
Spin-Phonon Coupling in A2BMnO6 (A = La, Pr, Nd, Sm, Gd; B = Co, Ni) Double-Perovskite Thin Films: Impact of the A-Site Cation Radius


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Experimental Details
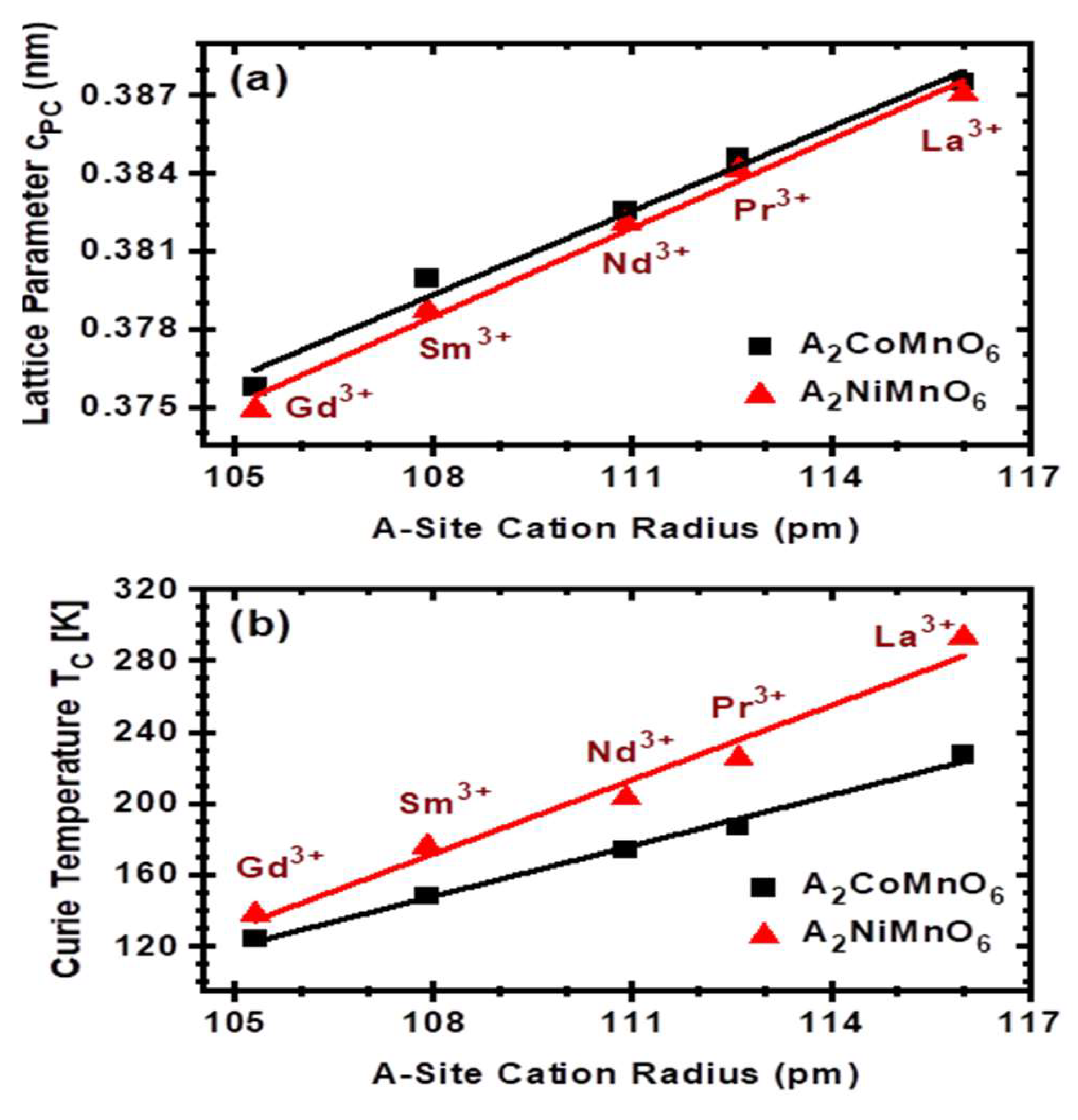
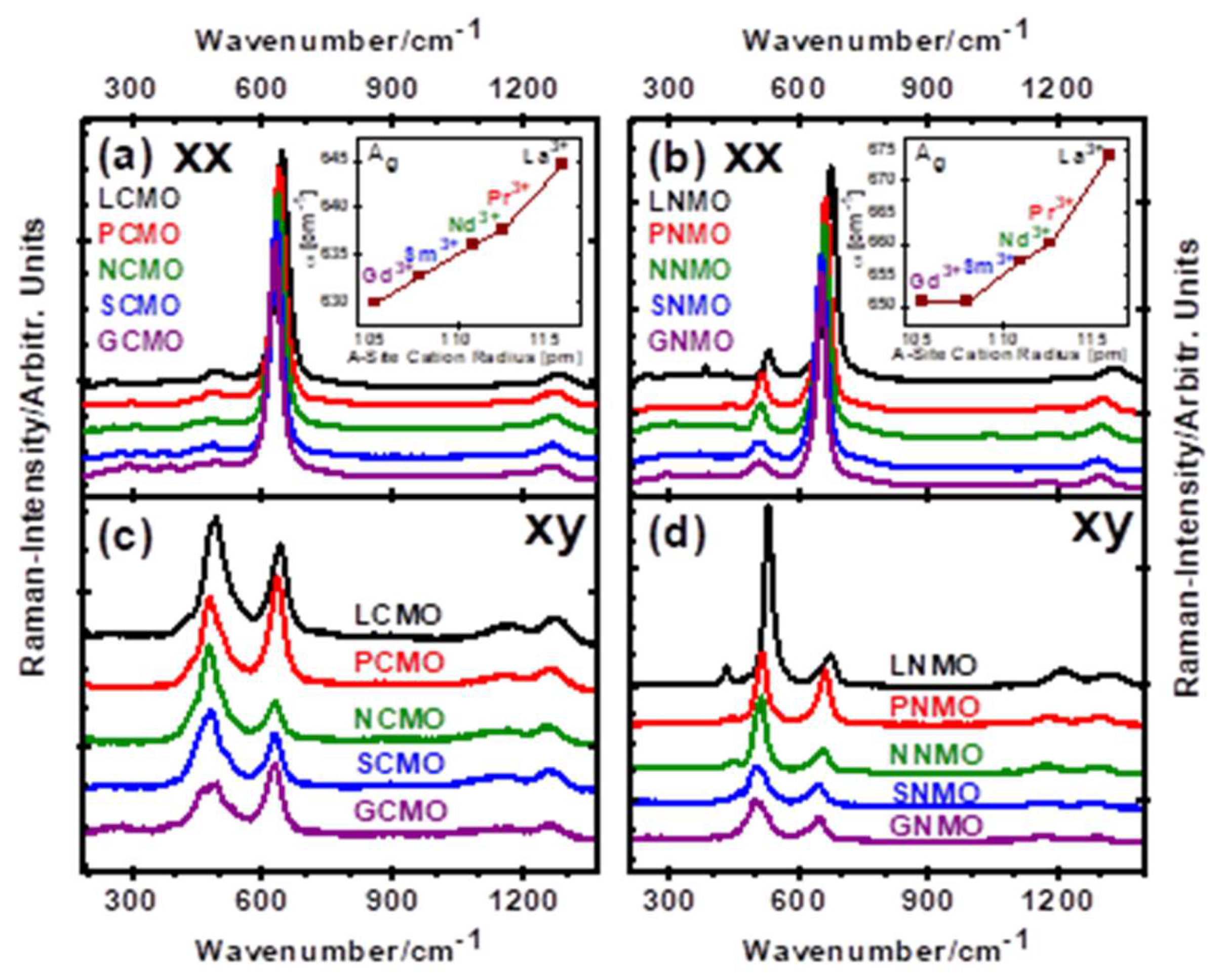
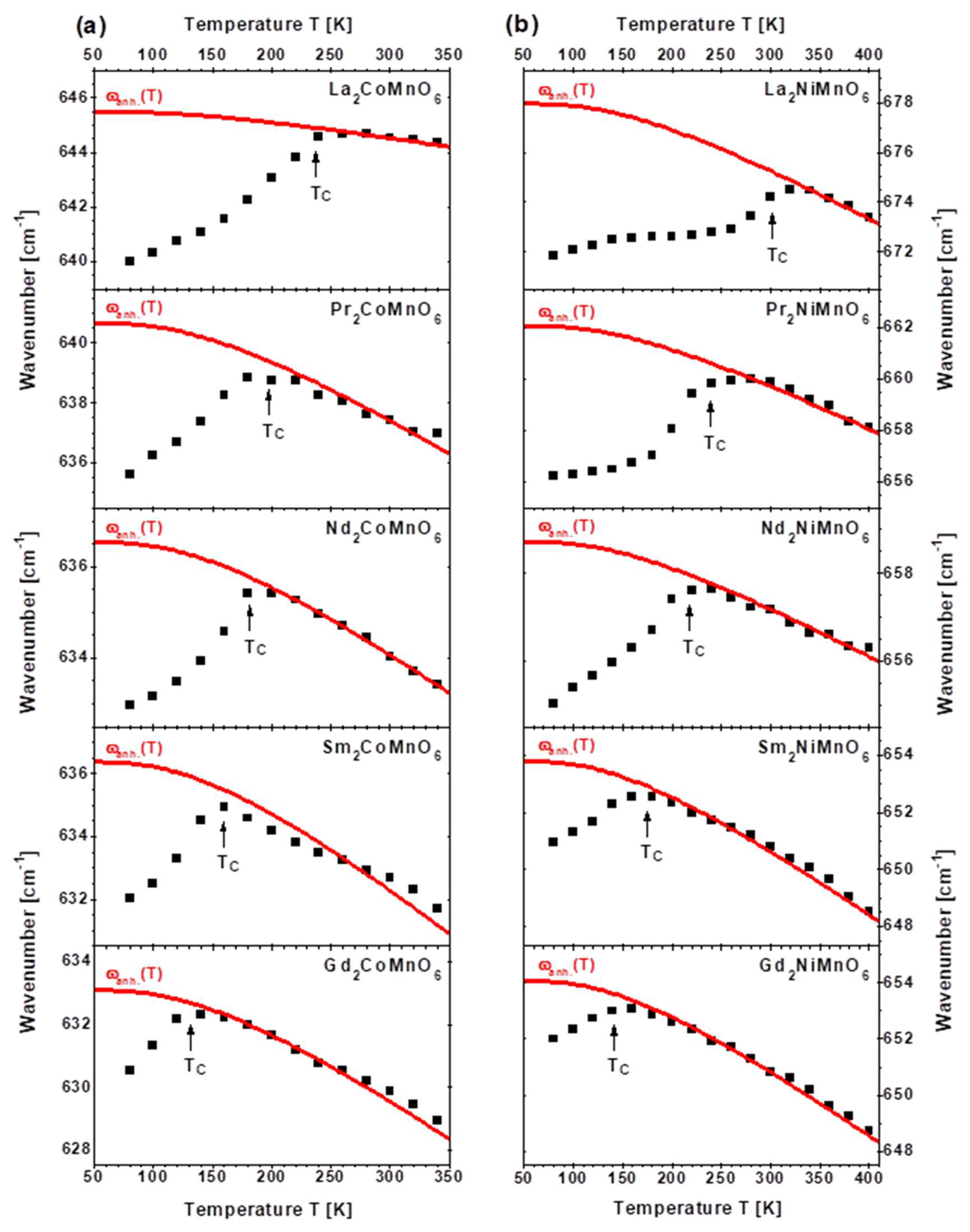
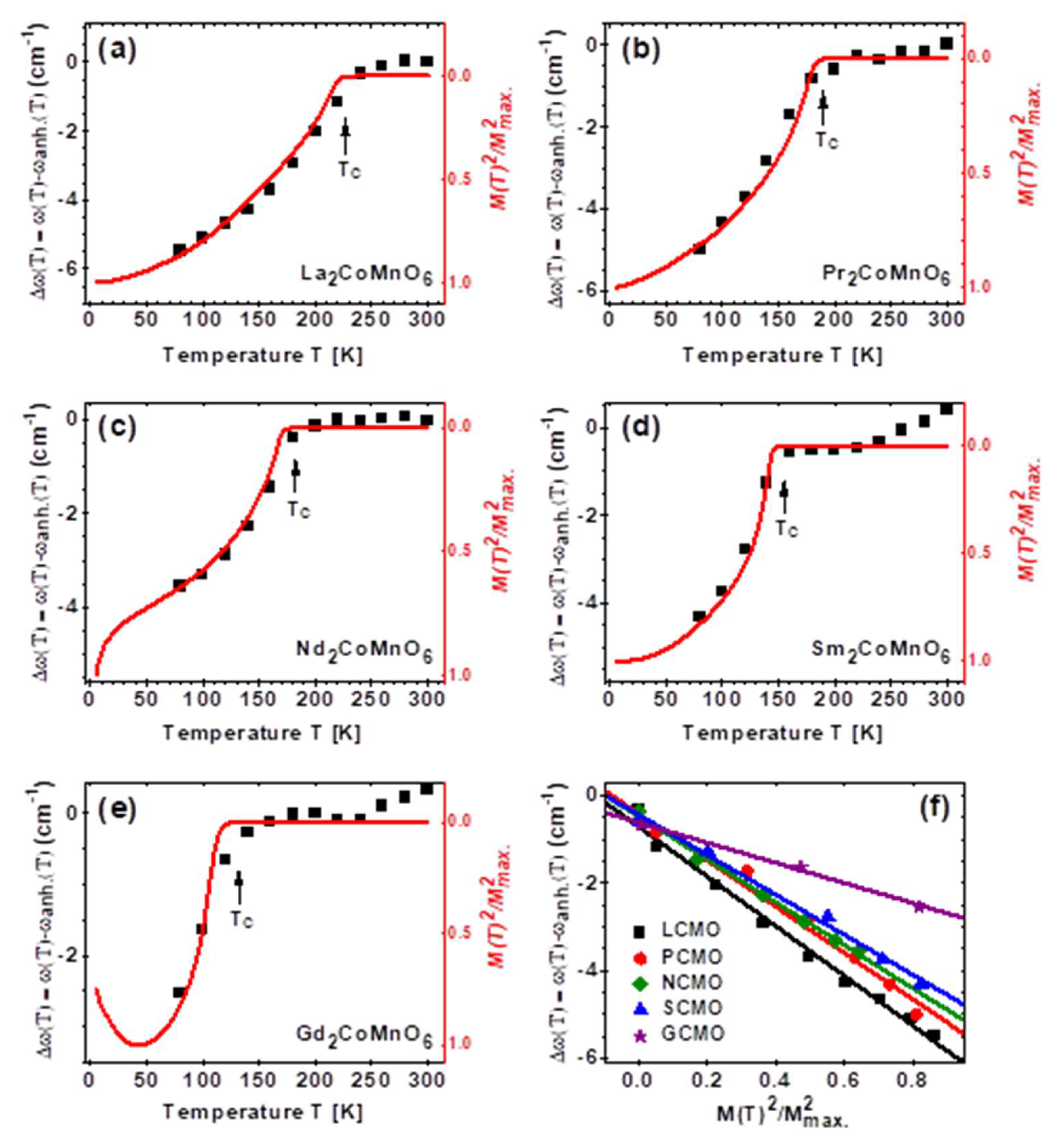
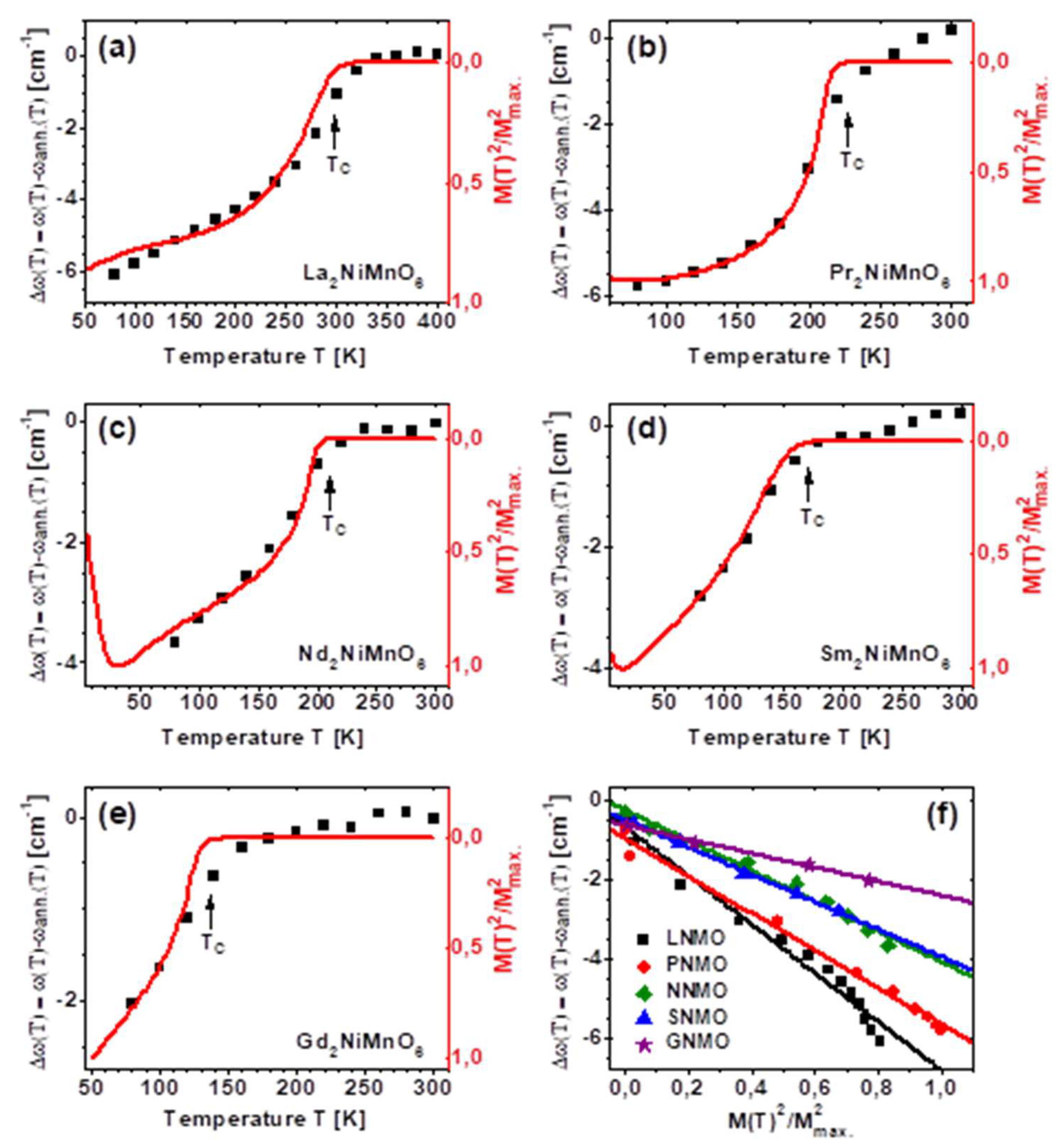
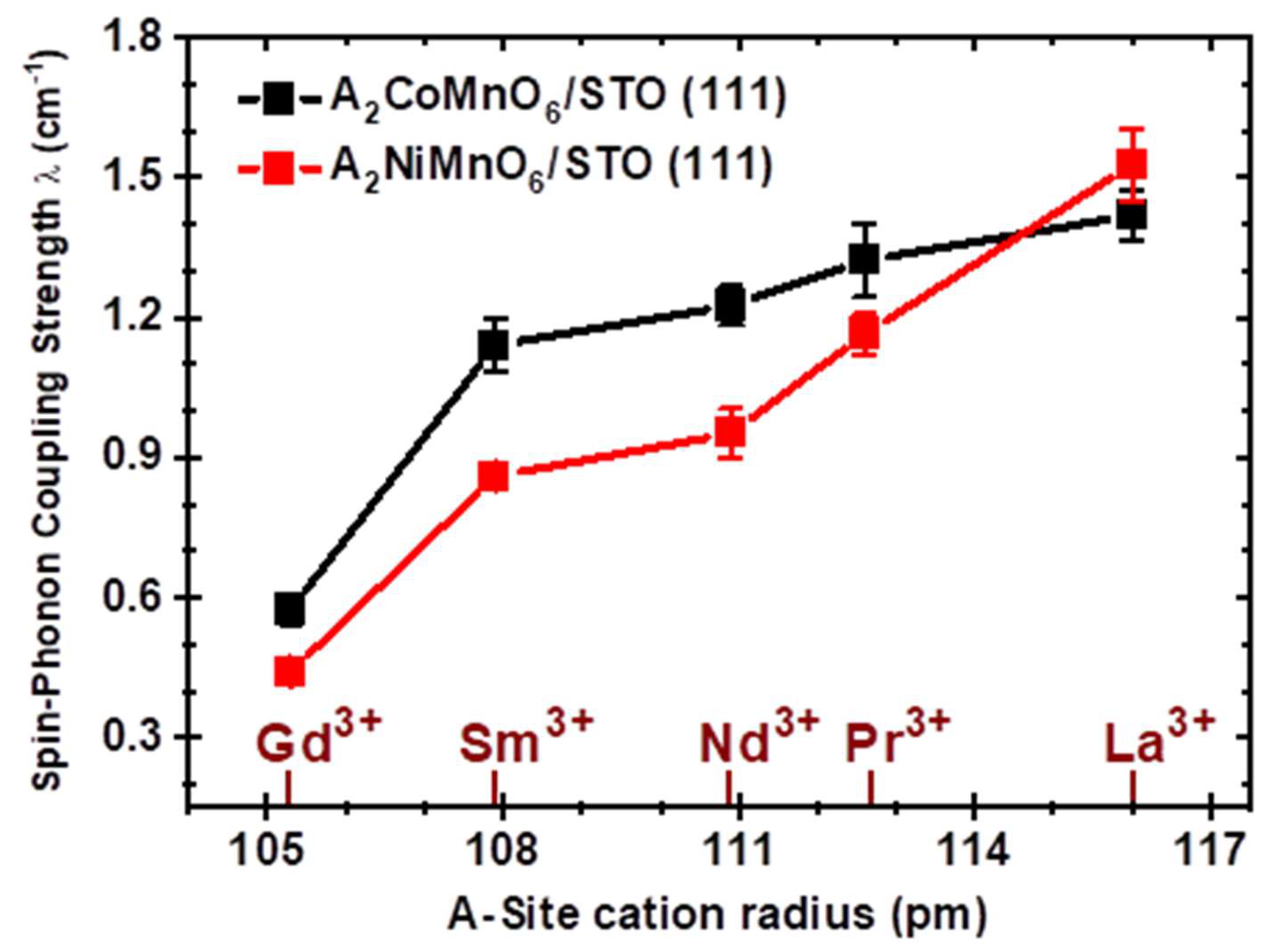
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Padhan, P.; Gupta, A. Functional Metal Oxides; Ogale, S.B., Venkatesan, T.V., Blamire, M.G., Eds.; Wiley VCH Springer: Weinheim, Germany, 2013; pp. 51–88. [Google Scholar]
- Vasala, S.; Karppinen, M. A2B′B″O6 perovskites: A review. Prog. Solid State Chem. 2015, 43, 1–36. [Google Scholar] [CrossRef]
- Shimakawa, Y.; Azuma, M.; Ichikawa, N. Multiferroic Compounds with Double-Perovskite Structures. Materials 2011, 4, 153–168. [Google Scholar] [CrossRef] [Green Version]
- Singh, M.P.; Truong, K.D.; Fournier, P. Magnetodielectric effect in double perovskite La2CoMnO6 thin films. Appl. Phys. Lett. 2007, 91, 042504. [Google Scholar] [CrossRef]
- Kim, M.K.; Moon, J.Y.; Choi, H.Y.; Oh, S.H.; Lee, N.; Choi, Y.J. Investigation of the magnetic properties in double perovskite R2CoMnO6 single crystals (R = Rare earth: La to Lu). J. Phys. Condens. Matter 2015, 27, 426002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bull, C.L.; McMillan, P.F. Raman scattering study and electrical properties characterization of elpasolite perovskites Ln2(BB′)O6 (Ln = La, Sm… Gd and B, B′ = Ni, Co, Mn). J. Solid State Chem. 2004, 177, 2323–2328. [Google Scholar] [CrossRef]
- Booth, R.; Fillman, R.; Whitaker, H.; Nag, A.; Tiwari, R.; Ramanujachary, K.; Gopalakrishnan, J.; Lofland, S. An investigation of structural, magnetic and dielectric properties of R2NiMnO6 (R = rare earth, Y). Mater. Res. Bull. 2009, 44, 1559–1564. [Google Scholar] [CrossRef]
- Dass, R.I.; Goodenough, J.B. Multiple magnetic phases of La2CoMnO6−δ (0 <~δ <~0.05). Phys. Rev. B 2003, 67, 014401. [Google Scholar]
- Dass, R.I.; Yan, J.-Q.; Goodenough, J.B. Oxygen stoichiometry, ferromagnetism, and transport properties of La2−xNiMnO6+δ. Phys. Rev. B 2003, 68, 064415. [Google Scholar] [CrossRef]
- Meyer, C.; Roddatis, V.; Ksoll, P.; Damaschke, B.; Moshnyaga, V. Structure, magnetism, and spin-phonon coupling in het-eroepitaxial La2CoMnO6/Al2O3 (0001) films. Phys. Rev. B 2018, 98, 134433. [Google Scholar] [CrossRef]
- Egoavil, R.; Hühn, S.; Jungbauer, M.; Gauquelin, N.; Béché, A.; Van Tendeloo, G.; Verbeeck, J.; Moshnyaga, V. Phase problem in the B-site ordering of La2CoMnO6: Impact on structure and magnetism. Nanoscale 2015, 7, 9835–9843. [Google Scholar] [CrossRef]
- Kleibeuker, J.E.; Choi, E.-M.; Jones, E.D.; Yu, T.-M.; Sala, B.; MacLaren, B.A.; Kepaptsoglou, D.; Hernandez-Maldonado, D.; Ramasse, Q.M.; Jones, L.; et al. Route to achieving perfect B-site ordering in double perovskite thin films. NPG Asia Mater. 2017, 9, e406. [Google Scholar] [CrossRef] [Green Version]
- Barón-González, A.J.; Frontera, C.; García-Muñoz, J.L.; Rivas-Murias, B.; Blasco, J. Effect of cation disorder on structural, magnetic and dielectric properties of La2MnCoO6 double perovskite. J. Phys. Condens. Matter 2011, 23, 496003. [Google Scholar] [CrossRef] [PubMed]
- Iliev, M.; Abrashev, M.V.; Litvinchuk, A.P.; Hadjiev, V.; Guo, H.; Gupta, A. Raman spectroscopy of ordered double perovskite La2CoMnO6 thin films. Phys. Rev. B 2007, 75, 104118. [Google Scholar] [CrossRef]
- Kumar, D.; Sathe, V. Raman spectroscopic study of structural transformation in ordered double perovskites La2CoMnO6 bulk and epitaxial film. Solid State Commun. 2015, 224, 10–14. [Google Scholar] [CrossRef]
- Kumar, D.; Kumar, S.; Sathe, V.G. Spin–phonon coupling in ordered double perovskites A2CoMnO6 (A = La, Pr, Nd) probed by micro-Raman spectroscopy. Solid State Commun. 2014, 194, 59–64. [Google Scholar] [CrossRef]
- Filho, R.B.M.; Barbosa, D.A.B.; Reichlova, H.; Marti, X.; de Menezes, A.S.; Ayala, A.P.; Paschol, C.W.A. Role of ra-re-earth ionic radii on the spin–phonon coupling in multiferroic ordered double perovskites. Mater. Res. Express 2015, 2, 075201. [Google Scholar] [CrossRef] [Green Version]
- Meyer, C.; Hühn, S.; Jungbauer, M.; Merten, S.; Damaschke, S.; Samwer, K.; Moshnyaga, V. Tip-enhanced Raman spectros-copy (TERS) on double perovskite La2CoMnO6 thin films: Field enhancement and depolarization effects. J. Raman Spectrosc. 2017, 48, 46–52. [Google Scholar] [CrossRef]
- Xie, C.; Shi, L.; Zhao, J.; Zhou, S.; Li, Y.; Yuan, X. Spin-phonon coupling in R2CoMnO6 (R = Pr, Nd, Sm) thin films under biax-ial compressive strain. J. Appl. Phys. 2016, 120, 155302. [Google Scholar] [CrossRef]
- Truong, K.D.; Singh, M.P.; Jandl, S.; Fournier, P. Influence of Ni/Mn cation order on the spin-phonon coupling in multifunc-tional La2NiMnO6 epitaxial films by polarized Raman spectroscopy. Phys. Rev. B 2009, 80, 134424. [Google Scholar] [CrossRef] [Green Version]
- Truong, K.D.; Laverdière, J.; Singh, M.P.; Jandl, S.; Fournier, P. Impact of Co∕Mn cation ordering on phonon anomalies in La2CoMnO6 double perovskites: Raman spectroscopy. Phys. Rev. B 2007, 76, 132413. [Google Scholar] [CrossRef]
- Kumar, D.; Kumar, S.; Sathe, V.G. Raman studies of ordered double perovskite thin film at high temperatures. NANOFORUM 2014 2015, 140030. [Google Scholar] [CrossRef]
- Jungbauer, M.; Hühn, S.; Egoavil, R.; Tan, H.; Verbeeck, J.; Van Tendeloo, G.; Moshnyaga, V. Atomic layer epitaxy of Rud-dlesden-popper SrO(SrTiO3)n films by means of metalorganic aerosol deposition. Appl. Phys. Lett. 2014, 104, 251603. [Google Scholar] [CrossRef] [Green Version]
- Moshnyaga, V.; Belenchuk, A.; Hühn, S.; Kalkert, C.; Jungbauer, M.; Lebedev, O.I.; Merten, S.; Choi, K.-Y.; Lemmens, P.; Damaschke, B.; et al. Intrinsic antiferromagnetic coupling underlies colossal magnetoresistance effect: Role of corre-lated polarons. Phys. Rev. B 2014, 89, 024420. [Google Scholar] [CrossRef]
- Norton, D.P. Synthesis and properties of epitaxial electronic oxide thin-film materials. Mater. Sci. Eng. R Rep. 2004, 43, 139–247. [Google Scholar] [CrossRef]
- Shannon, R.D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr. 1976, 32, 751–767. [Google Scholar] [CrossRef]
- Zhao, H.J.; Liu, X.Q.; Chen, X.M.; Bellaiche, L. Effects of chemical and hydrostatic pressures on structural, magnetic, and electronic properties of R2NiMnO6 (R = rare−earthion) double perovskites. Phys. Rev. B 2014, 90, 195147. [Google Scholar] [CrossRef]
- Chen, Y.; Peng, A.B.; Wang, B. Raman Spectra and Temperature-Dependent Raman Scattering of Silicon Nanowires. J. Phys. Chem. C 2007, 111, 5855–5858. [Google Scholar] [CrossRef]
- Granado, E.; García, A.; Sanjurjo, J.A.; Rettori, C.; Torriani, I.; Prado, F.; Sánchez, R.D.; Caneiro, A.; Oseroff, S.B. Magnetic ordering effects in the Raman spectra of La1−xMn1−xO3. Phys. Rev. B 1999, 60, 11879–11882. [Google Scholar] [CrossRef] [Green Version]
- Laverdière, J.; Jandl, S.; Mukhin, A.A.; Ivanonv, V.Y.; Ivanov, V.G.; Iliev, M.N. Spin-phonon coupling in orthorhombic RMnO3 (R = Pr, Nd, Sm, Eu, Gd, Tb, Dy, Ho, Y): A Raman study. Phys. Rev. B 2006, 73, 214301. [Google Scholar] [CrossRef]
- Pandey, P.K.; Choudhary, R.J.; Mishra, D.K.; Sathe, V.G.; Phase, D.M. Signature of spin-phonon coupling in Sr2CoO4 thin film: A Raman spectroscopic study. Appl. Phys. Lett. 2013, 102, 142401. [Google Scholar] [CrossRef]
- Jin, X.-W.; Lu, L.; Mi, S.-B.; Liu, M.; Jia, C.-L. Phase stability and B-site ordering in La2NiMnO6 thin films. Appl. Phys. Lett. 2016, 109, 31904. [Google Scholar] [CrossRef] [Green Version]
- Balkanski, M.; Wallis, R.F.; Haro, E. Anharmonic effects in light scattering due to optical phonons in silicon. Phys. Rev. B 1983, 28, 1928–1934. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meyer, C.; Ksoll, P.; Roddatis, V.; Moshnyaga, V. Spin-Phonon Coupling in A2BMnO6 (A = La, Pr, Nd, Sm, Gd; B = Co, Ni) Double-Perovskite Thin Films: Impact of the A-Site Cation Radius. Crystals 2021, 11, 747. https://doi.org/10.3390/cryst11070747
Meyer C, Ksoll P, Roddatis V, Moshnyaga V. Spin-Phonon Coupling in A2BMnO6 (A = La, Pr, Nd, Sm, Gd; B = Co, Ni) Double-Perovskite Thin Films: Impact of the A-Site Cation Radius. Crystals. 2021; 11(7):747. https://doi.org/10.3390/cryst11070747
Chicago/Turabian StyleMeyer, Christoph, Philipp Ksoll, Vladimir Roddatis, and Vasily Moshnyaga. 2021. "Spin-Phonon Coupling in A2BMnO6 (A = La, Pr, Nd, Sm, Gd; B = Co, Ni) Double-Perovskite Thin Films: Impact of the A-Site Cation Radius" Crystals 11, no. 7: 747. https://doi.org/10.3390/cryst11070747
APA StyleMeyer, C., Ksoll, P., Roddatis, V., & Moshnyaga, V. (2021). Spin-Phonon Coupling in A2BMnO6 (A = La, Pr, Nd, Sm, Gd; B = Co, Ni) Double-Perovskite Thin Films: Impact of the A-Site Cation Radius. Crystals, 11(7), 747. https://doi.org/10.3390/cryst11070747