Molecular Structures Polymorphism the Role of F…F Interactions in Crystal Packing of Fluorinated Tosylates

 and
and
Abstract
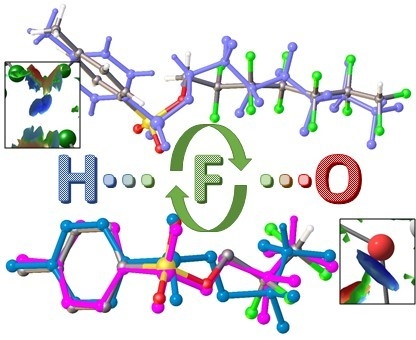
:
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Synthesis of Tosylates 1–4
2.3. Single Crystal Structure Analysis
2.4. Quantum Chemical Calculations
3. Results and Discussion
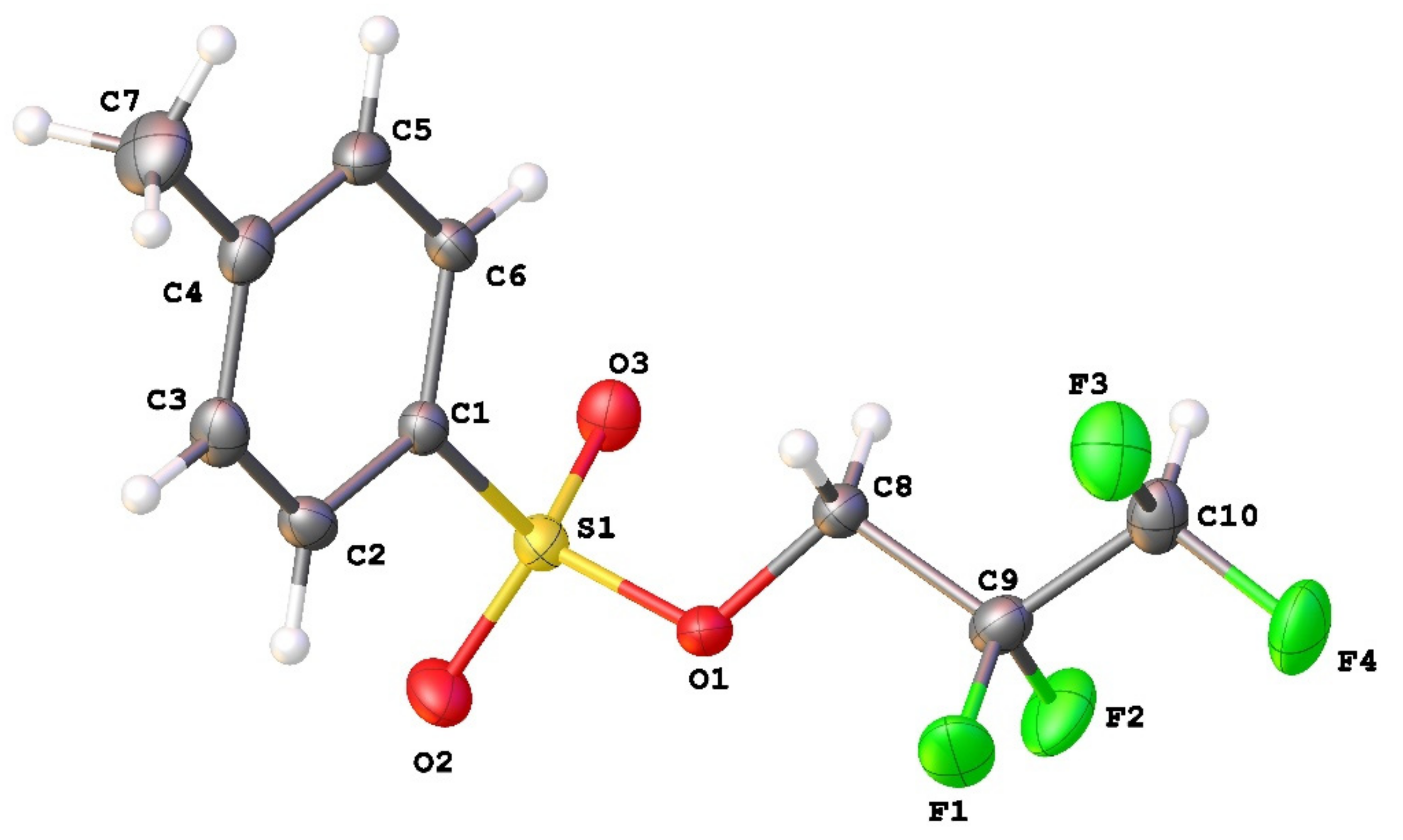
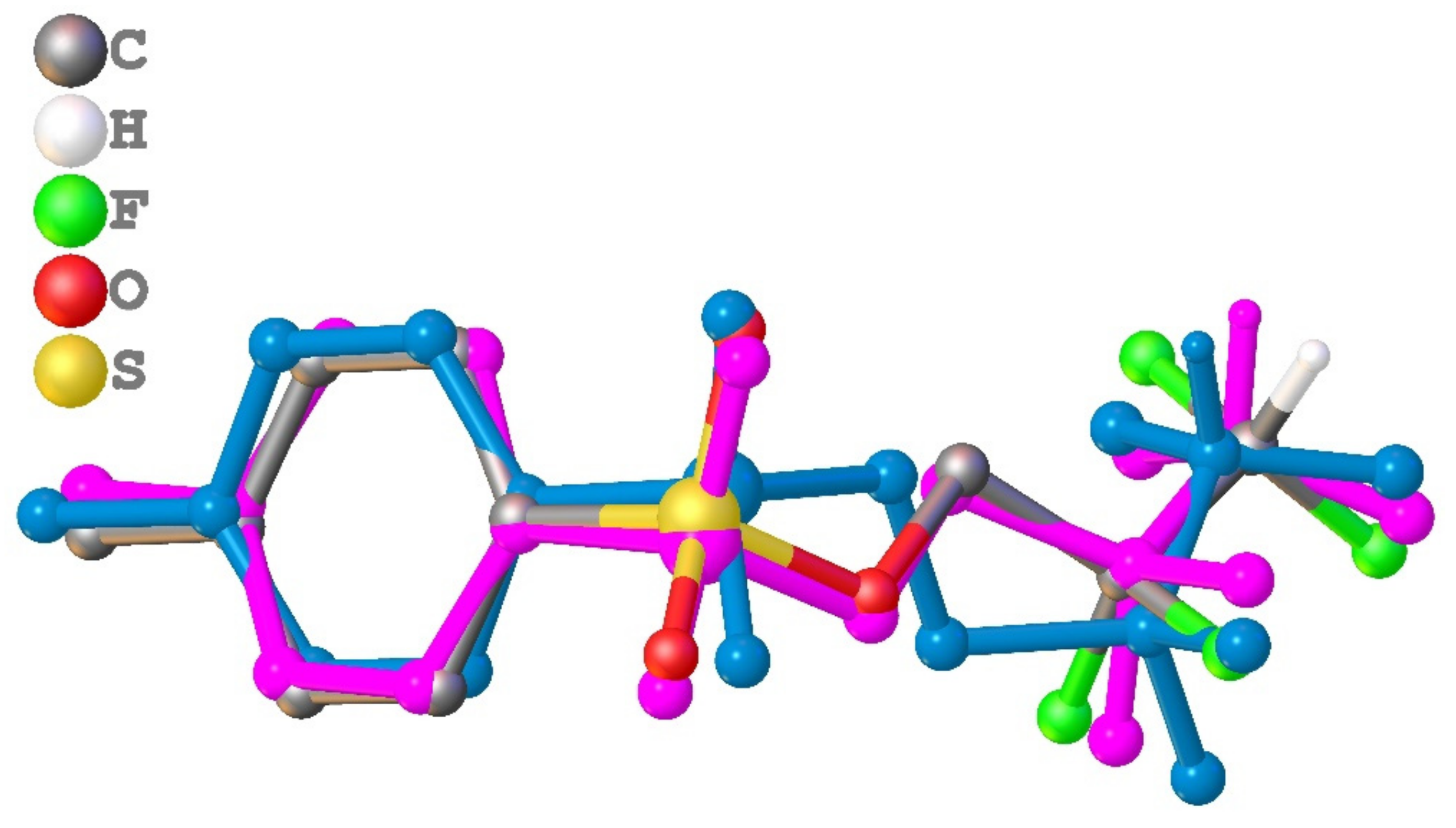
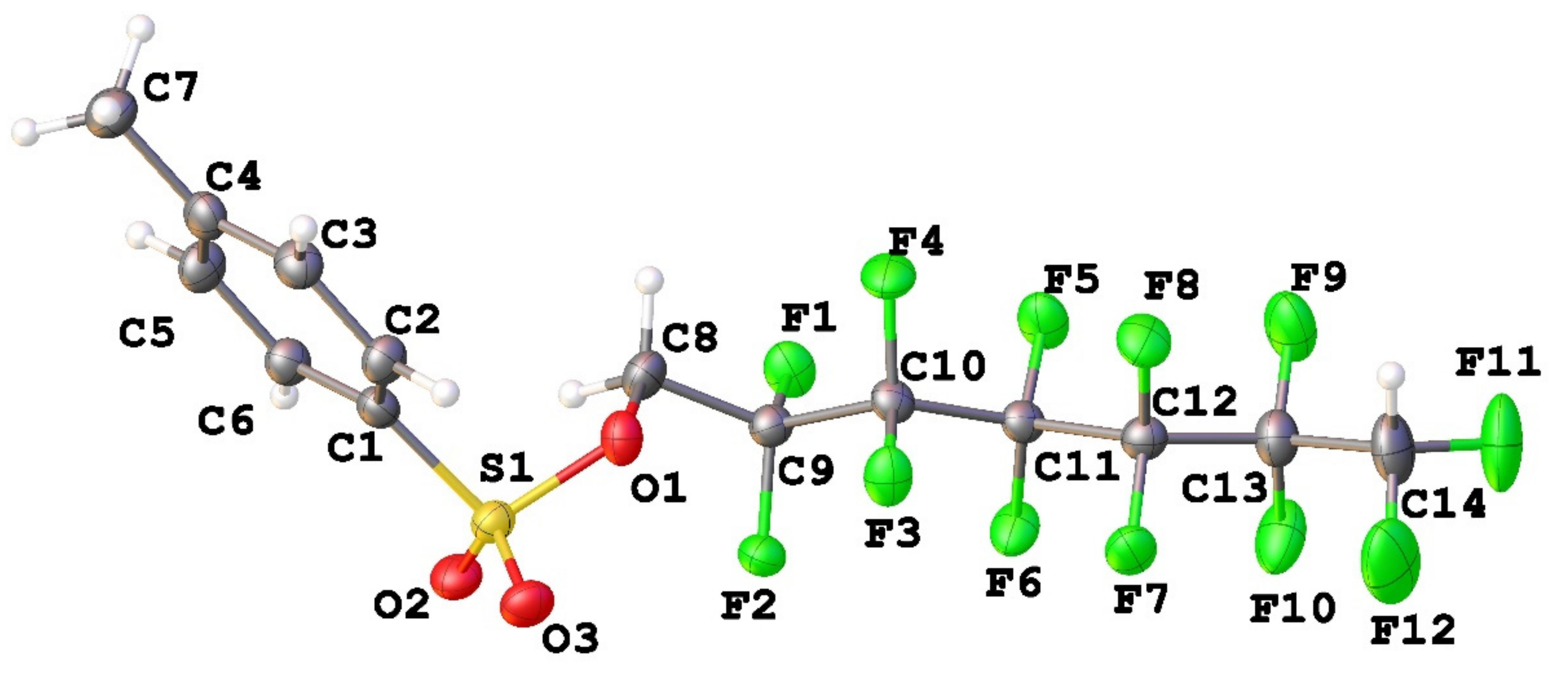
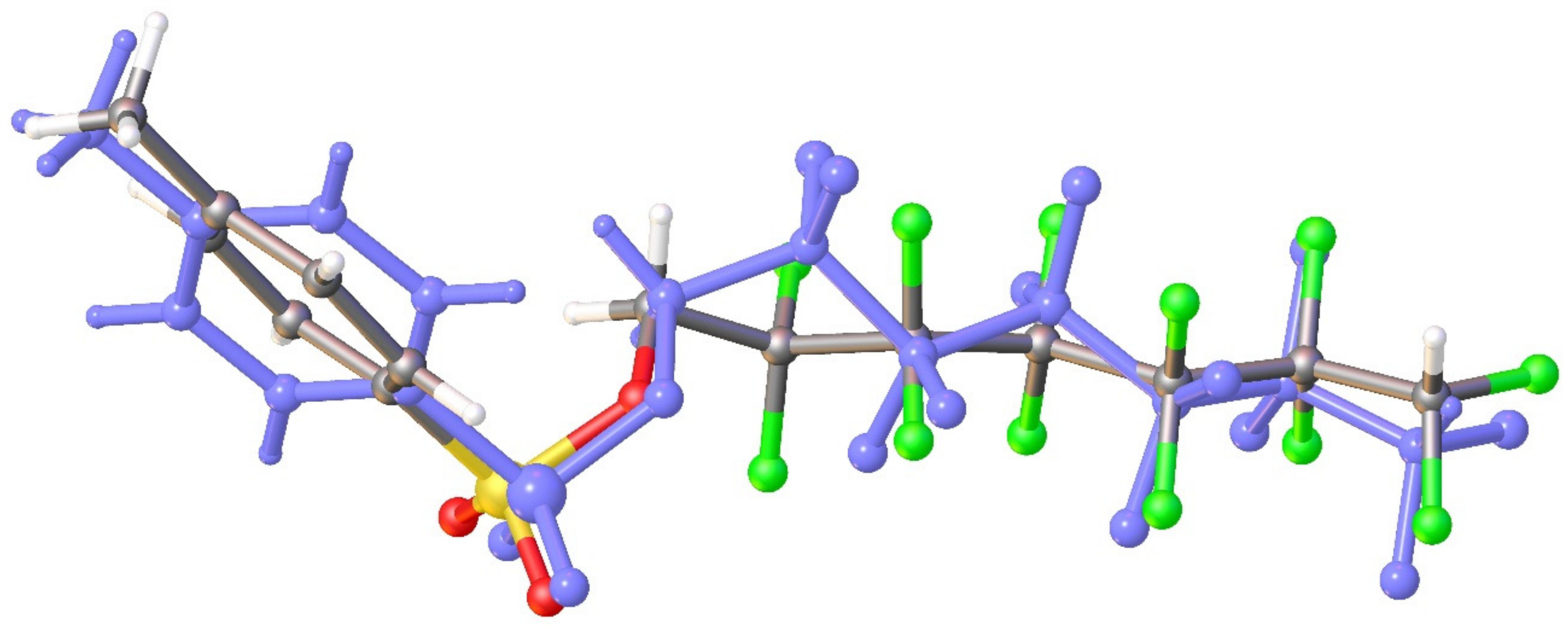
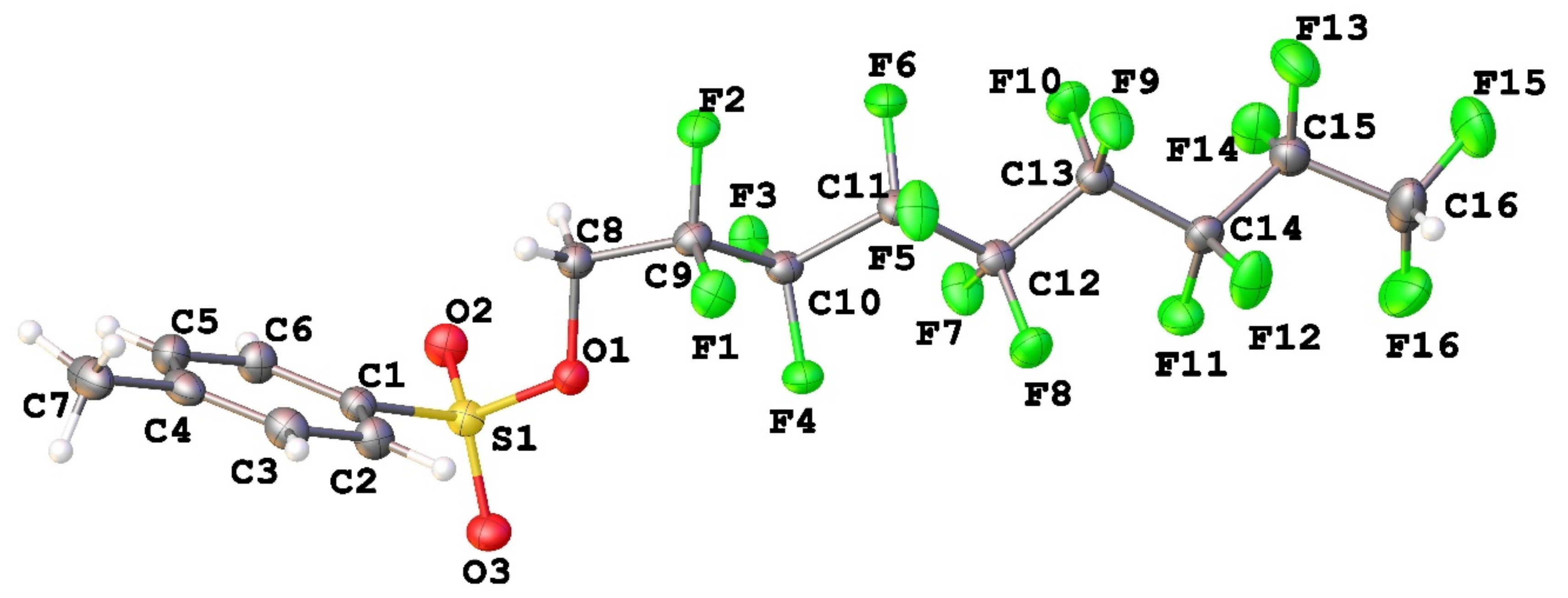
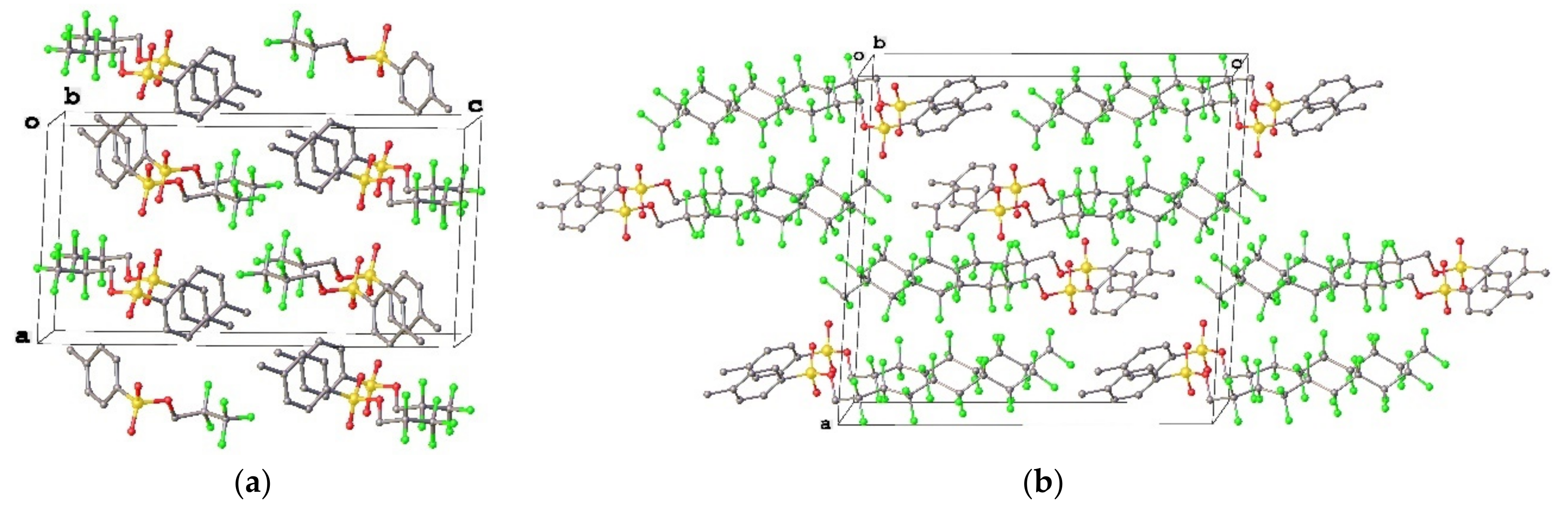
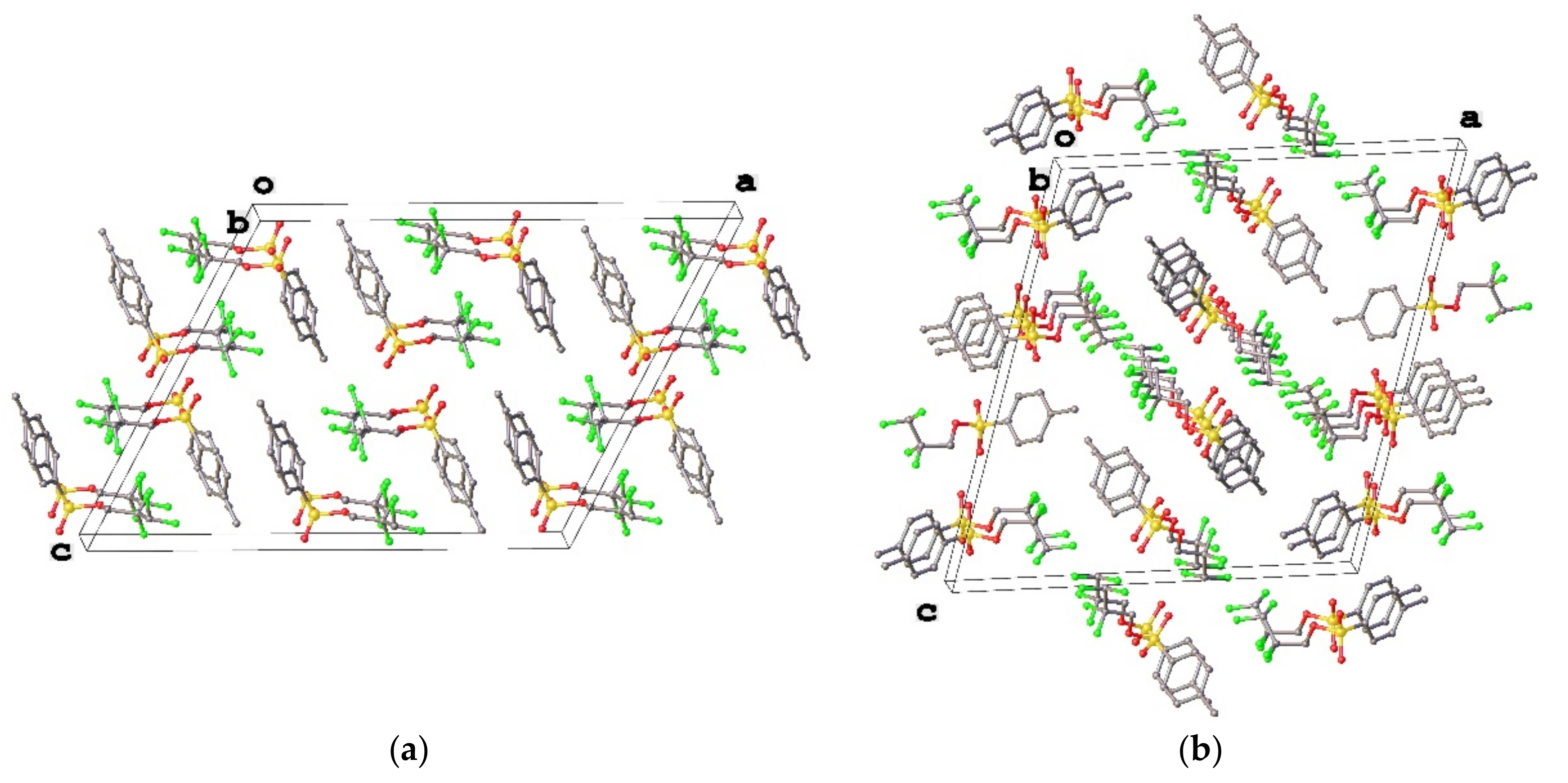
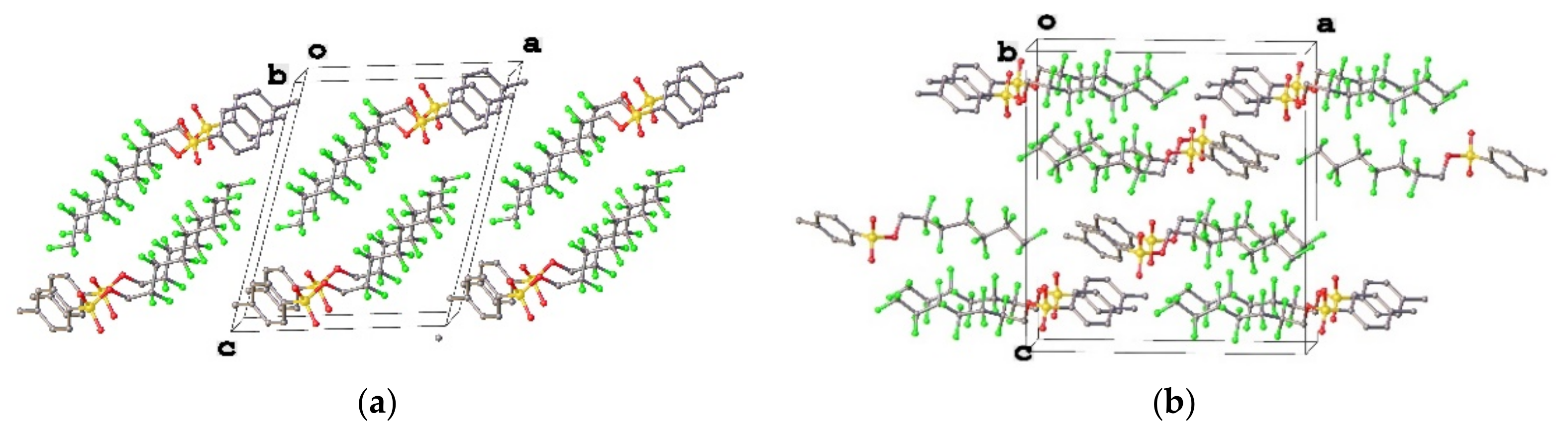
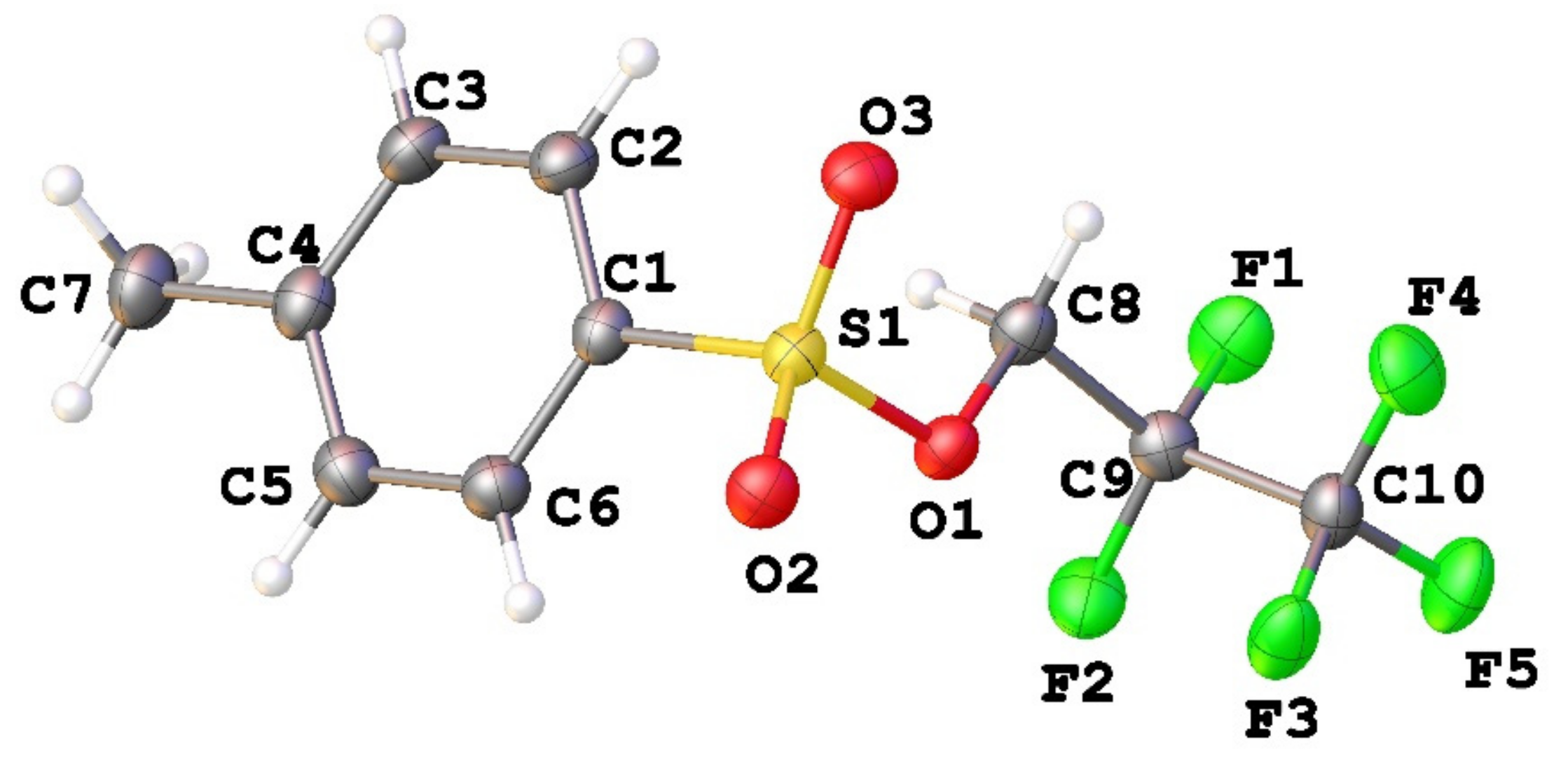
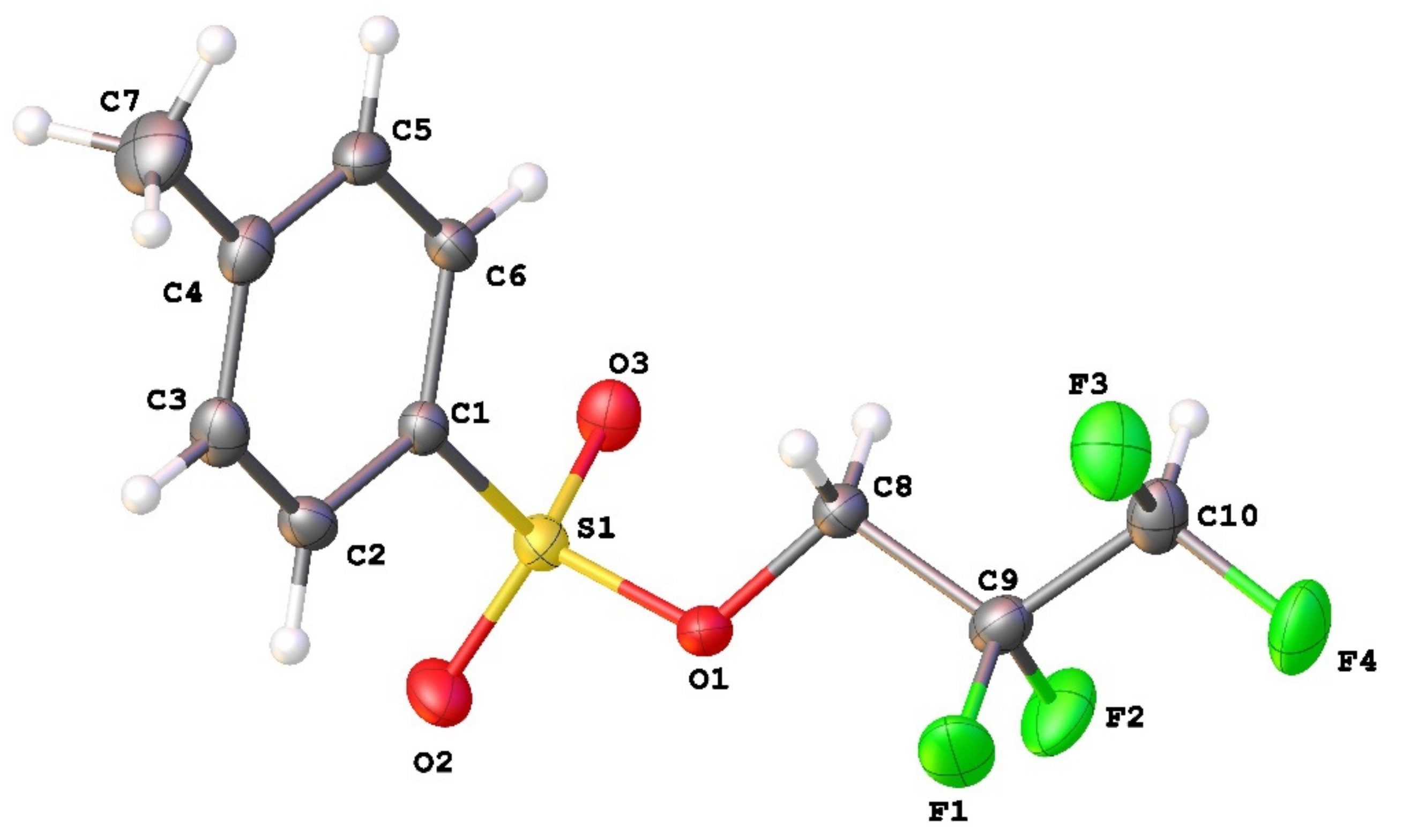
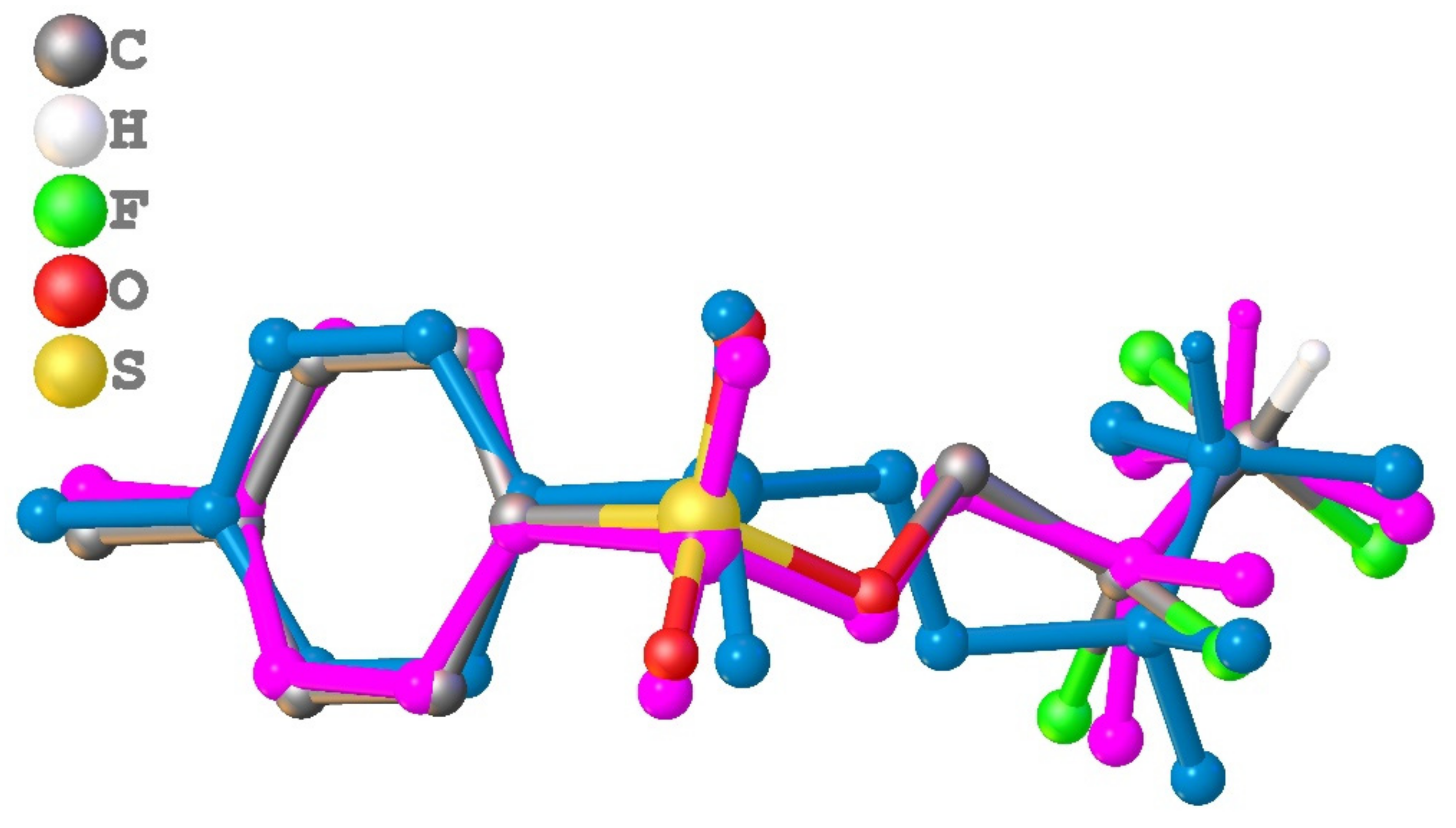
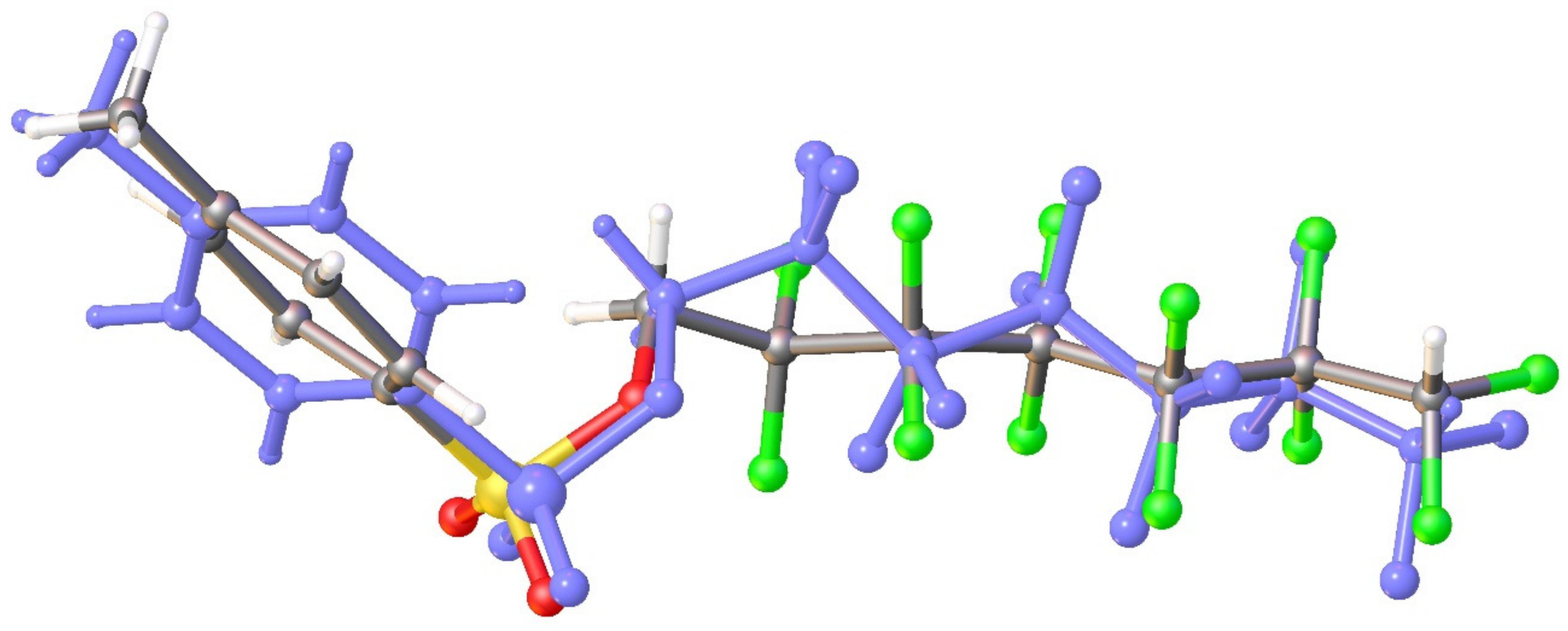
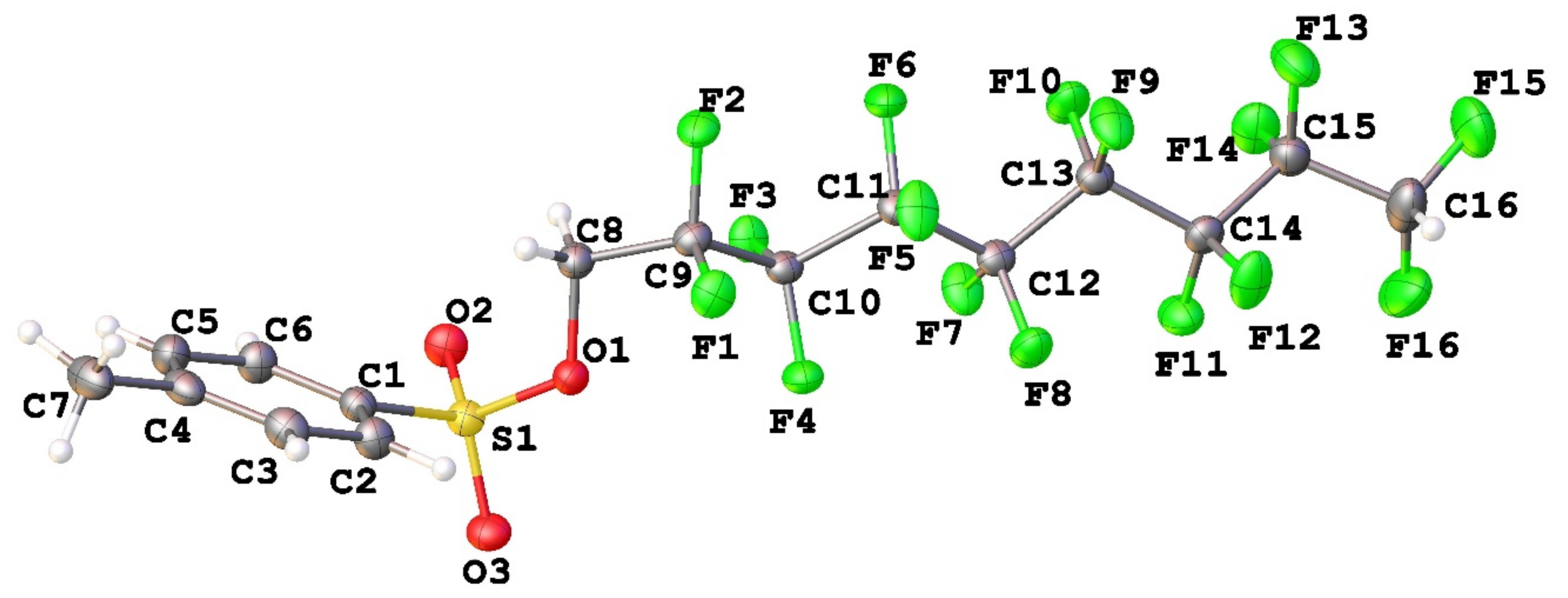
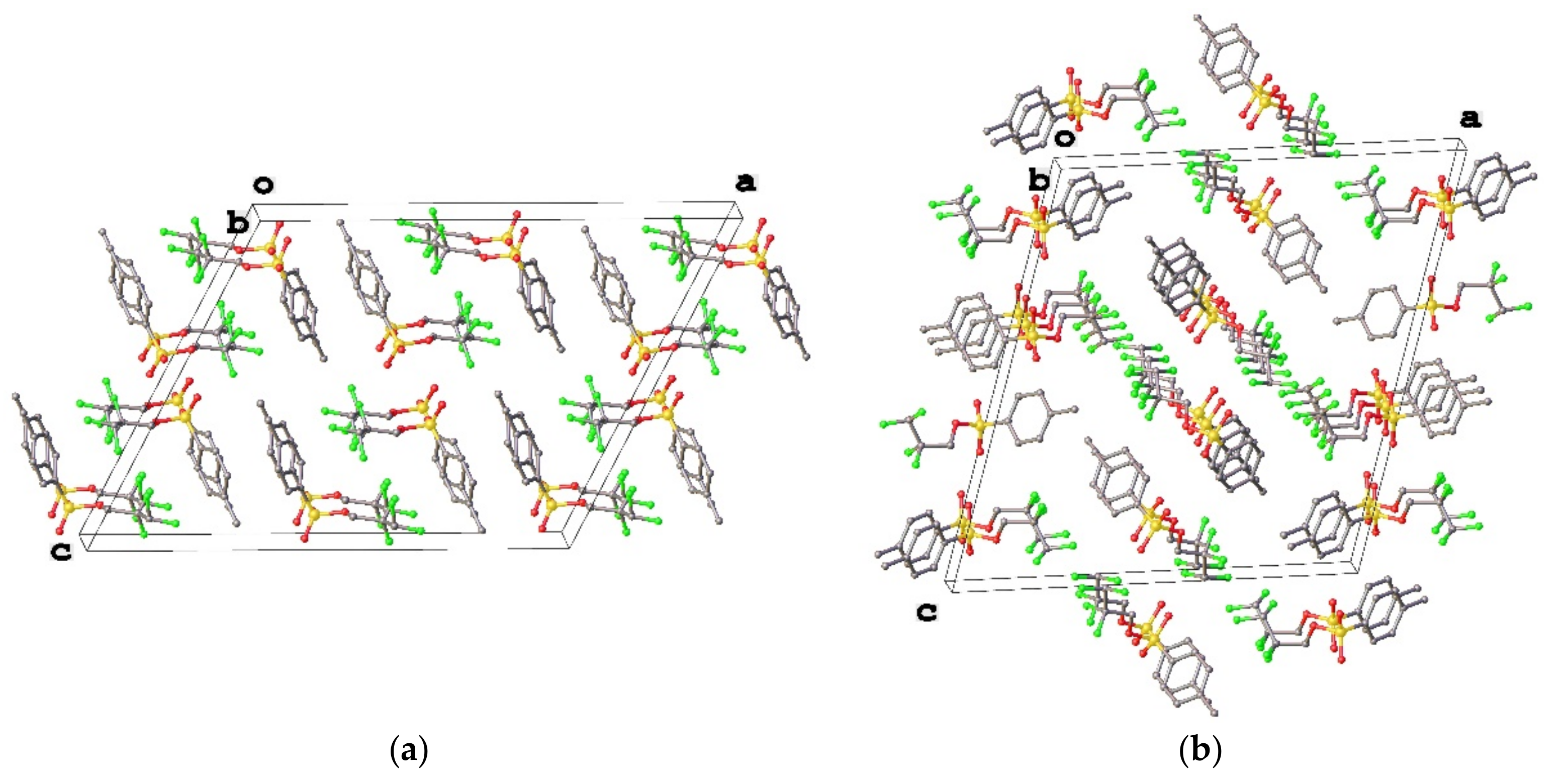
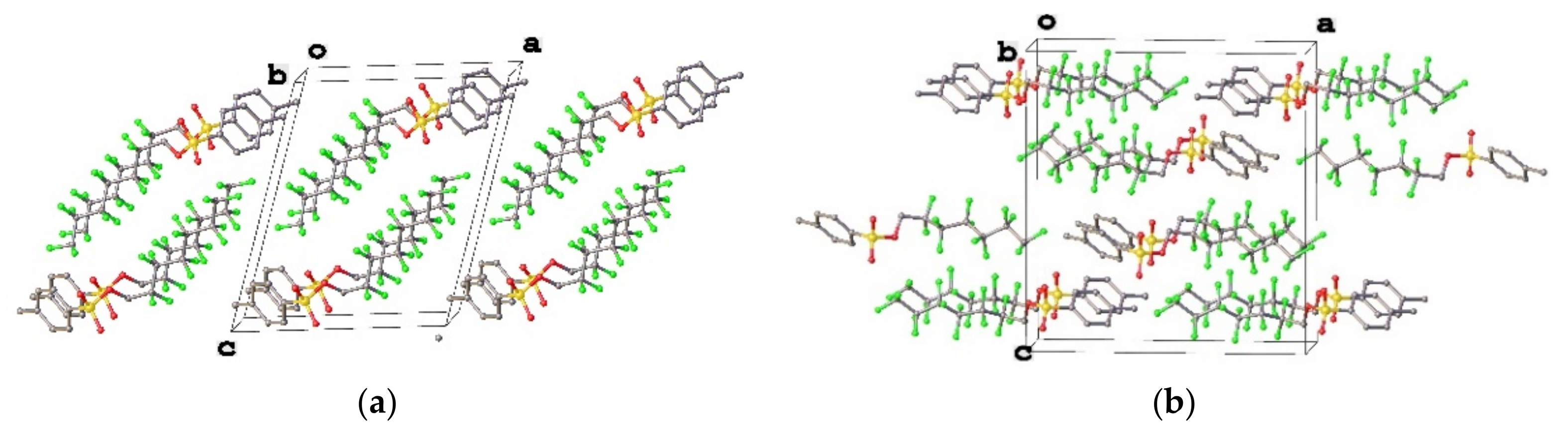
3.1. Geometry and Crystal Packing of 1–4
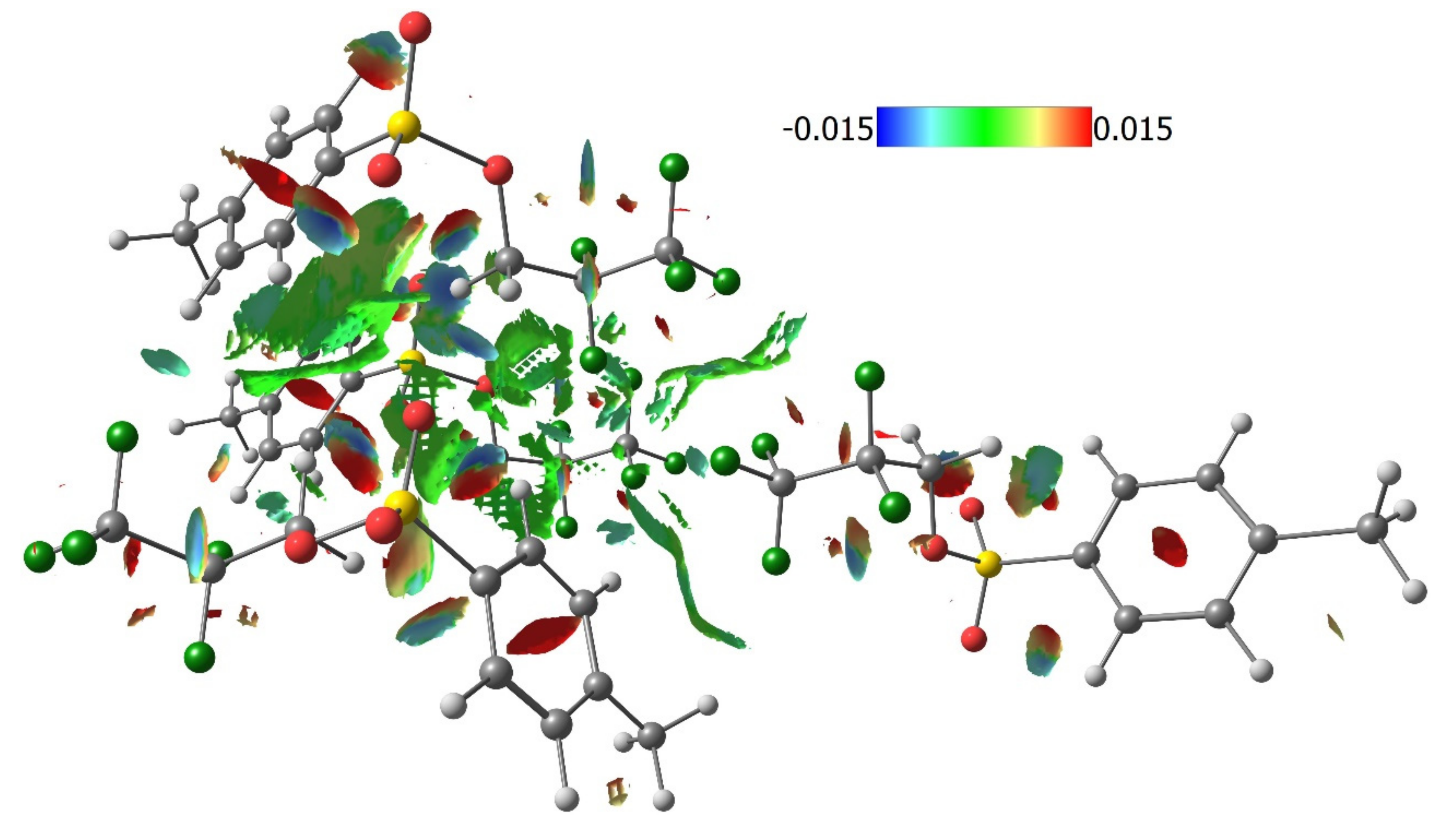
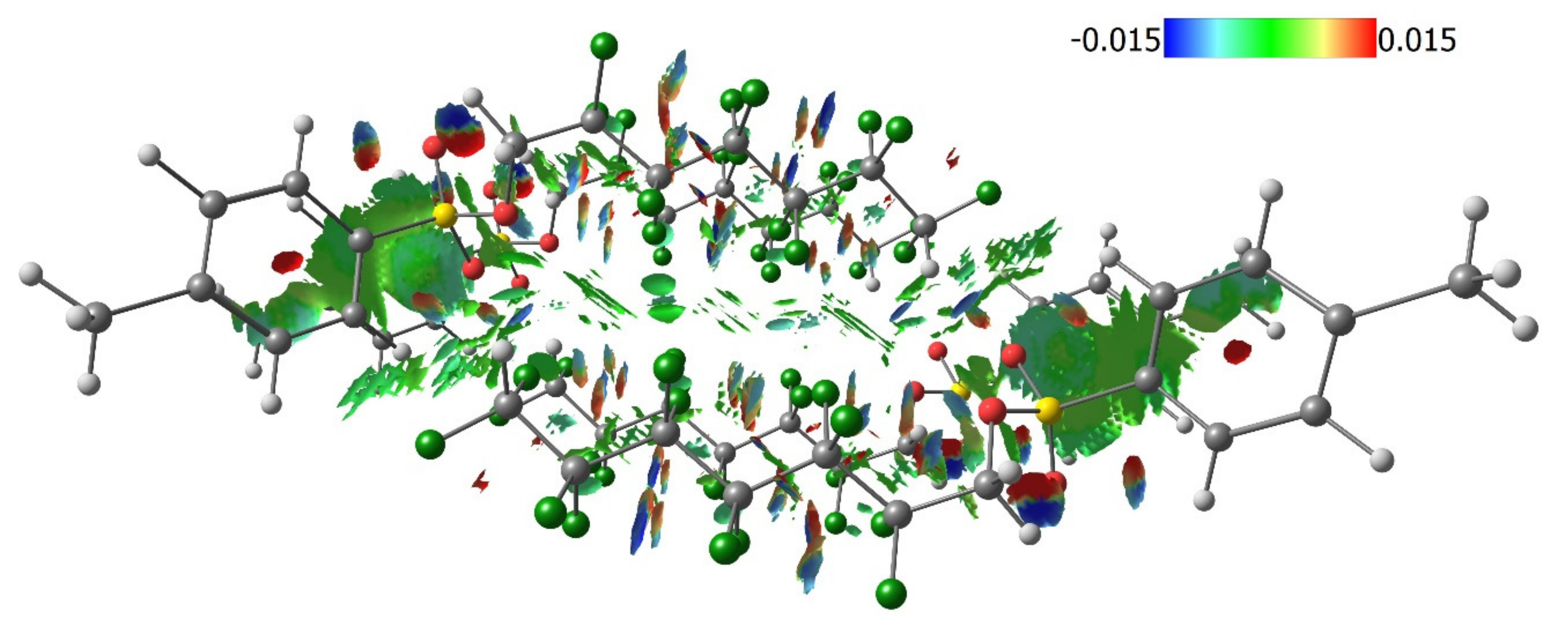
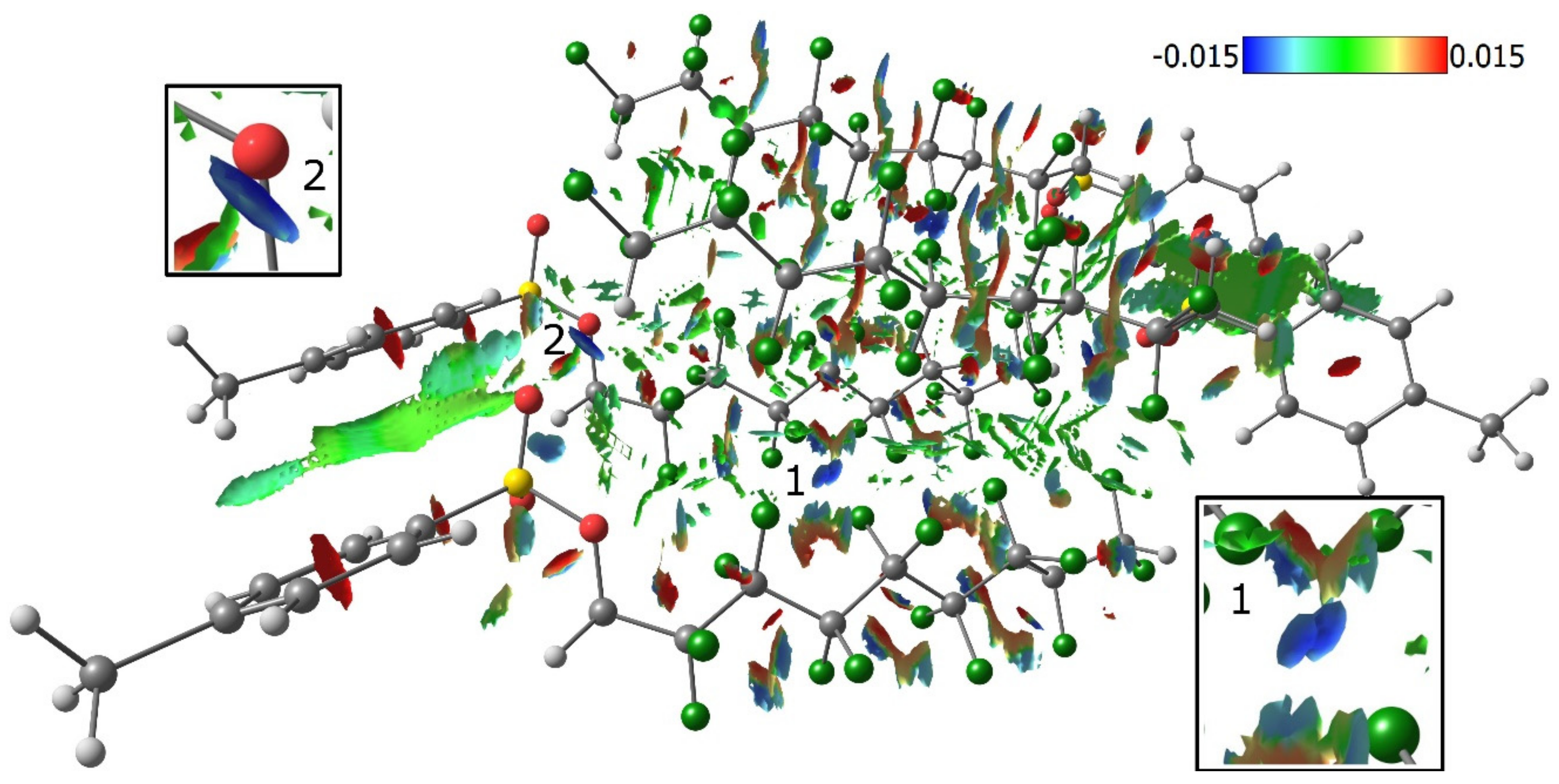
3.2. Quantum Chemical Calculations of Crystal Structures 1–4
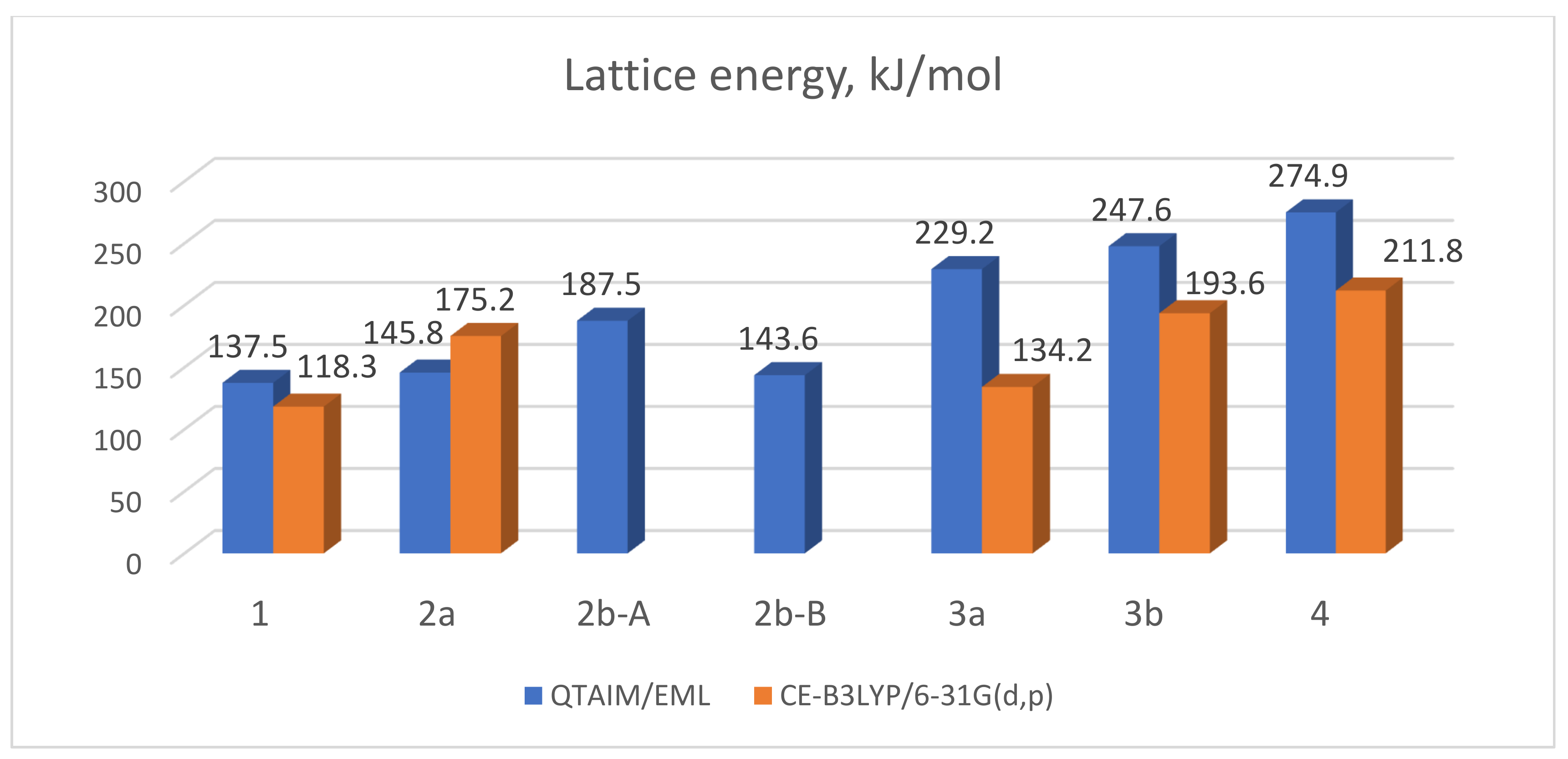
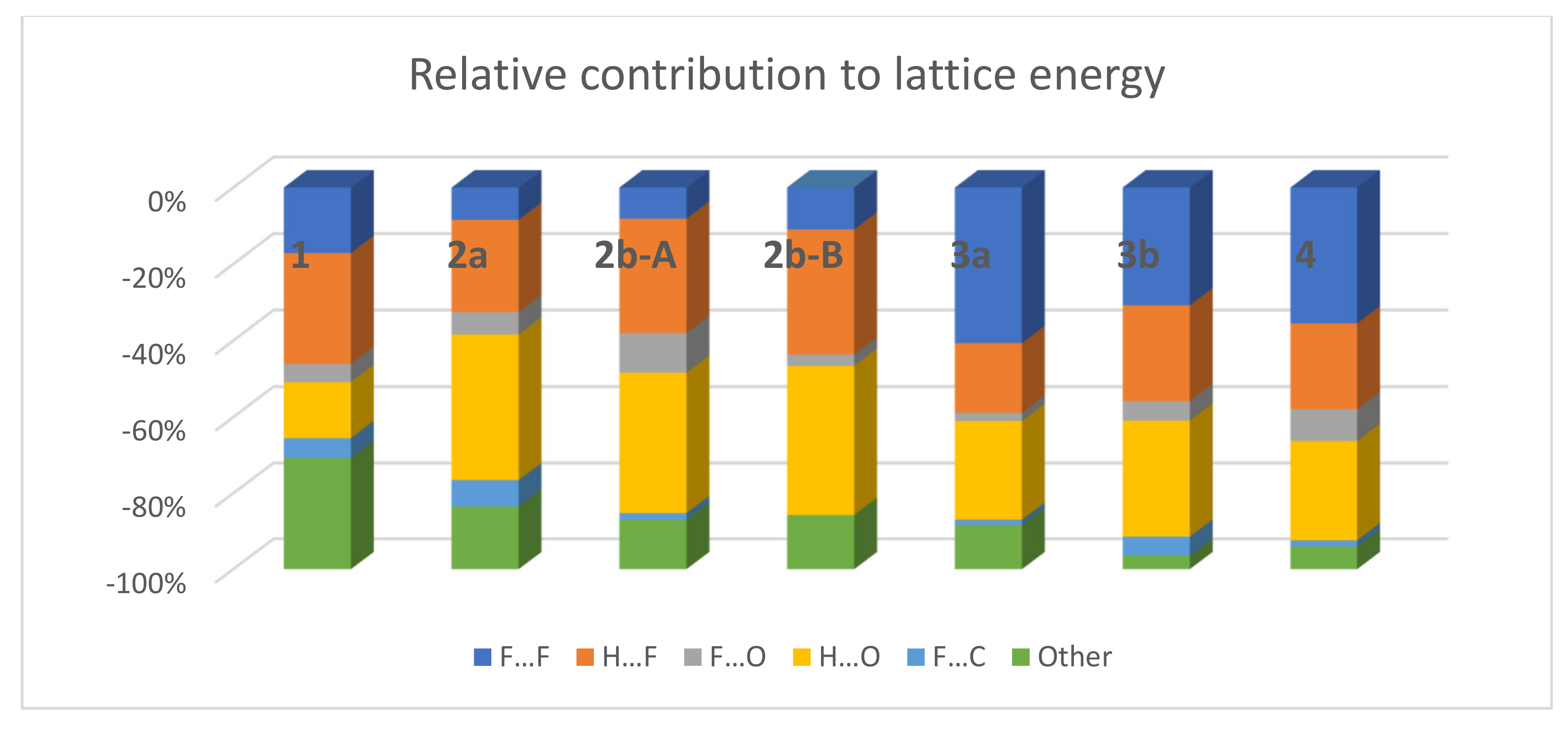
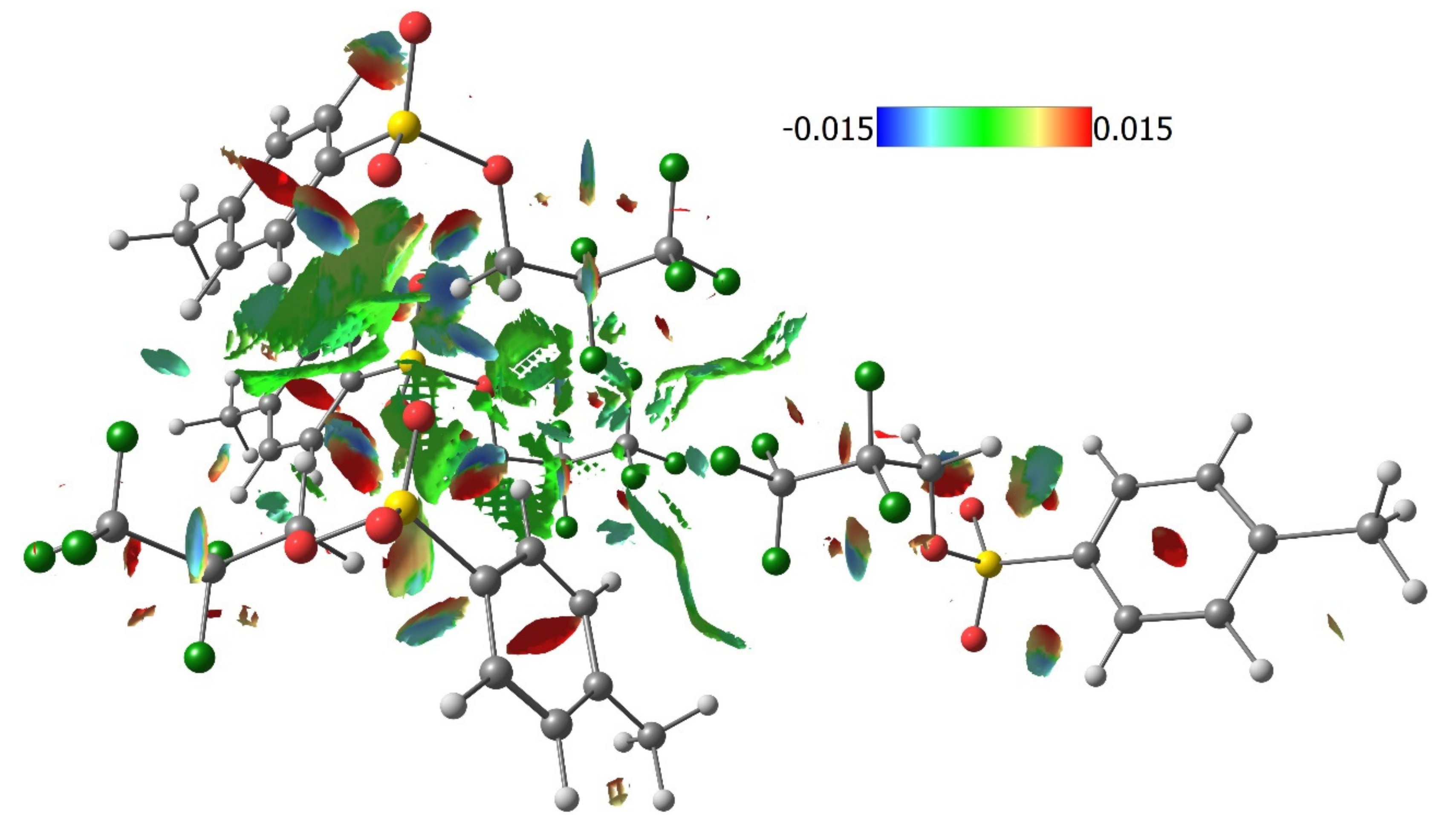
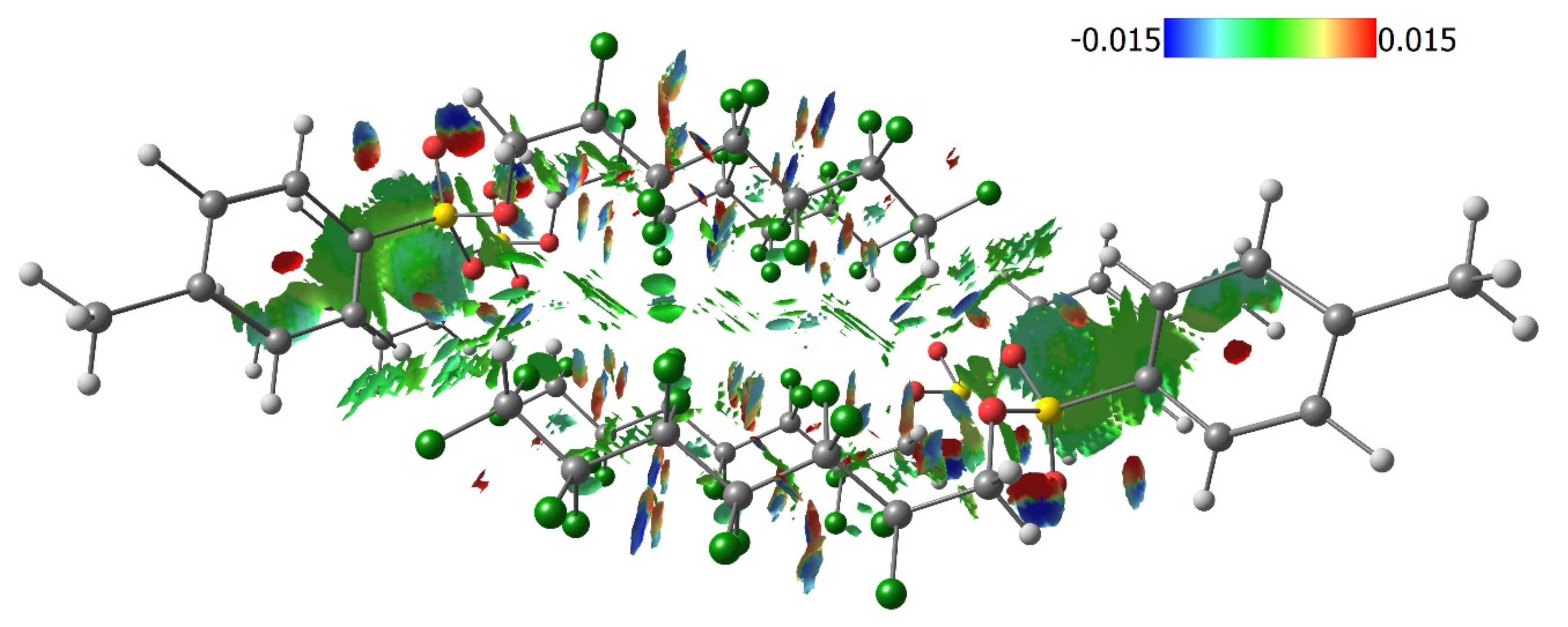
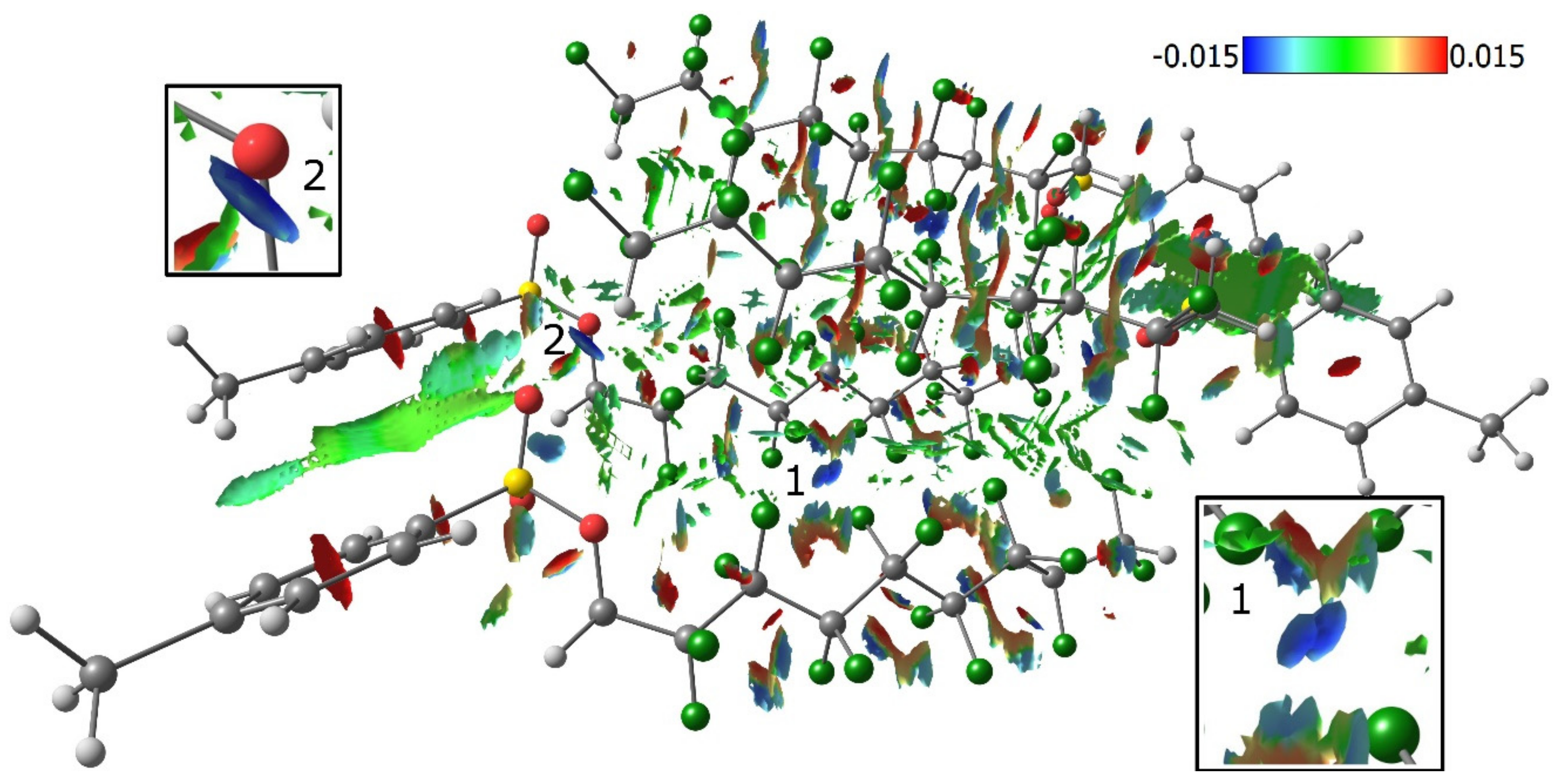
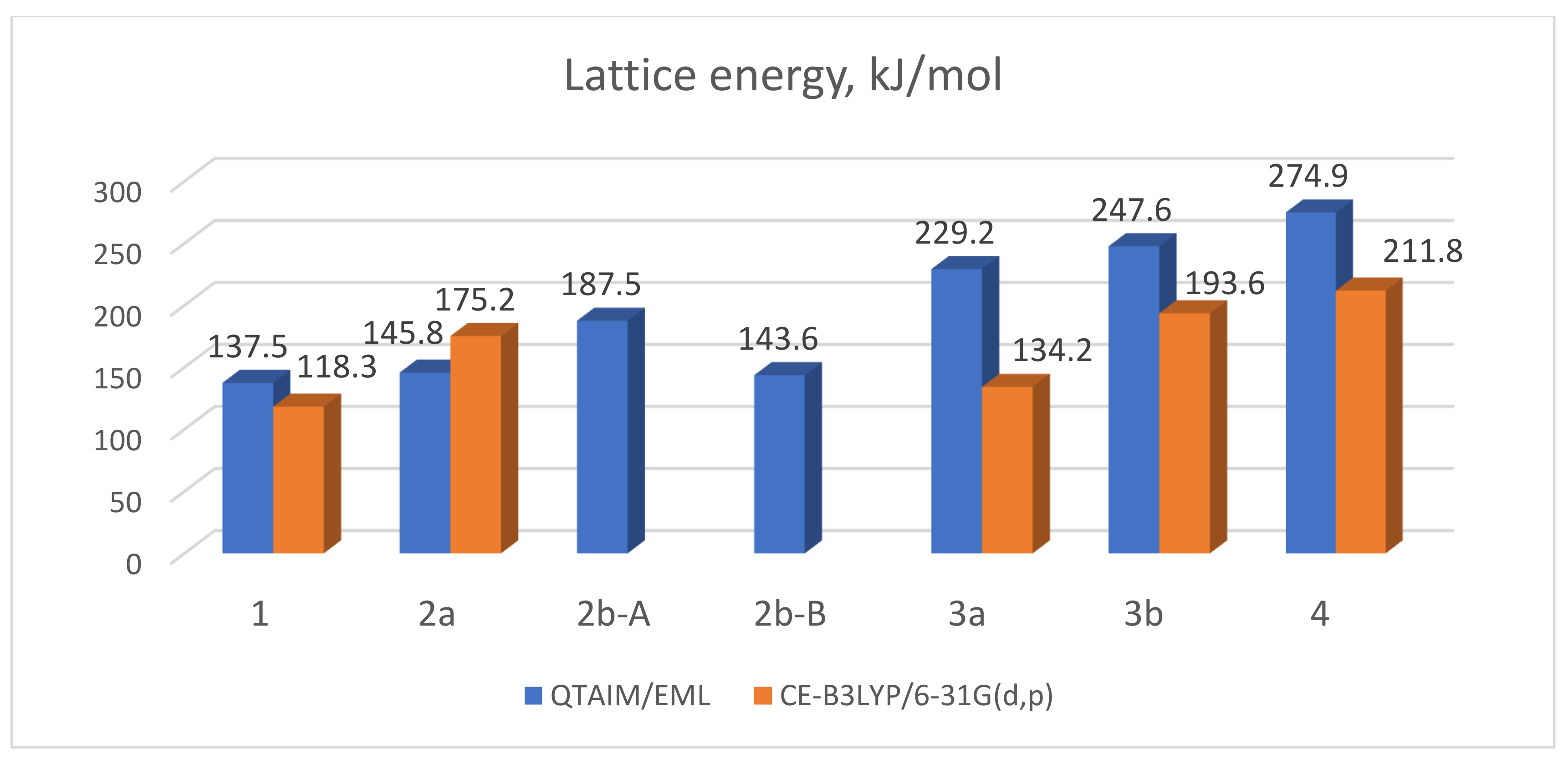
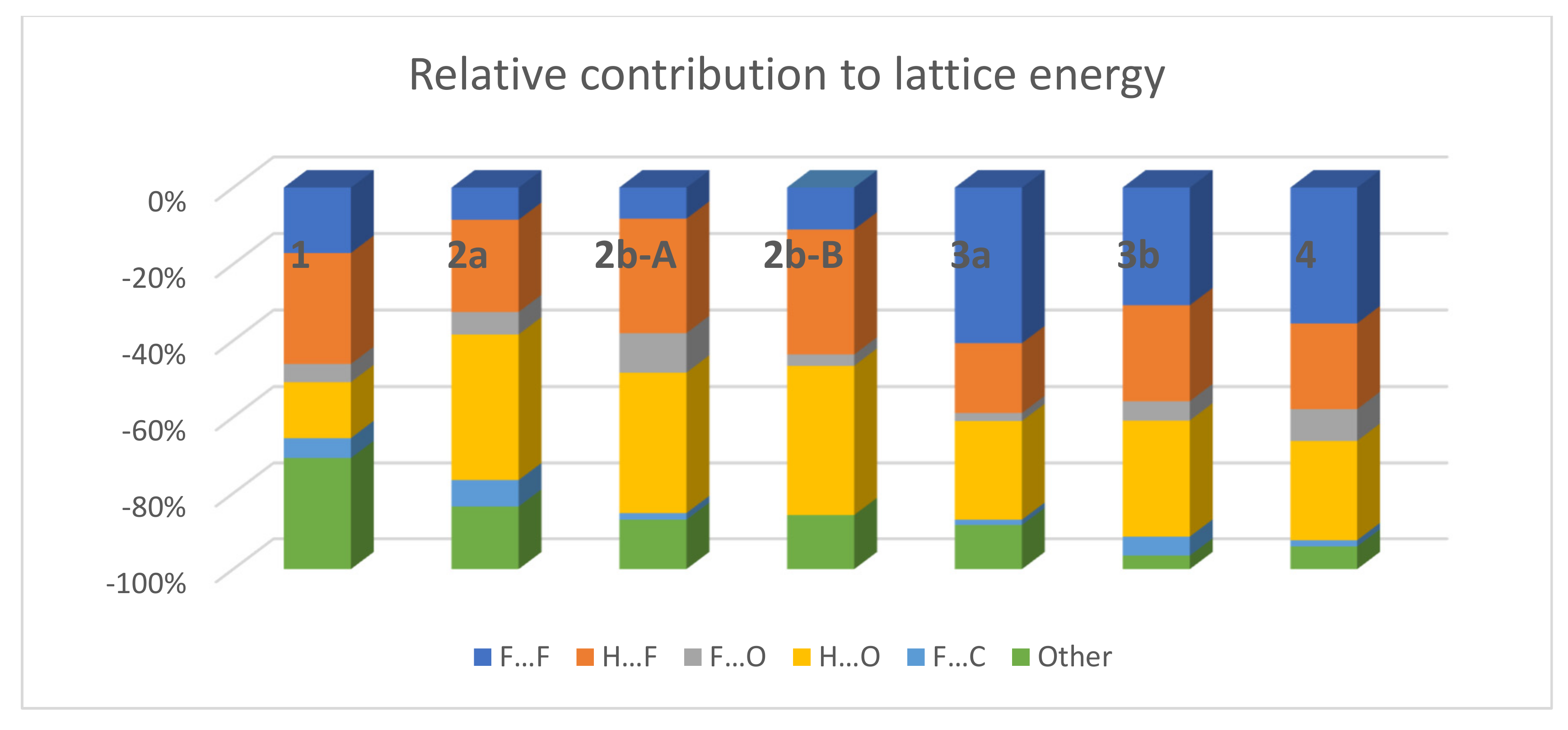
3.3. Lattice Energies and the Role of F…F Interactions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ahrens, L.; Bundschuh, M. Fate and effects of poly- and perfluoroalkyl substances in the aquatic environment: A Review: Fate and effects of polyfluoroalkyl and perfluoroalkyl substances. Environ. Toxicol. Chem. 2014, 33, 1921–1929. [Google Scholar] [CrossRef] [PubMed]
- Ospinal-Jiménez, M.; Pozzo, D.C. Structural analysis of protein denaturation with alkyl perfluorinated sulfonates. Langmuir 2012, 28, 17749–17760. [Google Scholar] [CrossRef]
- Rosholm, K.R.; Arouri, A.; Hansen, P.L.; González-Pérez, A.; Mouritsen, O.G. Characterization of fluorinated catansomes: A promising vector in drug-delivery. Langmuir 2012, 28, 2773–2781. [Google Scholar] [CrossRef] [PubMed]
- Krafft, M. Fluorocarbons and fluorinated amphiphiles in drug delivery and biomedical research. Adv. Drug Deliv. Rev. 2001, 47, 209–228. [Google Scholar] [CrossRef]
- Massi, L.; Guittard, F.; Géribaldi, S.; Levy, R.; Duccini, Y. Antimicrobial properties of highly fluorinated bis-ammonium salts. Int. J. Antimicrob. Agents 2003, 21, 20–26. [Google Scholar] [CrossRef]
- Luo, Z.; Shi, X.; Hu, Q.; Zhao, B.; Huang, M. Structural evidence of perfluorooctane sulfonate transport by human serum albumin. Chem. Res. Toxicol. 2012, 25, 990–992. [Google Scholar] [PubMed]
- Du, Z.; Deng, S.; Bei, Y.; Huang, Q.; Wang, B.; Huang, J.; Yu, G. Adsorption behavior and mechanism of perfluorinated compounds on various adsorbents—A Review. J. Hazard. Mater. 2014, 274, 443–454. [Google Scholar] [CrossRef] [PubMed]
- Wadekar, M.N.; Abezgaus, L.; Djanashvili, K.; Jager, W.F.; Mendes, E.; Picken, S.J.; Dganit, D. Supramolecular “leeks” of a fluorinated hybrid amphiphile that self-assembles into a disordered columnar phase. J. Phys. Chem. B 2013, 117, 2820–2826. [Google Scholar] [CrossRef] [PubMed]
- Prasanna, M.D.; Row, T.N.G. Weak interactions involving organic fluorine: analysis of structural motifs in flunazirine and haloperidol. J. Mol. Struct. 2001, 562, 55–61. [Google Scholar] [CrossRef]
- Choudhury, A.R.; Urs, U.K.; Guru Row, T.N.; Nagarajan, K. Weak interactions involving organic fluorine: a comparative study of the crystal packing in substituted isoquinolines. J. Mol. Struct. 2002, 605, 71–77. [Google Scholar] [CrossRef]
- Osuna, R.M.; Hernández, V.; Navarrete, J.T.L.; D’Oria, E.; Novoa, J.J. Theoretical evaluation of the nature and strength of the F···F intermolecular interactions present in fluorinated hydrocarbons. Theor. Chem. Acc. 2011, 128, 541–553. [Google Scholar] [CrossRef]
- Nayak, S.K.; Reddy, M.K.; Row, T.N.G.; Chopra, D. Role of hetero-halogen (F···X, X = Cl, Br, and I) or homo-halogen (X···X, X = F, Cl, Br, and I) interactions in substituted benzanilides. Cryst. Growth Des. 2011, 11, 1578–1596. [Google Scholar] [CrossRef]
- Prakash, G.K.S.; Wang, F.; Rahm, M.; Shen, J.; Ni, C.; Haiges, R.; Olah, G.A. On the nature of CH⋅⋅⋅FC interactions in hindered CF3C(sp3) bond rotations. Angew. Chem. Int. Ed. 2011, 50, 11761–11764. [Google Scholar] [CrossRef]
- Alkorta, I.; Elguero, J. Fluorine–fluorine interactions: NMR and AIM analysis. Struct. Chem. 2004, 15, 117–120. [Google Scholar] [CrossRef]
- Levina, E.O.; Chernyshov, I.Y.; Voronin, A.P.; Alekseiko, L.N.; Stash, A.I.; Vener, M.V. Solving the enigma of weak fluorine contacts in the solid state: A periodic dft study of fluorinated organic crystals. RSC Adv. 2019, 9, 12520–12537. [Google Scholar] [CrossRef]
- Omorodion, H.; Twamley, B.; Platts, J.A.; Baker, R.J. Further evidence on the importance of fluorous–fluorous interactions in supramolecular chemistry: a combined structural and computational study. Cryst. Growth Des. 2015, 15, 2835–2841. [Google Scholar] [CrossRef]
- Baker, R.J.; Colavita, P.E.; Murphy, D.M.; Platts, J.A.; Wallis, J.D. Fluorine–fluorine interactions in the solid state: an experimental and theoretical study. J. Phys. Chem. A 2012, 116, 1435–1444. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, Y.; Sakakura, Y.; Aso, N.; Okada, S.; Tanabe, Y. Practical and efficient methods for sulfonylation of alcohols using Ts(Ms)Cl/Et3N and Catalytic Me3H·HCl as combined base: Promising alternative to traditional pyridine. Tetrahedron 1999, 55, 2183–2192. [Google Scholar] [CrossRef]
- Banerjee, S.; Vidya, V.M.; Savyasachi, A.J.; Maitra, U. Perfluoroalkyl bile esters: A new class of efficient gelators of organic and aqueous–Organic media. J. Mater. Chem. 2011, 21, 14693–14705. [Google Scholar] [CrossRef]
- Cohen, W.V. Nucleophilic substitution in fluoroalkyl sulfates, sulfonates, and related compounds. J. Org. Chem. 1961, 26, 4021–4026. [Google Scholar] [CrossRef]
- Shilin, S.; Florensova, O.; Chernov, N.; Voronkov, M. Alpha, alpha, omega-trihydro-alpha-halo perfluoroalkanes. Zhurnal Obshchei Khimii 1991, 61, 1838–1840. [Google Scholar]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. 2015, A71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. 2015, C71, 3–8. [Google Scholar]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular-dynamics simulation of the liquid-metal amorphous-semiconductor transition in germanium. Phys. Rev. B 1994, 49, 14251–14269. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmuller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmuller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Moellmann, J.; Grimme, S. Importance of London dispersion effects for the packing of molecular crystals: A case study for intramolecular stacking in a Bis-thiophene derivative. Phys. Chem. Chem. Phys. 2010, 12, 8500. [Google Scholar] [CrossRef] [PubMed]
- Gonze, X.; Beuken, J.-M.; Caracas, R.; Detraux, F.; Fuchs, M.; Rignanese, G.-M.; Sindic, L.; Verstraete, M.; Zerah, G.; Jollet, F.; et al. First-principles computation of material properties: The ABINIT software project. Comput. Mater. Sci. 2002, 25, 478–492. [Google Scholar] [CrossRef]
- Otero-de-la-Roza, A.; Johnson, E.R.; Luaña, V. Critic2: A program for real-space analysis of quantum chemical interactions in solids. Comput. Phys. Commun. 2014, 185, 1007–1018. [Google Scholar] [CrossRef]
- Espinosa, E.; Molins, E.; Lecomte, C. Hydrogen bond strengths revealed by topological analyses of experimentally observed electron densities. Chem. Phys. Lett. 1998, 285, 170–173. [Google Scholar] [CrossRef]
- Mackenzie, C.F.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer model energies and energy frameworks: Extension to metal coordination compounds, organic salts, solvates and open-shell systems. IUCrJ 2017, 4, 575–587. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge structural database. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2016, 72, 171–179. [Google Scholar] [CrossRef]
- Rowland, R.S.; Taylor, R. Intermolecular nonbonded contact distances in organic crystal structures: comparison with distances expected from van der Waals Radii. J. Phys. Chem. 1996, 100, 7384–7391. [Google Scholar] [CrossRef]
- Desiraju, G.R.; Parthasarathy, R. The nature of Halogen...Halogen interactions: Are short halogen contacts due to specific attractive forces or due to close packing of nonspherical atoms? J. Am. Chem. Soc. 1989, 111, 8725–8726. [Google Scholar] [CrossRef]
- Bader, R.W.F. Atoms in Molecules: A Quantum Theory; Oxford University Press: New York, NY, USA, 1990. [Google Scholar]
- Levin, V.V.; Dilman, A.D.; Korlyukov, A.A.; Belyakov, P.A.; Struchkova, M.I.; Antipin, M.Yu.; Tartakovsky, V.A. Synthesis and structures of tris(pentafluorophenyl)silylamines. Russ. Chem. Bull. 2007, 56, 1394–1401. [Google Scholar] [CrossRef]
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Revealing noncovalent interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef]
- Saleh, G.; Gatti, C.; Lo Presti, L. Non-covalent Interaction via the reduced density gradient: independent atom model vs experimental multipolar electron densities. Comput. Theor. Chem. 2012, 998, 148–163. [Google Scholar] [CrossRef]
- Spackman, M.A. How reliable are intermolecular interaction energies estimated from topological analysis of experimental electron densities? Cryst. Growth Des. 2015, 15, 5624–5628. [Google Scholar] [CrossRef]
- Kuznetsov, M.L. Can halogen bond energy be reliably estimated from electron density properties at bond critical point? The case of the (A)nZ—Y•••X − (X, Y = F, Cl, Br) interactions. Int. J. Quantum Chem. 2019, 119, e25869. [Google Scholar] [CrossRef]
- Nelyubina, Y.V.; Korlyukov, A.A.; Lyssenko, K.A. Probing weak intermolecular interactions by using the invariom approach: A comparative study of s-tetrazine. Chem. Eur. J. 2014, 6978–6984. [Google Scholar] [CrossRef] [PubMed]
- Nelyubina, Y.V.; Korlyukov, A.A.; Lyssenko, K.A. Probing systematic errors in experimental charge density by multipole and invariom modeling: A twinned crystal of 1,10-phenanthroline hydrate. Mendeleev Commun. 2014, 24, 286–289. [Google Scholar] [CrossRef]
- Lyssenko, K.A.; Korlyukov, A.A.; Golovanov, D.G.; Ketkov, S.Yu.; Antipin, M.Yu. Estimation of the barrier to rotation of benzene in the (η6-C6H6)2Cr crystal via topological analysis of the electron density distribution function. J. Phys. Chem. A 2006, 110, 6545–6551. [Google Scholar] [CrossRef]
- Saha, A.; Rather, S.A.; Sharada, D.; Saha, B.K. ’C–X···X–C vs C–H···X–C, which one is the more dominant interaction in crystal packing (X = Halogen)? Cryst. Growth Des. 2018, 18, 6084–6090. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structural Parameters (Å and °) | Crystal Structure | |||||
|---|---|---|---|---|---|---|
| 1 | 2a | 2b | 3a | 3b | 4 | |
| S1-O1 | 1.5980(12) | 1.5896(12) | 1.5918(18) | 1.602(3) | 1.586(2) | 1.5900(14) |
| S1-O2 | 1.4238(14) | 1.4304(13) | 1.4284(19) | 1.438(3) | 1.425(2) | 1.4347(15) |
| S1-O3 | 1.4285(14) | 1.4276(13) | 1.4273(19) | 1.433(4) | 1.429(2) | 1.4298(15) |
| S1-C1 | 1.7474(17) | 1.7530(16) | 1.749(3) | 1.752(5) | 1.752(3) | 1.7584(19) |
| O1-C8 | 1.439(2) | 1.445(2) | 1.445(3) | 1.436(6) | 1.438(3) | 1.448(2) |
| C9-F | 1.353(2) | 1.356(2) | 1.358(3) | 1.360(5) | 1.356(3) | 1.357(2) |
| C-F * | - | - | - | 1.348(6) | 1.344(3) | 1.345(2) |
| C-Fterm | 1.326(2) | 1.356(2) | 1.354(3) | 1.338(8) | 1.347(5) | 1.350(4) |
| O1-S1-C1 | 103.65(8) | 103.97(7) | 104.04(11) | 103.6(2) | 103.08(12) | 103.32(8) |
| C8-O1-S1 | 116.09(11) | 116.91(10) | 117.77(16) | 119.9(3) | 116.98(16) | 117.07(11) |
| Interactions (Compound) | Type (Intramolecular/Intermolecular) | Experimental Distance | Calculated Distance | Energy, kJ/mol |
|---|---|---|---|---|
| F4…F4 (1) | intermolecular | 2.978(3) | 2.925 | −4.2 |
| F3…F3 (2a) | intermolecular | 2.750(2) | 2.785 | −5.9 |
| F4…F4A (2b) | intermolecular | 3.074(2) | 3.046 | −3.1 |
| F3…F7 (3a) | intramolecular | 2.633(5) | 2.649 | −12.2 |
| F3…F8 (3a) | intermolecular | 2.764(4) | 2.785 | −5.5 |
| F5…F9 (3a) | intramolecular | 2.582(5) | 2.620 | −4.6 |
| F4…F7 (3b) | intermolecular | 2.921(2) | 2.876 | −4.6 |
| F5…F8 (3b) | intermolecular | 2.543(3) | 2.572 | −10.5 |
| F4…F11 (4) | intermolecular | 2.9031(17) | 2.937 | −3.8 |
| F4…F12 (4) | intermolecular | 2.7953917) | 2.777 | −5.5 |
| F5…F7 (4) | intermolecular | 2.5112(17) | 2.507 | −13.0 |
| F14…F14 (4) | intermolecular | 2.942(3) | 2.895 | −5.0 |
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arkhipov, D.E.; Lyubeshkin, A.V.; Volodin, A.D.; Korlyukov, A.A. Molecular Structures Polymorphism the Role of F…F Interactions in Crystal Packing of Fluorinated Tosylates. Crystals 2019, 9, 242. https://doi.org/10.3390/cryst9050242
Arkhipov DE, Lyubeshkin AV, Volodin AD, Korlyukov AA. Molecular Structures Polymorphism the Role of F…F Interactions in Crystal Packing of Fluorinated Tosylates. Crystals. 2019; 9(5):242. https://doi.org/10.3390/cryst9050242
Chicago/Turabian StyleArkhipov, Dmitry E., Alexander V. Lyubeshkin, Alexander D. Volodin, and Alexander A. Korlyukov. 2019. "Molecular Structures Polymorphism the Role of F…F Interactions in Crystal Packing of Fluorinated Tosylates" Crystals 9, no. 5: 242. https://doi.org/10.3390/cryst9050242
APA StyleArkhipov, D. E., Lyubeshkin, A. V., Volodin, A. D., & Korlyukov, A. A. (2019). Molecular Structures Polymorphism the Role of F…F Interactions in Crystal Packing of Fluorinated Tosylates. Crystals, 9(5), 242. https://doi.org/10.3390/cryst9050242