Abstract
Cu-Cr-Zr alloy is a typical precipitation-strengthened alloy, and the interface stability between precipitates and the Cu matrix significantly influences the alloy’s strength and properties. However, research in this field is currently lacking. In light of this, we focus on the CuZr2 precipitate and investigate its surface and interface properties using the GGA/PBE method within density functional theory. The results indicate that the (100) and (010) surfaces of CuZr2 share the same atomic structure, with both being stoichiometric surfaces. By fitting the relationship between the total energy of surface supercells with varying numbers of atoms and the number of atomic layers, the surface energy values were accurately calculated. The (100) surface is a non-stoichiometric surface, featuring three surface terminations: Cu, Zr1, and Zr2, with the Zr2 termination being the most stable. Finally, based on experimental observations, the atomic structure of the CuZr2 (010)/Cu (110) interface was predicted. The calculated interfacial energy reveals that the lattice mismatch between the CuZr2 precipitate and the Cu matrix significantly affects interfacial stability.
1. Introduction
Cu-Cr-Zr alloy is one of the important materials in modern high-tech fields, widely used in rail transit, aerospace, nuclear fusion, and other areas [1,2,3,4]. As a crucial high-strength, high-conductivity Cu alloy, its optimal composition ratio depends on specific performance requirements. The classical proportion currently recognized by academia is Cu: >98.5%, Cr: 0.5–1.2%, and Zr: 0.03–0.3%. This composition can achieve a good balance among strength, conductivity, and workability through the necessary “solution-quenching-aging” heat treatment process [5,6,7,8,9].
However, with the continuous advancement of high-end manufacturing, Cu-Cr-Zr alloys need to adapt to even more demanding working environments. For example, under extreme conditions such as those in nuclear fusion applications, high temperatures (>450 °C) and neutron irradiation can lead to hardening of Cu-Cr-Zr materials and a decrease in ductility and toughness, as well as the coarsening of precipitates, ultimately resulting in the failure of Cu-Cr-Zr alloys during service [10,11]. Therefore, developing a new generation of high-strength, heat-resistant Cu-Cr-Zr alloys has become an urgent need in the industry.
Current research on the Cu-Cr-Zr alloy primarily focuses on the effects of alloy composition design, heat treatment, and processing manufacturing techniques on its microstructure and properties. For instance, Liu et al. designed and prepared Cu-1Cr-0.1Zr and Cu-1Cr-0.1Zr-0.069La alloy ingots and subjected them to a process involving solution treatment, warm rolling, cold rolling, pre-aging, cold rolling, and aging. The results showed that after warm rolling at 400 °C, a small amount of nano-sized fcc-structured Cr phase precipitated in the alloy matrix, while the primary Cr phase existed in spherical and rod-like forms at grain boundaries and within grains. The rare earth La encapsulated the outer layer of the Cr phase, forming a core–shell structure that inhibited the growth of the Cr phase [12]. Liu et al. employed a high-throughput experimental approach, simultaneously adjusting different alloy compositions alongside heat treatment and rolling processes, and obtained a Cu-Cr-Zr alloy with excellent comprehensive properties. This alloy exhibited a hardness (HV) greater than 170 and a high electrical conductivity of 85% IACS. After deformation, the Cu-Cr-Zr alloy underwent short-term aging at medium temperatures, leading to the formation of a large number of finely dispersed Cr-Zr and Cu-Zr nano-precipitates. Consequently, the corresponding copper alloy demonstrated outstanding mechanical and electrical properties [13]. Wu et al. performed high-deformation-rate cold rolling and aging heat treatment on a commercially composed alloy, achieving a Cu-Cr-Zr alloy with superior overall performance. Compared to its initial state, the hardness and tensile strength increased by 80%, and electrical conductivity improved by 15% [14]. Research by Hu et al. indicated that a two-stage aging heat treatment promotes the formation of solute-rich regions (G.P. zones) in Cu-Cr-Zr alloys, simultaneously enhancing strength, plasticity, and electrical conductivity [15].
The excellent properties of Cu-Cr-Zr alloys stem from their multi-scale microstructure. The fundamental reason lies in their nature as a typical precipitation-strengthened alloy, which can precipitate nanoscale second-phase particles such as Cr, Cu5Zr, and CuZr2 during the aging process. These precipitates can form stable interfaces with the Cu matrix, thereby hindering the movement of dislocations and grain boundaries and enhancing the strength of the alloy.
These nanoscale precipitates are, however, prone to coarsening at high temperatures, leading to interface instability and a decline in the mechanical properties of Cu-Cr-Zr alloys. Consequently, such alloys fail to meet the demands for long-term service under high-temperature conditions. Investigating the influence of alloying elements on interface stability is key to improving the high-temperature mechanical properties of the alloy. Yet, there is currently limited research at the atomic level on the interface structure of Cu-Cr-Zr alloys and the effect of solute atoms on interface stability. Moreover, there remains controversy over the types, composition, and structure of precipitates in Cu-Cr-Zr alloys, posing an obstacle to further research on interface structures. Until recently, the latest study reported the atomic structure, elasticity, and the influence of Cr atoms on the thermodynamic properties of the CuZr2 precipitate [16], but the effect of Cr atoms on interface stability remains unreported.
Herein, based on first-principles calculations, the present work provides the surface properties, interface structure, and stability of the CuZr2 precipitate at the atomic and electronic levels. It is hoped that this can offer theoretical references for further research on the impact of alloying atoms on the stability of the CuZr2/Cu interface.
2. Calculation Methods
All calculations in this work were performed using the Vienna Ab initio Simulation Package (VASP) (version number vasp5.3) [17], a software package based on density functional theory. The projector augmented wave (PAW) pseudopotential [18] was employed to describe the interaction between ion cores and electrons. The exchange–correlation interaction among electrons was treated using the Perdew–Burke–Ernzerhof (PBE) exchange–correlation functional within the generalized gradient approximation (GGA) [19]. For bulk calculations of the Cu and CuZr2 precipitate, Monkhorst–Pack k-point grids of 16 × 16 × 16 and 12 × 12 × 4 were used, respectively. The Monkhorst–Pack k-point grid densities for the (010) surface supercell, (100) surface supercell, and interface structure calculations were set to 2 × 8 × 1, 8 × 4 × 1, and 2 × 2 × 1, respectively. The convergence criterion for electronic iteration steps was set at 10−5 eV/atom, and the atomic force convergence criterion was set at 0.01 eV/Å. The energy cutoff for all calculations was set to 450 eV.
3. Results and Discussions
3.1. Surface Properties of CuZr2 Crystal
Figure 1 shows the unit cell structures of face-centered cubic Cu and body-centered tetragonal CuZr2 crystals. We first performed structural relaxation on the Cu and CuZr2 unit cells, including volume and atomic coordinates, and conducted bulk property tests. By fitting the Birch–Murnaghan equation of state, we obtained the lattice constants and bulk modulus for Cu and CuZr2, as summarized in Table 1. The results show reasonable agreement with reference data from the Inorganic Crystal Structure Database (ICSD) [20], experimental values [21,22,23], and other theoretical results [24]. This consistency validates the appropriateness of our computational parameter settings.
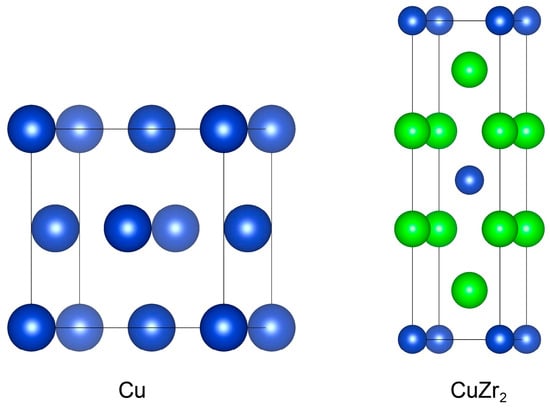
Figure 1.
Conventional unit cell of Cu and CuZr2. The blue and green colors represent Cu and Zr atoms, respectively.

Table 1.
The lattice parameters and bulk modules of Cu and CuZr2 crystals.
Before studying the interface structure, we first explored the surface properties of the CuZr2 crystal. Here, we focus only on the structures and properties of three low-index surfaces: (001), (010), and (001). Since the surface structures of CuZr2 (001) and (010) are identical, both are stoichiometric surfaces terminated with Cu-Zr. We take the (010) surface structure as an example to calculate its surface energy. Such calculation was performed using the constructed surface supercells with 5, 7, 9, and 11 layers, as shown in Figure 2. Please note that each supercell contains an implemented 15 Å vacuum region.
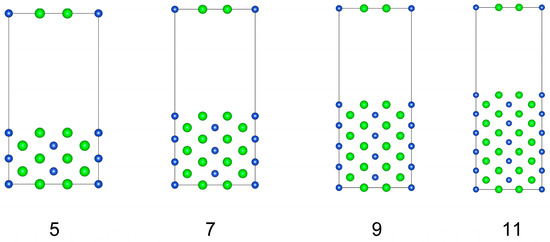
Figure 2.
Four models with different slabs of (010) surface of CuZr2 crystal.
For a given surface supercell, the surface energy can be expressed by the following equation:
where denotes the total energy of the supercell, and and represent the numbers of Cu and Zr atoms in the supercell. and are the chemical potentials of Cu and Zr in the surface state, and A is the base area of the surface supercell. It is evident that the chemical potentials of Cu and Zr atoms in the surface state are not equal to those in the bulk phase, and accurate values cannot be obtained solely through theoretical calculations.
However, when Cu and Zr in the surface state combine to form the CuZr2 crystal, their chemical potential must equal the chemical potential of the bulk . That is, the following condition must be satisfied:
Combining Equations (1) and (2), the following equation can be obtained:
For a stoichiometric surface supercell, . Therefore, Equation (3) can be simplified to the following equation:
Thus, according to Equation (4), the total energy of the surface supercell exhibits a linear relationship only with the number of Zr atoms (or equivalently, the total number of layers in the surface supercell). However, surface energy calculations using surface supercells with different numbers of atomic layers yield varying results. To avoid such discrepancies, we have constructed four (010) surface supercells with 5, 7, 9, and 11 atomic layers, as shown in Figure 2:
Based on the relationship between the total energy of different supercells and the number of atomic layers, their linear correlation was fitted, as presented in Figure 3. According to Equation (5), the intercept b of this fitted line was extracted, yielding a surface energy of 1.82 J/m2 for the Cu-Zr stoichiometric surface.
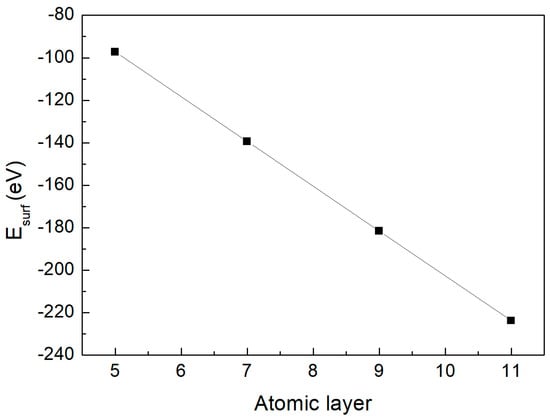
Figure 3.
The correlation between and slab thickness for (010) surface of the CuZr2 crystal.
The (001) surface of the CuZr2 crystal is a non-stoichiometric surface. We first constructed three surface supercells based on the consideration of center symmetry, as shown in Figure 4, using slab models with seven layers for Cu-terminated, nine layers for Zr1-terminated, and eleven layers for Zr2-terminated, respectively. And each supercell contains an implemented 15 Å vacuum region in order to avoid the interactions between periodic images. For non-stoichiometric surfaces, according to the report [25], the surface energies of different terminations on non-stoichiometric surfaces cannot be calculated as exact values but only as relative trends.
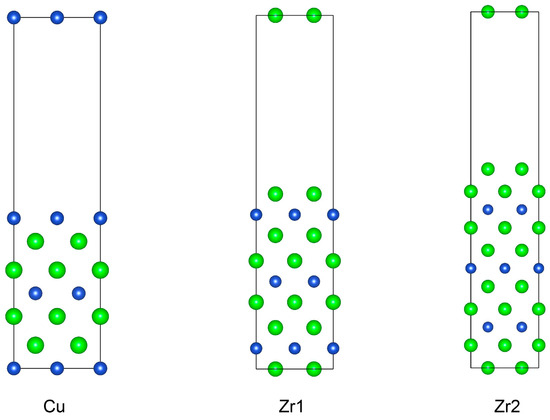
Figure 4.
Calculation models of three non-stoichiometric surfaces of CuZr2 (001) surface.
However, in the earlier literature, Brik and Heifets suggested that for compounds composed of more than three elements, precise calculation of surface energy values is possible [26,27]. Although the CuZr2 crystal is composed of only two elements, Cu and Zr, we attempt to apply this method to theoretically predict the surface energy of the non-stoichiometric (001) surface. The specific calculation formula is as follows:
where and are the cleaved energy and relaxed energy, respectively. Cleaved energy is defined as follows:
where represents the static energy for a surface supercell with termination. is the total energy of a CuZr2 unit cell. The 9 shows that the total number of all Cu and Zr atoms included in these three surface supercells is 9 times the atom numbers of a CuZr2 unit cell, and 6 indicates that three non-stoichiometric supercells have six surfaces in total.
Relaxed energy is given as follows:
where 2 denotes that each supercell has two surfaces.
Based on the above formula, we calculated the surface energies for three non-stoichiometric surfaces, with the final results presented in Table 2. It can be seen that among the non-stoichiometric (001) surfaces, the Zr1-terminated surface exhibits the highest surface energy, while the Zr2-terminated surface has the lowest value. The reason for this likely lies in the atomic structures of their surface and subsurface layers. In the Zr1-terminated surface, the subsurface layer consists of Cu atoms. Cu atoms have a smaller atomic radius, and there is mutual attraction between Cu and Zr, causing the outermost electrons of the Cu atoms in the subsurface layer to have a probability of appearing on the surface of the supercell. This increases the number of unpaired electrons, thereby leading to a higher surface energy. The surface energy of the Cu-terminated surface is close to that of the Zr1-terminated surface, as both have similar surface and subsurface layer structures, resulting in comparable surface energies. In contrast, the subsurface layer of the Zr2-terminated surface is still composed of Zr atoms. Zr atoms have a larger atomic radius, and the two layers of Zr atoms exhibit mutual repulsion, leading to the Zr atoms in the subsurface layer being further away from the supercell surface. As a result, their contribution to the surface energy is relatively smaller, giving rise to a lower surface energy value.
Table 2.
The calculated surface energies (J/m2) of CuZr2 crystal.
3.2. Interface Properties of Cu/CuZr2 Interface
Constructing the interface between the CuZr2 and Cu matrix requires consideration of the orientation relationship (OR), terminal structure, stacking mode, and interface strain. Existing studies have not explicitly provided the orientation relationship of the Cu/CuZr2 interface. However, experiments indicate that the preferred grain orientation of copper alloys after rolling, solid solution, and aging treatment is <220> [28]. When Liu et al. investigated the crystal structure of precipitates, such as CuZr2 in Cu-Cr-Zr alloys, the electron diffraction (SAED) patterns corresponded to the <0 1 −1> zone axis of the Cu matrix [13]. Given this, referencing the methodology used by Filho et al. [29], we select Cu (110)/CuZr2 (010) as the fundamental orientation relationship to construct the interface supercell.
First, considering the difference in elastic moduli between Cu and CuZr2, a compressive strain of approximately 2.8% and a tensile strain of 6% are applied to the CuZr2 crystal along the <001> and <300> basis vector directions, respectively, to achieve coherence with the Cu matrix. Second, taking full account of the centrosymmetry of the interface supercell and the types of terminal atomic structures at the interface, a sandwich structure model of Cu/CuZr2/Cu is employed to construct four different structural interface supercells, as shown in Figure 5. Here, the top and bottom five atomic layers represent the Cu matrix, while the middle seven atomic layers represent the CuZr2 precipitate phase. Relaxation calculations are performed on the constructed interface supercells, with the lowest-energy structure selected as the Cu (110)/CuZr2 (010) interface structure (as shown in Figure 5c).
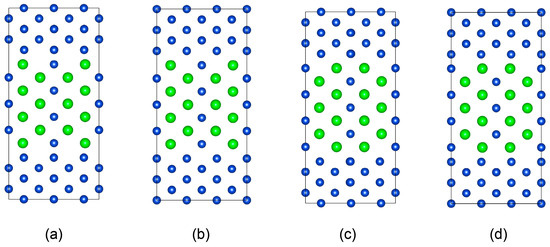
Figure 5.
All possible initial structures of Cu (110)/CuZr2 (010) interface. (a–d) are the interface structures with different atomic arrangement.
The strengthening effect of CuZr2 precipitates on Cu-Cr-Zr alloys is closely related to the stability of the Cu/CuZr2 interface structure. Here, we quantitatively investigate the stability of this structure by calculating the interfacial energy. The formula for calculating the interfacial energy is as follows [30]:
where denotes the total energy of the interface supercell, is the total number of atoms in the supercell, and is the phase fraction of Cu. and are the chemical potentials of Cu and CuZr2 phases at their pure bulk state.
The calculated interfacial energy from this formula comprises contributions from two aspects. One is the elastic strain energy consumed to overcome the lattice mismatch between the two phases and establish a coherent relationship, and the other is the chemical bonding energy resulting from the formation of new interfacial bonds. By averaging these energies over each atom near the interface, specific calculation formulas for both contributions can be derived below:
where is the interface energy per unit area, denotes the coherent strain energy per atom, and is the interface area.
Fitting lines for and can be obtained on the basis of the variation of and . By calculating the slope and intercept of these fitting lines, the strain-free interfacial energy and the interface strain energy can finally be determined.
As seen from Table 3, the coherent strain energy of the Cu (110)/CuZr2 (010) interface is 12.53 meV/atom, while the strain-free interfacial energy is 260 mJ/m2. Since there have been no relevant calculations of the interfacial energies for other precipitate phases in Cu-Cr-Zr alloys to date, the data provided here lacks comparability. However, recent studies by Zhang et al. have investigated the interfacial energies of L12-Al3X (X = Sc, Zr, Er) precipitates in Al alloys using the same method [31,32]. For comparative analysis, we provided the interfacial energy values of the most stable Al (100)/L12-Al3X (100) interface in Table 3. It can be observed that the coherent strain energy at the Cu (110)/CuZr2 (010) interface is significantly higher than that of L12-Al3X. This is likely related to the lattice mismatch during interface formation.
Table 3.
The interface energies of Cu (110)/CuZr2 (010) and Al3X (100)/Al (100) configurations.
To further illustrate this, we present the lattice mismatch values for the Cu (110)/CuZr2 (010) and Al (100)/L12-Al3X (100) interfaces in Table 4. It is evident that the lattice mismatch between L12 precipitates and the Al matrix is relatively small compared to that of the Cu (110)/CuZr2 (010) interface. This smaller mismatch results in better interface stability, which naturally corresponds to lower interfacial energies.
Table 4.
The interface strain in Cu (110)/CuZr2 (010) and Al3X (100)/Al (100) configurations.
4. Conclusions
The surface energies and interface structures of three low-index surfaces of the CuZr2 precipitate in Cu-Cr-Zr alloys were investigated at the atomic level. The results are as follows. The (100) and (010) surfaces of CuZr2 have identical atomic structures and are both stoichiometric surfaces. By fitting the relationship between the total energy of different surface supercells and the number of atomic layers, the surface energy was accurately calculated to be 1.82 J/m2. The (001) surface is a non-stoichiometric surface, with three different surface terminations: Cu, Zr1, and Zr2. The Zr2 termination exhibits the smallest surface energy, approximately 1.72 J/m2. The final atomic arrangement structure of the Cu (110)/CuZr2 (010) interface was determined. The interface strain energy and the chemical bonding energy, excluding strain, were subsequently calculated. The results showed that, in comparison with the precipitates with L12 structure in Al alloys, the greater the lattice mismatch between the matrix and the precipitate, the larger the interfacial energy value.
Author Contributions
Methodology, H.-W.H.; software, X.-F.C. and J.-G.Y.; formal analysis, H.-W.H.; investigation, H.-W.H.; curation, J.-G.Y. and X.-F.C.; writing—original draft preparation, H.-W.H.; writing—review and editing, X.-F.C.; supervision, J.-G.Y.; project administration, J.-G.Y.; funding acquisition, H.-W.H. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the School-Enterprise Cooperation Fund Project of Yantai Nanshan University (No. 2025KJ02010).
Data Availability Statement
The original contributions presented in this study are included in the article. Further inquiries can be directed to the corresponding author.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Ma, M.; Xiao, Z.; Meng, X.; Li, Z.; Gong, S.; Dai, J.; Jiang, H.; Jinag, Y.; Lei, Q.; Wei, H. Effects of trace calcium and strontium on microstructure and properties of Cu-Cr alloys. J. Mater. Sci. Technol. 2022, 12, 11–23. [Google Scholar] [CrossRef]
- Lei, Q.; Yang, Y.H.; Xiao, Z.; Jiang, Y.; Gong, S.; Zhou, L. Research Progress and Prospect of High-strength, High-conductivity and High-heat-resistant Copper Alloys. Mater. Rep. 2021, 35, 9. [Google Scholar]
- Li, J.; Ding, H.; Li, B. Study on the variation of properties of Cu-Cr-Zr alloy by different rolling and aging sequence. Mater. Sci. Eng. A 2021, 802, 140570. [Google Scholar] [CrossRef]
- Mishnev, R.; Shakhova, I.; Belyakov, A.; Kaibyshev, R. Deformation microstructures, strengthening mechanisms, and electrical conductivity in a Cu-Cr-Zr alloy. Mater. Sci. Eng. A 2015, 629, 29–40. [Google Scholar] [CrossRef]
- Lü, C.; Li, H.; Li, Y.; Zhong, F.; Li, H.; Zheng, W.; Ma, Y.; Gao, Z.; Yang, J.; He, Y. Optimization of microstructure and mechanical properties of electron beam welded CuCrZr alloys. Chin. J. Nonferrous Met. 2023, 33, 2170–2184. [Google Scholar] [CrossRef]
- Vinogradov, A.; Suzuki, Y.; Ishida, T.; Kitagawa, K.; Kopylov, V.I. Effect of Chemical Composition on Structure and Properties of Ultrafine Grained Cu-Cr-Zr Alloys Produced by Equal-Channel Angular Pressing. Mater. Trans. 2004, 45, 2187–2191. [Google Scholar] [CrossRef]
- Sun, Y.H.; Louzguine-Luzgin, D.V.; Ketov, S.; Greer, A.L. Pure shear stress reversal on a Cu-based bulk metallic glass reveals a Bauschinger-type effect. J. Alloys Compd. 2014, 615, S75–S78. [Google Scholar] [CrossRef]
- Purcek, G.; Yanar, H.; Demirtas, M.; Alemdag, Y.; Shangina, D.V.; Dobatkin, S.V. Optimization of strength, ductility and electrical conductivity of Cu-Cr-Zr alloy by combining multi-route ECAP and aging. Mater. Sci. Eng. A 2016, 649, 114–122. [Google Scholar] [CrossRef]
- Yang, W.; Liu, F.; Liu, H.; Wang, H.F.; Chen, Z.; Yang, G.C. Glass forming ability in Cu–Zr binary alloy: Effect of nucleation mode. J. Alloys Compd. 2009, 484, 702–707. [Google Scholar] [CrossRef]
- Ocelík, V.; De Oliveira, U.; De Boer, M.; De Hosson, J.T.M. Thick Co-based coating on cast iron by side laser cladding: Analysis of processing conditions and coating properties. Surf. Coat. Technol. 2007, 201, 5875–5883. [Google Scholar] [CrossRef]
- Barabash, V.; Peacock, A.; Fabritsiev, S.; Kalinin, G.M. Materials challenges for ITER and beyond. J. Nucl. Mater. 2007, 367, 21–32. [Google Scholar] [CrossRef]
- Liu, J.; Tang, X.; Wang, S.; Xiao, Y.; Zhang, R.; Song, H.; Zhang, S. Effect of La-microalloying and Warm Rolling on Microstructure and Properties of Cu-Cr-Zr Alloy. Rare Met. Mater. Eng. 2025, 54, 2126–2135. [Google Scholar]
- Liu, Y.; Han, T.; Liu, Y.; Zheng, C.; Li, X.; Qiao, L.; Li, Y.; Li, J.; Meng, X. Relationship between Microstructure and Properties of High-Strength High-Conductivity Cu-Cr-Zr Alloy. Spec. Cast. Nonferrous Alloys 2025, 8, 1148–1159. [Google Scholar]
- Wu, X.; Zhang, J.; Zhao, J.; Meng, Q.; Zhang, W.; Pan, F.; Liu, Y.; Ximin, L.; Liu, Y.; Liu, X.; et al. Enhancing the strength and conductivity of commercialized Cu-Cr-Zr plate through simple high strain cold rolling and aging. Mater. Charact. 2023, 205, 113213. [Google Scholar] [CrossRef]
- Hu, J.; Tian, Y.; Yu, H.; Ling, G.; Li, S.; Jiang, M.; Li, H.; Qin, G. Optimizing strength and electrical conductivity of Cu-Cr-Zr alloy by two-stage aging treatment. Mater. Lett. 2022, 315, 131937–131941. [Google Scholar] [CrossRef]
- Wang, K.; Xu, H.Y.; Zheng, X.; Zhang, H.F. First-principles study of electronic structure, elastic properties and hardness of Cr-doped CuZr2. Acta Phys. Sin. 2025, 74, 137101. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmuller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Inorganic Crystal Structure Database (ICSD); Collection Code: 52265; FIZ Karlsruhe—Leibniz Institute for Information Infrastructure: Karlsruhe, Germany, 2023.
- Meyers, M.A.; Chawla, K.K. Mechanical Behavior of Materials; Cambridge University Press: Cambridge, UK, 2008. [Google Scholar]
- Kane, R.H.; Giessen, B.C.; Grant, N.J. New metastable phases in binary tin alloy systems. Acta Metall. 1966, 14, 605–609. [Google Scholar] [CrossRef]
- Pan, J.; Chan, K.C.; Chen, Q.; Liu, L. Enhanced plasticity by introducing icosahedral medium-range order in ZrCuNiAl metallic glass. Intermetallics 2012, 24, 79–83. [Google Scholar]
- Du, J.; Wen, B.; Melnik, R.; Kawazoe, Y. Phase stability, elastic and electronic properties of Cu–Zr binary system intermetallic compounds: A first-principles study. J. Alloys Compd. 2014, 588, 96–102. [Google Scholar] [CrossRef]
- Sun, S.P.; Li, X.P.; Wang, H.J.; Jiang, H.F.; Lei, W.N.; Jiang, Y.; Yi, D.Q. First-principles investigations on the electronic properties and stabilities of low-index surfaces of L12-Al3Sc intermetallic. Appl. Surf. Sci. 2014, 288, 609–615. [Google Scholar] [CrossRef]
- Brik, M.G.; Ma, C.G.; Krasnenko, V. First-principles calculations of the structural, electronic and optical properties of CuGaS2 and CuInS2 after the substitution of Zn for Cu. Surf. Sci. 2013, 608, 146–151. [Google Scholar]
- Heifets, E.; Eglitis, R.I.; Kotomin, E.A.; Maier, J.; Borstel, G. Ab initio modeling of the (001) surface of cubic and Tetragonal barium titanate. Phys. Rev. B 2001, 64, 235417. [Google Scholar] [CrossRef]
- Kuai, Z.; Li, Z.; Liu, B.; Chen, Y.; Lu, S.; Bai, P. Effect of heat treatment on CuCrZr alloy fabricated by selective laser melting: Microstructure evolution, mechanical properties and fracture mechanism. J. Mater. Res. Technol. 2023, 23, 2658–2671. [Google Scholar] [CrossRef]
- Filho, M.A.M.; Farmer, W.; Hsiao, C.L.; Dos Santos, R.B.; Hultman, L.; Birch, J.; Ankit, K.; Gueorguiev, G.K. Density Functional Theory-Fed Phase Field Model for Semiconductor Nanostructures: The Case of Self-Induced Core–Shell InAlN Nanorods. Cryst. Growth Des. 2024, 24, 4717–4727. [Google Scholar] [CrossRef]
- Mao, Z.G.; Chen, W.; Seidman, D.N.; Wolverton, C. First-principles study of the nucleation and stability of ordered precipitates in ternary Al-Sc-Li alloys. Acta Mater. 2011, 59, 3012–3023. [Google Scholar] [CrossRef]
- Zhang, C.; Jiang, Y.; Cao, F.; Hu, T.; Wang, Y.; Yin, D. Formation of coherent, core-shelled nano-particles in dilute Al-Sc-Zr alloys from the first-principles. J. Mater. Sci. Technol. 2019, 35, 930–938. [Google Scholar]
- Zhang, C.M.; Yin, D.F.; Jiang, Y.; Wang, Y.R. Precipitation of L12-phase nano-particles in dilute Al-Er-Zr alloys from the first-principles. Comput. Mater. Sci. 2019, 162, 171–177. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.