controlled radical polymerization. Nat. Rev. Chem. 2021, 5, 859–869. [Google Scholar] [CrossRef]
- Yadav, S.; Ramesh, K.; Kumar, P.; Jo, S.H.; Yoo, S.I.; Gal, Y.-S.; Park, S.-H.; Lim, K.T. Near-infrared light-responsive shell-crosslinked micelles of poly(d,l-lactide)-b-poly((furfuryl methacrylate)-co-(N-acryloylmorpholine)) prepared by Diels-Alder reaction for the triggered release of doxorubicin. Materials 2021, 14, 7913. [Google Scholar] [CrossRef]
- Lei, J.Q.; Song, Y.J.; Li, D.; Lei, M.H.; Tan, R.; Liu, Y.Q.; Zheng, H. pH-sensitive and charge-reversal Daunorubicin-conjugated polymeric micelles for enhanced cancer therapy. J. Appl. Polym. Sci. 2022, 139, 51535. [Google Scholar] [CrossRef]


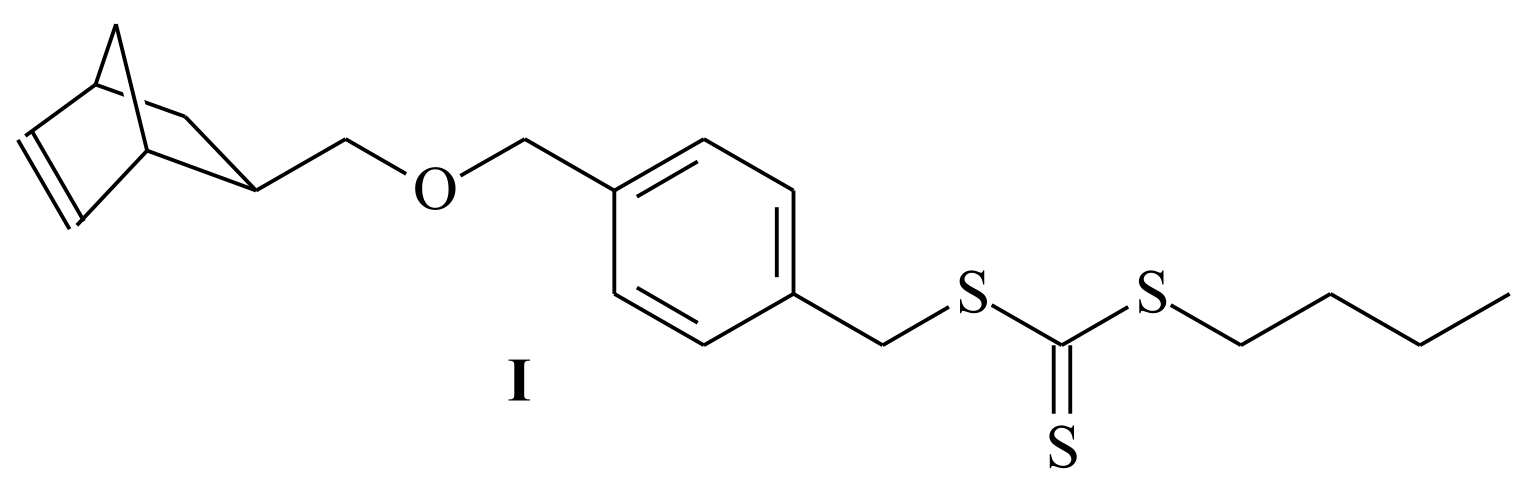

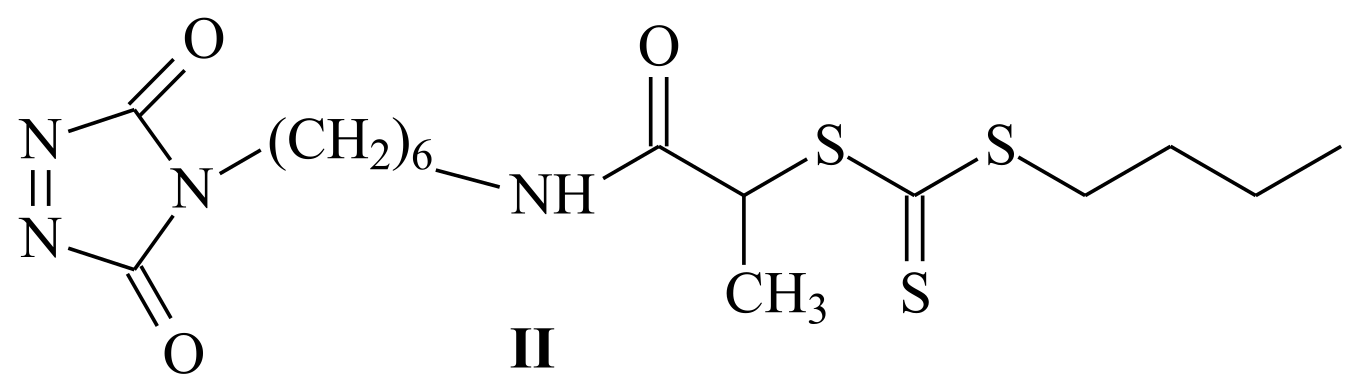
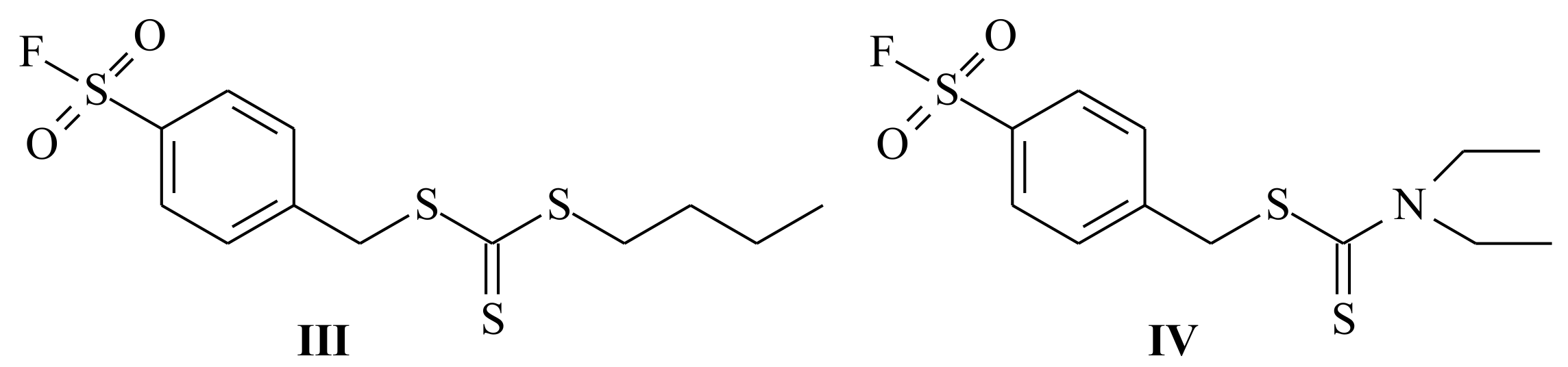


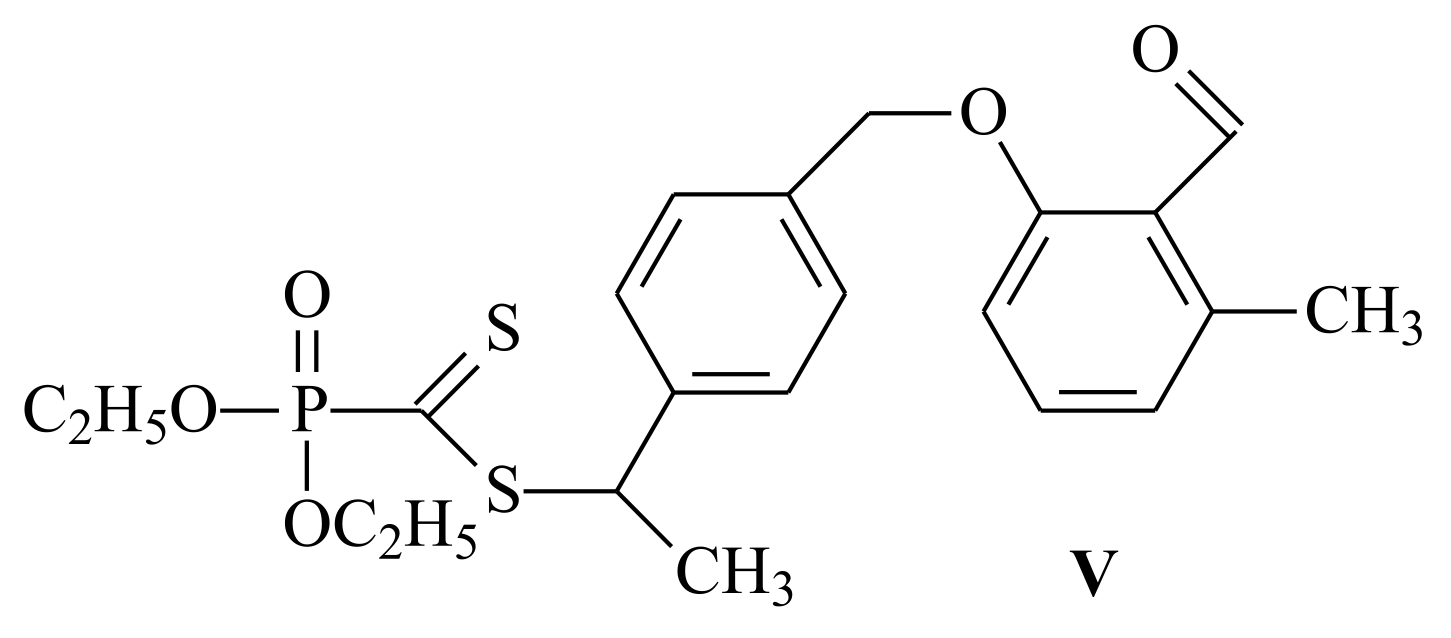


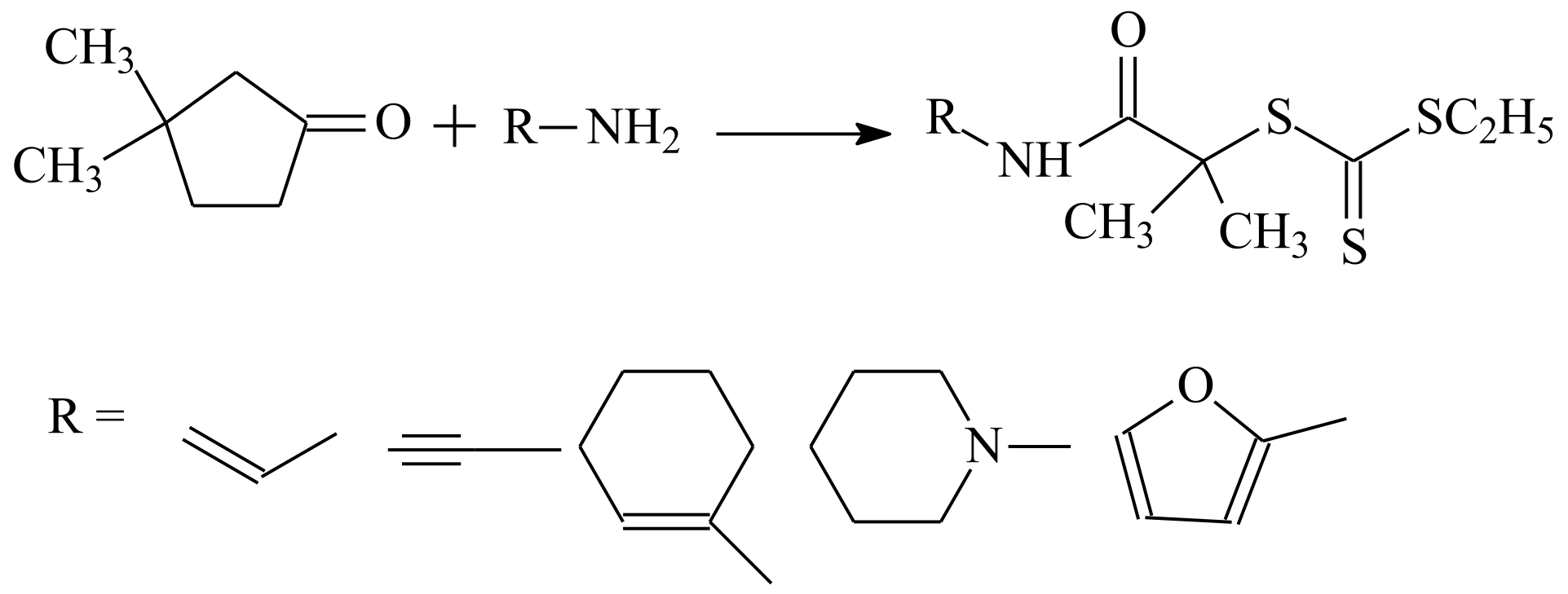


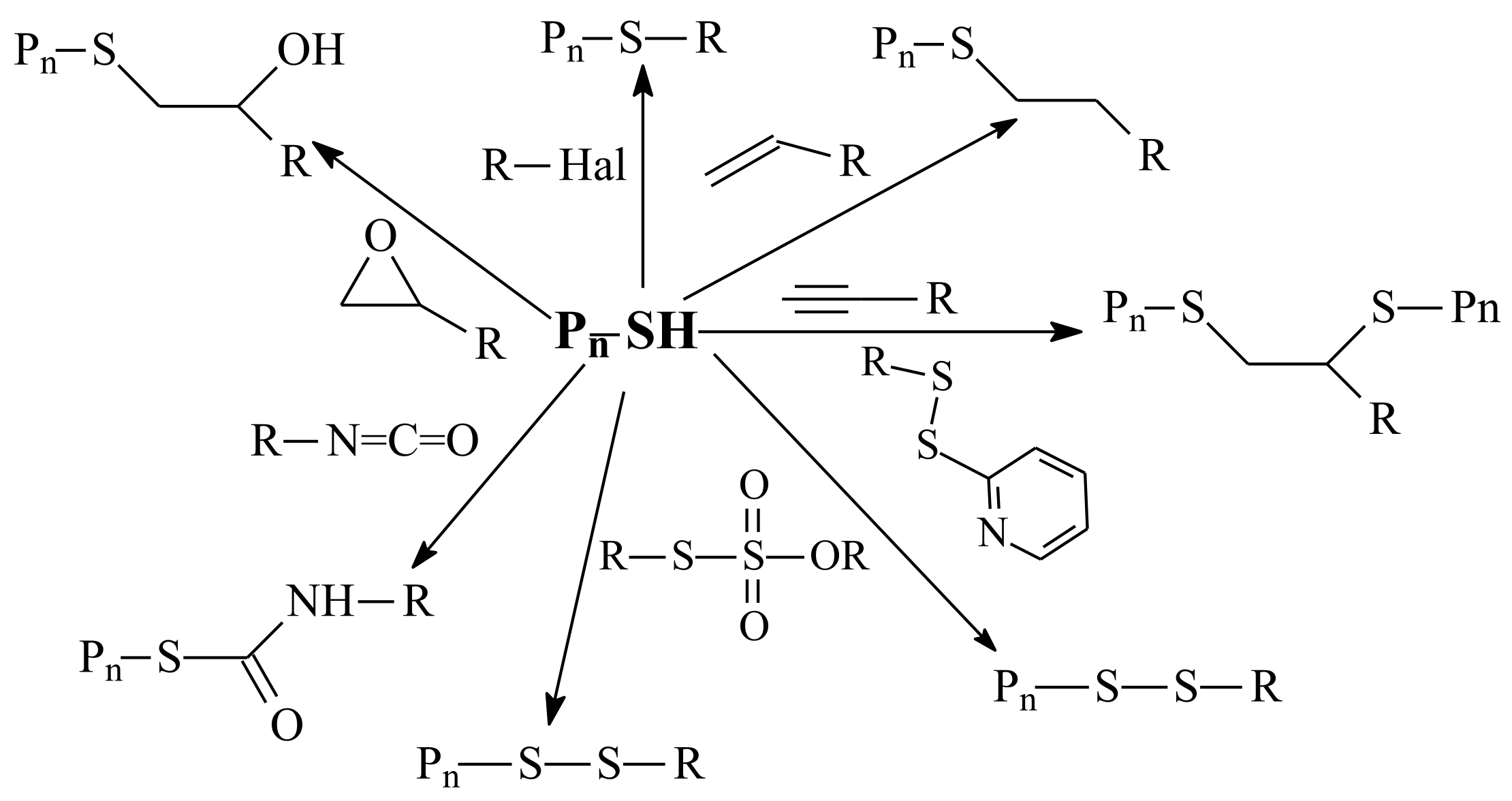
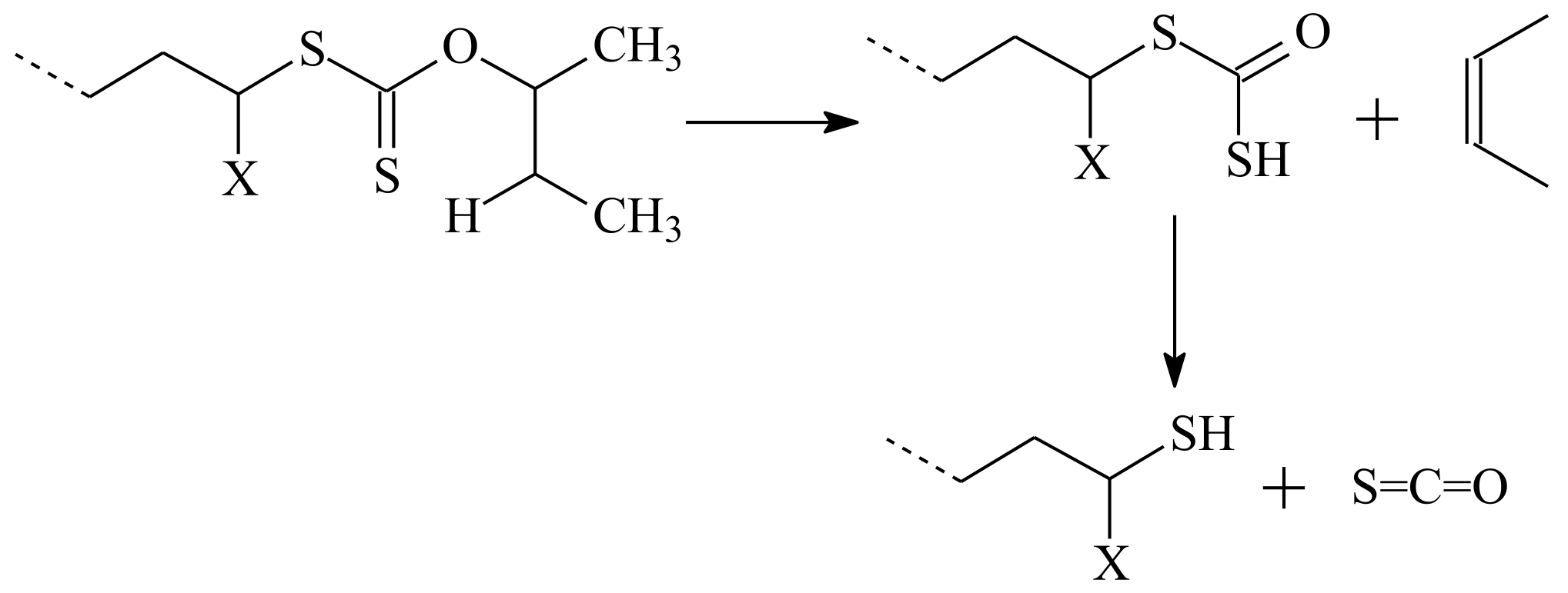
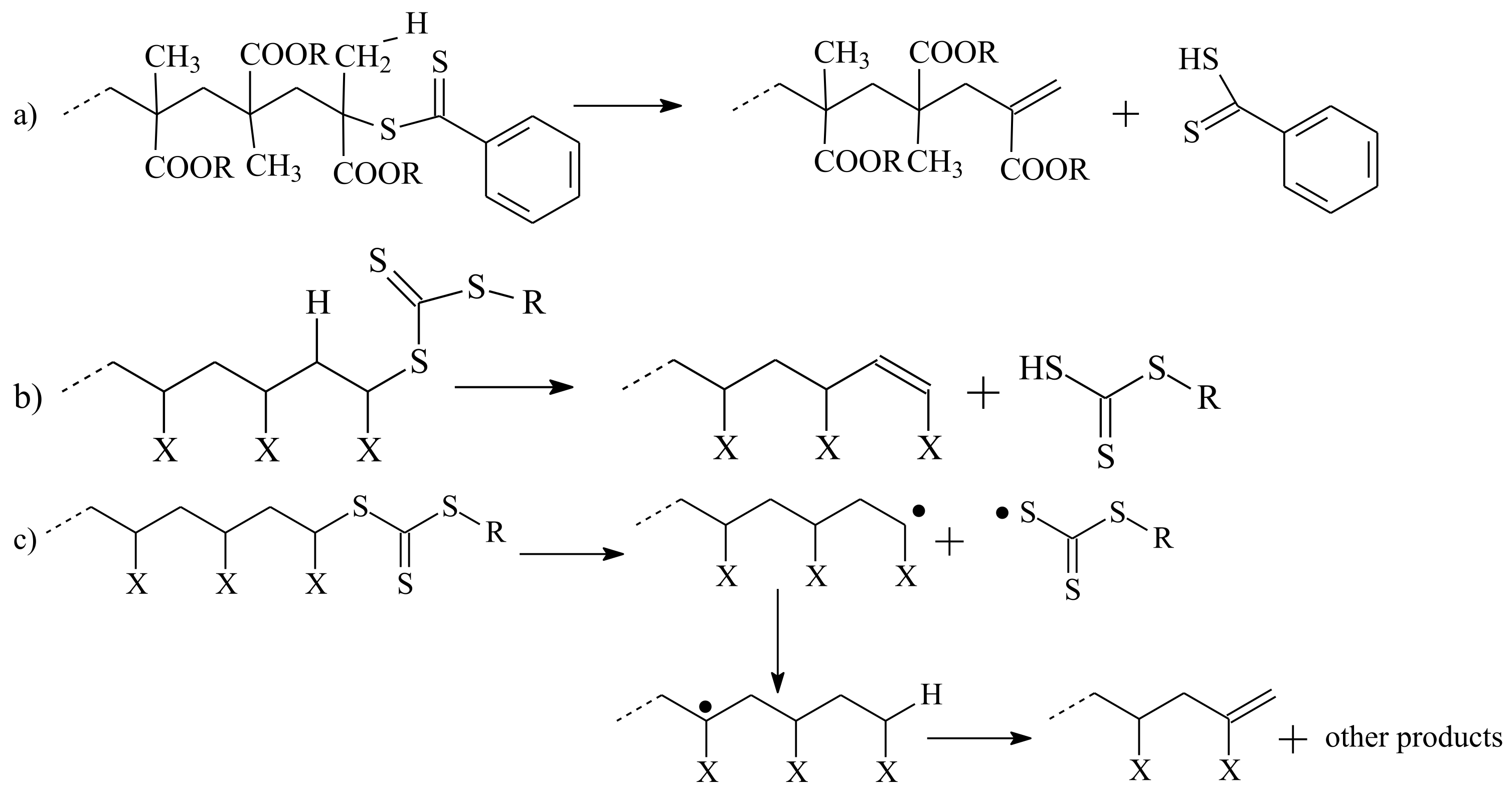
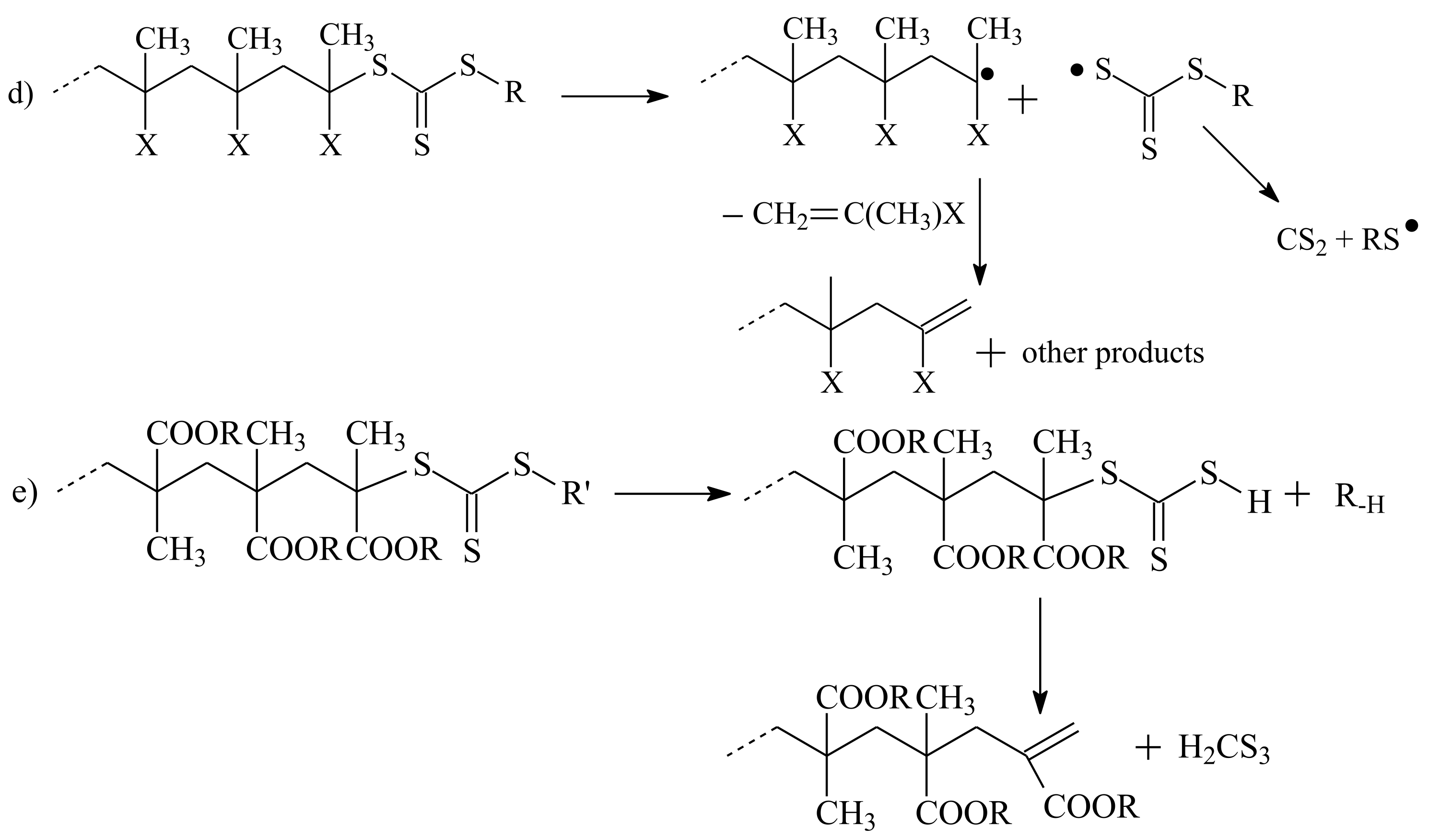
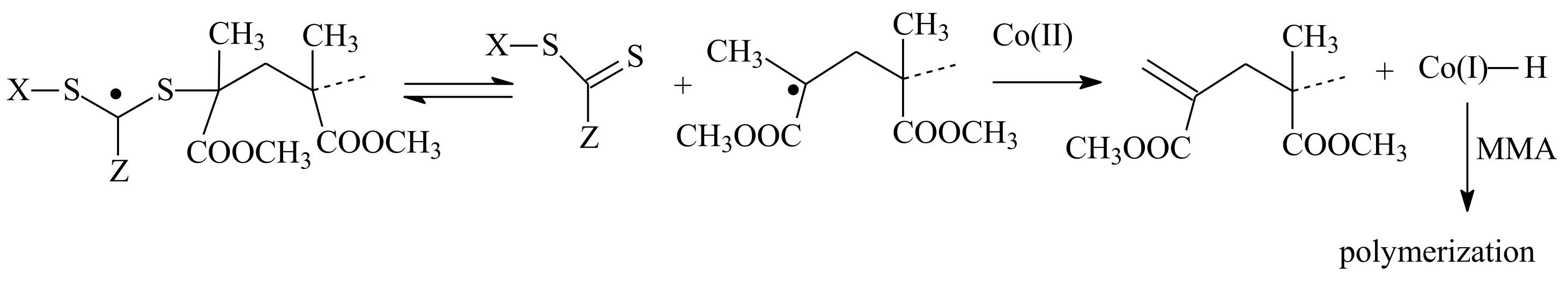

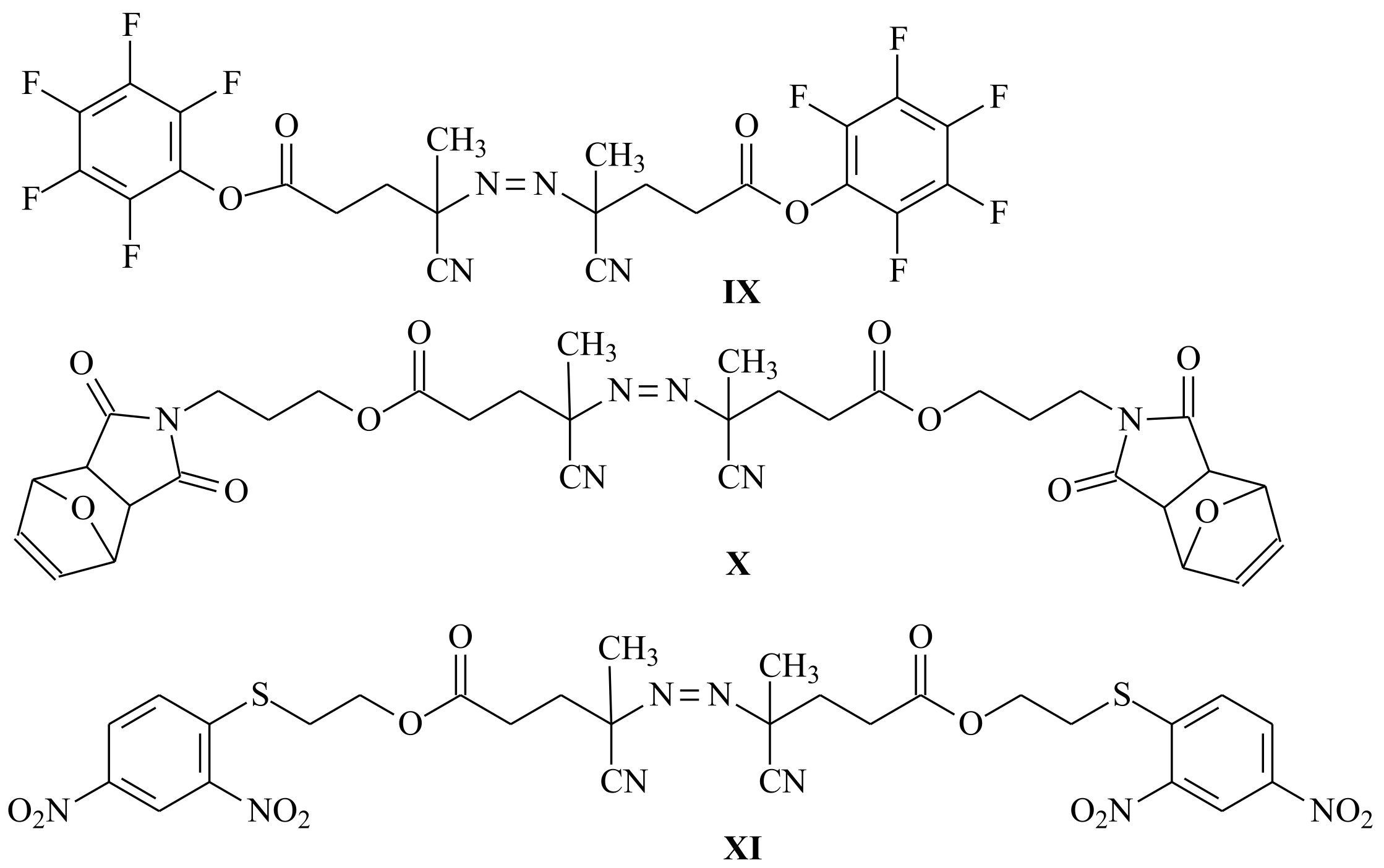
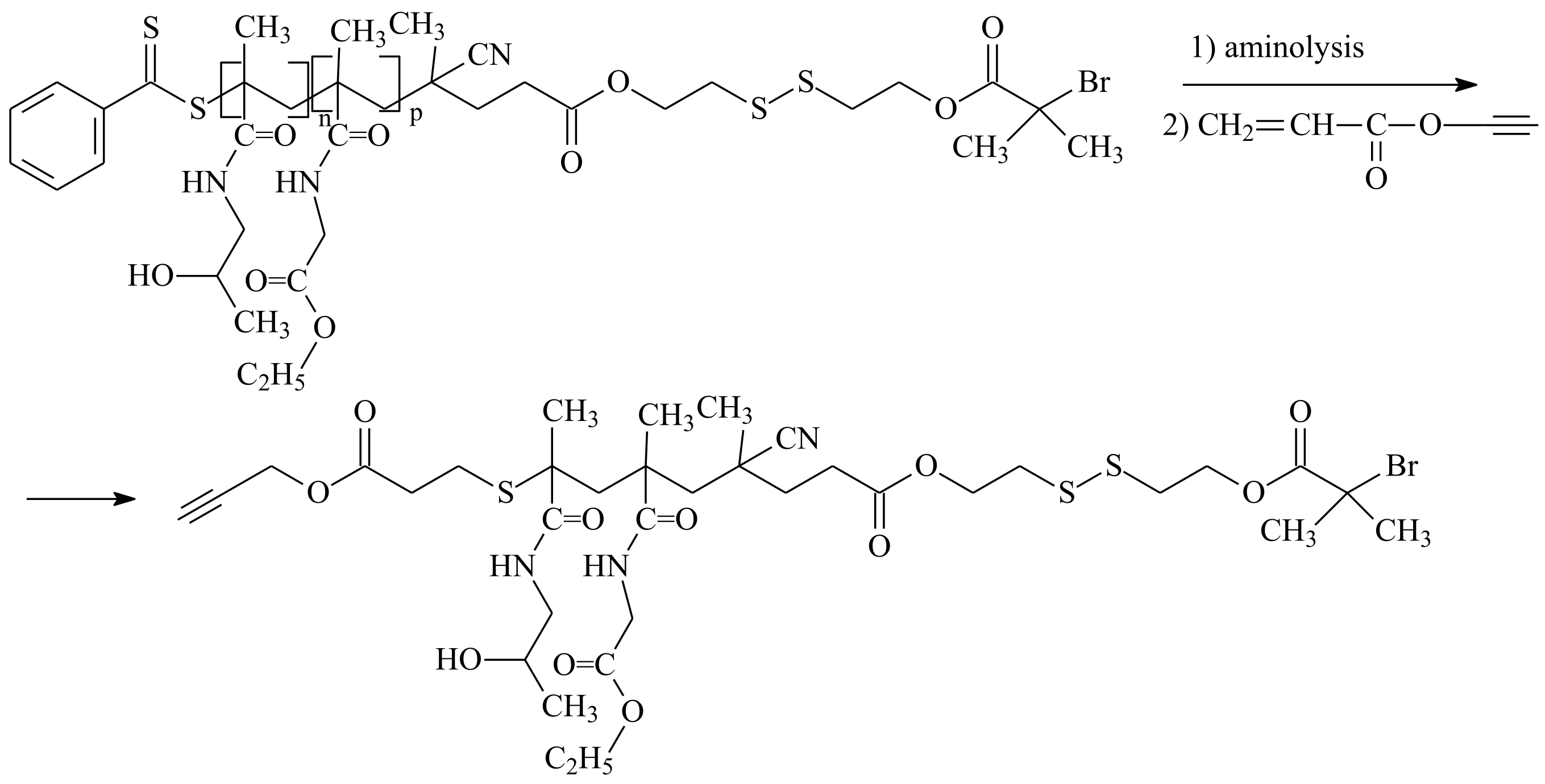
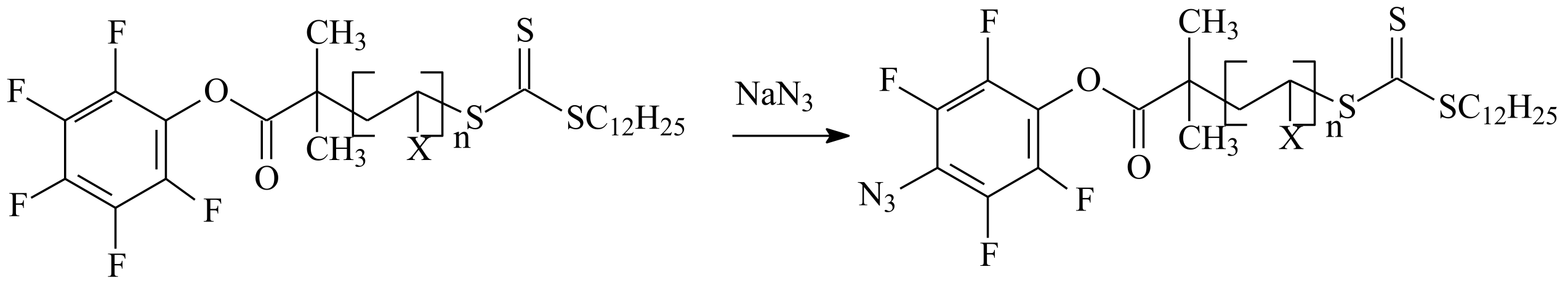
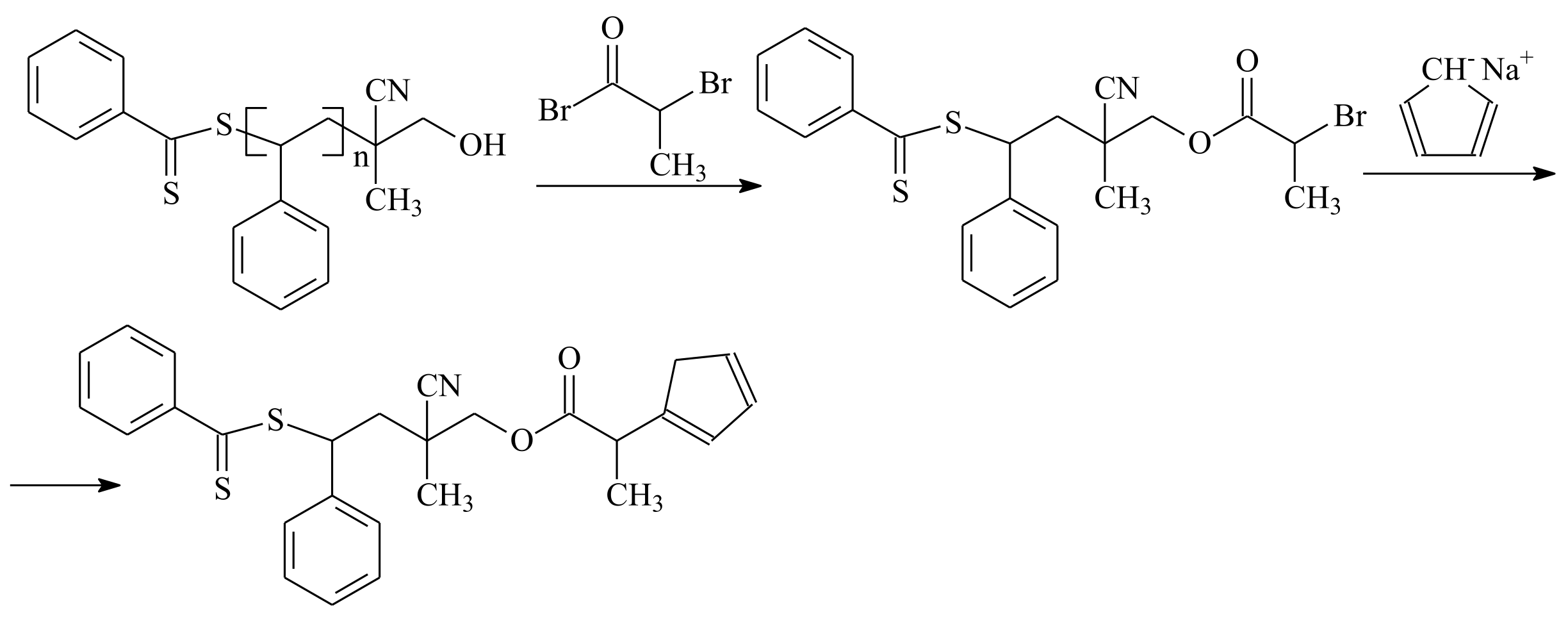
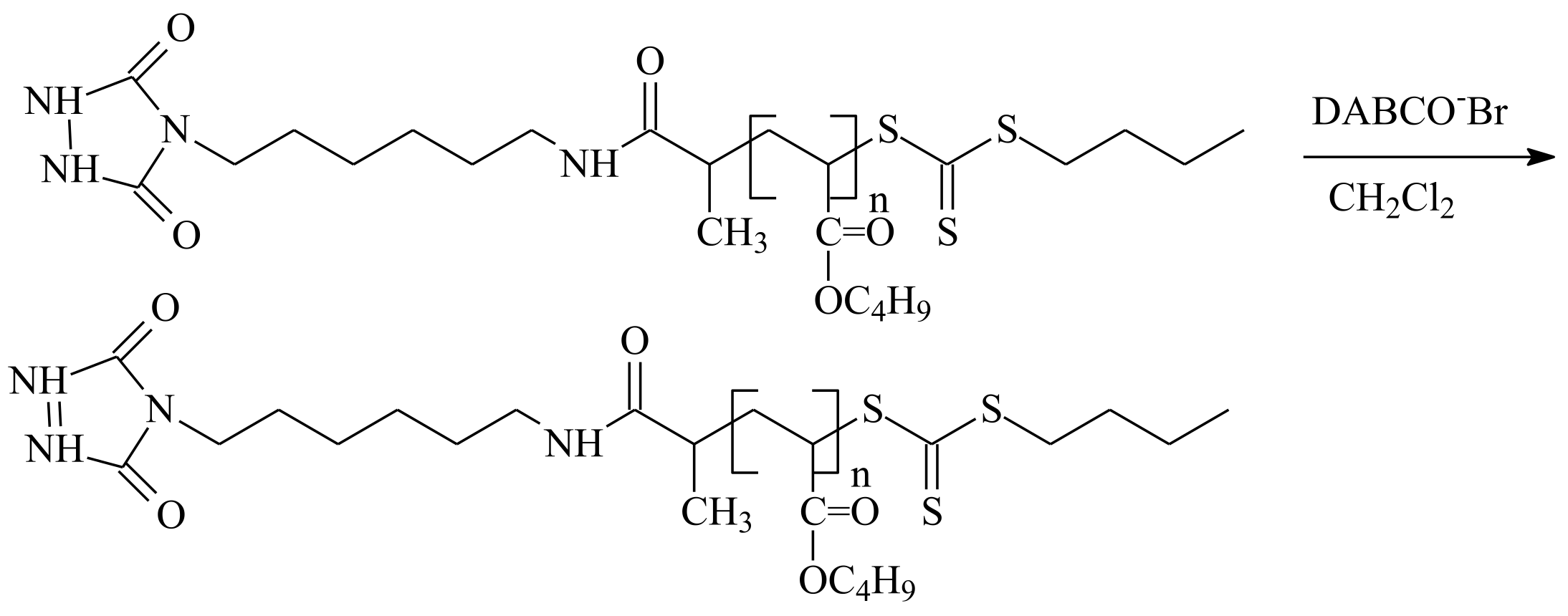
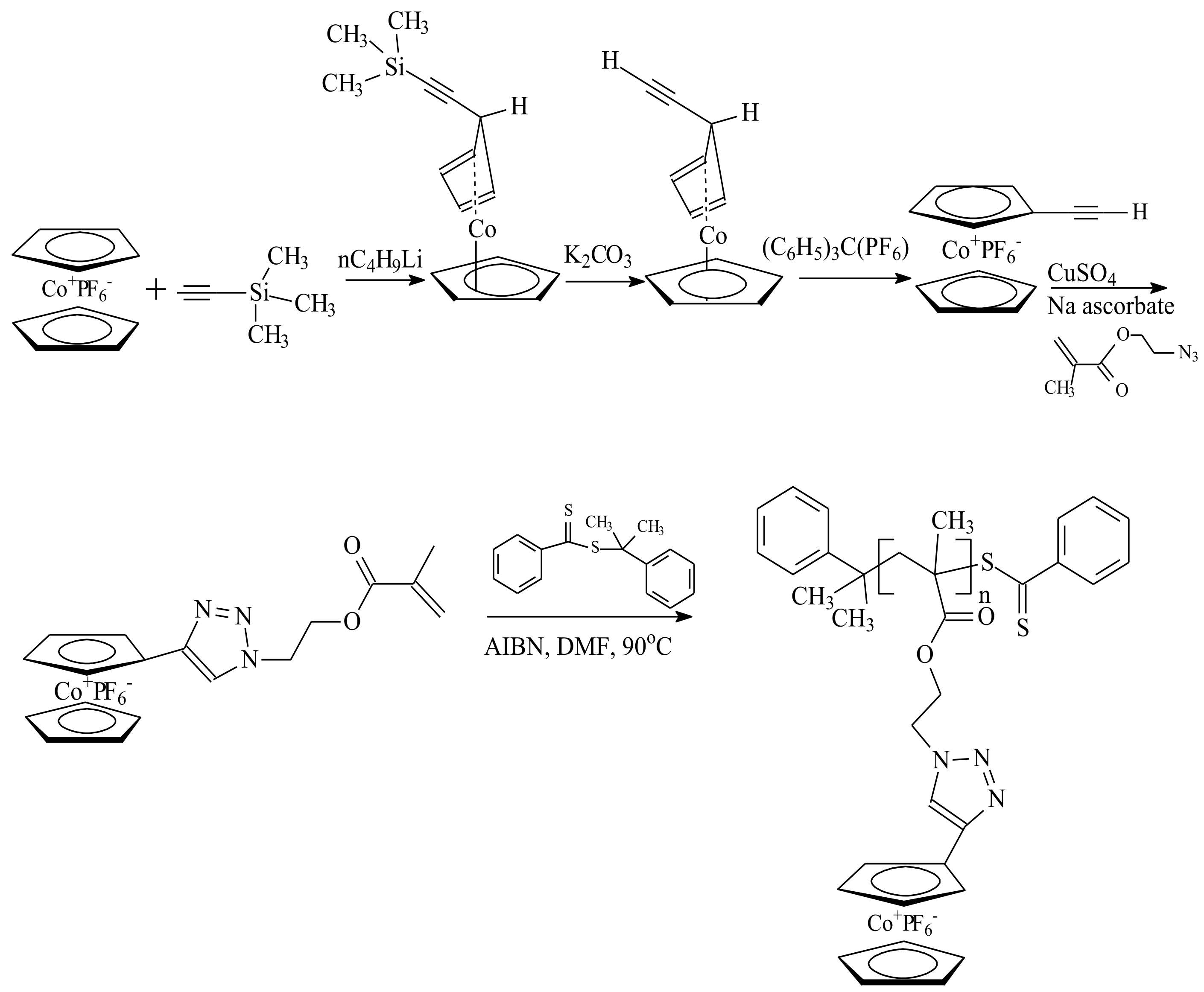
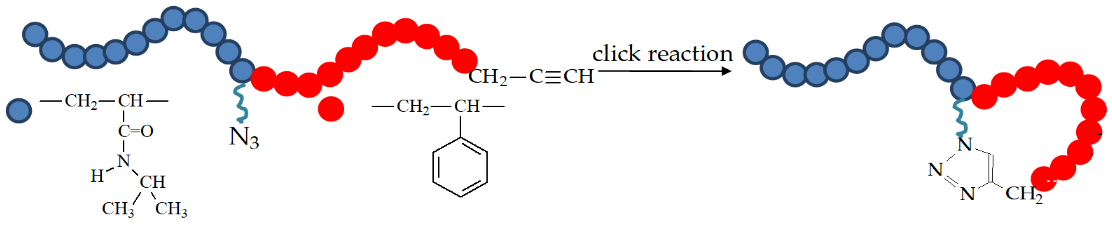
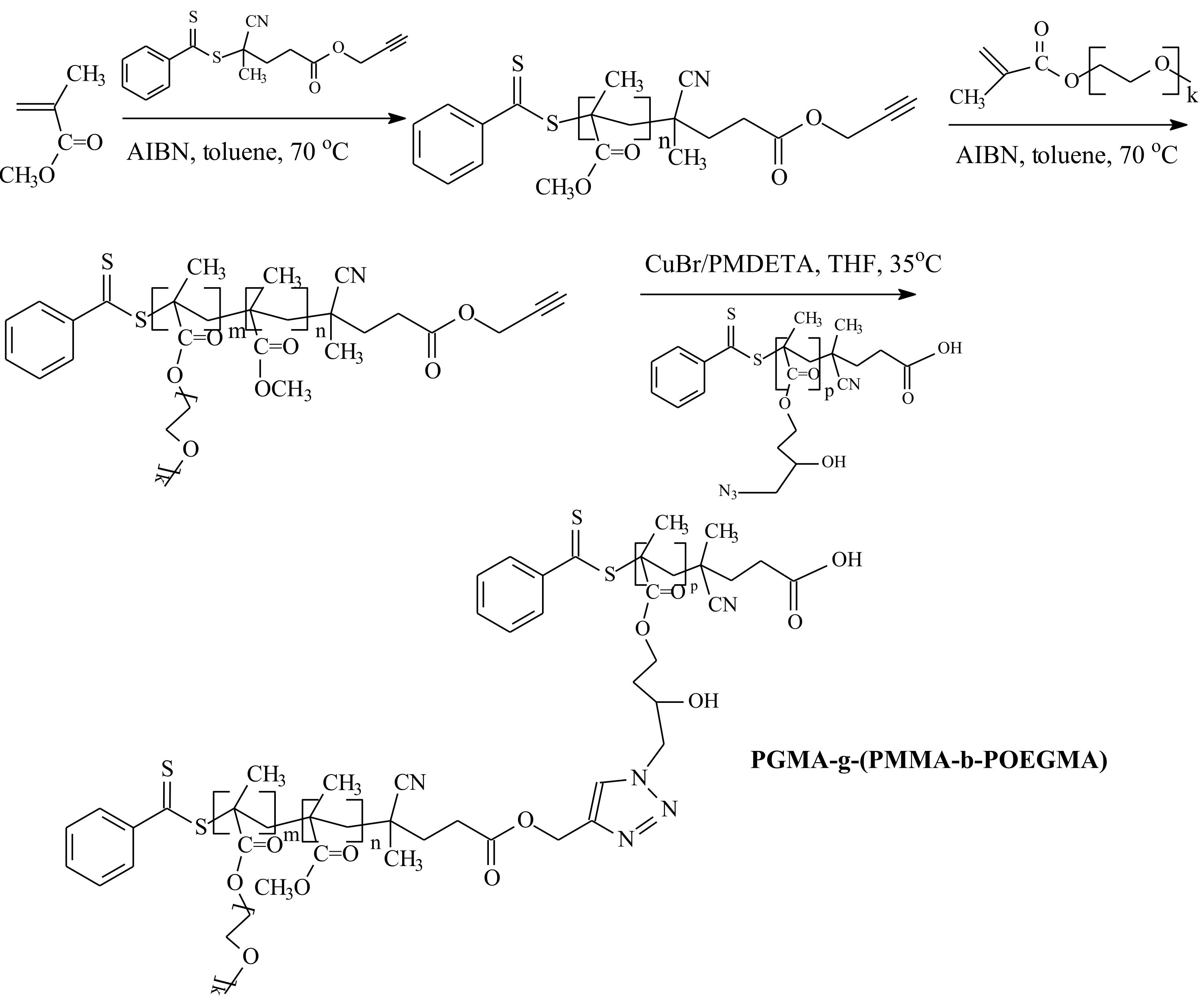
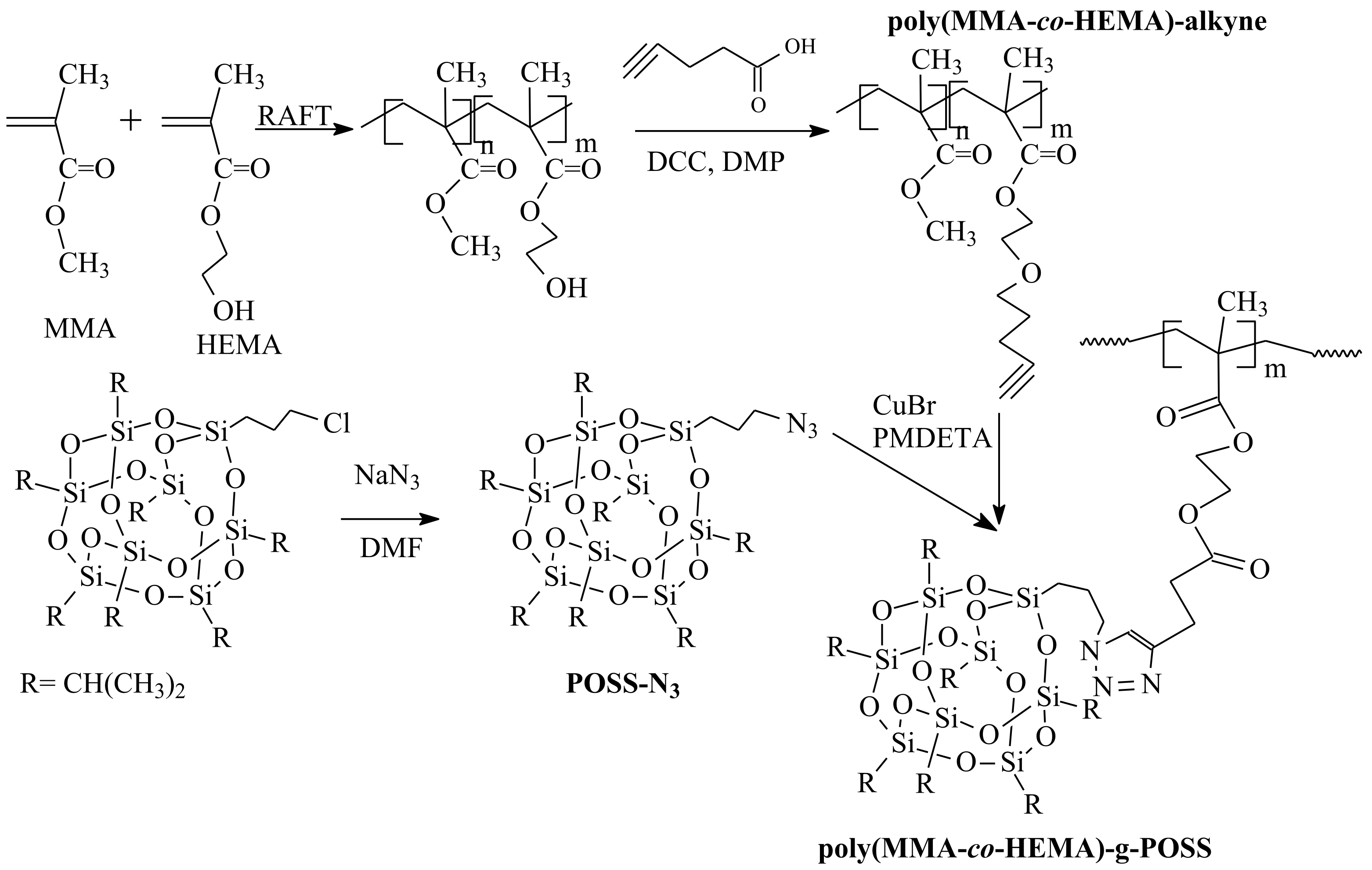
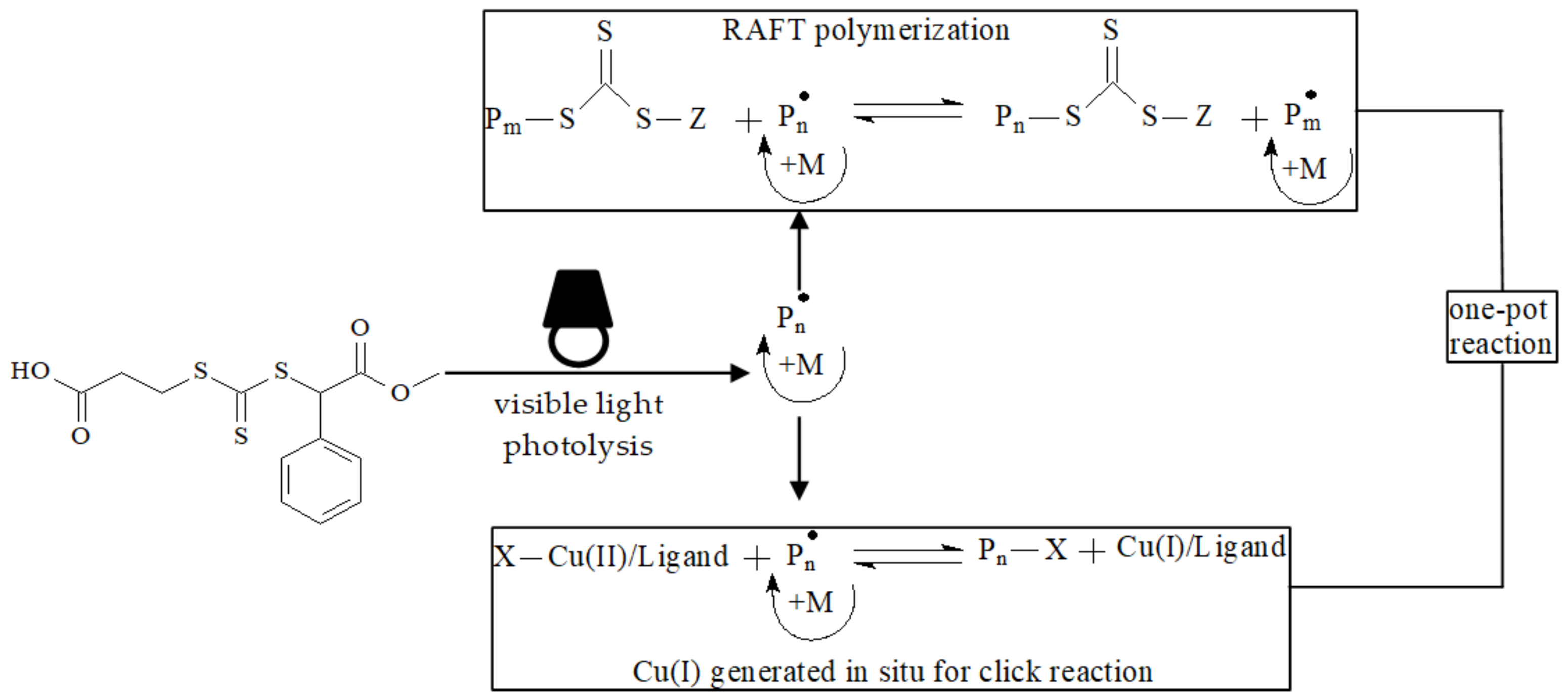
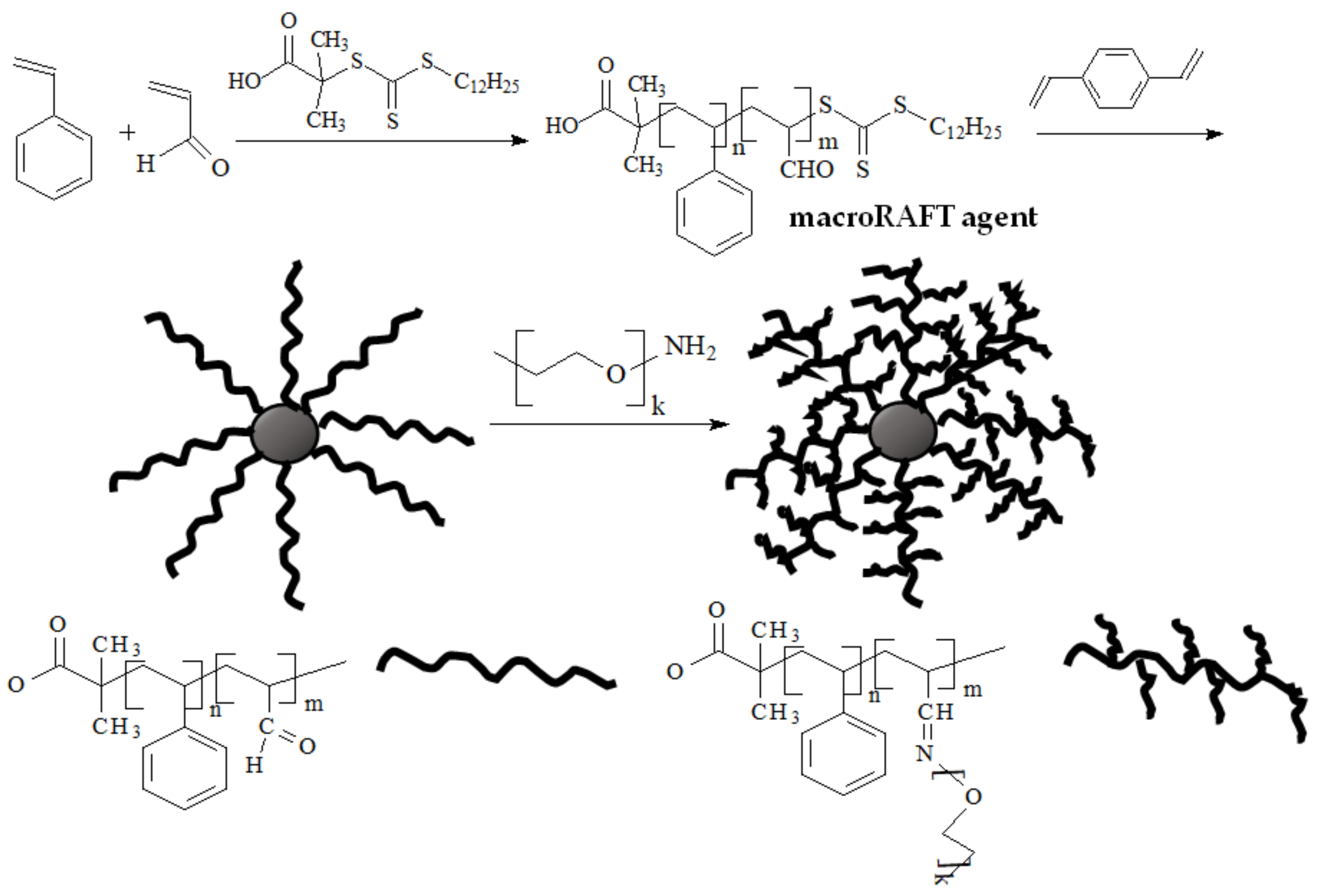
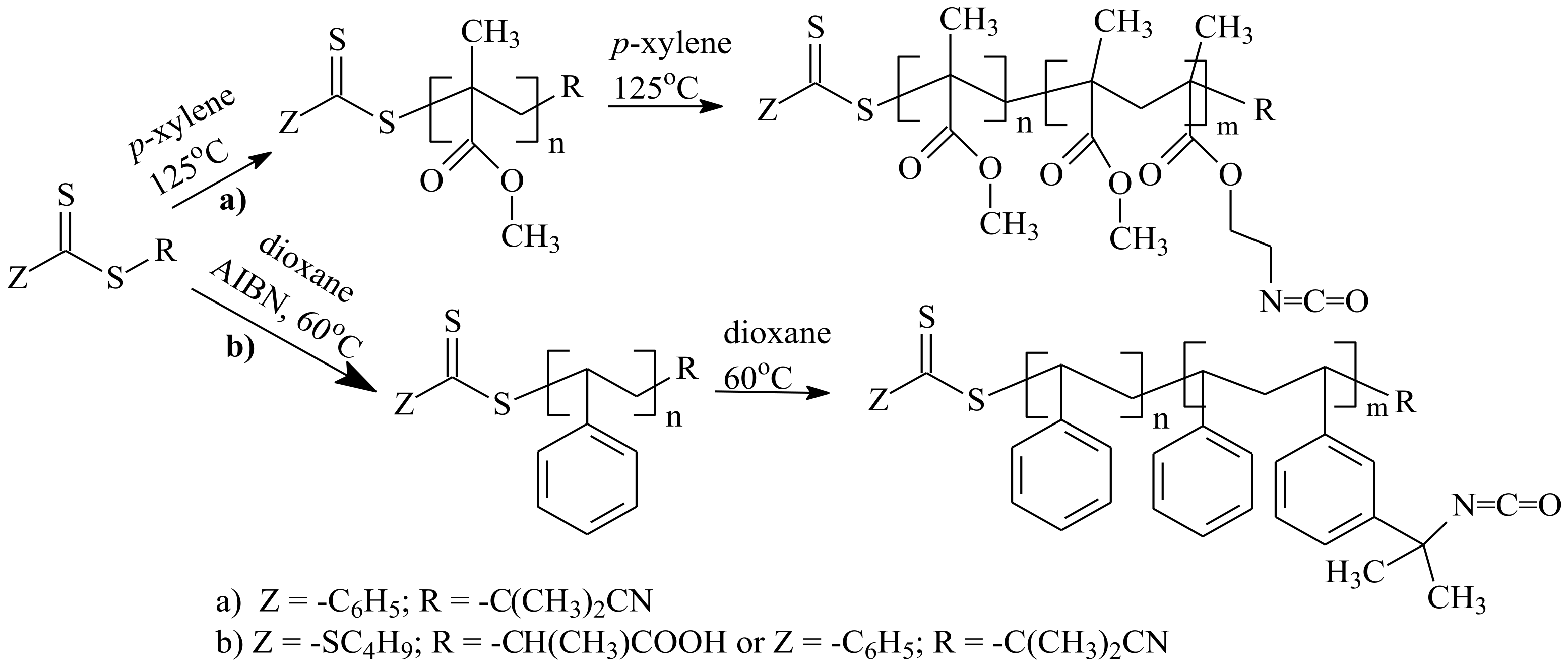
| RAFT Agent | Reactant | Monomer |
|---|---|---|
| Dithiobenzoate | NaBH4/ LiB(C2H5)3H Amines Hydrazine hydrate Alkali NaN3 | NIPAM [128,129], MMA [124], HPMA [130], St [131,132], StSO3Na [126] NIPAM [133], MMA [124,134,135], St [136], PEGA [137], AN [138], AA [139], LMA [140], DMAEMA [139], MA [141], St/2VP [142] St [117], St/StOAc [119], MA [117] MAA [130] BA [115], NIPAM [115] |
| Trithiocarbonate | NaBH4 Amines Hydrazine hydrate Alkali | NIPAM [143], MMA [124] NIPAM [109,144,145], St [146] St [117,121], NIPAM [117], MMA [121] BA [147], NIPAM [148] |
| Xanthate | NaBH4 Amines Hydrazine hydrate | NVP [149,150] NVP [150] NVP [151] |
| Dithiocarbamate | Alkali | NIPAM [152] |
| R’ Group | T, °C |
|---|---|
 | 180 |
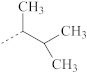 | 180 |
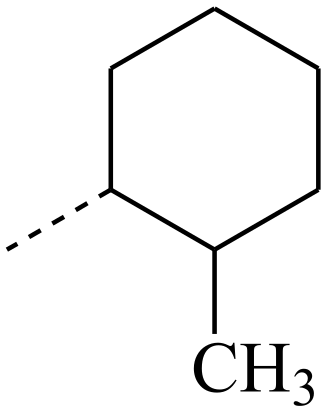 | 180 |
 | 130 |
| Z group | Monomer | Ta), °C | Reference |
|---|---|---|---|
| C12H25S | Styrene, 4-vinylpyridine | 165 | [179] |
| Styrene, butyl acrylate | 100–200 | [168] | |
| Methyl acrylate | 220 | [174] | |
| Methyl methacrylate | 100–120 | [177] | |
| PnS | Styrene, butyl acrylate | 100–200 | [168] |
| Styrene, butyl acrylate Styrene/butyl acrylate | 140–200 | [170] |
| Polymer | “Switchable” Group | T a), °C | Temperature Range b), °C |
|---|---|---|---|
| PS | 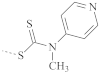 | 224 | 205–235 |
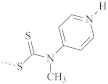 | 185 | 170–215 | |
| PMMA |  | 136 | 120–150 |
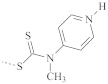 | 121 | 110–135 | |
| PNIPAM | 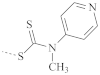 | 232 | 210–240 |
 | 197 | 165–210 | |
| PVAc |  | 306 | 290–315 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chernikova, E.V.; Kudryavtsev, Y.V. RAFT-Based Polymers for Click Reactions. Polymers 2022, 14, 570. https://doi.org/10.3390/polym14030570
Chernikova EV, Kudryavtsev YV. RAFT-Based Polymers for Click Reactions. Polymers. 2022; 14(3):570. https://doi.org/10.3390/polym14030570
Chicago/Turabian StyleChernikova, Elena V., and Yaroslav V. Kudryavtsev. 2022. "RAFT-Based Polymers for Click Reactions" Polymers 14, no. 3: 570. https://doi.org/10.3390/polym14030570
APA StyleChernikova, E. V., & Kudryavtsev, Y. V. (2022). RAFT-Based Polymers for Click Reactions. Polymers, 14(3), 570. https://doi.org/10.3390/polym14030570