Abstract
Rising atmospheric CO2 levels demand efficient and sustainable carbon capture solutions. Direct air capture (DAC) via crystallizing hydrogen-bonded frameworks such as carbonate salts has emerged as a promising approach. This review explores the potential of crystal engineering, in tandem with advanced quantum crystallography techniques and computational modeling, to unlock the full potential of DAC materials. We examine the critical role of hydrogen bonding and other noncovalent interactions within a family of bis-guanidines that governs the formation of carbonate salts with high CO2 capture capacity and low regeneration energies for utilization. Quantum crystallography and charge density analysis prove instrumental in elucidating these interactions. A case study of a highly insoluble carbonate salt of a 2,6-pyridine-bis-(iminoguanidine) exemplifies the effectiveness of these approaches. However, challenges remain in the systematic and precise determination of hydrogen atom positions and atomic displacement parameters within DAC materials using quantum crystallography, and limitations persist in the accuracy of current energy estimation models for hydrogen bonding interactions. Future directions lie in exploring diverse functional groups, designing advanced hydrogen-bonded frameworks, and seamlessly integrating experimental and computational modeling with machine learning. This synergistic approach promises to propel the design and optimization of DAC materials, paving the way for a more sustainable future.
1. Introduction
The balance of the planetary climate is facing a significant challenge from the calamitous increase in global temperatures due to the increased emissions of greenhouse gases, most importantly CO2 [1,2,3]. Addressing climate change requires global efforts to mitigate greenhouse gas emissions through transitioning to renewable energy sources, improving energy efficiency, implementing sustainable practices in industries and agriculture, conserving ecosystems, and adopting policies to reduce carbon footprints. Direct air capture (DAC), a technology that removes CO2 directly from ambient air, holds the potential to reverse the accumulation of CO2 in the atmosphere, enabling its widespread adoption across various sectors to reduce greenhouse gas emissions [4]. Yet, designing effective DAC materials remains a challenge. The low atmospheric concentration of CO2 (currently 415 ppm) requires sorbents that bind CO2 strongly and selectively against other components in the air (nitrogen, water, etc.). Technical and economic hurdles, including high energy requirements and a lack of incentives for negative emissions, contribute to DAC’s higher estimated cost of USD 94 to USD 232 per ton of captured CO2 [5].
An ideal sorbent needs to selectively separate low concentrations of CO2 while allowing other gases to pass through and release CO2 with a low energy input when required. There are currently few viable CO2 sorbents for DAC. Recently, crystal engineering approaches have been proven effective in the design of metal–organic and hydrogen-bonded frameworks (HBFs) and have emerged as leading contenders for DAC material design [6]. The extended networks of strong yet tunable hydrogen bonds provide the driving force for the formation of carbonate salts. Understanding the precise geometry and strength of hydrogen bonds facilitates the design of optimized materials for efficient carbon capture and utilization [7].
Neutron and X-ray scattering methods, such as neutron diffraction, neutron spectroscopy, and X-ray diffraction (XRD), contribute to understanding the atomic-level structure and dynamics of materials used in carbon capture, including those potentially applicable to DAC [7,8,9]. This review article introduces the field of quantum crystallography (QC) [10], a burgeoning field capable of accurately deciphering the geometrical and energetic parameters of hydrogen-bonded materials using experimental data from single-crystal X-ray and neutron diffraction. Figure 1 illustrates that Bader’s [11] quantum theory of atoms in molecules (QTAIM) can be employed to analyze the electron density distribution and identify bond critical points (BCPs). A comparison of experimental and theoretical hydrogen bond parameters can be used to validate computational models and improve their accuracy. These techniques help analyze the arrangement of atoms and molecules within these materials, providing insights into their behavior and interactions relevant to CO2 capture. The greatest advantage of neutron studies is that neutrons interact with atomic nuclei and hence provide detailed information about the material’s atomic structure, including accurate positions of hydrogen atoms [12]. Accurate hydrogen atom positions are relevant in understanding molecular architecture and interactions between specific molecular fragments [13]. It is well known that hydrogen atoms often participate in crucial bonding patterns and can influence molecular properties, e.g., reactivity [14], polarity [15], or biological activity [16]. Thus, precise information about their positions is vital for understanding chemical reactions [17], designing drugs [18,19], or predicting material behaviors [20,21,22]. While neutron diffraction offers highly accurate information about crystal structures [23,24], it can be limited by insufficient crystal size and the availability of neutron sources. An appealing alternative lies in utilizing single-crystal XRD data. However, this technique does not allow for the exact determination of hydrogen atom positions experimentally. Due to the weak scattering power of hydrogen atoms in XRD experiments, their positions are typically inferred closer to their parent atoms, resulting in shorter X–H distances than those obtained from neutron experiments. Fortunately, this discrepancy can be corrected using QC, and computational modeling, to gain a comprehensive understanding of the structures and functionalities of materials for carbon capture, including those explored for DAC applications [23,25,26,27,28].
Utilizing QC for the study of DAC materials presents a promising avenue, particularly in addressing the challenges associated with refining hydrogen atom parameters against X-ray diffraction data. Recent advancements in quantum crystallography, as highlighted in this article, introduce three distinct approaches: Hirshfeld atom refinement (HAR), HAR coupled to extremely localized molecular orbitals, and X-ray wavefunction refinement (XWR) [29,30,31]. New developments in these methods enable a comprehensive examination of hydrogen bond parameters, facilitating the validation of computational models and enhancing their precision. The accurately determined topological properties of the electron density from QC studies can be used to characterize the nature of the hydrogen bonding in the crystal structure for a precise understanding of the relationship between hydrogen bonding and the DAC function. The field of QC has the potential to serve as a tool for guiding the design of optimized materials for efficient carbon capture and utilization. This review aims to shed light on how single-crystal neutron and XRD methods have found application in the study of the family of bis-guanidines that form carbonate salts as promising DAC materials. However, due to the limited availability of works, this review also intends to demonstrate the potency of using single-crystal neutron and XRD methods in the field of the family of (bis)carbonate salts tested for DAC materials, highlighting their complementarity, especially in accurately locating hydrogen atoms and their importance in the structure–property relationships. Thus, an overview of possible solutions for obtaining accurate hydrogen positions, where neutron data are not available, is also provided. This review investigates crystal structures beyond simple geometrical analysis using the quantum crystallography approach.
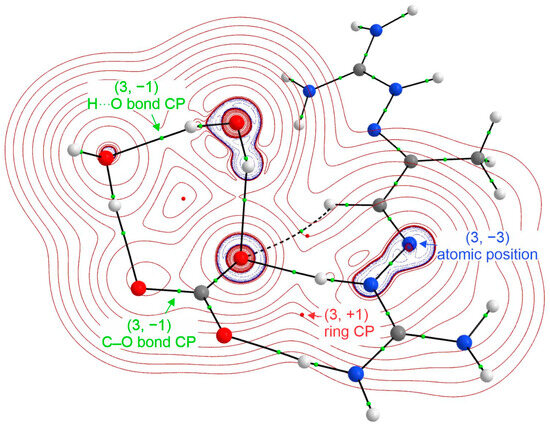
Figure 1.
Molecular graph and 2D Laplacian (eÅ−5) obtained with QTAIM for the carbon–guanidinium complex. Green and red dots correspond to the critical points. Red and blue lines denote positive and negative values, respectively. The (3, +1) critical point associated is with the ring position, the (3, −1) critical point is associated either with the covalent or noncovalent bond, and (3, −3) is the atomic position.
We will discuss how QC can be applied to the study of DAC by crystallization of hydrogen-bonded frameworks, as outlined in a recent article by Gianopoulos et al. [32]. The relationship between hydrogen bond strengths and CO2 binding energies can be investigated, providing insights into how structural modifications influence CO2 capture efficiency. We will also highlight the status and challenges of QC and DAC and suggest future directions for this interdisciplinary research field.
2. Family of Bis-Iminoguanidine Carbonate Salts Used for DAC Materials
The family of bis-iminoguanidines (BIGs) has been discovered to be effective in DAC, and there have been significant advancements and diversification in recent years [33]. In 2017, Custelcean and Seipp et al. [34] proposed a simple aqueous guanidine sorbent, specifically 2,6-pyridine-bis(iminoguanidine) (PyBIG), that effectively captures CO2 from the ambient air and bonds it as a crystalline carbonate salt through guanidinium hydrogen bonding (Figure 2). This innovative approach was a milestone as a promising method for addressing the challenge of CO2 capture from the atmosphere. The authors suggested that the electron-withdrawing pyridine ring in PyBIG enhances the acidity of the guanidinium groups, leading to stronger binding and more effective separation of oxoanions. Using guanidine sorbents for CO2 capture represented a significant step towards the development of efficient and sustainable methods to reduce atmospheric CO2 levels.
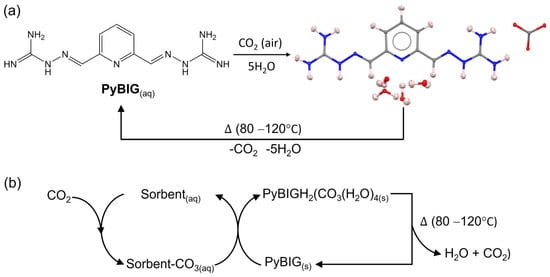
Figure 2.
(a) DAC cycle with aqueous PyBIG as the sorbent, involving crystallization of PyBIGH2(CO3)(H2O)4 (single-crystal neutron structure shown: C, grey; H, pink; N, blue; O, red), followed by CO2 release and PyBIG regeneration by mild heating of the carbonate crystals. (b) Two-stage DAC cycle combining CO2 absorption by an aqueous sorbent with crystallization of PyBIGH2(CO3)(H2O)4 and sorbent regeneration, followed by CO2 release and PyBIG regeneration by heating of the carbonate crystals. Reproduced from [35] with permission from Springer Nature, 2018.
Williams et al. [36] demonstrated an efficient CO2 scrubbing process using a glyoxal-bis(iminoguanidine) (GBIG) compound for CO2 capture from flue gas mixtures. The synthesis of GBIG involves the imine condensation of glyoxal with aminoguanidinium salts. Upon CO2 absorption, the aqueous GBIG sorbent leads to the crystallization of a relatively insoluble bicarbonate salt, forming “anti-electrostatic” hydrogen-bonded (HCO3−)2 dimers stabilized by three hydrogen bonds from the iminoguanidinium cations and by two from the water molecules. The CO2 was released by mild heating of the bicarbonate crystals, resulting in quantitative regeneration of GBIG, which could be recycled multiple times. The CO2 capture cycle was tested with a flue gas simulant, showing excellent performance over ten consecutive cycles. The regeneration energy of GBIG is 24% lower than that of monoethanolamine (MEA) [37], a benchmark industrial sorbent, due to the solid-state regeneration process, avoiding energy-intensive heating and evaporation of aqueous solutions. The study provided experimental and computational evidence for a CO2 release mechanism and offered the prospect of an energy-efficient and cost-effective carbon-capture technology. Unfortunately, no DAC activity was found when GBIG was used to capture CO2 from the air [8]. The study by Custelcean et al. [8] also mentioned the structurally analogous meta-benzene-bis(iminoguanidine) (m-BBIG); although it was able to capture CO2 from the air, it formed a poorly crystalline carbonate solid whose crystal structure has so far remained elusive.
Next, the DAC of CO2 using aqueous peptides and solid BIGs was tested [38]. The process involves the absorption of CO2 by small peptides such as GlyGly, which are then converted into carbonate and bicarbonate anions. The GBIG compound removes H+ and HCO3− ions from the solution and crystallizes as a bicarbonate salt, thus regenerating the peptide absorbent. This crystallization-based regeneration eliminates the need to heat the aqueous peptide, extend its lifetime, and reduce regeneration energy. The DAC cycle concludes with the mild heating of the bicarbonate crystals. This process releases CO2 and efficiently regenerates the GBIG with minimal energy. The unique feature of GBIG among BIG compounds is its formation of a bicarbonate salt with a specific crystal structure, which contributes to its stability and efficiency in capturing CO2 from air [38]. The combination of aqueous peptides and GBIG is essential for an efficient DAC process, as the peptide solution quickly reacts with CO2 from the atmosphere, resulting in the formation of the stable crystalline structure GBIGH2(HCO3)2(H2O)2. The relatively low pKa of the peptide is critical for lowering the solution pH and enabling efficient DAC with GBIG. However, most amino acids are less effective due to their higher pKa values.
Kasturi et al. [39] investigated the thermodynamics and kinetics of the regeneration of glycine using a GBIG compound. The study discovered that regenerating glycine with GBIG releases heat, unlike the endothermic process involved in the thermally driven regeneration of aqueous glycine solvent. The kinetics of the system were experimentally measured, and the reaction of the protonation of GBIG by glycine, along with the subsequent crystallization of GBIGH22+(HCO3−)2(H2O)2, were identified as the controlling steps of the overall regeneration process. The study also showcased the effectiveness of an intensified process, wherein CO2 was captured from a gas phase by an amino acid within a liquid solution in the presence of GBIG. This configuration led to significantly improved CO2 capture compared to a system without GBIG. The findings underscored the importance of additional research in identifying optimal combinations of amino acids and guanidine compounds to further enhance DAC performance.
The GBIG compound was further examined and modified. For example, the synthesis and evaluation of a modified GBIG chemical sorbent, namely 2,5-furan-bis(iminoguanidine) (FuBIG), was investigated [40]. Firstly, the study reported the determination of two binding modes with nine hydrogen bonds between FuBIG, CO2 (as CO32−), and H2O using single-crystal XRD analysis. Secondly, the stepwise and overall thermodynamic and kinetic parameters for CO2 absorption and heat release were obtained through van’t Hoff analysis, thermogravimetric analysis (TGA), differential scanning calorimetry (DSC), and in situ reaction analysis. The study revealed that the absorption of CO2 in an aqueous solution of FuBIG is highly advantageous, as indicated by the overall enthalpy value (ΔH7) of −116.10 kJ/mol and an overall equilibrium constant (K7) of 5.97 × 104 for the CO2 absorption reaction. The release of CO2 from FuBIGH2(CO3) (H2O)4 required relatively less energy, with an enthalpy value (ΔH8) of 209.31 kJ/mol. The study proposed a simple and intuitive symbol for evaluating CO2 sorbents, the Rs value, representing the ratio of sorbent solubility to the solubility of carbonate salts. The Rs values for FuBIG, PyBIG, and GBIG were reported as 43.12, 7.97, and 1.60, respectively, at 25 °C. The study also demonstrated that the CO2 absorption process followed second-order reaction kinetics with a rate constant (k) of 4.8102 × 10−4 L/mol at 25 °C, and the kinetic characteristics for the release of CO2 and H2O from the FuBIG carbonate salt aligned with the geometric phase-boundary model based on isothermal TGA analysis. The FuBIG carbonate salt releases CO2 spontaneously in DMSO, suggesting a near-zero-energy technique for DAC. Density functional theory (DFT) calculations further elucidated this process of spontaneous CO2 release in DMSO. The study also highlighted the favorable biocompatibility of the biomass-derived CO2 sorbent of FuBIG based on acute toxicity and embryo toxicity assays in a zebrafish model.
The GBIG system was structurally modified by substituting specific hydrogen atoms with methyl groups, demonstrating that crystal engineering can effectively control the DAC of CO2 with GBIG [8]. The research findings indicated that relatively minor modifications in the molecular structure of BIGs led to significant differences in the crystal structures and aqueous solubilities of the resulting compounds. Introducing methyl groups resulted in a substantial increase in aqueous solubility, with factors of 96 and 14 for methyl-glyoxal-bis(iminoguanidine) (MGBIG) and diacetyl bis(iminoguanidine) (DABIG), respectively, compared to the original GBIG. The study also highlighted the impact of π-stacking interactions, conformational flexibility, and steric hindrance from the methyl groups on the crystal structures and solubilities of the compounds. Furthermore, the study compared the crystal structures of neutral ligands and their corresponding carbonate salts, revealing differences in solubilities and thermodynamic driving forces for DAC reactions. The inclusion of water molecules in the carbonate crystals is identified as a critical factor, providing stability to the carbonate anions and influencing the regeneration energies for the compounds. The study revealed essential design principles for crystalline BIG solids favoring DAC chemistry. It emphasized the importance of maximizing the aqueous solubility of neutral BIG solids while minimizing the solubility of their carbonate counterparts. Additionally, the study discussed the potential for computational methodologies to predict optimal crystal structures and thermodynamics for DAC performance and the need for process design and development to translate fundamental science into practical DAC technology.
The effects of amino acids and small oligopeptides crystallizing MGBIG carbonate from aqueous solutions on DAC’s efficacy were also tested [41]. The study revealed that sarcosine, a secondary amino acid, significantly improves DAC by crystallizing MGBIG-CO3, leading to a six-fold increase in the amount of CO2 extracted from the air when added to an aqueous solution of MGBIG. This synergy between aqueous MGBIG and sarcosine offers the prospect of an effective DAC process.
Separating and thermally regenerating the precipitated guanidine carbonate salt could lead to an overall low-temperature, low-energy direct air capture process. Recently, Jang et al. [42] evaluated the feasibility of microwave heating of the crystalline solid of MGBIG carbonate for efficient CO2 desorption and found that microwave heating effectively regenerates MGBIG carbonate, resulting in a significant reduction in electrical energy consumption. These findings suggest that microwave regeneration may be an energy-efficient method for fast regeneration of solid sorbents used for direct air capture.
Also, the regeneration energy required for CO2-loaded aqueous potassium sarcosinate (K-SAR) solvent and crystalline MGBIG sorbent was evaluated using DSC, TGA, and Fourier transform infrared spectroscopy (FTIR) in a recent study [43]. The findings revealed that for aqueous K-SAR, the sensible heat amounts to 1.5 GJ/tCO2, and the desorption enthalpy is estimated at 3.68 GJ/tCO2. The study also determined that the total regeneration energy required for MGBIG is approximately 7.0 GJ/tCO2. The study also observed CO₂ and H2O release during the thermal regeneration using FTIR measurements. The regeneration of CO2-loaded aqueous K-SAR solvent and crystalline MGBIG sorbent requires moderate temperatures lower than those required for other DAC solvents/sorbents. These findings imply that leveraging low-cost heat sources, like geothermal energy, could potentially reduce the overall cost of DAC.
3. DAC Studied by Single-Crystal Neutron Diffraction
As we already mentioned, the crystal structures of DAC can be studied by neutron diffraction using either powder or single-crystal samples. Studying accurate hydrogen positions solely from powder neutron diffraction can be challenging due to several limitations associated with this technique. Powder neutron diffraction provides valuable information about the average atomic positions within a crystalline sample, but determining the precise positions of light atoms like hydrogen is inherently difficult. In contrast, single-crystal neutron diffraction provides higher-resolution data compared to powder diffraction. It allows for the determination of precise atomic positions and their thermal vibrations within a crystal structure, as demonstrated by high-resolution single crystal structures from TOPAZ beamline at the Spallation Neutron Source [24]. Moreover, preferred orientation effects might occur in powder diffraction, hindering precise structural determination, and single crystals help to avoid these effects. On the other hand, obtaining a proper size, or a single crystal itself, may be challenging, and thus, specific single-crystal neutron structures for DAC materials are not widely available.
However, ongoing research in the field of materials science and carbon capture continues to leverage single-crystal neutron diffraction techniques to advance the understanding and development of efficient materials for capturing CO2 from the atmosphere. Recently, the structure–property relationships have been elucidated for a series of bis-iminoguanidine (BIG) carbonate salts (Figure 3) [8,34]. The analysis of single crystals of BIGs involved both X-ray and neutron diffraction techniques (Figure 3, Figure 4, Figure 5, Figure 6 and Figure 7). The papers emphasized the importance of hydrogen bonding in determining the structural arrangement and stability of the bicarbonate salt formed during CO2 absorption by specific BIGs. The authors discussed the role of hydrogen bonds in the formation of the BIG bicarbonate salt, specifically highlighting the unique nature of the hydrogen bonding network in the investigated crystalline structures. Both single-crystal X-ray and neutron diffraction analyses revealed the formation of hydrogen-bonded carbonate-BIG dimers. Water molecules also link the crystal networks into extended clusters.
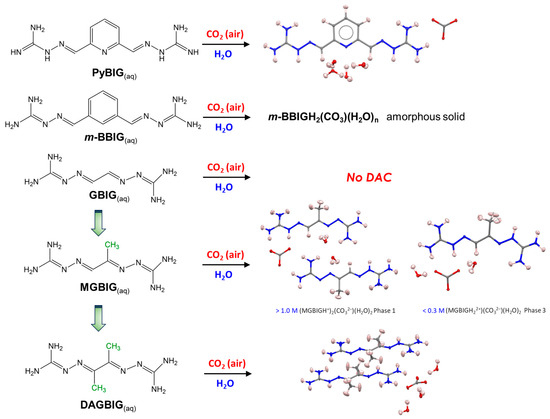
Figure 3.
CO2 capture effectiveness of various bis(iminoguanidine) systems (BIGs) studied by Custelcean et al. The capture of CO2 from the air has been confirmed for 2,6-pyridine-bis(iminoguanidine) (PyBIG), meta-benzene-bis(iminoguanidine) (m-BBIG), and modified glyoxal bis(iminoguanidine) compounds methylglyoxal bis(iminoguanidine) (MGBIG) and diacetyl bis(iminoguanidine) (DABIG). Partially reproduced from [8] with permission from Jon Wiley and Sons, 2020.
Interestingly, the DAC chemistry for MBIG is concentration-dependent. At concentrations > 1.0 M, MGBIG primarily crystallizes as (MGBIGH+)2(CO32−)(H2O)2 (P1). Some high-concentration (>0.75 M) batches exhibit a secondary phase, (MGBIGH+)2(MGBIGH22+)(CO32−)2(H2O)6 (P2). Upon lowering the initial concentration below 0.3 M, a third phase forms, characterized by the composition (MGBIGH22+)(CO32−)(H2O)2 (P3). Figure 4a shows the carbonate binding site consisting of eight hydrogen bonds from guanidinium and guanidine groups for the P1 structure, and for P3, it shows the carbonate binding site consisting of six hydrogen bonds from guanidinium groups (N–H⋅⋅⋅O contact distances of 1.654–1.886 Å) and three hydrogen bonds from water molecules (O–H⋅⋅⋅O contact distances of 1.737–1.912 Å). Similar to P3, the crystal structure of DABIG (Figure 5) also contains extended [CO3(H2O)32-]n clusters linked by four carbonate–water and water–water hydrogen bonds with O–H⋅⋅⋅O contact distances ranging between 1.635 and 1.846 Å. Each carbonate anion accepts five guanidinium hydrogen bonds with N–H⋅⋅⋅O contact distances ranging between 1.735 and 1.853 Å. The quasi-planar DABIGH+ and DABIGH22+ cations alternate in the crystal and form stacks, with the distance between the mean planes measuring 3.248 Å. The overall crystal packing comprised cationic stacks flanking anionic carbonate–water clusters, interconnected via guanidinium–carbonate, guanidinium–water, and guanidine–water hydrogen bonds. The heightened conformational flexibility and steric hindrance resulted from the presence of methyl groups in MGBIG and DABIG. In stark contrast to the neutral ligands, the crystal structures of MGBIG and DABIG carbonate salts exhibit notably planar BIG cations tightly stacked within the crystals (Figure 5d). A recurring structural characteristic in these carbonate crystals is the incorporation of a minimum of two water molecules per carbonate anion, forming extended carbonate–water clusters. Consequently, the solubility of the carbonate salts of DABIG and MGBIG is significantly reduced compared to their corresponding neutral ligands. This substantial reduction in solubility provides a substantial thermodynamic driving force for the corresponding DAC reactions [8].
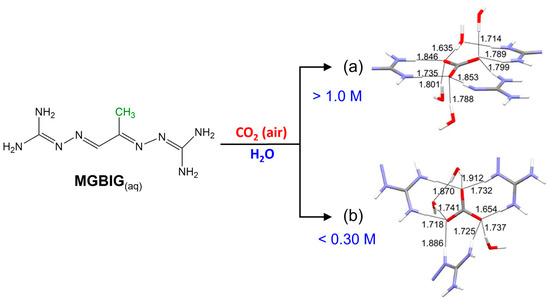
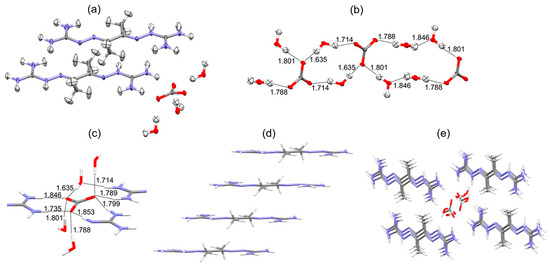
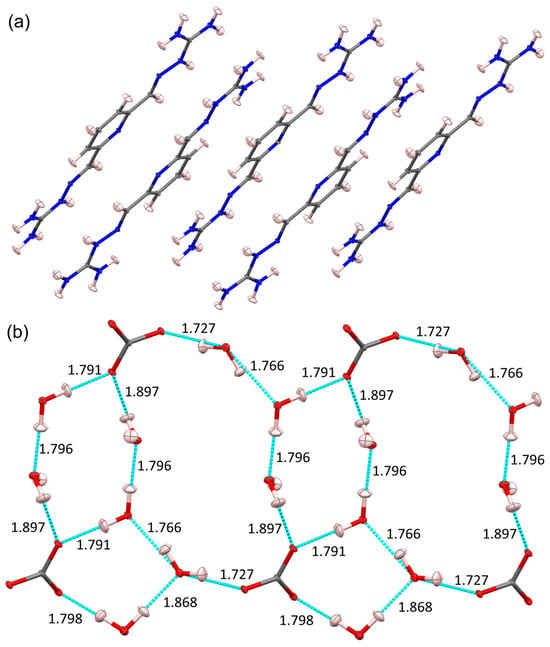
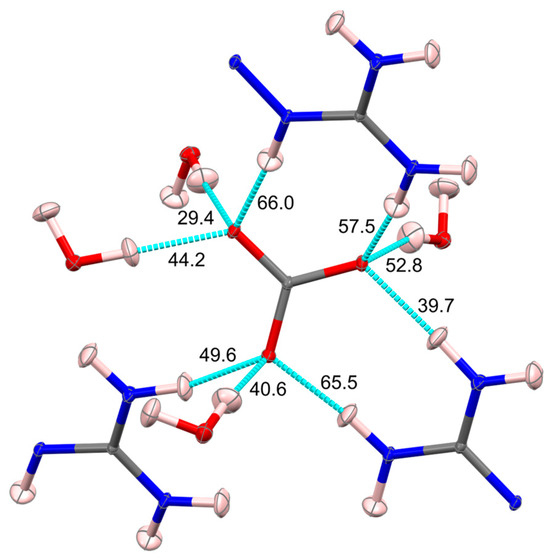
Figure 4.
Crystal structures of the two crystalline phases obtained from DAC with aqueous MGBIG: (a) (MGBIGH+)2(CO32−)(H2O)2 (phase 1); (b) (MGBIGH22+)(CO32−)(H2O)2 (phase 3). The contact distances of the H-bonds involved in carbonate binding were determined by single-crystal neutron diffraction. Partially reproduced from Ref. [8] with permission from Jon Wiley and Sons, 2020.
Figure 5.
Single-crystal neutron diffraction structure of (DABIGH+)(DABIGH22+)0.5(CO32−)(H2O)3. (a) ORTEP representation; (b) hydrogen-bonded [CO3(H2O)32−]n clusters with O–H⋅⋅⋅H contact distances in Å; (c) carbonate binding site consisting of 5 hydrogen bonds from guanidinium groups and 4 hydrogen bonds from water molecules, with contact distances in Å; (d) stacking of the DABIGH+ and DABIGH22+ cations; (e) crystal packing consisting of cationic stacks that flank anionic [CO3(H2O)32−]n clusters. Reproduced from [8] with permission from Jon Wiley and Sons, 2020.
Figure 6.
(a) The stacking of PyBIGH22+ cations in PyBIGH2(CO3)(H2O)4 based on the neutron structure, with displacement ellipsoids at the 20% probability level; (b) scheme of the hydrogen bonds in the anion–water ribbons based on the neutron structure, with displacement ellipsoids at the 20% probability level and distances in Å. Partially reproduced from [32] with permission from the International Union of Crystallography, 2019.
Figure 7.
The hydrogen bonds for the carbonate anion of PyBIG salt in PyBIGH2(CO3)(H2O)4. Carbonate is bound by five guanidinium and four water hydrogen bonds, with estimated HB energies in kJ/mol. Partially reproduced from [32] with permission from the International Union of Crystallography, 2019.
The X-ray and neutron studies indicated a complex network of hydrogen bonding interactions involving carbonate anions, BIG cations, and water molecules. The presence of these specific hydrogen bonds contributes to the stability of the bicarbonate salt. Interestingly, the carbonate in the studied systems, namely PyBIG, MGBIG (P3), and DABIG, which exhibit potential for DAC technology, is consistently associated with nine hydrogen bonds (Table 1).

Table 1.
Number of hydrogen bonds found for the carbonate anion in the studied systems.
Therefore, it seems that a detailed understanding of hydrogen bonding aids in comprehending the compound’s behavior during the CO2 capture and release cycles, contributing to its efficiency and recyclability in the carbon capture process. The combination of single-crystal neutron and XRD methods, with DFT calculations, provides a powerful approach for studying DAC materials, allowing for the accurate localization of hydrogen atoms and providing complementary information crucial for understanding the structure–property relationships and energy calculations. However, an experimental cross-validation method for theoretical findings, going beyond the geometrical analysis, is indeed necessary.
4. DAC Studied by High-Resolution Single-Crystal X-ray Diffraction Quantum Crystallography Studies
One of the possible ways to experimentally determine the interaction energy between molecular fragments is through the topological analysis of the electron density distribution in crystals. As previously discussed, the distinctive hydrogen bond network within the BIG family system necessitates a comprehensive analysis, particularly concerning its significance in understanding crystal stability and preferences for DAC. Modern crystallography approaches can help to obtain that information in experimental high-resolution XRD data.
According to the best of our knowledge, the analysis of experimental electron density for the aforementioned DAC materials from high-resolution XRD data has been documented in only one paper [32]. The study investigated the DAC of CO2 and focused on topological analysis of the experimental electron density using the QTAIM and the approach proposed by Espinosa et al. [44,45]. This method assumes that the energy of intermolecular interaction for hydrogen bonds (EHB) is equal to half of the local electronic potential energy density determined at the bond critical point of a hydrogen bond. The advantages and disadvantages of the Espinosa approach are described later in the “Interactions energy from X-ray electron density study” section.
The compound under study is the highly insoluble carbonate salt mentioned earlier, involving a 2,6-pyridine-bis(iminoguanidine) molecule coordinated with CO32− and four water molecules [(PyBIGH)2(CO3)(H2O)4]. The article discussed the crystallographic analysis and characterization of this specific compound, examining its molecular structure and properties. The QTAIM method delves into the electron density and likely sheds light on the chemical bonding and interactions within the crystal structure. Particularly, the electron density analysis provides an accurate characterization of the bonding situation in the studied crystals, revealing the delocalized nature of the bonding in the planar cation. It also identifies a strong hydrogen bonding network, encompassing anion–water ribbons and connections between anionic ribbons and cationic stacks, significantly contributing to the extremely low aqueous solubility of this salt (Figure 6).
Water molecules form hydrogen bonds with carbonate anions and guanidinium cations, which are crucial in stabilizing the crystal structure (Figure 7). Apart from strong hydrogen bonds, the analysis identified supplementary weaker interactions, some of which are characterized as π–π interactions (the values are all less than 5 kJ mol−1). These interactions contributed to the overall stability of the crystal lattice. The conclusion also mentioned that the electrostatic contribution to the lattice energy remains relatively modest, attributed to charge transfer and delocalization.
Overall, the primary takeaway is the comprehensive understanding of the molecular interactions, including the role of hydrogen bonding, water molecules, and supplementary interactions, determining this carbonate salt’s stability and extremely low aqueous solubility. The study provided valuable insights into the electronic features that contribute to the compound’s properties, which are relevant in understanding and potentially manipulating insoluble carbonate salts or similar compounds.
5. Accurate Hydrogen Atom Positions from Quantum Crystallography Studies
As we mentioned above, the accurate location of hydrogen atoms is crucial to understanding the structure–property relationship for DAC materials. In practice, this is true for any material, pharmaceutical cocrystal, or biological structure. When dealing with single-crystal XRD data, it is well known that the positions of hydrogen atoms are less reliable than those obtained from neutron studies. Also, information about their thermal parameters, going beyond the isotropic treatment, is not accessible, especially when only the independent atom model (IAM) is applied. Fortunately, there are methods that enable the estimation of both the positions and anisotropic displacement parameters (ADPs) of hydrogen atoms when neutron data are unavailable.
First, Allen and Bruno [46] proposed a method for estimating hydrogen atom positions in a molecule using neutron diffraction data. Their approach involves utilizing positions of heavier atoms obtained from neutron data diffraction and applying certain empirical rules or statistical models to predict hydrogen atom positions relative to these heavier atoms based on the coordinates stored in the Cambridge Structural Database (CSD) [47]. These positions are calculated (Table 2) and can be accessed in the paper [46]. Table 2 displays the selected mean X–H distances (Å) obtained from single-crystal neutron diffraction, as proposed by Allen and Bruno, alongside those available from studies conducted for DAC. These values underscore that an XRD study, lacking an advanced model or hydrogen atom treatment, may not accurately determine H-atom positions.
Table 2.
Comparison of mean X–H distances (Å) of organic compounds from single-crystal neutron diffraction and the mean and median X–H distances of DAC compounds.
Furthermore, Madsen et al. [48] emphasized the importance of including ADPs for H atoms when modeling high-resolution XRD data. Specifically, they showed that the static model electron density for covalent bonds is affected while only the isotropic description of H atoms is applied. Consequently, the SHADE3 server [49] was designed as a computational tool enabling the estimation of internal mean square displacements (MSDs) of the H atoms by utilizing normal-mode frequencies of high-frequency vibrations. For the vibrating k atom, the MSD matrix Batom(k) is defined as follows:
where:
- N is the number of atoms in the unit cell;
- N is the number of cells in the crystal;
- e(k|jq) represents the kth component of a normalized complex eigenvector e(jq);
- ωj is the frequency of mode j;
- mk is the mass of atom k;
- Ej(q) is the energy of the mode.
Hoser et al. [50] tested various approaches for treating hydrogen atoms in experimental charge density studies within the framework of multipolar refinement [51]. The authors emphasized the importance of an appropriate refinement strategy when dealing with different types of hydrogen bonds in molecular structures. They highlighted that solely standardizing X–H distances based on average neutron data without additional improvements in atomic displacement parameters (ADPs) and X–H directions is not sufficient for accurately determining the topological parameters of the electron density. The authors suggested employing a mixed refinement strategy, involving high-order refinement of heavy atoms, low-angle refinement of H atoms. and elongation of the X–H distance to the average neutron bond lengths plus estimated anisotropic atomic displacement parameters for H atoms. Otherwise, significant differences in geometric and topological properties based on different models of H atoms were observed (Figure 8). These differences not only were notable in properties mapped for strong or weak hydrogen bonds but also impacted the obtained integrated properties of the non-H atoms, especially for those covalently bonded to the hydrogen atoms.
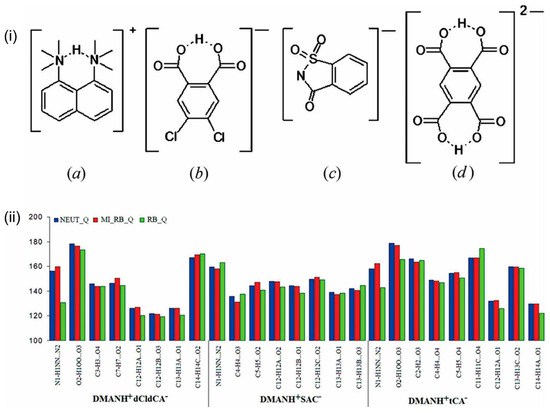
Figure 8.
(i) Schemes of (a) the 1,8-bis(dimethylamino)naphthalene (DMANH+) cation and organic anions from (b) 4,5-dichlorophthalic acid (DMANH+dCldCA−), (c) o-benzoic sulfimide dihydrate (DMANH+SAC−), and (d) 1,2,4,5-benzenetetracarboxylic acid (DMANH+tCA−) salts. (ii) Illustration of the hydrogen bond structural parameter D—H⋯A angle (°). Only the three most representative cases of refinement are shown. A typical s.u. for the bond angle is 0.03°. Reproduced from [50] with permission from the International Union of Crystallography, 2009.
The method recommended by Hoser et al. has found successful application in numerous crystallographic studies, particularly those involving high-resolution data modeled within the framework of multipolar refinement [52,53,54,55,56,57,58,59].
Another method for obtaining ADPs for hydrogen atoms is the NoMoRe method, developed by Hoser and Madsen [60,61]. NoMoRe is a normal-mode refinement approach used for crystal structures. It involves refining lattice-dynamical models against diffraction data by refining ab initio calculated normal-mode frequencies against X-ray or neutron diffraction data. The program allows users to specify the temperature of the X-ray or neutron data collection and the frequencies to be optimized. Initially, all normal-mode coordinates and their frequencies are derived from calculations performed in the CRYSTAL program, with each frequency assigned a scaling factor of 1.0. The program then calculates the atomic displacement parameters (ADPs) for all atoms, including hydrogen atoms, and refines only the coordinates using SHELXL. In subsequent refinement steps, selected frequencies are optimized by refining frequency scaling factors against the diffraction data to minimize wR2. The optimization is performed using the quasi-Newton Broyden–Fletcher–Goldfarb–Shannon algorithm (BFGS) implemented in the Python programming package Scipy. The NoMoRe model has been successfully used to study the stability of pyrazinamide polymorphs [61] or hydrogen positions of transition metal hydride complexes [62].
However, when the collection of high-resolution data is not possible, a useful approach might involve the reconstruction of electron density within pseudoatom databases, such as ELMAM [63,64], ELMAM2 [65], MATTS [66] (previously UBDB [67,68]), and Invariom [69,70] which store multipolar parameters for specific atom types. The idea of using pseudoatom databanks is based on the transferability of properties assigned to specific atom types between different molecules or molecular environments. For instance, the properties assigned to a carbon atom in one molecule would be applicable to a carbon atom in another molecule, a concept known as transferable atom refinement (TAAM). Studies have demonstrated that this approach can accurately reproduce the positions of hydrogen atoms and their ADPs [71]. Importantly, TAAM refinement is not limited to small molecules but can also be applied to protein structures.
Another alternative to dealing with hydrogen atoms is the application of QC methods, which are based on the wavefunction [29,30,72,73] and hold promise in accurate hydrogen atom positions. In QC, a sophisticated method, among others, is Hirshfeld atom refinement (HAR), which uses aspherical atomic scattering factors calculated on-fly by HF/DFT methods, which are then refined against experimental data [29]. HAR was successfully utilized for small and big molecules [74,75], yielding H-atom positions almost as accurate as those obtained from the neutron data [76,77,78,79,80,81]. It was also shown that within HAR, the H-atom ADPs can be refined even for low-resolution data [82]. HAR can also be combined with the libraries of extremely localized molecular orbitals (ELMOs) [31,83,84], leading to the HAR-ELMO method [74], providing a benefit over IAM refinements and improving the representation of intermolecular interactions in HAR [76]. This approach allows for the fast, accurate, and precise refinement of crystal structures of various compounds, including polypeptides, proteins, and molecules containing heavy elements, at a significantly reduced computational cost compared to traditional methods [76,85,86].
6. Interaction Energies from X-ray Electron Density Study
Comprehending the distinctive properties and challenges inherent in molecular and ionic systems is pivotal in enhancing the performance of DAC materials. Intermolecular interactions significantly shape these materials’ structural and chemical properties, influencing their efficiency, selectivity, and CO2 capture kinetics. Among others, amine and hydroxide functional groups have been explored for CO2 capture due to their ability to chemically react with and capture CO2 molecules. X-ray and neutron studies have confirmed that the O/N—H···O interactions contribute to the adsorption of CO2 molecules onto surfaces containing amine functional groups [8]. These interactions strengthen the binding of CO2 to the amine-containing materials, thereby improving the capture efficiency. Moreover, these interactions can facilitate the selective capture of CO2 over other gases in a mixture. It is worth noticing that CO2 is a linear molecule with a dipole moment, meaning it has partial positive and negative charges at different ends. Amines, for example, have lone pairs of electrons on the nitrogen atom, allowing them to react readily with CO2 through a process called chemical sorption or chemisorption. This process sometimes leads to the formation of highly insoluble HBF crystals, capturing CO2, and forms stable HBF–carbamate compounds. Thus, if a part of a molecule possesses charged or polarized regions, the electrostatic forces play a role in CO2 capture processes too.
Analyzing these interactions at the subatomic level, going beyond simple structural analysis, yields crucial insights into the mechanisms governing CO2 adsorption and desorption, pivotal for developing efficient DAC technologies. Moreover, a profound understanding of intermolecular interactions is indispensable in the design and optimization of DAC materials. It serves as a guiding force in the development of novel sorbents with enhanced CO2 capture capabilities. Hence, delving into these interactions stands as a cornerstone in propelling the advancement of effective and sustainable DAC technologies.
Crystallography offers several methods and software tools for analyzing intermolecular interaction for CO2 capturing from XRD data (Figure 9). Indeed, it is possible to compute interaction energy from the experimental XRD data, based on the electron density analysis, within the Exact Potential and Multipole Moments (EPMM) method or Espinosa–Molins–Lecomte (EML) approach [51]. It serves as a critical starting point for computational methods and theoretical models that can estimate or calculate these energies too (periodic DFT [87], dispersion-corrected DFT [88], or ab initio molecular orbital calculations [89,90]). Combining experimental data with computational approaches allows for a deeper understanding of the nature and strength of interatomic interactions within materials and serves as a cross-validation. For more curious readers, a detailed description of these methods with examples is provided below. In the context of DAC studies, the intermolecular interactions present in the (PyBIGH)2(CO3)(H2O)4 system [32] were recently studied from the electron density point of view within the EML approach. The hydrogen bonds analyzed varied in strength from modest (14 kJ/mol) to strong (66 kJ/mol). The strongest hydrogen bonds were associated with the carbonate anion, accepting numerous interactions from guanidinium groups and water molecules. Correlating hydrogen bond energies with observed H···O contact distances showed an exponential relationship, as anticipated. The cumulative energy from all hydrogen bonds and weaker interactions amounted to 449.9 kJ/mol, with a significant fraction (37.1%) derived from hydrogen bonding to water molecules. This strong hydrogen bonding between carbonate and water contributes substantially (441.9 kJ/mol) to the stability and low aqueous solubility of (PyBIGH2)−(CO3)(H2O)4 crystals, partially compensating for the considerable free energy of dehydration of the anion. Additionally, the lattice energy, comprising electrostatic and other interactions, likely contributed to the crystals’ low solubility.
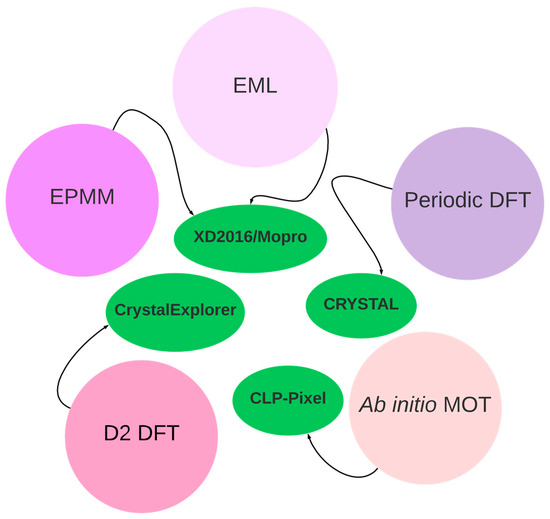
Figure 9.
Scheme of the available software and methods in crystallography for studying intermolecular interaction energies. Explanation of abbreviations: Exact Potential and Multipole Moments (EPMM) method, Espinosa–Molins–Lecomte (EML) approach, density functional theory (DFT), dispersion-corrected DFT (D2 DFT), and ab initio molecular orbital theory (MOT).
The EPMM method [91], which can be implemented in the XDPROP module of the XD2016 [92] software or MoproViewer module of the MoPro (Molecular Properties) [93,94] software, stands out. It is a computational approach used for the calculation of intermolecular interaction energies and involves the evaluation of the exact Coulomb integral in the inner region (≤4.5 Å) and combines it with a Buckingham-type multipole moment approximation for long-range interatomic interactions [95,96]. The EPMM method is particularly valuable for accurately assessing electrostatic contributions to intermolecular interactions. It provides a detailed understanding of the forces governing molecular associations [66], whether based on experimental electron density or theoretical electron densities, such as those obtained from pseudoatom databanks. This method has found extensive application across diverse research areas. For instance, it has been used in studying glycopeptide antibiotics and their complexes, allowing for the calculation of interaction energies based on charge densities reconstructed with a databank [97]. Additionally, the EPMM method has been employed in the study of aminoglycoside–RNA complexes, demonstrating its ability to reproduce Coulombic intermolecular interaction energies determined by ab initio methods [98]. Furthermore, the EPMM method has been utilized in the study of proteins, where atom–atom electrostatic energies are computed by means of a multipole expansion with regular spherical harmonics, providing insights into the electrostatic interactions in proteins [99,100,101]. Additionally, the EPMM method has been applied to the study of self-assembling systems, such as organotrifluoroborates, where it has been used to assess intermolecular interactions and the participation of organic fluorine in the formation of these interactions [102]. Moreover, the EPMM method has been employed in the study of bis(4-chlorophenylacetate)bis(pyridine-4-carboxamide)zinc(II) complexes, where it has been used in conjunction with Hirshfeld surface analysis to investigate intermolecular interactions and energy frameworks [103]. This demonstrates the versatility of the EPMM method in providing valuable insights into the nature of intermolecular interactions in diverse chemical systems [104].
The energy density obtained from crystallographic electron density has been a subject of interest in various studies [105,106,107,108]. It has been demonstrated by Espinosa et al. [44,45] that relationships between the topology of the electron density in the hydrogen bond region and some energetic properties, such as the local potential (Vr) and kinetic (Gr) energy densities at the hydrogen bond critical point (BCP), exist, and this is called the EML approach:
This indicates that the electron density in the hydrogen bond region (X—H ··· O, where X = C, N, O) can provide valuable insights into the energetic properties associated with the hydrogen bond critical point. Therefore, the electron density obtained from X-ray crystallography can be used to analyze and understand the energetic aspects of intermolecular interactions, particularly in the context of hydrogen bonding [44]. For example, this approach has been used to investigate the distinct characteristics of neutral and ionic hydrogen bonding in Schiff bases [109]. The study focused on the differences in geometrical parameters and electron density distribution between these two types of hydrogen bonds. Specifically, the research highlighted the unique behavior of ionic hydrogen bonds, where the corresponding interaction lines appear to be curved in the vicinity of the hydrogen atoms. This work contributed to a deeper understanding of the nature of hydrogen bonding in Schiff bases, shedding light on their structural and chemical properties by using an analysis of the properties of the electron density [11,110]. As one can notice, the Espinosa approach should be applied only for the specific hydrogen bonds that are intermolecular interactions. This approach assumes a stabilizing character of the hydrogen bond interactions (the value of the Vr is always negative), which is not always true, especially in the case of intramolecular interactions [111]. This observation was also raised by Gatti et al. [112], Nikolaienko et al. [113], and Kuznetsov [114]. Further works, for example, by Mata et al. [115] showed that the coefficient applied to the relation between EHB and Vr should be equal to 0.31 rather than 0.50. Also, other modifications to the Espinosa equation were proposed. For example, Afonin et al. [116] proposed the following formula:
Jablonski and Monaco [111] proposed a constant value of 3.4 kcal/mol added to the EML equation while dealing with the C—H ··· O intramolecular hydrogen bond.
Another approach involves using X-ray crystallography to obtain experimental coordinates and then employing theoretical methods to estimate interaction energies. The Pixel method, initially developed by Gavezzotti [117,118,119,120], employs a semi-empirical approach to represent molecules in a crystal structure using electron and nuclear density blocks divided into small cubic volume elements known as pixels. The method calculates molecular electron densities ab initio using Gaussian [121] at the MP2/6-31G** level of theory [122]. The electrostatic energy between molecules is then computed by applying Coulomb’s law to pairs of pixels from each molecule and summing the values. The method has been applied to various systems, including the quantitative investigation of synthons for crystal engineering and the elucidation of intermolecular interactions and lattice energies for polymorphs of 5-methyl-2-(2-nitro-phenyl)-amino-3-thio-phene-carbo-nitrile (ROY) at high pressure [123]. The Pixel method is implemented in the Pixel-C module of the CLP-Pixel package and is sensitive to H-atom positions, requiring “normalized” distances for accurate results. The method has been compared to periodic density functional theory and is similar in estimating sublimation enthalpies of organic solids but at a fraction of the computing time [124]. Moreover, the Pixel method has been used to explain the effect of chemical substitution on halogen bonding [125], identify the features of racemic and homochiral polymorphs [126], and rationalize the metastable form of glycolide [127]. The method is also used to calculate lattice energies and intermolecular interaction energies, and the results of each calculation are stored in plain text files. The Pixel method is a valuable tool for the screening of many molecular crystals and is generally applicable throughout the periodic table, offering a cost-effective approach for predicting crystal polymorphism and studying crystal growth processes. Recently, the MrPIXEL procedure was proposed [128]. It enables Pixel calculations to be carried out with minimal user intervention from the graphical interface of Mercury [129], a software distributed with the Cambridge Structural Database (CSD) [47]. The integration of MrPIXEL with the Mercury interface provides a user-friendly and automated approach for conducting Pixel calculations, offering convenience and ease of use for researchers working with crystallographic data. Furthermore, the CrystalExplorer17.5 [88] software has a feature for calculating pairwise interaction energies within a crystal. It allows for the visualization and analysis of crystal structures, providing insights into intermolecular interactions, lattice energies, and molecular properties [130,131,132,133]. The software has been utilized in diverse research areas, including the investigation and description of synthons for crystal engineering, the elucidation of intermolecular interactions and lattice energies for polymorphs, and the identification of metastable forms of organic compounds [134].
Finally, experimental geometries obtained from diffraction studies can be optimized in CRYSTAL software [87] within periodic DFT to analyze the electronic structure and properties of crystalline materials. This method facilitates the exploration of various phenomena, including lattice energies and crystal stability, and the prediction of crystal structures. Periodic DFT calculations have been shown to be valuable in elucidating the crystal structures of organic compounds, predicting phase stability, and understanding the properties of molecular crystals. Additionally, periodic DFT calculations have been employed to explore the energy landscape, investigate the freezing transition of vortex-line liquids, and study the spectroscopic properties of materials. The non-empirical nature of DFT allows for precise exploration of the energy landscape, making it a versatile and powerful tool for understanding the properties and behavior of crystalline materials. This approach is particularly significant and can serve as a cross-validation method.
7. Challenges
The recognition of the critical role of chemical engineering aspects in the development of DAC technology has recently been highlighted [7]. The development of DAC systems is complex, involving design, optimization, scale-up, manufacturing, and testing of efficient air–solid or air–liquid contactors, crystallizers, and solid–liquid separator steps. Also, CO2 desorption processes are analyzed to minimize temperature requirements and maximize the purity of the CO2 product. The economic feasibility and environmental impact of DAC technologies are evaluated through rigorous technoeconomic and lifecycle analysis. Furthermore, the large-scale deployment of crystalline organic materials for DAC applications is limited. Therefore, understanding and improving the fundamental DAC chemistry is a critical first step in the development of viable DAC technologies.
First, the determination of hydrogen atom positions and their anisotropic displacement parameters (ADPs) in molecular and ionic systems is a significant challenge in X-ray crystallography. This is particularly relevant in the context of molecular/ionic systems, where the accurate determination of atomic positions is crucial for understanding the structural and chemical properties of the system. Despite various proposed solutions to determine accurate information about hydrogen atom positions and ADPs, there is still a lack of systematic and statistical studies regarding the accuracy of determined H-atom ADPs within quantum crystallography. Current studies have shown that accurate hydrogen atom positions can be obtained using wavefunction-based methods [79,80]. However, it has been highlighted that the determination of H-atom ADPs depends on the level of theory used during wavefunction calculations. Including electron correlation effects is necessary to avoid non-positive defined thermal parameters [76]. Moreover, it has been shown that the distributions of the dynamic structure factors showing the effects of electron correlation and treatment of H-atom ADPs have similar shapes [135], which supports the observation made by Malaspina et al. [76]. In the context of data quality, it is essential to ensure that the data used for these studies are of high quality and accurately represent the experimental observations. Data quality and data quality assessment challenges have been recognized in the context of the big data era. The continuous improvement of data quality in crystallography has been identified as a challenging task, emphasizing the importance of maintaining high data quality standards for scientific research. Accurate determination of H-atom ADPs in crystallography is influenced by various experimental errors such as X-ray absorption, extinction, sample decay, anharmonic vibrations of atoms, and anomalous dispersion. These errors can lead to systematic errors in XRD data, affecting the precision and accuracy of the results [136]. Although sophisticated XRD experiments, data reduction, and electron density modeling are required to minimize these errors, there remains a gap in crystallography regarding the influence of these experimental errors on the accurate determination of H-atom ADPs. The discrepancy observed in the results of X-ray absorption spectroscopy (XAS) and XRD highlights the need for further investigation into the impact of experimental errors on the determination of ADPs of H atoms [137]. Therefore, filling this gap is of great importance to ensure the reliability and accuracy of crystallographic studies.
Secondly, special attention must be paid while dealing with the intermolecular interaction energies. As we have shown in Section 6, the EML approach for intermolecular interactions has been discussed in the literature, and some studies have highlighted important caveats associated with its use [138]. Although the EML relationship has been used to calculate the interaction energies for various intermolecular interactions [139,140,141], there are indications that this approach may not work as expected in certain cases. The EML relationship has been applied to estimate the interaction energies between molecules in ionic systems, but there are instances in which the calculated energies differ significantly from those predicted solely from net molecular charge [142]. Furthermore, the EML approach may not provide a general relation between interaction energies and charge penetration contributions to the exact electrostatic energies (resulting from EPMM), as observed by Kumar et al. [142]. However, the relationship between EML energies and charge penetration contributions has been noted as a significant observation, suggesting a quantitative link between structural analysis and interactions based on whole molecular charge densities. However, further investigations are necessary to fully understand the physics behind the observed phenomenological relationship between EML energies and charge penetration. Therefore, while the EML approach has been widely used to estimate interaction energies, its limitations and the need for additional understanding of its applicability in different contexts are evident from the literature. On the other hand, recent theoretical work has shown that the ionic character of the molecules has no influence on the nature of the hydrogen bond, as both neutral and ionic complexes exhibit similar electrostatic forces in the hydrogen bond region. Additionally, energy decomposition methods indicate that the hydrogen bond contribution to the dissociation profile resembles that of the neutral complexes [143].
This suggests that more complex studies of ionic systems should be conducted, probably supported by the machine learning approach, as proposed by Bauer et al. [144]. Their work presents machine learning models for hydrogen bond acceptor (HBA) and hydrogen bond donor (HBD) strengths. Large and diverse training data generated by first-principles interaction free energies were utilized to develop these models. This approach is significant as it leverages machine learning to predict hydrogen bond strengths, offering potential applications in various fields such as chemistry, biology, and solvation. The use of first-principles interaction free energies as training data enhances the accuracy and reliability of the machine learning models, providing valuable insights into the nature of hydrogen bond interactions. This publication contributes to the advancement of predictive models for hydrogen bond strengths, offering potential implications for understanding intermolecular interactions and their impact on various chemical and biological processes.
The competition and reaction with atmospheric moisture significantly affect the performance of physisorbents in DAC, underlining the crucial role of detailed structural studies in understanding these interactions at the atomic level. DAC materials also pose several challenges for diffraction studies. The limited-resolution data present a significant obstacle to accurately characterizing these materials [145]. Optimizing solid-supported amine-based materials for DAC requires a comprehensive understanding of their atomic and molecular structures to enhance their CO2 uptake and sorption kinetics at ambient temperature. However, obtaining high-resolution and high-quality XRD data at this temperature can be extremely challenging or even unfeasible, which is crucial for accurately estimating the interaction energy from experimental data. Furthermore, the performance of quantum crystallography in the context of ionic systems has not been entirely clear, indicating a need for further research and development in this area. Investigating DAC materials not only is challenging from the experimental point of view but also requires the use of sophisticated crystallographic software. While significant progress has been made in this field recently, there is still a gap in systematic studies for polymeric structures, ionic systems, and larger complexes, especially under periodic conditions.
8. Conclusions
In conclusion, DAC technology development involves complex chemical engineering processes. The large-scale deployment of crystalline organic materials for DAC applications is limited, emphasizing the critical importance of understanding and improving the fundamental DAC chemistry as a first step in developing viable DAC technologies.
In X-ray crystallography, accurately determining the positions and anisotropic displacement parameters (ADPs) of hydrogen atoms is crucial for understanding the structural and chemical properties of molecular and ionic systems. Although various solutions have been proposed to determine accurate information about hydrogen atom positions and ADPs, there is still a lack of systematic and statistical studies regarding the accuracy of determined H-atom ADPs within quantum crystallography. The influence of experimental errors such as X-ray absorption, extinction, sample decay, anharmonic vibrations of atoms, and anomalous dispersion on the accurate determination of H-atom ADPs remains a gap in crystallography, highlighting the need for further investigation to ensure the reliability and accuracy of crystallographic studies.
The EML approach for intermolecular interactions has been discussed recently in the literature, with some studies highlighting important caveats associated with its use [44,45]. Although the EML relationship has been used to calculate interaction energies for various intermolecular interactions, there are indications that this approach may not work as expected in some instances. The limitations of the EML approach and the need for an additional understanding of its applicability in different contexts are evident from the literature. Additionally, recent theoretical work has shown that the ionic character of molecules has no influence on the nature of the hydrogen bond, emphasizing the need for more complex studies of ionic systems, potentially supported by machine learning approaches.
Moreover, the diffraction studies of DAC materials pose several challenges, including limited resolution and the amount of diffraction data compared to the number of atoms in the system, as well as competition and reaction with atmospheric moisture, which significantly affect the performance of physisorbents in DAC. The effectiveness of solid-supported amine-based materials for DAC requires a thorough understanding of their atomic and molecular structures to optimize their CO2 uptake and sorption kinetics at ambient temperature, highlighting the need for detailed structural studies to understand these interactions at the atomic level.
In summary, the development of DAC technology, the accurate determination of hydrogen atom positions and ADPs in crystallography, and the challenges of diffraction studies of DAC materials all require further research and development to address the existing gaps and limitations. These areas present opportunities to advance scientific understanding and technological innovation, with potential implications for various fields such as chemistry, biology, and environmental sustainability.
Author Contributions
Conceptualization, S.P. and X.W.; writing—original draft preparation, S.P.; writing—review and editing, S.P. and X.W.; visualization, S.P.; supervision, X.W.; project administration, X.W.; funding acquisition, X.W. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Neutron Sciences Directorate Science Initiatives at Oak Ridge National Laboratory (ORNL); ORNL is managed by UT-Battelle, LLC, under contract DE-AC05-00OR22725 with the U.S. Department of Energy (DOE).
Data Availability Statement
No new data were generated.
Acknowledgments
This research used resources at the Spallation Neutron Source, a Department of Energy (DOE) Office of Science User Facility operated by ORNL. S.P. and X.W. acknowledge helpful discussions with R. Custelcean and J. Einkauf. This manuscript has been authored by UT-Battelle, LLC, under contract DE-AC05-00OR22725 with the U.S. Department of Energy (DOE).
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| Abbreviation | Description |
| ADPs | Anisotropic Displacement Parameters |
| BCPs | Bond Critical Points |
| BIGs | Bis-iminoguanidines |
| CSD | Cambridge Structural Database |
| DABIG | Diacetyl bis(iminoguanidine) |
| DAC | Direct Air Capture |
| DFT | Density Functional Theory |
| DSC | Differential Scanning Calorimetry |
| ELMOs | Extremely Localized Molecular Orbitals |
| EML | Espinosa–Molins–Lecomte approach |
| EPMM | Exact Potential and Multipole Moments |
| FTIR | Fourier Transform Infrared Spectroscopy |
| FuBIG | 2,5-Furan-bis(iminoguanidine) |
| GBIG | Glyoxal-bis(iminoguanidine) Compound |
| HAR | Hirshfeld Atom Refinement |
| HBA | Hydrogen Bond Acceptor |
| HBD | Hydrogen Bond Donor |
| HBFs | Hydrogen-Bonded Frameworks |
| IAM | Independent Atom Model |
| m-BBIG | Meta-benzene-bis(iminoguanidine) |
| MEA | Monoethanolamine |
| MGBIG | Methyl-glyoxal-bis(iminoguanidine) |
| MSDs | Mean Square Displacements |
| NoMoRe | Normal-Mode Refinement |
| P1 | Phase 1 of the MGBIG |
| P2 | Phase 2 of the MGBIG |
| P3 | Phase 3 of the MGBIG |
| PyBIG | 2,6-Pyridine-bis(iminoguanidine) |
| TAAM | Transferable Atom Refinement |
| QC | Quantum Crystallography |
| QTAIM | Quantum Theory of Atoms in Molecules |
| TGA | Thermogravimetric Analysis |
| XAS | X-ray Absorption Spectroscopy |
| XRD | X-ray Diffraction |
| XWR | X-ray Wavefunction Refinement |
References
- Mansfield, L.A.; Nowack, P.J.; Kasoar, M.; Everitt, R.G.; Collins, W.J.; Voulgarakis, A. Predicting Global Patterns of Long-Term Climate Change from Short-Term Simulations Using Machine Learning. Npj Clim. Atmos. Sci. 2020, 3, 44. [Google Scholar] [CrossRef]
- Abbass, K.; Qasim, M.Z.; Song, H.; Murshed, M.; Mahmood, H.; Younis, I. A Review of the Global Climate Change Impacts, Adaptation, and Sustainable Mitigation Measures. Environ. Sci. Pollut. Res. 2022, 29, 42539–42559. [Google Scholar] [CrossRef] [PubMed]
- Erans, M.; Sanz-Pérez, E.S.; Hanak, D.P.; Clulow, Z.; Reiner, D.M.; Mutch, G.A. Direct Air Capture: Process Technology, Techno-Economic and Socio-Political Challenges. Energy Environ. Sci. 2022, 15, 1360–1405. [Google Scholar] [CrossRef]
- Sanz-Pérez, E.S.; Murdock, C.R.; Didas, S.A.; Jones, C.W. Direct Capture of CO2 from Ambient Air. Chem. Rev. 2016, 116, 11840–11876. [Google Scholar] [CrossRef] [PubMed]
- Keith, D.W.; Holmes, G.; St. Angelo, D.; Heidel, K. A Process for Capturing CO2 from the Atmosphere. Joule 2018, 2, 1573–1594. [Google Scholar] [CrossRef]
- Yusov, A.; Dillon, A.M.; Ward, M.D. Hydrogen Bonded Frameworks: Smart Materials Used Smartly. Mol. Syst. Des. Eng. 2021, 6, 756–778. [Google Scholar] [CrossRef]
- Custelcean, R. Direct Air Capture of CO2 via Crystal Engineering. Chem. Sci. 2021, 12, 12518–12528. [Google Scholar] [CrossRef]
- Custelcean, R.; Williams, N.J.; Wang, X.; Garrabrant, K.A.; Martin, H.J.; Kidder, M.K.; Ivanov, A.S.; Bryantsev, V.S. Dialing in Direct Air Capture of CO2 by Crystal Engineering of Bisiminoguanidines. ChemSusChem 2020, 13, 6381–6390. [Google Scholar] [CrossRef]
- Liu, S.; Chen, Y.; Yue, B.; Wang, C.; Qin, B.; Chai, Y.; Wu, G.; Li, J.; Han, X.; da-Silva, I.; et al. Regulating Extra-Framework Cations in Faujasite Zeolites for Capture of Trace Carbon Dioxide. Chem. Eur. J. 2022, 28, e202201659. [Google Scholar] [CrossRef]
- Grabowsky, S.; Genoni, A.; Bürgi, H.-B. Quantum Crystallography. Chem. Sci. 2017, 8, 4159–4176. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; International Series of Monographs on Chemistry; Oxford University Press: Oxford, NY, USA, 1994; ISBN 978-0-19-855865-1. [Google Scholar]
- Axe, J.D. Neutron Scattering: Progress and Prospects. Science 1991, 252, 795–802. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.L.; White, N.G. Hydrogen Atoms in Supramolecular Chemistry: A Structural Perspective. Where Are They, and Why Does It Matter? Chem. Soc. Rev. 2023, 52, 6254–6269. [Google Scholar] [CrossRef]
- Bhadra, M.; Lee, J.Y.C.; Cowley, R.E.; Kim, S.; Siegler, M.A.; Solomon, E.I.; Karlin, K.D. Intramolecular Hydrogen Bonding Enhances Stability and Reactivity of Mononuclear Cupric Superoxide Complexes. J. Am. Chem. Soc. 2018, 140, 9042–9045. [Google Scholar] [CrossRef]
- Nichols, D.A.; Hargis, J.C.; Sanishvili, R.; Jaishankar, P.; Defrees, K.; Smith, E.W.; Wang, K.K.; Prati, F.; Renslo, A.R.; Woodcock, H.L.; et al. Ligand-Induced Proton Transfer and Low-Barrier Hydrogen Bond Revealed by X-ray Crystallography. J. Am. Chem. Soc. 2015, 137, 8086–8095. [Google Scholar] [CrossRef]
- Oksanen, E.; Chen, J.C.-H.; Fisher, S.Z. Neutron Crystallography for the Study of Hydrogen Bonds in Macromolecules. Mol. J. Synth. Chem. Nat. Prod. Chem. 2017, 22, 596. [Google Scholar] [CrossRef] [PubMed]
- Pflug, C.; Rudolph, D.; Schleid, T.; Kohlmann, H. Hydrogenation Reaction Pathways and Crystal Structures of La2H2Se, La2H3Se and La2H4Se. Eur. J. Inorg. Chem. 2022, 2022, e202101095. [Google Scholar] [CrossRef]
- Anderson, A.C. The Process of Structure-Based Drug Design. Chem. Biol. 2003, 10, 787–797. [Google Scholar] [CrossRef]
- Manzoni, F.; Wallerstein, J.; Schrader, T.E.; Ostermann, A.; Coates, L.; Akke, M.; Blakeley, M.P.; Oksanen, E.; Logan, D.T. Elucidation of Hydrogen Bonding Patterns in Ligand-Free, Lactose- and Glycerol-Bound Galectin-3C by Neutron Crystallography to Guide Drug Design. J. Med. Chem. 2018, 61, 4412–4420. [Google Scholar] [CrossRef]
- Zhao, C.; Chen, L.; Che, Y.; Pang, Z.; Wu, X.; Lu, Y.; Liu, H.; Day, G.M.; Cooper, A.I. Digital Navigation of Energy–Structure–Function Maps for Hydrogen-Bonded Porous Molecular Crystals. Nat. Commun. 2021, 12, 817. [Google Scholar] [CrossRef]
- Arnold, J.E.; Day, G.M. Crystal Structure Prediction of Energetic Materials. Cryst. Growth Des. 2023, 23, 6149–6160. [Google Scholar] [CrossRef]
- Sato, T.; Mochizuki, T.; Ikeda, K.; Honda, T.; Otomo, T.; Sagayama, H.; Yang, H.; Luo, W.; Lombardo, L.; Züttel, A.; et al. Crystal Structural Investigations for Understanding the Hydrogen Storage Properties of YMgNi4-Based Alloys. ACS Omega 2020, 5, 31192–31198. [Google Scholar] [CrossRef] [PubMed]
- Hathwar, V.R.; Sist, M.; Jørgensen, M.R.V.; Mamakhel, A.H.; Wang, X.; Hoffmann, C.M.; Sugimoto, K.; Overgaard, J.; Iversen, B.B. Quantitative Analysis of Intermolecular Interactions in Orthorhombic Rubrene. IUCrJ 2015, 2, 563–574. [Google Scholar] [CrossRef] [PubMed]
- Jørgensen, M.R.V.; Hathwar, V.R.; Sist, M.; Wang, X.; Hoffmann, C.M.; Briseno, A.L.; Overgaard, J.; Iversen, B.B. Accurate Atomic Displacement Parameters from Time-of-Flight Neutron-Diffraction Data at TOPAZ. Acta Crystallogr. Sect. Found. Adv. 2014, 70, 679–681. [Google Scholar] [CrossRef]
- Grosjean, A.; Spackman, P.R.; Edwards, A.J.; Tolborg, K.; Vosegaard, E.S.; Koutsantonis, G.A.; Iversen, B.B.; Spackman, M.A. Insights into Host–Guest Binding in Hydroquinone Clathrates: Single-Crystal X-ray and Neutron Diffraction, and Complementary Computational Studies on the Hydroquinone-CO2 Clathrate. Cryst. Growth Des. 2021, 21, 3477–3486. [Google Scholar] [CrossRef]
- Carrington, E.J.; Vitórica-Yrezábal, I.J.; Brammer, L. Crystallographic Studies of Gas Sorption in Metal–Organic Frameworks. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2014, 70, 404–422. [Google Scholar] [CrossRef] [PubMed]
- Takamizawa, S.; Nakata, E.; Yokoyama, H.; Mochizuki, K.; Mori, W. Carbon Dioxide Inclusion Phases of a Transformable 1D Coordination Polymer Host [Rh2(O2CPh)4(Pyz)]n. Angew. Chem. Int. Ed. 2003, 42, 4331–4334. [Google Scholar] [CrossRef] [PubMed]
- Evans, H.A.; Mullangi, D.; Deng, Z.; Wang, Y.; Peh, S.B.; Wei, F.; Wang, J.; Brown, C.M.; Zhao, D.; Canepa, P.; et al. Aluminum Formate, Al(HCOO)3: An Earth-Abundant, Scalable, and Highly Selective Material for CO2 Capture. Sci. Adv. 2022, 8, eade1473. [Google Scholar] [CrossRef]
- Capelli, S.C.; Bürgi, H.-B.; Dittrich, B.; Grabowsky, S.; Jayatilaka, D. Hirshfeld Atom Refinement. IUCrJ 2014, 1, 361–379. [Google Scholar] [CrossRef]
- Jayatilaka, D. Wave Function for Beryllium from X-ray Diffraction Data. Phys. Rev. Lett. 1998, 80, 798–801. [Google Scholar] [CrossRef]
- Meyer, B.; Genoni, A. Libraries of Extremely Localized Molecular Orbitals. 3. Construction and Preliminary Assessment of the New Databanks. J. Phys. Chem. A 2018, 122, 8965–8981. [Google Scholar] [CrossRef]
- Gianopoulos, C.G.; Chua, Z.; Zhurov, V.V.; Seipp, C.A.; Wang, X.; Custelcean, R.; Pinkerton, A.A. Direct Air Capture of CO 2—Topological Analysis of the Experimental Electron Density (QTAIM) of the Highly Insoluble Carbonate Salt of a 2,6-Pyridine-Bis(Iminoguanidine), (PyBIGH2)(CO3)(H2O)4. IUCrJ 2019, 6, 56–65. [Google Scholar] [CrossRef]
- Custelcean, R. Direct Air Capture with Bis-Iminoguanidines: From Discovery to Commercialization. Chem 2021, 7, 2848–2852. [Google Scholar] [CrossRef]
- Seipp, C.A.; Williams, N.J.; Kidder, M.K.; Custelcean, R. CO2 Capture from Ambient Air by Crystallization with a Guanidine Sorbent. Angew. Chem. Int. Ed. 2017, 56, 1042–1045. [Google Scholar] [CrossRef] [PubMed]
- Brethomé, F.M.; Williams, N.J.; Seipp, C.A.; Kidder, M.K.; Custelcean, R. Direct Air Capture of CO2 via Aqueous-Phase Absorption and Crystalline-Phase Release Using Concentrated Solar Power. Nat. Energy 2018, 3, 553–559. [Google Scholar] [CrossRef]
- Williams, N.J.; Seipp, C.A.; Brethomé, F.M.; Ma, Y.-Z.; Ivanov, A.S.; Bryantsev, V.S.; Kidder, M.K.; Martin, H.J.; Holguin, E.; Garrabrant, K.A.; et al. CO2 Capture via Crystalline Hydrogen-Bonded Bicarbonate Dimers. Chem 2019, 5, 719–730. [Google Scholar] [CrossRef]
- Ma, C.; Pietrucci, F.; Andreoni, W. Capture and Release of CO2 in Monoethanolamine Aqueous Solutions: New Insights from First-Principles Reaction Dynamics. J. Chem. Theory Comput. 2015, 11, 3189–3198. [Google Scholar] [CrossRef] [PubMed]
- Custelcean, R.; Garrabrant, K.A.; Agullo, P.; Williams, N.J. Direct Air Capture of CO2 with Aqueous Peptides and Crystalline Guanidines. Cell Rep. Phys. Sci. 2021, 2, 100385. [Google Scholar] [CrossRef]
- Kasturi, A.; Gabitto, J.; Tsouris, C.; Custelcean, R. Carbon Dioxide Capture with Aqueous Amino Acids: Mechanistic Study of Amino Acid Regeneration by Guanidine Crystallization and Process Intensification. Sep. Purif. Technol. 2021, 271, 118839. [Google Scholar] [CrossRef]
- Zhang, Q.; Jiang, Y.; Li, Y.; Song, X.; Luo, X.; Ke, Z.; Zou, Y. Design, Synthesis, and Physicochemical Study of a Biomass-Derived CO2 Sorbent 2,5-Furan-Bis(Iminoguanidine). iScience 2021, 24, 102263. [Google Scholar] [CrossRef]
- Stamberga, D.; Thiele, N.A.; Custelcean, R. Synergistic Direct Air Capture of CO2 with Aqueous Guanidine/Amino Acid Solvents. MRS Adv. 2022, 7, 399–403. [Google Scholar] [CrossRef]
- Jang, G.G.; Kasturi, A.; Stamberga, D.; Custelcean, R.; Keum, J.K.; Yiacoumi, S.; Tsouris, C. Ultra-Fast Microwave Regeneration of CO2 Solid Sorbents for Energy-Efficient Direct Air Capture. Sep. Purif. Technol. 2023, 309, 123053. [Google Scholar] [CrossRef]
- Kasturi, A.; Gug Jang, G.; Stamberga, D.; Custelcean, R.; Yiacoumi, S.; Tsouris, C. Determination of the Regeneration Energy of Direct Air Capture Solvents/Sorbents Using Calorimetric Methods. Sep. Purif. Technol. 2023, 310, 123154. [Google Scholar] [CrossRef]
- Espinosa, E.; Souhassou, M.; Lachekar, H.; Lecomte, C. Topological Analysis of the Electron Density in Hydrogen Bonds. Acta Crystallogr. B 1999, 55, 563–572. [Google Scholar] [CrossRef] [PubMed]
- Espinosa, E.; Molins, E.; Lecomte, C. Hydrogen Bond Strengths Revealed by Topological Analyses of Experimentally Observed Electron Densities. Chem. Phys. Lett. 1998, 285, 170–173. [Google Scholar] [CrossRef]
- Allen, F.H.; Bruno, I.J. Bond Lengths in Organic and Metal-Organic Compounds Revisited: X—H Bond Lengths from Neutron Diffraction Data. Acta Crystallogr. B 2010, 66, 380–386. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. B 2016, 72, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Madsen, A.Ø.; Sørensen, H.O.; Flensburg, C.; Stewart, R.F.; Larsen, S. Modeling of the Nuclear Parameters for H Atoms in X-ray Charge-Density Studies. Acta Crystallogr. A 2004, 60, 550–561. [Google Scholar] [CrossRef]
- Madsen, A.Ø.; Hoser, A.A. SHADE3 Server: A Streamlined Approach to Estimate H-Atom Anisotropic Displacement Parameters Using Periodic Ab Initio Calculations or Experimental Information. J. Appl. Crystallogr. 2014, 47, 2100–2104. [Google Scholar] [CrossRef]
- Hoser, A.A.; Dominiak, P.M.; Woźniak, K. Towards the Best Model for H Atoms in Experimental Charge-Density Refinement. Acta Crystallogr. A 2009, 65, 300–311. [Google Scholar] [CrossRef]
- Coppens, P. X-ray Charge Densities and Chemical Bonding; International Union of Crystallography: Chester, UK, 1997; ISBN 978-0-19-535694-6. [Google Scholar]
- Pawlędzio, S.; Makal, A.; Plażuk, D.; Woźniak, K. Experimental Charge Density of Ferrocenyl Derivative of β-Lactam. J. Mol. Struct. 2020, 1217, 128274. [Google Scholar] [CrossRef]
- Korlyukov, A.A.; Malinska, M.; Vologzhanina, A.V.; Goizman, M.S.; Trzybinski, D.; Wozniak, K. Charge Density View on Bicalutamide Molecular Interactions in the Monoclinic Polymorph and Androgen Receptor Binding Pocket. IUCrJ 2020, 7, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Tchoń, D.; Makal, A.; Gutmann, M.; Woźniak, K. Doxycycline Hydrate and Doxycycline Hydrochloride Dihydrate—Crystal Structure and Charge Density Analysis. Z. Krist. Cryst. Mater. 2018, 233, 649–661. [Google Scholar] [CrossRef]
- Cole, J.M.; Goeta, A.E.; Howard, J.a.K.; McIntyre, G.J. X-ray and Neutron Diffraction Studies of the Non-Linear Optical Compounds MBANP and MBADNP at 20 K: Charge-Density and Hydrogen-Bonding Analyses. Acta Crystallogr. B 2002, 58, 690–700. [Google Scholar] [CrossRef]
- Mallinson, P.R.; Smith, G.T.; Wilson, C.C.; Grech, E.; Wozniak, K. From Weak Interactions to Covalent Bonds: A Continuum in the Complexes of 1,8-Bis(Dimethylamino)Naphthalene. J. Am. Chem. Soc. 2003, 125, 4259–4270. [Google Scholar] [CrossRef]
- Dominiak, P.M.; Makal, A.; Mallinson, P.R.; Trzcinska, K.; Eilmes, J.; Grech, E.; Chruszcz, M.; Minor, W.; Woźniak, K. Continua of Interactions between Pairs of Atoms in Molecular Crystals. Chem. Weinh. Bergstr. Ger. 2006, 12, 1941–1949. [Google Scholar] [CrossRef] [PubMed]
- Munshi, P.; Madsen, A.Ø.; Spackman, M.A.; Larsen, S.; Destro, R. Estimated H-Atom Anisotropic Displacement Parameters: A Comparison between Different Methods and with Neutron Diffraction Results. Acta Crystallogr. A 2008, 64, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Wozniak, K.; Mallinson, P.R.; Smith, G.T.; Wilson, C.C.; Grech, E. Role of C-H···O Hydrogen Bonds in the Ionic Complexes of 1,8-Bis(Dimethylamino)Naphthalene. J. Phys. Org. Chem. 2003, 16, 764–771. [Google Scholar] [CrossRef]
- Hoser, A.A.; Madsen, A.Ø. Dynamic Quantum Crystallography: Lattice-Dynamical Models Refined against Diffraction Data. I. Theory. Acta Crystallogr. Sect. Found. Adv. 2016, 72, 206–214. [Google Scholar] [CrossRef]
- Hoser, A.A.; Rekis, T.; Madsen, A.Ø. Dynamics and Disorder: On the Stability of Pyrazinamide Polymorphs. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2022, 78, 416–424. [Google Scholar] [CrossRef]
- Woińska, M.; Hoser, A.A.; Chodkiewicz, M.L.; Woźniak, K. Enhancing Hydrogen Positions in X-ray Structures of Transition Metal Hydride Complexes with Dynamic Quantum Crystallography. IUCrJ 2024, 11, 45–56. [Google Scholar] [CrossRef]
- Pichon-Pesme, V.; Jelsch, C.; Guillot, B.; Lecomte, C. A Comparison between Experimental and Theoretical Aspherical-Atom Scattering Factors for Charge-Density Refinement of Large Molecules. Acta Crystallogr. A 2004, 60, 204–208. [Google Scholar] [CrossRef] [PubMed]
- Zarychta, B.; Pichon-Pesme, V.; Guillot, B.; Lecomte, C.; Jelsch, C. On the Application of an Experimental Multipolar Pseudo-Atom Library for Accurate Refinement of Small-Molecule and Protein Crystal Structures. Acta Crystallogr. A 2007, 63, 108–125. [Google Scholar] [CrossRef] [PubMed]
- Domagała, S.; Jelsch, C. Optimal Local Axes and Symmetry Assignment for Charge-Density Refinement. J. Appl. Crystallogr. 2008, 41, 1140–1149. [Google Scholar] [CrossRef]
- Rybicka, P.M.; Kulik, M.; Chodkiewicz, M.L.; Dominiak, P.M. Multipolar Atom Types from Theory and Statistical Clustering (MATTS) Data Bank: Impact of Surrounding Atoms on Electron Density from Cluster Analysis. J. Chem. Inf. Model. 2022, 62, 3766–3783. [Google Scholar] [CrossRef] [PubMed]
- Dominiak, P.M.; Volkov, A.; Li, X.; Messerschmidt, M.; Coppens, P. A Theoretical Databank of Transferable Aspherical Atoms and Its Application to Electrostatic Interaction Energy Calculations of Macromolecules. J. Chem. Theory Comput. 2007, 3, 232–247. [Google Scholar] [CrossRef] [PubMed]
- Koritsanszky, T.; Volkov, A.; Coppens, P. Aspherical-Atom Scattering Factors from Molecular Wave Functions. 1. Transferability and Conformation Dependence of Atomic Electron Densities of Peptides within the Multipole Formalism. Acta Crystallogr. A 2002, 58, 464–472. [Google Scholar] [CrossRef]
- Dittrich, B.; Koritsánszky, T.; Luger, P. A Simple Approach to Nonspherical Electron Densities by Using Invarioms. Angew. Chem. Int. Ed. 2004, 43, 2718–2721. [Google Scholar] [CrossRef]
- Dittrich, B.; Hübschle, C.B.; Luger, P.; Spackman, M.A. Introduction and Validation of an Invariom Database for Amino-Acid, Peptide and Protein Molecules. Acta Crystallogr. D Biol. Crystallogr. 2006, 62, 1325–1335. [Google Scholar] [CrossRef]
- Bąk, J.M.; Domagala, S.; Dittrich, B.; Jelsch, C.; Hübschle, C.; Dominiak, P.M. Verification of Structural and Electrostatic Properties Obtained by the Use of Different Pseudoatom Databases. Acta Crystallogr. Sect. Found. Crystallogr. 2011, B67, 141–153. [Google Scholar] [CrossRef]
- Jayatilaka, D.; Dittrich, B. X-ray Structure Refinement Using Aspherical Atomic Density Functions Obtained from Quantum-Mechanical Calculations. Acta Crystallogr. Sect. A 2008, 64, 383–393. [Google Scholar] [CrossRef]
- Jayatilaka, D.; Grimwood, D.J. Wavefunctions Derived from Experiment. I. Motivation and Theory. Acta Crystallogr. Sect. A 2001, 57, 76–86. [Google Scholar] [CrossRef]
- Malaspina, L.A.; Wieduwilt, E.K.; Bergmann, J.; Kleemiss, F.; Meyer, B.; Ruiz-López, M.F.; Pal, R.; Hupf, E.; Beckmann, J.; Piltz, R.O.; et al. Fast and Accurate Quantum Crystallography: From Small to Large, from Light to Heavy. J. Phys. Chem. Lett. 2019, 10, 6973–6982. [Google Scholar] [CrossRef] [PubMed]
- Chodkiewicz, M.L.; Woińska, M.; Woźniak, K. Hirshfeld Atom like Refinement with Alternative Electron Density Partitions. IUCrJ 2020, 7, 1199–1215. [Google Scholar] [CrossRef] [PubMed]
- Malaspina, L.A.; Genoni, A.; Jayatilaka, D.; Turner, M.J.; Sugimoto, K.; Nishibori, E.; Grabowsky, S. The Advanced Treatment of Hydrogen Bonding in Quantum Crystallography. J. Appl. Crystallogr. 2021, 54, 718–729. [Google Scholar] [CrossRef] [PubMed]
- Malaspina, L.A.; Hoser, A.A.; Edwards, A.J.; Woińska, M.; Turner, M.J.; Price, J.R.; Sugimoto, K.; Nishibori, E.; Bürgi, H.-B.; Jayatilaka, D.; et al. Hydrogen Atoms in Bridging Positions from Quantum Crystallographic Refinements: Influence of Hydrogen Atom Displacement Parameters on Geometry and Electron Density. CrystEngComm 2020, 22, 4778–4789. [Google Scholar] [CrossRef]
- Woinska, M.; Jayatilaka, D.; Dittrich, B.; Flaig, R.; Luger, P.; Woźniak, K.; Dominiak, P.M.; Grabowsky, S. Validation of X-Ray Wavefunction Refinement. Chemphyschem Eur. J. Chem. Phys. Phys. Chem. 2017, 18, 3334–3351. [Google Scholar] [CrossRef] [PubMed]
- Woińska, M.; Jayatilaka, D.; Spackman, M.A.; Edwards, A.J.; Dominiak, P.M.; Woźniak, K.; Nishibori, E.; Sugimoto, K.; Grabowsky, S. Hirshfeld Atom Refinement for Modelling Strong Hydrogen Bonds. Acta Crystallogr. Sect. A 2014, 70, 483–498. [Google Scholar] [CrossRef]
- Woińska, M.; Grabowsky, S.; Dominiak, P.M.; Woźniak, K.; Jayatilaka, D. Hydrogen Atoms Can Be Located Accurately and Precisely by X-ray Crystallography. Sci. Adv. 2016, 2, e1600192. [Google Scholar] [CrossRef]
- Woinska, M.; Wanat, M.; Taciak, P.; Pawinski, T.; Minor, W.; Wozniak, K. Energetics of Interactions in the Solid State of 2-Hydroxy-8-X-Quinoline Derivatives (X = Cl, Br, I, S-Ph): Comparison of Hirshfeld Atom, X-ray Wavefunction and Multipole Refinements. IUCrJ 2019, 6, 868–883. [Google Scholar] [CrossRef]
- Wanat, M.; Malinska, M.; Hoser, A.A.; Woźniak, K. Further Validation of Quantum Crystallography Approaches. Molecules 2021, 26, 3730. [Google Scholar] [CrossRef]
- Genoni, A. X-Ray Constrained Extremely Localized Molecular Orbitals: Theory and Critical Assessment of the New Technique. J. Chem. Theory Comput. 2013, 9, 3004–3019. [Google Scholar] [CrossRef] [PubMed]
- Ernst, M.; Genoni, A.; Macchi, P. Analysis of Crystal Field Effects and Interactions Using X-ray Restrained ELMOs. J. Mol. Struct. 2020, 1209, 127975. [Google Scholar] [CrossRef]
- Wieduwilt, E.K.; Macetti, G.; Scatena, R.; Macchi, P.; Genoni, A. Extending Libraries of Extremely Localized Molecular Orbitals to Metal Organic Frameworks: A Preliminary Investigation. Crystals 2021, 11, 207. [Google Scholar] [CrossRef]
- Wieduwilt, E.K.; Macetti, G.; Malaspina, L.A.; Jayatilaka, D.; Grabowsky, S.; Genoni, A. Post-Hartree-Fock Methods for Hirshfeld Atom Refinement: Are They Necessary? Investigation of a Strongly Hydrogen-Bonded Molecular Crystal. J. Mol. Struct. 2020, 1209, 127934. [Google Scholar] [CrossRef]
- Dovesi, R.; Orlando, R.; Civalleri, B.; Roetti, C.; Saunders, V.R.; Zicovich-Wilson, C.M. CRYSTAL: A Computational Tool for the Ab Initio Study of the Electronic Properties of Crystals. Z. Krist. Cryst. Mater. 2009, 220, 571–573. [Google Scholar] [CrossRef]
- Spackman, P.R.; Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer: A Program for Hirshfeld Surface Analysis, Visualization and Quantitative Analysis of Molecular Crystals. J. Appl. Crystallogr. 2021, 54, 1006–1011. [Google Scholar] [CrossRef] [PubMed]
- Gavezzotti, A. Calculation of Lattice Energies of Organic Crystals: The PIXEL Integration Method in Comparison with More Traditional Methods. Z. Krist. Cryst. Mater. 2005, 220, 499–510. [Google Scholar] [CrossRef]
- Gavezzotti, A. Efficient Computer Modeling of Organic Materials. The Atom–Atom, Coulomb–London–Pauli (AA-CLP) Model for Intermolecular Electrostatic-Polarization, Dispersion and Repulsion Energies. New J. Chem. 2011, 35, 1360–1368. [Google Scholar] [CrossRef]
- Volkov, A.; King, H.F.; Coppens, P.; Farrugia, L.J. On the Calculation of the Electrostatic Potential, Electric Field and Electric Field Gradient from the Aspherical Pseudoatom Model. Acta Crystallogr. Sect. A 2006, 62, 400–408. [Google Scholar] [CrossRef]
- Volkov, A.; Macchi, C.; Farrugia, L.J.; Gatti, P.; Mallinson, P.R.; Richter, T.; Koritsanszky, T. XD2016—A Computer Program Package for Multipole Refinement, Topological Analysis of Charge Densities and Evaluation of Intermolecular Energies from Experimental or Theoretical Structure Factors; University at Buffalo: Amherst, NY, USA, 2016. [Google Scholar]
- Jelsch, C.; Guillot, B.; Lagoutte, A.; Lecomte, C. Advances in Protein and Small-Molecule Charge-Density Refinement Methods Using MoPro. J. Appl. Crystallogr. 2005, 38, 38–54. [Google Scholar] [CrossRef]
- Guillot, B.; Viry, L.; Guillot, R.; Lecomte, C.; Jelsch, C. Refinement of Proteins at Subatomic Resolution with MOPRO. J. Appl. Crystallogr. 2001, 34, 214–223. [Google Scholar] [CrossRef]
- Volkov, A.; Li, X.; Koritsanszky, T.; Coppens, P. Ab Initio Quality Electrostatic Atomic and Molecular Properties Including Intermolecular Energies from a Transferable Theoretical Pseudoatom Databank. J. Phys. Chem. A 2004, 108, 4283–4300. [Google Scholar] [CrossRef]
- Volkov, A.; Koritsanszky, T.; Coppens, P. Combination of the Exact Potential and Multipole Methods (EP/MM) for Evaluation of Intermolecular Electrostatic Interaction Energies with Pseudoatom Representation of Molecular Electron Densities. Chem. Phys. Lett. 2004, 391, 170–175. [Google Scholar] [CrossRef]
- Li, X.; Volkov, A.V.; Szalewicz, K.; Coppens, P. Interaction Energies between Glycopeptide Antibiotics and Substrates in Complexes Determined by X-ray Crystallography: Application of a Theoretical Databank of Aspherical Atoms and a Symmetry-Adapted Perturbation Theory-Based Set of Interatomic Potentials. Acta Crystallogr. D Biol. Crystallogr. 2006, 62, 639–647. [Google Scholar] [CrossRef] [PubMed]
- Kulik, M.; Goral, A.M.; Jasiński, M.; Dominiak, P.M.; Trylska, J. Electrostatic Interactions in Aminoglycoside-RNA Complexes. Biophys. J. 2015, 108, 655–665. [Google Scholar] [CrossRef] [PubMed]
- Budniak, U.A.; Karolak, N.K.; Kulik, M.; Młynarczyk, K.; Górna, M.W.; Dominiak, P.M. The Role of Electrostatic Interactions in IFIT5-RNA Complexes Predicted by the UBDB+EPMM Method. J. Phys. Chem. B 2022, 126, 9152–9167. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Mills, M.J.L.; Popelier, P.L.A. Multipolar Electrostatics for Proteins: Atom–Atom Electrostatic Energies in Crambin. J. Comput. Chem. 2014, 35, 343–359. [Google Scholar] [CrossRef] [PubMed]
- Malińska, M.; Jarzembska, K.N.; Goral, A.M.; Kutner, A.; Woźniak, K.; Dominiak, P.M. Sunitinib: From Charge-Density Studies to Interaction with Proteins. Acta Crystallogr. D Biol. Crystallogr. 2014, 70, 1257–1270. [Google Scholar] [CrossRef]
- Falcicchio, A.; Lill, S.O.N.; Perna, F.M.; Salomone, A.; Coppi, D.I.; Cuocci, C.; Stalke, D.; Capriati, V. Organotrifluoroborates as Attractive Self-Assembling Systems: The Case of Bifunctional Dipotassium Phenylene-1,4-Bis(Trifluoroborate). Dalton Trans. 2015, 44, 19447–19450. [Google Scholar] [CrossRef]
- Özbek, F.E.; Sertçelik, M.; Yüksek, M. Hirshfeld Surface Analysis and Interaction Energy Calculations of Bis(4-Chlorophenylacetate)Bis(Pyridine-4-Carboxamide)Zinc(II). Cauc. J. Sci. 2020, 7, 83–91. [Google Scholar] [CrossRef]
- Kumar, P.; Bojarowski, S.A.; Jarzembska, K.N.; Domagała, S.; Vanommeslaeghe, K.; MacKerell, A.D.; Dominiak, P.M. A Comparative Study of Transferable Aspherical Pseudoatom Databank and Classical Force Fields for Predicting Electrostatic Interactions in Molecular Dimers. J. Chem. Theory Comput. 2014, 10, 1652–1664. [Google Scholar] [CrossRef] [PubMed]
- Munshi, P.; Guru Row, T.N. Topological Analysis of Charge Density Distribution in Concomitant Polymorphs of 3-Acetylcoumarin, A Case of Packing Polymorphism. Cryst. Growth Des. 2006, 6, 708–718. [Google Scholar] [CrossRef]
- Munshi, P.; Guru Row, T.N. Intra- and Intermolecular Interactions in Small Bioactive Molecules: Cooperative Features from Experimental and Theoretical Charge-Density Analysis. Acta Crystallogr. B 2006, 62, 612–626. [Google Scholar] [CrossRef] [PubMed]
- Bilal, A.; Mehmood, A.; Noureen, S.; Lecomte, C.; Ahmed, M. Crystal Engineering of a Co-Crystal of Antipyrine and 2-Chlorobenzoic Acid: Relative Energetic Contributions Based on Multipolar Refinement. CrystEngComm 2022, 24, 7758–7770. [Google Scholar] [CrossRef]
- Pawlędzio, S.; Makal, A.; Trzybiński, D.; Woźniak, K. Crystal Structure, Interaction Energies and Experimental Electron Density of the Popular Drug Ketoprophen. IUCrJ 2018, 5, 841–853. [Google Scholar] [CrossRef] [PubMed]
- Dominiak, P.M.; Grech, E.; Barr, G.; Teat, S.; Mallinson, P.; Woźniak, K. Neutral and Ionic Hydrogen Bonding in Schiff Bases. Chem. Eur. J. 2003, 9, 963–970. [Google Scholar] [CrossRef]
- Koch, U.; Popelier, P.L.A. Characterization of C-H-O Hydrogen Bonds on the Basis of the Charge Density. J. Phys. Chem. 1995, 99, 9747–9754. [Google Scholar] [CrossRef]
- Jabłoński, M.; Monaco, G. Different Zeroes of Interaction Energies as the Cause of Opposite Results on the Stabilizing Nature of C–H···O Intramolecular Interactions. J. Chem. Inf. Model. 2013, 53, 1661–1675. [Google Scholar] [CrossRef]
- Gatti, C.; May, E.; Destro, R.; Cargnoni, F. Fundamental Properties and Nature of CH··O Interactions in Crystals on the Basis of Experimental and Theoretical Charge Densities. The Case of 3,4-Bis(Dimethylamino)-3-Cyclobutene-1,2-Dione (DMACB) Crystal. J. Phys. Chem. A 2002, 106, 2707–2720. [Google Scholar] [CrossRef]
- Nikolaienko, T.Y.; Bulavin, L.A.; Hovorun, D.M. Bridging QTAIM with Vibrational Spectroscopy: The Energy of Intramolecular Hydrogen Bonds in DNA-Related Biomolecules. Phys. Chem. Chem. Phys. 2012, 14, 7441–7447. [Google Scholar] [CrossRef]
- Kuznetsov, M.L. Relationships between Interaction Energy and Electron Density Properties for Homo Halogen Bonds of the [(A)nY–X···X–Z(B)m] Type (X = Cl, Br, I). Molecules 2019, 24, 2733. [Google Scholar] [CrossRef] [PubMed]
- Mata, I.; Alkorta, I.; Espinosa, E.; Molins, E. Relationships between Interaction Energy, Intermolecular Distance and Electron Density Properties in Hydrogen Bonded Complexes under External Electric Fields. Chem. Phys. Lett. 2011, 507, 185–189. [Google Scholar] [CrossRef]
- Afonin, A.V.; Vashchenko, A.V.; Sigalov, M.V. Estimating the Energy of Intramolecular Hydrogen Bonds from 1H NMR and QTAIM Calculations. Org. Biomol. Chem. 2016, 14, 11199–11211. [Google Scholar] [CrossRef] [PubMed]
- Gavezzotti, A. Calculation of Intermolecular Interaction Energies by Direct Numerical Integration over Electron Densities. I. Electrostatic and Polarization Energies in Molecular Crystals. J. Phys. Chem. B 2002, 106, 4145–4154. [Google Scholar] [CrossRef]
- Gavezzotti, A. Towards a Realistic Model for the Quantitative Evaluation of Intermolecular Potentials and for the Rationalization of Organic Crystal Structures. Part, I. Philosophy. CrystEngComm 2003, 5, 429–438. [Google Scholar] [CrossRef]
- Gavezzotti, A. Quantitative Ranking of Crystal Packing Modes by Systematic Calculations on Potential Energies and Vibrational Amplitudes of Molecular Dimers. J. Chem. Theory Comput. 2005, 1, 834–840. [Google Scholar] [CrossRef]
- Gavezzotti, A. The Lines-of-Force Landscape of Interactions between Molecules in Crystals; Cohesive versus Tolerant and ‘collateral Damage’ Contact. Acta Crystallogr. B 2010, 66, 396–406. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 16, Revision, C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Frisch, M.J.; Head-Gordon, M.; Pople, J.A. A Direct MP2 Gradient Method. Chem. Phys. Lett. 1990, 166, 275–280. [Google Scholar] [CrossRef]
- Funnell, N.P.; Bull, C.L.; Ridley, C.J.; Capelli, S. Structural Behaviour of OP-ROY at Extreme Conditions. CrystEngComm 2019, 21, 4473–4483. [Google Scholar] [CrossRef]
- Maschio, L.; Civalleri, B.; Ugliengo, P.; Gavezzotti, A. Intermolecular Interaction Energies in Molecular Crystals: Comparison and Agreement of Localized Møller-Plesset 2, Dispersion-Corrected Density Functional, and Classical Empirical Two-Body Calculations. J. Phys. Chem. A 2011, 115, 11179–11186. [Google Scholar] [CrossRef]
- Carlucci, L.; Gavezzotti, A. A Quantitative Measure of Halogen Bond Activation in Cocrystallization. Phys. Chem. Chem. Phys. 2017, 19, 18383–18388. [Google Scholar] [CrossRef] [PubMed]
- Dunitz, J.D.; Gavezzotti, A. Proteogenic Amino Acids: Chiral and Racemic Crystal Packings and Stabilities. J. Phys. Chem. B 2012, 116, 6740–6750. [Google Scholar] [CrossRef]
- Hutchison, I.B.; Bull, C.L.; Marshall, W.G.; Parsons, S.; Urquhart, A.J.; Oswald, I.D.H. Compression of Glycolide-H4 to 6 GPa. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2017, 73, 1151–1157. [Google Scholar] [CrossRef]
- Reeves, M.G.; Wood, P.A.; Parsons, S. MrPIXEL: Automated Execution of Pixel Calculations via the Mercury Interface. J. Appl. Crystallogr. 2020, 53, 1154–1162. [Google Scholar] [CrossRef] [PubMed]
- Macrae, C.F.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Shields, G.P.; Taylor, R.; Towler, M.; van de Streek, J. Mercury: Visualization and Analysis of Crystal Structures. J. Appl. Crystallogr. 2006, 39, 453–457. [Google Scholar] [CrossRef]
- Spackman, M.A.; Jayatilaka, D. Hirshfeld Surface Analysis. CrystEngComm 2009, 11, 19–32. [Google Scholar] [CrossRef]
- McKinnon, J.J.; Jayatilaka, D.; Spackman, M.A. Towards Quantitative Analysis of Intermolecular Interactions with Hirshfeld Surfaces. Chem. Commun. 2007, 0, 3814–3816. [Google Scholar] [CrossRef]
- Spackman, M.A.; McKinnon, J.J. Fingerprinting Intermolecular Interactions in Molecular Crystals. CrystEngComm 2002, 4, 378–392. [Google Scholar] [CrossRef]
- Turner, M.J.; Thomas, S.P.; Shi, M.W.; Jayatilaka, D.; Spackman, M.A. Energy Frameworks: Insights into Interaction Anisotropy and the Mechanical Properties of Molecular Crystals. Chem. Commun. 2015, 51, 3735–3738. [Google Scholar] [CrossRef]
- Ziemniak, M.; Pawlędzio, S.; Zawadzka-Kaźmierczuk, A.; Dominiak, P.M.; Trzybiński, D.; Koźmiński, W.; Zieliński, R.; Fokt, I.; Priebe, W.; Woźniak, K.; et al. X-Ray Wavefunction Refinement and Comprehensive Structural Studies on Bromo-Substituted Analogues of 2-Deoxy-D-Glucose in Solid State and Solution. RSC Adv. 2022, 12, 8345–8360. [Google Scholar] [CrossRef]
- Pawlędzio, S.; Malinska, M.; Kleemiss, F.; Grabowsky, S.; Woźniak, K. Influence of Modelling Disorder on Hirshfeld Atom Refinement Results of an Organo-Gold(I) Compound. IUCrJ 2022, 9, 497–507. [Google Scholar] [CrossRef]
- Fugel, M.; Jayatilaka, D.; Hupf, E.; Overgaard, J.; Hathwar, V.R.; Macchi, P.; Turner, M.J.; Howard, J.A.K.; Dolomanov, O.V.; Puschmann, H.; et al. Probing the Accuracy and Precision of Hirshfeld Atom Refinement with HARt Interfaced with Olex2. IUCrJ 2018, 5, 32–44. [Google Scholar] [CrossRef]
- Köhler, C.; Lübben, J.; Krause, L.; Hoffmann, C.; Herbst-Irmer, R.; Stalke, D. Comparison of Different Strategies for Modelling Hydrogen Atoms in Charge Density Analyses. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2019, 75, 434–441. [Google Scholar] [CrossRef] [PubMed]
- Spackman, M.A. How Reliable Are Intermolecular Interaction Energies Estimated from Topological Analysis of Experimental Electron Densities? Cryst. Growth Des. 2015, 15, 5624–5628. [Google Scholar] [CrossRef]
- Khalilov, L.M.; Mescheryakova, E.S.; Bikmukhametov, K.S.; Makhmudiyarova, N.N.; Shangaraev, K.R.; Tulyabaev, A.R. Twist-Chair Conformation of the Tetraoxepane Ring Remains Unchanged in Tetraoxaspirododecane Diamines. Acta Crystallogr. Sect. C Struct. Chem. 2020, 76, 276–286. [Google Scholar] [CrossRef] [PubMed]
- Kowsalya, P.S.; Bhuvanesh, N.S.P.; Neelakantan, M.A. Chemical Reactivity and Quantifying the Intra- and Intermolecular Interactions in Zwitterionic Compounds. ChemistrySelect 2018, 3, 2045–2052. [Google Scholar] [CrossRef]
- Matta, C.F.; Castillo, N.; Boyd, R.J. Extended Weak Bonding Interactions in DNA: π-Stacking (Base−Base), Base−Backbone, and Backbone−Backbone Interactions. J. Phys. Chem. B 2006, 110, 563–578. [Google Scholar] [CrossRef]
- Kumar, P.; Cabaj, M.K.; Dominiak, P.M. Intermolecular Interactions in Ionic Crystals of Nucleobase Chlorides—Combining Topological Analysis of Electron Densities with Energies of Electrostatic Interactions. Crystals 2019, 9, 668. [Google Scholar] [CrossRef]
- Alkorta, I.; Mata, I.; Molins, E.; Espinosa, E. Charged versus Neutral Hydrogen-Bonded Complexes: Is There a Difference in the Nature of the Hydrogen Bonds? Chem. Eur. J. 2016, 22, 9226–9234. [Google Scholar] [CrossRef]
- Bauer, C.A.; Schneider, G.; Göller, A.H. Machine Learning Models for Hydrogen Bond Donor and Acceptor Strengths Using Large and Diverse Training Data Generated by First-Principles Interaction Free Energies. J. Cheminform. 2019, 11, 59. [Google Scholar] [CrossRef]
- Xu, X.; Song, C.; Andresen, J.M.; Miller, B.G.; Scaroni, A.W. Novel Polyethylenimine-Modified Mesoporous Molecular Sieve of MCM-41 Type as High-Capacity Adsorbent for CO2 Capture. Energy Fuels 2002, 16, 1463–1469. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).