
Study of New Infrared Non-Linear Optical Crystal BaGa4Se7 Based on First Principle


Abstract
:1. Introduction
2. Theoretical Model and Computational Method
2.1. Theoretical Model
2.2. Computational Details
3. Results and Discussion
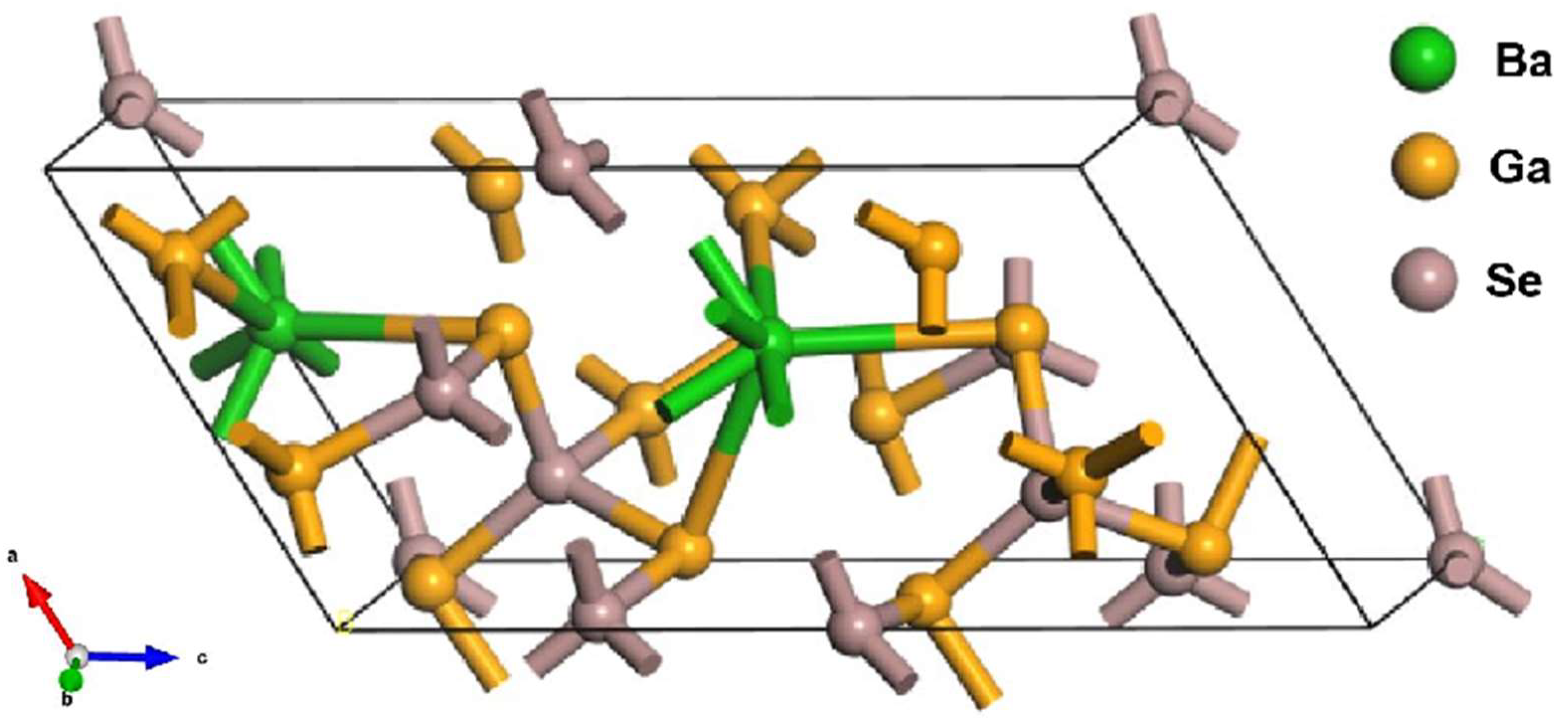
3.1. Geometric Properties
3.2. Electronic Structure
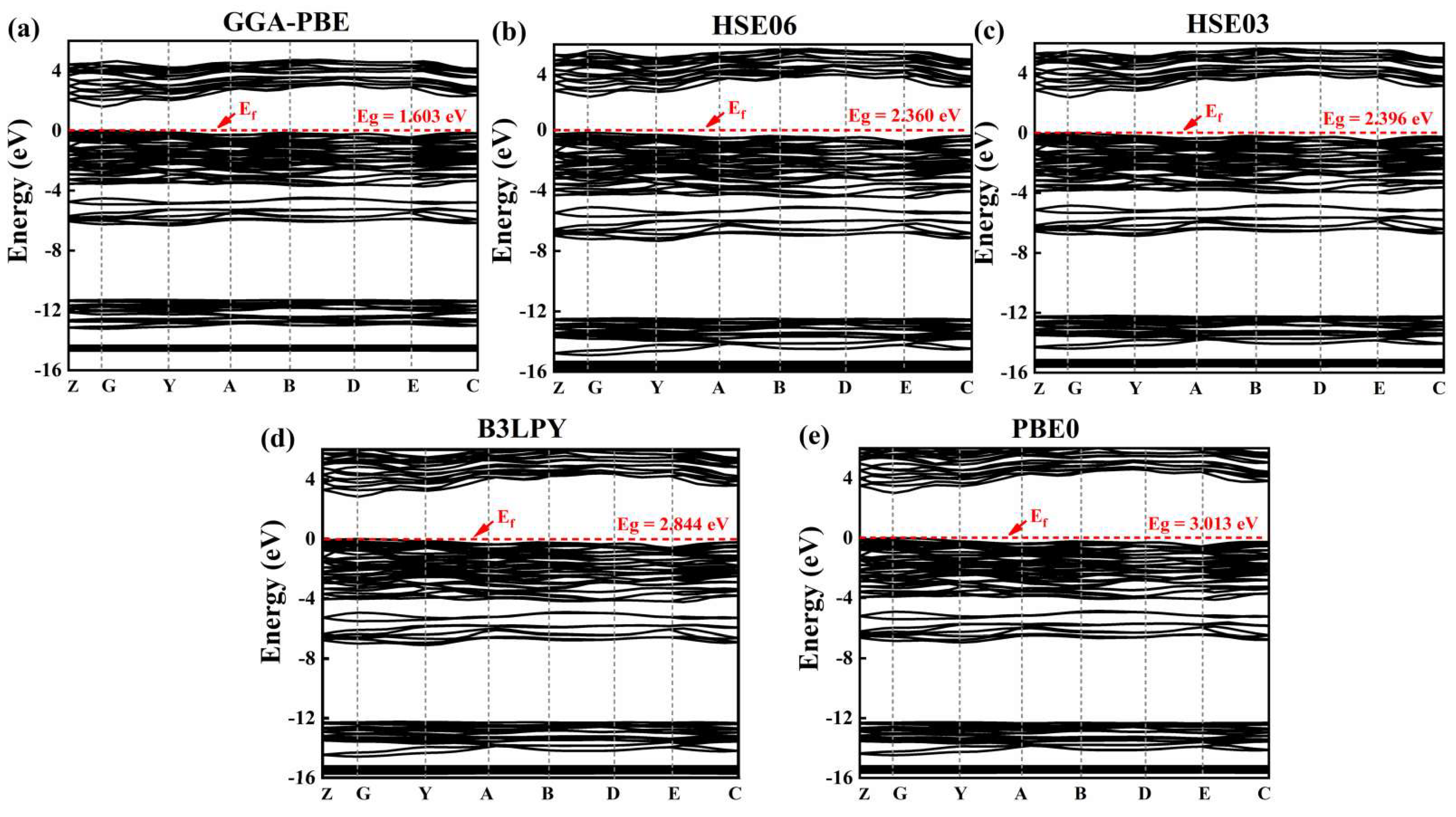
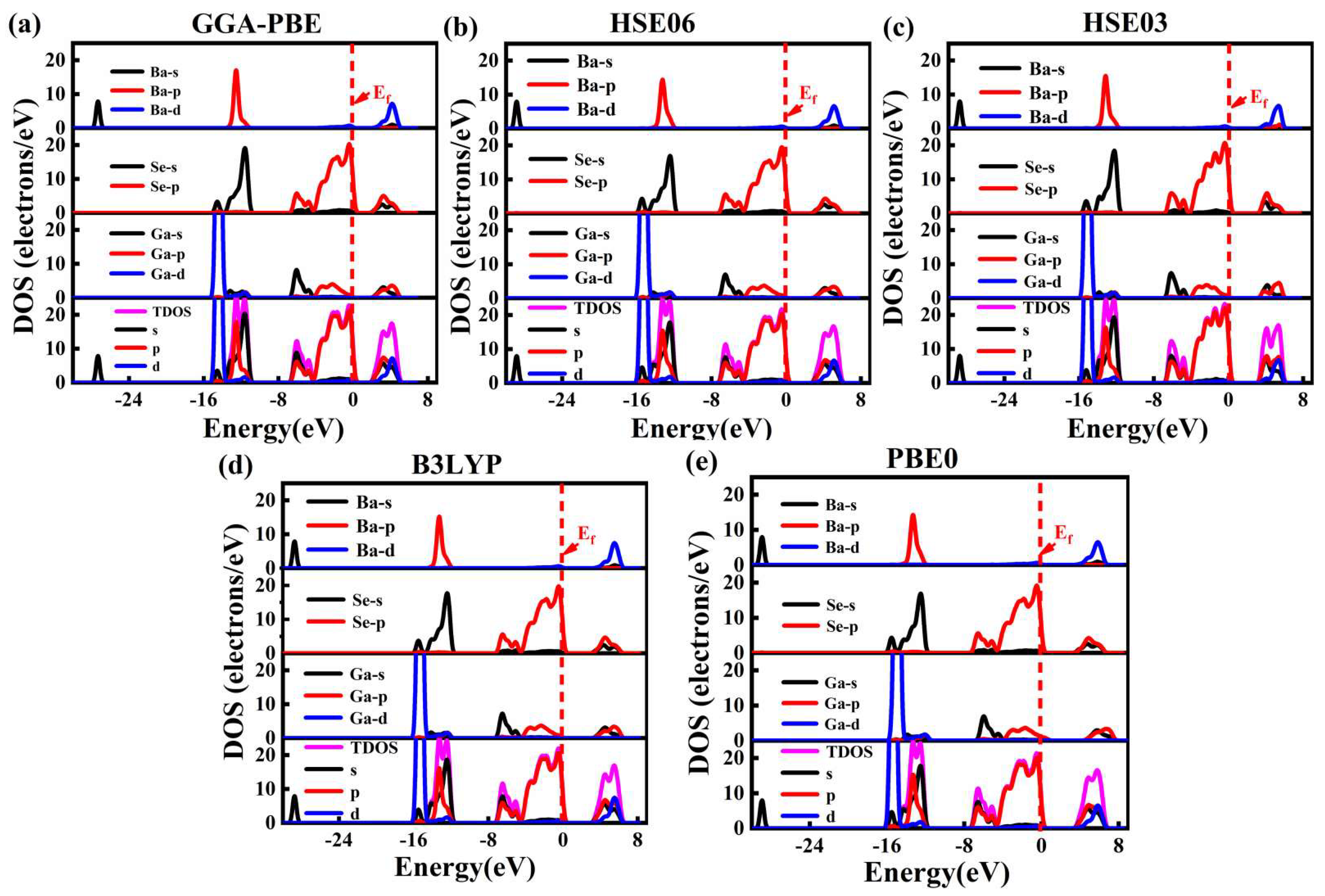
3.2.1. Band Structure and Density of States
3.2.2. Differential Charge Density
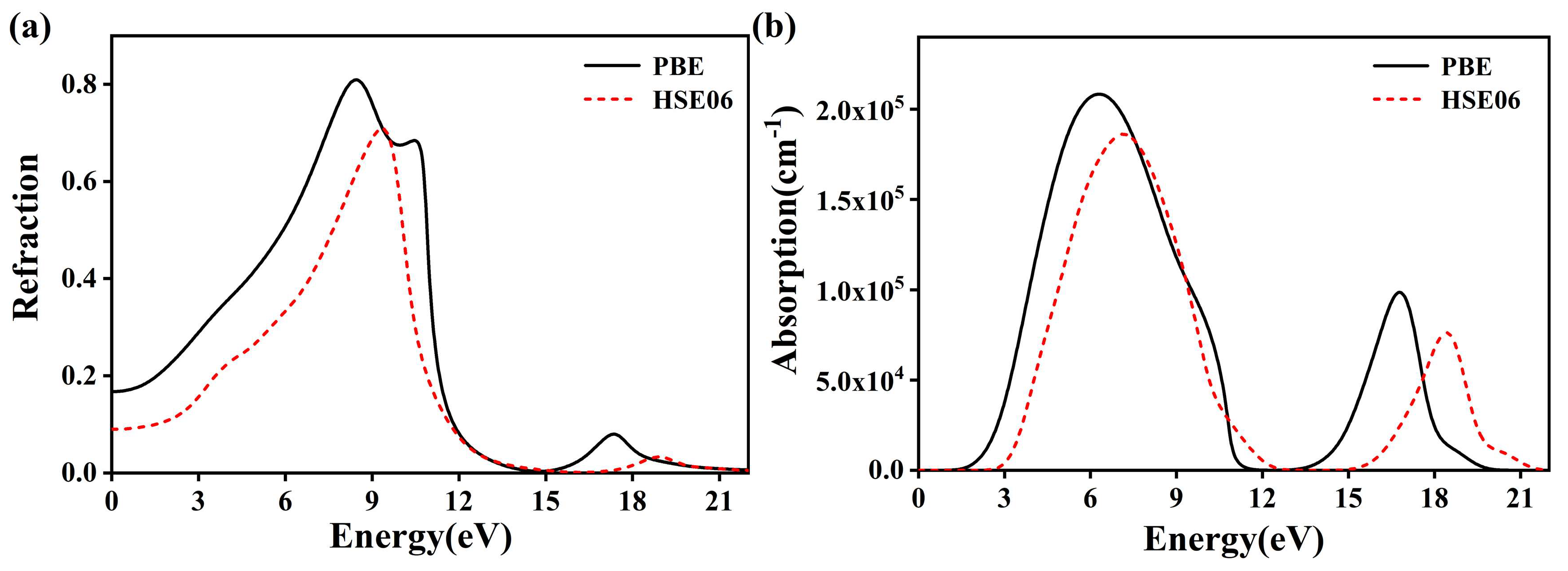
3.3. Optical Properties
3.3.1. BaGa4Se7 Complex Dielectric Function
3.3.2. BaGa4Se7 Reflection and Absorption Spectrum
3.3.3. BaGa4Se7 Complex Refractive Index
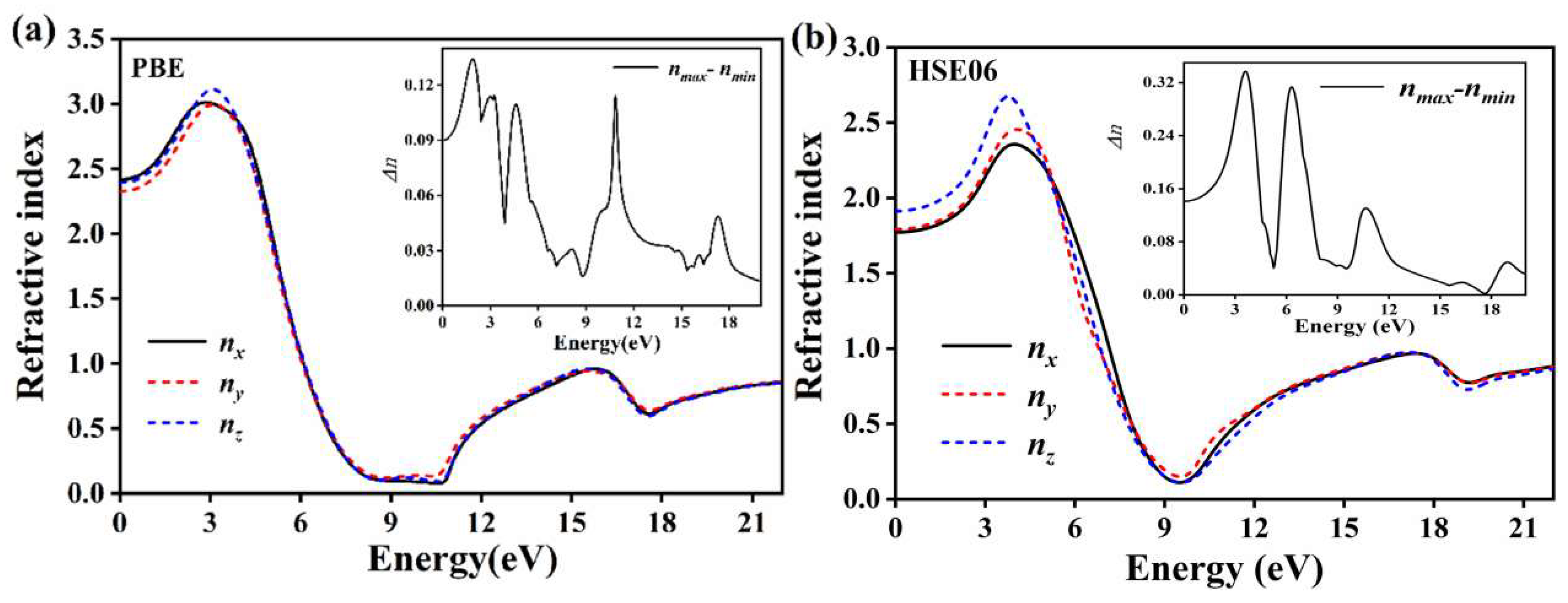
3.3.4. BaGa4Se7 Refractive Index and Birefringence
3.3.5. BaGa4Se7 Energy Loss Function
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Boyd, G.D.; Kasper, H.M.; Mcfee, J.H. Linear and nonlinear optical properties of some ternary selenides. IEEE J. Quantum Electron. 1972, 8, 900–908. [Google Scholar] [CrossRef]
- Wagner, S.; Shay, J.L.; Migliorato, P.; Kasper, H.M. CuInSe2/CdS heterojunction photovoltaic detectors. Appl. Phys. Lett. 1974, 25, 434. [Google Scholar] [CrossRef]
- Burland, D.M.; Miller, R.D.; Walsh, C.A. Second-order nonlinearity in poled polymer systems. Chem. Rev. 1994, 94, 31–75. [Google Scholar] [CrossRef]
- Zhang, G.D.; Wang, S.P.; Tao, X.T. Research Progress of Infrared Nonlinear Optical Crystals. J. Synth. Cryst. 2012, 41, 7–17. [Google Scholar]
- Havva, B.O.; Haci, O.; Engin, D. A new quaternary semiconductor compound (Ba2Sb4GeS10): Ab initio study. Philos. Mag. 2017, 97, 549–560. [Google Scholar]
- Yao, J.Y.; Yin, W.L.; Feng, K. Growth and characterization of BaGa4Se7 crystal. J. Cryst. Growth 2012, 346, 1–4. [Google Scholar] [CrossRef]
- Eisenmann, B.; Jakowski, M.; Schafer, H. Zur Kenntnis BaAl4S7 and BaGa4S7. Rev. Chim. Miner. 1983, 20, 329–337. [Google Scholar]
- Badikov, V.; Badikov, D.; Shevyrdyaeva, G. BaGa4S7: Wide-bandgap phase-matchable nonlinear crystal for the mid-infrared. J. Opt. Soc. Am. 2011, 190, 4330–4410. [Google Scholar]
- Badikov, V.; Badikov, D.; Shevyrdyaeva, G. Phase-matching properties of BaGa4S7 and BaGa4Se7:wide-bandgap nonlinear crystals for the mid-infrared. Phy. Stat. Sol. 2011, 5, 3103. [Google Scholar]
- Zhai, N.; Li, C.; Xu, B.; Bai, L.; Yao, J.; Zhang, G.; Hu, Z.; Wu, Y. Temperature-Dependent Sellmeier Equations of IR Nonlinear Optical Crystal BaGa4Se7. Crystals 2017, 7, 62. [Google Scholar] [CrossRef] [Green Version]
- Yao, J.; Mei, D.; Bai, L. BaGa4Se7: A new congruent-melting IR nonlinear optical material. Inorg. Chem. 2010, 49, 9212–9216. [Google Scholar] [CrossRef] [PubMed]
- Yelisseyev, A.P.; Lobanov, S.I.; Krinitsin, P.G.; Isaenko, L.I. The optical properties of the nonlinear crystal BaGa4Se7. Opt. Mater. 2019, 99, 109564. [Google Scholar] [CrossRef]
- Kolker, D.B.; Kostyukova, N.Y.; Boyko, A.A. Widely tunable (2.6–10.4 μm) BaGa4Se7 optical parametric oscillator pumped by a Q-switched Nd:YLiF4 laser. J. Phys. Commun. 2018, 2, 035–039. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perdew, J.P.; Ruzsinszky, A.; Csonka, G.I.; Vydrov, O.A.; Scuseria, G.E. Restoring the density-gradient expansion for exchange in solids and surfaces. Phys. Rev. Lett. 2008, 100, 136–406. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.; Dai, R.; Zhang, L.; Wang, W.; Zhang, F.C.; Zhang, W.B. Theoretical study of a novel infrared nonlinear optical crystal of BaGa4S7. J. Nanoelectron. Optoelectron. 2020, 15, 1–11. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Hammer, B.; Hansen, L.B.; Nørskov, J.K. Improved adsorption energetics within density-functional theory using revised Perdew-Burke-Ernzerhof functionals. Phys. Rev. B 1999, 59, 7413. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef]
- Liu, N.N.; Song, R.B.; Sun, H.Y.; Du, D.W. First-principles calculation of electronic structure and thermodynamic properties of Mg2Sn. Acta Phy. Sin. 2008, 57, 7145–7150. [Google Scholar]
- Huang, K. (Ed.) Solid Physic; Higher Education Press: Beijing, China, 2002; p. 439. [Google Scholar]
- Fang, R.C. (Ed.) Solid State Pectroscopy; University of Science and Technology of China Press: Hefei, China, 2001. [Google Scholar]
- Shen, X.C. (Ed.) Spectra and Optical Properties of Semiconductors, 2nd ed.; Science Press: Beijing, China, 2002. [Google Scholar]
- Yang, C.Y.; Zhang, R.; Zhang, L.M.; Ke, X.W. Electronic structure and optical properties of 0.5NdAlO3-0.5CaTiO3 from first-principles calculation. Acta Phys. Sin. 2012, 61, 077–702. [Google Scholar]
- Feng, J.; Xiao, B.; Chen, J.C. Electronic and optical properties of CuInSe2 from ab-initio calculations. Acta Phys. Sin. 2007, 56, 5990–5995. [Google Scholar] [CrossRef]
- Li, X.Z.; Xie, Q.; Chen, Q.; Zhao, F.J.; Cui, D.M. The study on the electronic structure and optical properties of OsSi2. Acta Phys. Sin. 2010, 59, 2016–2021. [Google Scholar]
- Zhou, H.G.; Wen, X.W.; Fang, Z.X.; Li, Y.; Ding, K.N.; Huang, X.; Zhang, Y.F. Electronic structures and optical properties of AgGa(S1−xSex)2 solid solutions. Chem. Phys. 2013, 29, 920–928. [Google Scholar]
- Wang, L.; Men, Y.B. Comparison study of CsLiB6O10 and β-BaB2O4 as nonlinear media for optical parametric oscillators. Appl. Optics. 2003, 42, 2720–2723. [Google Scholar] [CrossRef]
- Chen, W.D.; Cousin, J. Continuous-wave mid-infrared laser sources based on difference frequency generation. Comptes Rendus Phys. 2007, 8, 1129–1150. [Google Scholar] [CrossRef]
- Hazama, H.; Takatani, Y. Integrated ultraviolet and tunable mid-infrared laser source for analyses of protein. Proc. Soc. Photo-Opt. Instrum. Eng. 2007, 6455, 45507. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Theoretical Value | Experimental Value [11] | Relative Errors |
|---|---|---|---|
| a/Å | 7.739 | 7.625 | 0.014 |
| b/Å | 6.635 | 6.511 | 0.019 |
| c/Å | 14.936 | 14.702 | 0.015 |
| α = γ(°) | 90 | 90 | 0.000 |
| β(°) | 121.048 | 121.240 | 0.001 |
| Method | Parameter | Z | G | Y | A | B | D | E | C |
|---|---|---|---|---|---|---|---|---|---|
| GGA | EV (eV) | −0.084 | 0 | −0.125 | −0.256 | −0.197 | −0.258 | −0.449 | −0.170 |
| EC (eV) | 2.070 | 1.600 | 2.050 | 2.840 | 3 | 3.100 | 2.920 | 2.370 | |
| HSE06 | EV (eV) | −0.084 | 0 | −0.198 | −0.368 | −0.237 | −0.294 | −0.540 | −0.251 |
| EC (eV) | 2.942 | 2.360 | 2.860 | 3.711 | 3.849 | 3.960 | 3.850 | 3.201 | |
| HSE03 | EV (eV) | −0.086 | 0 | −0.203 | −0.378 | −0.241 | −0.297 | −0.528 | −0.242 |
| EC (eV) | 2.960 | 2.396 | 2.840 | 3.660 | 3.850 | 3.940 | 3.760 | 3.170 | |
| B3LYP | EV (eV) | 0.098 | 0 | −0.151 | −0.374 | −0.277 | −0.359 | −0.600 | −0.212 |
| EC (eV) | 3.310 | 2.840 | 3.240 | 4.140 | 4.250 | 4.410 | 4.220 | 1.360 | |
| PBE0 | EV (eV) | −0.094 | 0 | −0.217 | −0.409 | −0.263 | −0.314 | −0.560 | −0.260 |
| EC (eV) | 3.500 | 3.010 | 3.470 | 4.310 | 4.510 | 4.600 | 4.420 | 3.820 |
| Method | Theoretical Value (eV) | Experimental Value (eV) | Relative Error /% |
|---|---|---|---|
| GGA | 1.603 | 2.640 | 0.392 |
| HSE03 | 2.396 | 2.640 | 0.092 |
| HSE06 | 2.360 | 2.640 | 0.106 |
| B3LYP | 2.844 | 2.640 | 0.080 |
| PBE0 | 3.013 | 2.640 | 0.141 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, X.; Dai, R.; Zhang, L.; Ning, J.; Zhang, F.; Yan, J. Study of New Infrared Non-Linear Optical Crystal BaGa4Se7 Based on First Principle. Crystals 2022, 12, 143. https://doi.org/10.3390/cryst12020143
Huang X, Dai R, Zhang L, Ning J, Zhang F, Yan J. Study of New Infrared Non-Linear Optical Crystal BaGa4Se7 Based on First Principle. Crystals. 2022; 12(2):143. https://doi.org/10.3390/cryst12020143
Chicago/Turabian StyleHuang, Xie, Rong Dai, Lei Zhang, Jing Ning, Fuchun Zhang, and Junfeng Yan. 2022. "Study of New Infrared Non-Linear Optical Crystal BaGa4Se7 Based on First Principle" Crystals 12, no. 2: 143. https://doi.org/10.3390/cryst12020143
APA StyleHuang, X., Dai, R., Zhang, L., Ning, J., Zhang, F., & Yan, J. (2022). Study of New Infrared Non-Linear Optical Crystal BaGa4Se7 Based on First Principle. Crystals, 12(2), 143. https://doi.org/10.3390/cryst12020143