Abstract
Enrofloxacin is a poorly soluble antibacterial drug of the fluoroquinolones class used in veterinary medicine. The main purpose of this work was to investigate the structural and pharmaceutical properties of new enrofloxacin salts. Enrofloxacin anhydrate and its organic salts with tartaric acid, nicotinic acid and suberic acid formed as pure crystalline anhydrous solids. All the crystals were grown from a mixed solution by slow evaporation at room temperature. These products were then characterized by field-emission scanning electron microscopy, powder X-ray diffraction, Fourier transform infrared spectroscopy and differential scanning calorimetry. Further, X-ray single crystal diffraction analysis was used to study the crystal structure. The intermolecular interactions and packing arrangements in the crystal structures were studied, and the solubility of these salts in water was determined using high-performance liquid chromatography. The results show that the new salts of enrofloxacin developed in this study exhibited excellent water solubility.
1. Introduction
Crystal engineering through multicomponent crystals has attracted substantial attention in the pharmaceutical field in recent years [1,2] because of the possibility of improved solubility and bioavailability of newly designed drug compounds (polymorphs, salt and cocrystals) [3,4]. The formation of the salt/cocrystal is based on the crystal engineering concepts [5]. The salt/cocrystal formation provides an enormous scope for the manipulation and modification of crucial pharmaceutical physical properties such as the dissolution rate, solubility, thermodynamic stability and bioavailability [6,7]. Proton transfer is a decisive factor that distinguishes salts from cocrystals: In salt formation, proton transfer and ionization occur, while these do not occur in the formation of cocrystals [8]. Salts and cocrystals have been employed in the pharmaceutical industry because of their excellent solubility [9]. Although cocrystallization has many exciting advantages, salt formation still represents a widely accepted method to obtain higher solubility of the drug [10].
Most of the active pharmaceutical ingredients (APIs) available in the current market cause formulation difficulties because of poor water solubility, which may lead to poor oral bioavailability [11,12]. Hence, improving the solubility and bioavailability of APIs without changing their stability and other characteristics has become a challenging task. Notably, every crystal structure is the result of mutual balance between numerous noncovalent interactions, but the hydrogen bond remains an important factor in supramolecular assembly [13]. The design of supramolecular heterosynthons derived from organic salts was hypothesized on the basis of the supramolecular synthon strategy in the context of crystal engineering [14,15]. Recently, this strategy has been effectively adopted in the field of pharmaceutical crystallization [16].
Enrofloxacin (1-cyclopropyl-7-(4-ethylpiperazin-1-yl)-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylate) shows a wide spectrum of antibacterial activity, and it belongs to the class of fluoroquinolone antibiotics [17]. As an important synthetic bacteriostatic drug, it has been widely used in stock raising. In good clinical trials, its effectiveness in treating uncomplicated and complicated urinary tract infections, urethral and cervical gonococcal infections, respiratory tract infections, and skin and tissue infections has been proven [18]. It exhibits concentration-dependent antibacterial activity [19]. Enrofloxacin exists in a zwitterionic form within a neutral aqueous solution due to the acid/base interaction between the basic nitrogen of the piperazine and the carboxylic acid group [20,21]. Therefore, in water at pH ≈ 7, enrofloxacin exhibits a low solubility (0.45 mg/mL) [21]. In addition, low solubility of enrofloxacin is one of the unfavourable properties in formulation [22]. Therefore, a method to improve its solubility without compromising performance has been sought. A weak organic acid can be used as an organic counterionic component for salt formation [23,24]. These acids can potentially present the formation of salts with multiple stoichiometries due to the containment of carboxylic group. This approach is widely used in pharmaceutical industries to enhance solubility, bioavailability and controlled release of drugs [25]. Therefore, we adopted crystal engineering concepts to select pharmaceutically acceptable organic counterions to form salts with enrofloxacin.
In this study, we prepared enrofloxacin anhydrate and salt trihydrate with tartaric acid and salt solvate with nicotinic acid and suberic acid, and we analysed the crystal structures of these compounds. The obtained crystal compounds were characterized by field-emission scanning electron microscopy (FESEM), Fourier transform infrared spectroscopy (FT-IR), powder X-ray diffraction (PXRD) and differential scanning calorimetry (DSC). All the crystal structure data were successfully resolved by single-crystal X-ray diffraction (SCXRD), and the crystal conformations and packing arrangements were studied in detail. Finally, the solubility of the new phases in water was also determined by high-performance liquid chromatography (HPLC). The new salts were found to exhibit significantly improved solubility and were therefore suitable for use in drug formulation.
2. Experimental
2.1. Materials
Enrofloxacin (C19H22FN3O3, 98%) was obtained from Nanjing Kangmanlin Biomedical Technology Co. Ltd. (Nanjing, China). These organic acids (tartaric acid (C4H6O6,99.5%), nicotinic acid (C6H5NO2,99.5%), suberic acid (C6H10O4,99%)) were purchased from Shanghai Macklin Biochemical Co., Ltd. (Shanghai, China). The chromatography grade organic solvents (methanol (MeOH), acetonitrile (ACN), triethylamine (TEA), phosphate) were purchased from Tianjin Kemio Chemical Reagent Co., Ltd. (Tianjin, China). All of the analytical-grade organic solvents (ethanol (EtOH), dichloromethane (DCM), N,N-Dimethylformamide (DMF)) were obtained from Tianjin Damao Chemical Reagent Factory (Tianjin, China) and these were used without further purification. Purified water was prepared using the Millipore Milli-Q system.
2.2. Crystallization of Enrofloxacin Anhydrate
Enrofloxacin Anhydrate (1): Enrofloxacin (230 mg, 0.64 mmol) was added to 4 mL of an ethanol/dichloromethane mixed solvent (1:1 v/v) for crystallization. The 1 crystals in the suspension were then filtered and dried in air (yield: 77%).
2.3. Preparation of Salts and Crystallization
Enrofloxacin and various acids in equal molar ratios were dissolved in a mixed solvent in a 50-mL conical flask, and this mixture was stirred at 50 °C until a completely clear solution was obtained. The solution was then allowed to slowly evaporate at room temperature to obtain single crystals of the products. Diffraction-quality crystals were obtained within 1–5 days. Each sample was scaled up 20 times for the solubility determination.
Enrofloxacin Tartrate Trihydrate (2): A 1:1 mixture of enrofloxacin (460 mg, 1.28 mmol) and tartaric acid (180 mg, 1.28 mmol) was added to 12 mL of a water/ethanol/dichloromethane mixed solvent (4:7:3 v/v/v) for crystallization. Then, 2 was obtained by filtration and drying in air (yield: 76%).
Enrofloxacin Nicotinat-EtOH Salt Solvate (3): A 1:1 mixture of enrofloxacin (230 mg, 0.64 mmol) and nicotinic acid (83.7 mg, 0.64 mmol) was added to 5 mL of an ethanol/dichloromethane mixed solvent (3:2 v/v) for crystallization. The resulting 3 crystals were filtered and dried in air (yield: 86%).
Enrofloxacin Suberate-2EtOH Salt Solvate (4): A 1:1 mixture of enrofloxacin (230 mg, 0.64 mmol) and suberic acid (110 mg, 0.64 mmol) was added to 4 mL of an ethanol/dichloromethane mixed solvent (1:1 v/v) for crystallization. Crystals were then obtained by filtration and drying (yield: 77%).
2.4. Field-Emission Scanning Electron Microscopy (FESEM)
The surface morphology of the samples was investigated by FESEM (Heidelberg, Germany) using a Zeiss Sigma 300.
2.5. Powder X-ray Diffraction (PXRD)
PXRD measurements of the new compounds were carried out on a Rigaku-Ultima IV X-ray powder diffractometer (Tokyo, Japan) using Cu Kα radiation (λ = 1.54178 Å) at 40 kV and 40 mA. Samples were analysed in the 2θ range from 5° to 80° with a scanning rate of 8°/min. These data were collected at room temperature and analysed using Jade 6.0 software (Livermore, CA, USA).
2.6. Fourier Transform Infrared (FT-IR)
FT–IR spectra were collected using a Bruker Vertex 70 spectrometer and measured in the range of 4000−400 cm−1 with an RT-DLaTGS detector (Ettlingen, Germany). The KBr diffuse-reflectance mode was used (sample concentration: 1 mg in 100 mg of KBr) for recording the IR spectra of these samples. Data were analysed using the OPUS software (San Francisco, CA, USA).
2.7. Single Crystal X-ray Diffraction (SCXRD)
X-ray diffraction data were collected using a Rigaku Oxford Diffraction SuperNova diffractometer (Oxford, UK) equipped with a monochromator mirror for Cu Kα (λ = 1.54184 Å) radiation at 150 K. Data reduction was carried out using the CrysAlisPro software, and absorption correction was implemented in the SCALE3 ABSPACK scaling algorithm. The structure was solved (direct methods) and refined (using least squares minimization) with Olex2 [26] using SHELX [27] structure solution programs. All the non-H atoms were refined anisotropically. In addition, all the figures, including the packing and molecular structure diagrams, were drawn using Olex2 and PLATON [28]. Table 1 gives the pertinent crystal lographic data, and Table 2 gives the hydrogen-bond parameters.

Table 1.
Crystallographic Data for Compounds 1–4.
Table 2.
Hydrogen Bond Geometry Parameters in Compounds 1–4.
2.8. Differential Scanning Calorimetry (DSC)
Thermal analyses of these samples were performed on a DSC (Q200 V24.10 Build 122) instrument (New Castle, DE, USA): 2–3 mg of the crystals was placed in standard aluminium pans and scanned at 20 °C/min in the range 25–280 °C under a nitrogen gas flow of 50 mL/min. Data were analysed using the Universal Analysis 2000 software (New Castle, DE, USA).
2.9. Solubility Analyses
The solubility of these products was determined according to the shake-flask method [29]. Excess amounts (100 mg) of the crystals were added to the screw-capped glass vials containing 2.5 mL of ultrapure water, and the resulting suspensions was shaken at room temperature. After 24 h, the suspensions were filtered through 0.22-μm polycarbonate filters, and the compounds’ concentrations were determined using the Agilent 1260 Infinity II HPLC system (Palo Alto, CA, USA) equipped with a 1260 Multi λ Fluorescence detector with the wavelength of the excited and emitted spectra of crystals being 280 nm and 450 nm, respectively. The C18 HPLC column (Poroshell 120 EC-C18, 4.6 mm × 100 mm, 2.7 μm) was employed, and acetonitrile, methanol and triethylamine phosphate (pH = 2.5) were used as the mobile phase (1:24:75, v:v::v) with a flow rate of 1 mL/min. Each solubility test was performed in triplicate.
3. Results and Discussion
3.1. Field-Emission Scanning Electron Microscopy (FESEM)
The FESEM (Heidelberg, Germany) images provide information about the crystal morphology of several new crystals (Figure 1). Needle-shaped or long rod-shaped forms are seen for the products 1 and 2, whereas 3 and 4 displayed rectangular blocks and fragmentary crystals.

Figure 1.
Field-emission scanning electron microscopy (FESEM) photos of compounds 1–4.
3.2. PXRD Analysis
PXRD enables the study and characterization of novel crystalline materials [30]. PXRD patterns of the four crystal samples are shown in Figure 2. The patterns for 1 were different from those of the starting material and from the previously published PXRD patterns for the polymorphic form [31]. The purity of all products was confirmed by comparing the PXRD patterns with the simulated patterns obtained from the single crystal data (see the Supplementary Materials, Figures S1−S4). No apparent peaks corresponding to impurities were observed, so the obtained powder compounds were considered to be of high chemical purity. Further, these PXRD patterns of enrofloxacin compounds 2–4 differ significantly from the patterns of individual APIs, proving the formation of the new crystalline phases.
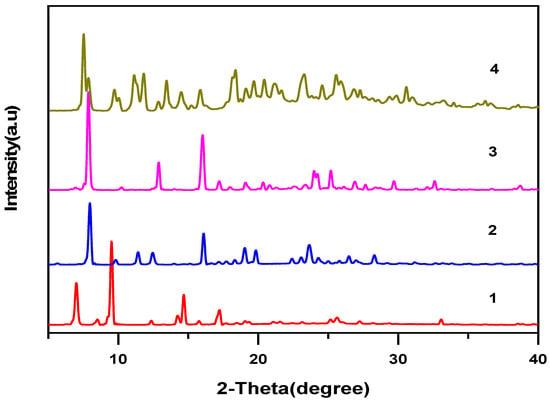
Figure 2.
Powder X-ray diffraction (PXRD) patterns of compounds 1–4.
3.3. Spectroscopic Characterization
The FTIR analysis results for the products are given in Figure 3. The absorption band of crystalline enrofloxacin was located at 1737 cm−1 because of the carbonyl stretching of its unionized carboxylic acid C=O group [31]. The C=O band in 1 was at 1736.4 cm−1, which indicates that the enrofloxacin exists as a neutral molecule. Slight peak shifts were also seen in compounds 2–4, in particular, for the carboxylic acid C=O stretching, which shifted to 1728.8 cm−1, 1728.5 cm−1 and 1729.3 cm−1, respectively. This can be attributed to intermolecular interactions such as the formation of hydrogen bonds [32]. The terminal amino group of the piperazine ring was protonated in the process of crystallization, and this is proved by the presence of broad bands from 2600 cm−1 to 3000 cm−1. However, confirming this is difficult because the broad IR absorption bands possibly overlap with others, e.g., in the case of the C−H stretching [33]. However, Karanam et al. reported FTIR analysis results that showed that enrofloxacin salts were protonated in the process of crystallization [34]. The medium-intensity broad band in the region of 3300–3500 cm−1 is attributed to the O−H stretching of the water molecule 2 and ethanol molecule 3–4. The existence of enrofloxacin as a neutral molecular and in the ionic state in the crystal structures was confirmed by SCXRD analysis.
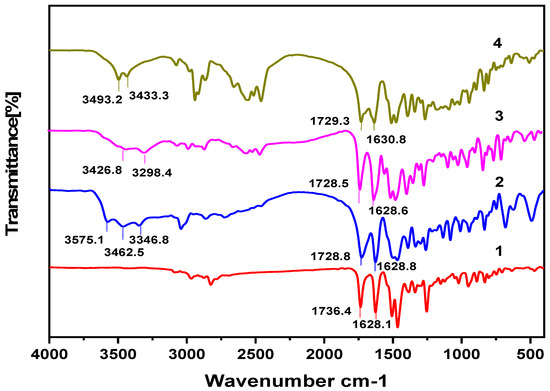
Figure 3.
Fourier transform infrared (FTIR) spectrum of compounds 1–4.
3.4. Crystal Structure Analysis
3.4.1. Crystallization of Enrofloxacin Anhydrate (1)
Single crystals suitable for X-ray diffraction were obtained to determine the structures of the anhydrous form with a neutral molecule (no proton transfer from carboxylic acid to piperazinyl N atom; Figure 4a). The enrofloxacin anhydrate crystallized in the monoclinic P21/n space group with one molecule in the asymmetric unit. In addition, the piperazine ring in enrofloxacin usually exhibits a stable chair conformation with a torsion angle of C12–N2–C15–C16 = 158.25°. It has the same crystal structure compared with the previous published [34]. The carboxylic acid group participated in intramolecular O−H···O hydrogen bonding with the carbonyl oxygen atom of the quinolone moiety. The crystal structure was stacked with π···π (3.598 Å) interactions and was further stabilized by weak C−H···O and C−H···F hydrogen bonds Table 2 in enrofloxacin (Figure 4b).
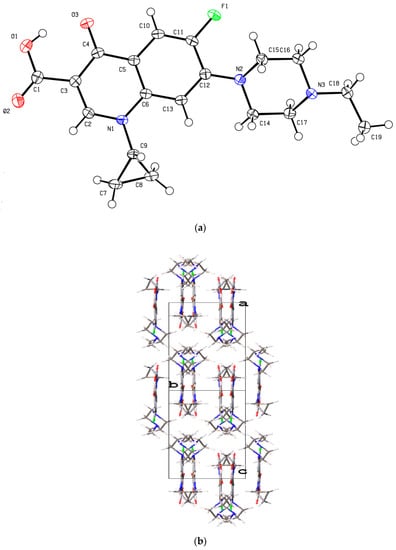
Figure 4.
(a) The molecular structure diagram of the product 1 (ellipsoids were drawn at the 50% probability level). (b) Molecular packing projections for product 1 along the [101] direction.
3.4.2. Enrofloxacin Tartrate Trihydrate (2)
The novel enrofloxacin salt trihydrate 2 crystallized in the triclinic space group. The 1:1 salt structure of enrofloxacin tartrate trihydrate contains an enrofloxacin cation, a tartrate anion and three H2O molecules in the asymmetric unit (Figure 5a). One of the carboxylic acid groups of tartaric acid transferred one proton to the piperazinyl-ring N atom of the enrofloxacin molecule, resulting in a tartrate anion and enrofloxacin cation in the crystal structure (Figure 5b). The carboxylic acid group of enrofloxacin was involved in intramolecular O−H···O hydrogen bonding with the quinolone oxygen atom. The product 2 displayed a unique hydrogen-bonding pattern with the formation of multicomponent crystals. The enrofloxacin cation interacted with the tartrate ion via N+−H···O interactions to form the crystal structure, rather than forming hydrogen bonds with ionized oxygen atoms. Further, one of the water molecules connected the enrofloxacin cation and tartrate anion via O−H···O interactions (Figure 5c).
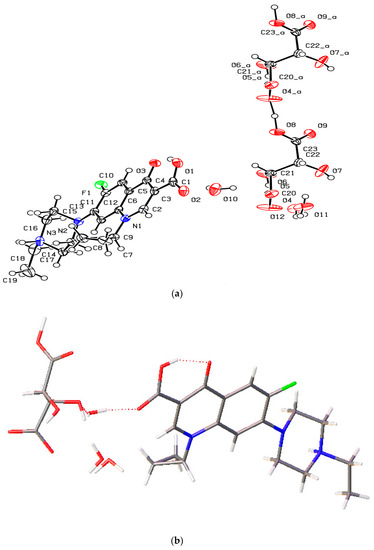
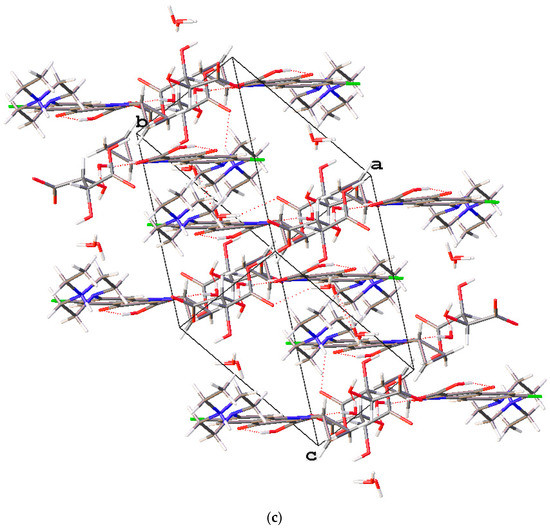
Figure 5.
(a) The molecular structure diagram of the salt trihydrate 2 (ellipsoids were drawn at the 50% probability level). (b) Interaction between enrofloxacin and tartaric acid molecules in the crystal via O−H···O hydrogen bonds. (c) Molecular packing projections for salt trihydrate 2 along the [111] direction.
3.4.3. Enrofloxacin Nicotinate-EtOH Salt Solvate (3)
The novel enrofloxacin salt solvate 3 had a triclinic system and space group . Its crystal contained an enrofloxacin cation, a nicotinate anion and an EtOH molecule in the asymmetric unit (Figure 6a). The carboxylic acid group of 3 was involved in intramolecular O−H···O hydrogen bonding with the quinolone oxygen atom. The nicotinic acid was ionized by proton transfer to the enrofloxacin molecule to form N+−H···O−, while the EtOH molecule formed the O−H···O hydrogen bond with the carboxylic acid C=O group of nicotinate (Figure 6b). The quinolone moieties of the enrofloxacin molecules stacked via π···π (3.538 Å) interactions (Figure 6c).
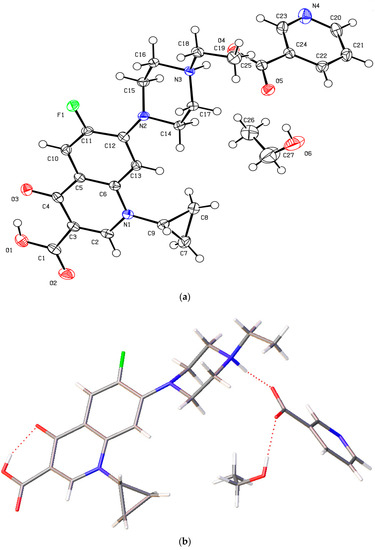
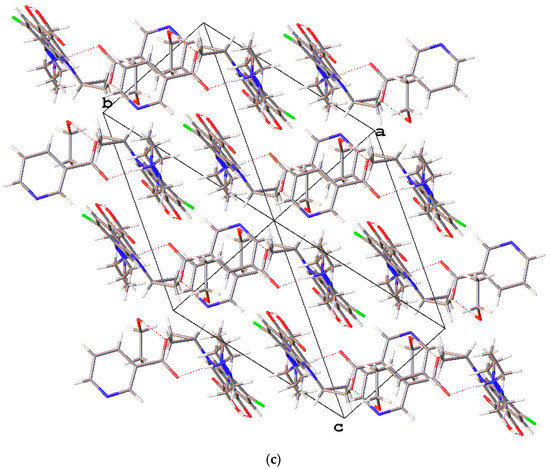
Figure 6.
(a) The molecular structure diagram of the salt solvate 3 (ellipsoids were drawn at the 50% probability level). (b) Interaction between enrofloxacin and nicotinic acid molecules in the crystal via N+−H···O− and O−H···O hydrogen bonds. (c) Molecular packing projections for salt solvate 3 along the [111] direction.
3.4.4. Enrofloxacin Suberate-2EtOH Salt Solvate (4)
The novel enrofloxacin salt solvate 4 had a triclinic system and space group with an enrofloxacin cation, a suberate anion and two EtOH molecules in the asymmetric unit. The carboxylic acid and cyclopropyl groups of the enrofloxacin molecule were observed to exhibit disorder in this crystal structure (Figure 7a). One of the carboxylic acid groups of the suberic acid transferred one proton to the piperazinyl-ring N atom of the enrofloxacin molecule, thereby forming a suberate anion and enrofloxacin cations in the crystal structure (Figure 7b). The carboxylic acid group of 4 was involved in intramolecular O−H···O hydrogen bonding with the quinolone oxygen atom. Enrofloxacin interactsedwith the suberate ion via N+−H···O− interactions in the crystal structure, while the EtOH molecule formed the O−H···O hydrogen bond with the carboxylic acid C=O group of the suberate ion. The quinolone moieties of the enrofloxacin molecule stacked via π···π (3.872 Å) interactions (Figure 7c).
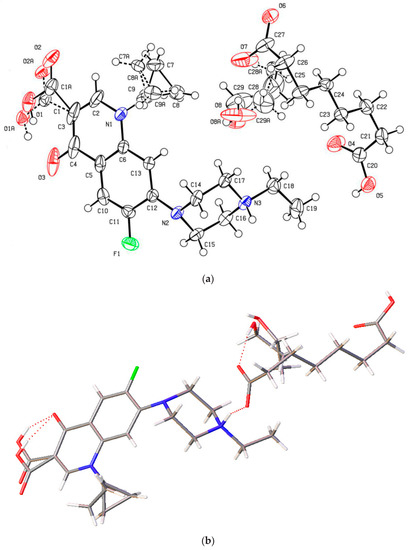
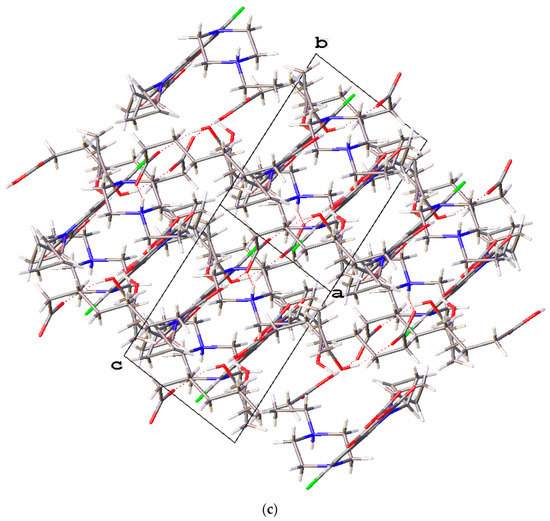
Figure 7.
(a) The molecular structure diagram of the salt solvate 4 (ellipsoids were drawn at the 50% probability level). (b) Interaction between enrofloxacin and suberic acid molecules in the crystal by N+−H···O− and O−H···O hydrogen bonds. (c) Molecular packing projections for salt solvate 4 along the [011] direction.
3.5. Thermal Analysis
DSC curves showing the thermal behaviour of the products 1–4 are shown in Figure 8. The endothermic peak for melting of 1 was found at 228 °C. In the case of 2, the endothermic melting peak was at 228 °C, and this was followed by a phase transition. The endothermic transitions between 75 °C and 125 °C in the DSC thermograms for 2 showed the loss of water molecules from the crystal structure. In the DSC thermogram for 3, several steps of small endothermic transitions at about 120–180 °C due to solvent loss were observed, followed by a four-step endothermic melting transition of the salt solvate Further, 4 was found to melt at 112 °C.
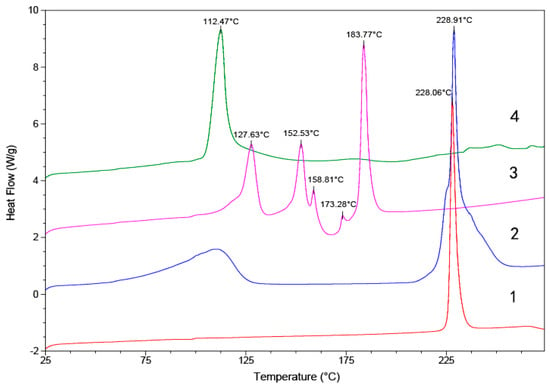
Figure 8.
Differential scanning calorimetry (DSC) diagrams of compounds 1–4.
3.6. Results for Solubility Study
The solubility data are presented in Table 3. The solubility of commercially available enrofloxacin in water was 0.14 mg/mL, which is similar to the previously reported values [21]. The solubilities of 1 and the commercially available enrofloxacin are similar. As expected, the enrofloxacin salts showed a considerable improvement in solubility: 2–4 were found to be 57- to 406-times more soluble than pure enrofloxacin. Correlating the solubility with crystal structures is difficult because the limited experimental/calculated data on crystal packing, etc. However, it can be speculated that the solubility enhancement in the case of the salts was a result of greater ionization.
Table 3.
Solubility Data for Compounds 1–4.
4. Discussion
Enrofloxacin anhydrate was crystallized, and the crystal structure was determined. Further, new salts formed using tartaric acid, nicotinic acid and suberic acid have been reported for the first time. The novel salts were prepared efficiently via evaporation using a mixed solvent has been found to be highly rewarding. These compounds formed a layered structure, and these layers were stacked via hydrogen bonds and π···π interactions. In the structure of salts, the piperazinyl moieties of enrofloxacin interacted with the carboxylate ions. These carboxylate ions connected the H2O and EtOH molecules formed a stacking structure. The enrofloxacin salts prepared in this study showed a 57- to 406-fold higher solubility than the starting material.
Supplementary Materials
The following are available online at https://www.mdpi.com/2073-4352/10/8/646/s1. The PXRD patterns with the simulated patterns obtained from the single crystal data.CIF files giving crystal data can be obtained free of charge from the Cambridge Crystallographic Data Centre via the Internet www.ccdc.cam.ac.uk/data_request/cif.
Author Contributions
Writing–original draft, H.P.; Conceptualization, Y.-B.S.; Formal analysis, Y.Y., and L.-Z.C.; Funding acquisition, B.-H.F.; Investigation, J.-W.Z.; Methodology, M.-J.X.; Software, H.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Natural Science Foundation of China (No. 31872522).
Conflicts of Interest
There are no conflict to declare.
References
- Vishweshwar, P.; McMahon, J.A.; Peterson, M.L.; Hickey, M.B.; Shattock, T.R.; Zaworotko, M.J. Crystal engineering of pharmaceutical co-crystals from polymorphic active pharmaceutical ingredients. Chem. Commun. 2005, 36, 4601. [Google Scholar] [CrossRef] [PubMed]
- Maeno, Y.; Fukami, T.; Kawahata, M.; Yamaguchi, K.; Tagami, T.; Ozeki, T.; Suzuki, T.; Tomono, K. Novel pharmaceutical cocrystal consisting of paracetamol and trimethylglycine, a new promising cocrystal former. Int. J. Pharmaceut. 2014, 473, 179–186. [Google Scholar] [CrossRef]
- Blagden, N.; de Matas, M.; Gavan, P.T.; York, P. Crystal engineering of active pharmaceutical ingredients to improve solubility and dissolution rates. Adv. Drug Deliv. Rev. 2007, 59, 617–630. [Google Scholar] [CrossRef] [PubMed]
- McNamara, D.P.; Childs, S.L.; Giordano, J.; Iarriccio, A.; Cassidy, J.; Shet, M.S.; Mannion, R.; O’Donnell, E.; Park, A. Use of a Glutaric Acid Cocrystal to Improve Oral Bioavailability of a Low Solubility API. Pharm. Res. Dordr. 2006, 23, 1888–1897. [Google Scholar] [CrossRef]
- Jones, W.; Motherwell, W.D.S.; Trask, A.V. Pharmaceutical Cocrystals: An Emerging Approach to Physical Property Enhancement. MRS Bull. 2006, 11, 875–879. [Google Scholar] [CrossRef]
- Thakuria, R.; Delori, A.; Jones, W.; Lipert, M.P.; Roy, L.; Rodríguez-Hornedo, N. Pharmaceutical cocrystals and poorly soluble drugs. Int. J. Pharm. 2013, 453, 101–125. [Google Scholar] [CrossRef] [PubMed]
- Darwish, S.; Zeglinski, J.; Krishna, G.R.; Shaikh, R.; Khraisheh, M.; Walker, G.M.; Croker, D.M. A New 1:1 Drug-Drug Cocrystal of Theophylline and Aspirin: Discovery, Characterization, and Construction of Ternary Phase Diagrams. Cryst. Growth Des. 2018, 18, 7526–7532. [Google Scholar] [CrossRef]
- Aakeröy, C.B.; Fasulo, M.E.; Desper, J. Cocrystal or Salt: Does It Really Matter? Mol. Pharm. 2007, 4, 317–322. [Google Scholar] [CrossRef]
- Basavoju, S.; Boström, D.; Velaga, S.P. Pharmaceutical Cocrystal and Salts of Norfloxacin. Cryst. Growth Des. 2006, 6, 2699–2708. [Google Scholar] [CrossRef]
- Berge, S.M.; Bighley, L.D.; Monkhouse, D.C. Pharmaceutical salts. J. Pharm. Sci. 1977, 66, 1–19. [Google Scholar] [CrossRef]
- Datta, S.; Grant, D.J.W. Crystal structures of drugs: Advances in determination, prediction and engineering. Nat. Rev. Drug Discov. 2004, 3, 42–57. [Google Scholar] [CrossRef] [PubMed]
- Meyer, M.C. Drug Product Selection—Part 2: Scientific basis of bioavailability and bioequivalence testing. Am. Pharm. 1991, NS31, 47–52. [Google Scholar] [CrossRef]
- Fleischman, S.G.; Kuduva, S.S.; McMahon, J.A.; Moulton, B.; Bailey Walsh, R.D.; Rodríguez-Hornedo, N.; Zaworotko, M.J. Crystal Engineering of the Composition of Pharmaceutical Phases: Multiple-Component Crystalline Solids Involving Carbamazepine. Cryst. Growth Des. 2003, 3, 909–919. [Google Scholar] [CrossRef]
- Childs, S.L.; Chyall, L.J.; Dunlap, J.T.; Smolenskaya, V.N.; Stahly, B.C.; Stahly, G.P. Crystal Engineering Approach to Forming Cocrystals of Amine Hydrochlorides with Organic Acids. Molecular Complexes of Fluoxetine Hydrochloride with Benzoic, Succinic, and Fumaric Acids. J. Am. Chem. Soc. 2004, 126, 13335–13342. [Google Scholar] [CrossRef]
- Bis, J.A.; Zaworotko, M.J. The 2-Aminopyridinium-carboxylate Supramolecular Heterosynthon: A Robust Motif for Generation of Multiple-Component Crystals. Cryst. Growth Des. 2005, 5, 1169–1179. [Google Scholar] [CrossRef]
- Douroumis, D.; Ross, S.A.; Nokhodchi, A. Advanced methodologies for cocrystal synthesis. Adv. Drug Deliv. Rev. 2017, 117, 178–195. [Google Scholar] [CrossRef]
- Efthimiadou, E.K.; Katsaros, N.; Karaliota, A.; Psomas, G. Synthesis, characterization, antibacterial activity, and interaction with DNA of the vanadyl-enrofloxacin complex. Bioorg. Med. Chem. Lett. 2007, 17, 1238–1242. [Google Scholar] [CrossRef]
- Fan, J.; Sun, D.; Yu, H.; Kerwin, S.M.; Hurley, L.H. Self-Assembly of a Quinobenzoxazine-Mg2+ Complex on DNA: A New Paradigm for the Structure of a Drug-DNA Complex and Implications for the Structure of the Quinolone Bacterial Gyrase-DNA Complex. J. Med. Chem. 1995, 38, 408–424. [Google Scholar] [CrossRef]
- TerHune, T.N.; Skogerboe, T.L.; Shostrom, V.K.; Weigel, D.J. Comparison of pharmacokinetics of danofloxacin and enrofloxacin in calves challenged with Mannheimia haemolytica. Am. J. Vet. Res. 2005, 66, 342–349. [Google Scholar] [CrossRef]
- Barrón, D.; Irles, A.; Barbosa, J. Prediction of electrophoretic mobilities in non-aqueous capillary electrophoresis: Optimal separation of quinolones in acetonitrile–water media. J. Chromatogr. A 2000, 871, 367–380. [Google Scholar] [CrossRef]
- Lizondo, M.; Pons, M.; Gallardo, M.; Estelrich, J. Physicochemical properties of enrofloxacin. J. Pharm. Biomed. 1997, 15, 1845–1849. [Google Scholar] [CrossRef]
- Romañuk, C.B.; Manzo, R.H.; Linck, Y.G.; Chattah, A.K.; Monti, G.A.; Olivera, M.E. Characterization of the Solubility and Solid-State Properties of Saccharin Salts of Fluoroquinolones. J. Pharm. Sci. 2009, 10, 3788–3801. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Jiang, L.; Mei, X. Supramolecular structures and physicochemical properties of norfloxacin salts. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2014, 70, 750–760. [Google Scholar] [CrossRef] [PubMed]
- Surov, A.O.; Manin, A.N.; Voronin, A.P.; Drozd, K.V.; Simagina, A.A.; Churakov, A.V.; Perlovich, G.L. Pharmaceutical salts of ciprofloxacin with dicarboxylic acids. Eur. J. Pharm. Sci. 2015, 77, 112–121. [Google Scholar] [CrossRef]
- Banerjee, R.; Bhatt, P.M.; Ravindra, N.V.; Desiraju, G.R. Saccharin Salts of Active Pharmaceutical Ingredients, Their Crystal Structures, and Increased Water Solubilities. Cryst. Growth Des. 2005, 5, 2299–2309. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT-Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Spek, A.L. Structure validation in chemical crystallography. Acta Cryst. 2009, D65, 148–155. [Google Scholar]
- Glomme, A.; März, J.; Dressman, J.B. Comparison of a Miniaturized Shake-Flask Solubility Method with Automated Potentiometric Acid/Base Titrations and Calculated Solubilities. J. Pharm. Sci. 2005, 94, 1–16. [Google Scholar] [CrossRef]
- Shaibat, M.A.; Casabianca, L.B.; Wickramasinghe, N.P.; Guggenheim, S.; de Dios, A.C.; Ishii, Y. Characterization of Polymorphs and Solid-State Reactions for Paramagnetic Systems by13 C Solid-State NMR and ab Initio Calculations. J. Am. Chem. Soc. 2007, 129, 10968–10969. [Google Scholar] [CrossRef]
- Mesallati, H.; Umerska, A.; Tajber, L. Fluoroquinolone Amorphous Polymeric Salts and Dispersions for Veterinary Uses. Pharmaceutics 2019, 11, 268. [Google Scholar] [CrossRef] [PubMed]
- Löbmann, K.; Laitinen, R.; Grohganz, H.; Strachan, C.; Rades, T.; Gordon, K.C. A theoretical and spectroscopic study of co-amorphous naproxen and indomethacin. Int. J. Pharm. 2013, 453, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Gunasekaran, S.; Anita, B. Spectral investigation and normal coordinate analysis of piperazine. Indian J. Pure Appl. Phys. 2008, 46, 833–838. [Google Scholar]
- Karanam, M.; Choudhury, A.R. Structural Landscape of Pure Enrofloxacin and Its Novel Salts: Enhanced Solubility for Better Pharmaceutical Applicability. Cryst. Growth Des. 2013, 13, 1626–1637. [Google Scholar] [CrossRef]











© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).