N-Acetyl Indole Linked to a Fused Triazolo/Thiadiazole Scaffold: Synthesis, Single Crystal X-Ray Structure, and Molecular Insight


 ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. General
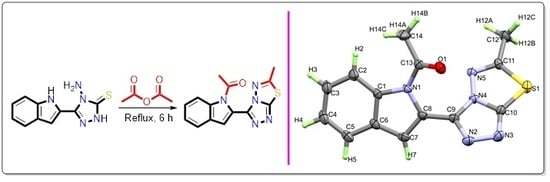
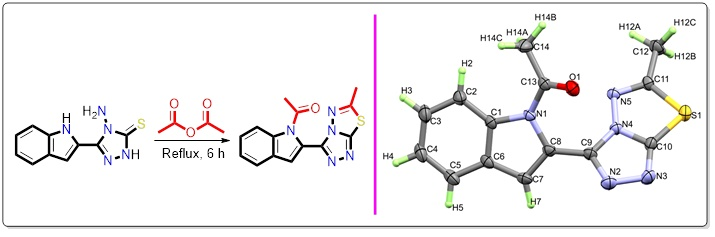
2.2. Synthesis of 1-(2-(6-methyl-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazol-3-yl)-1H-indol-1-yl)ethan-1-one
2.3. Single-Crystal X-Ray Diffraction Analysis
2.4. Computational Methods
3. Results and Discussion
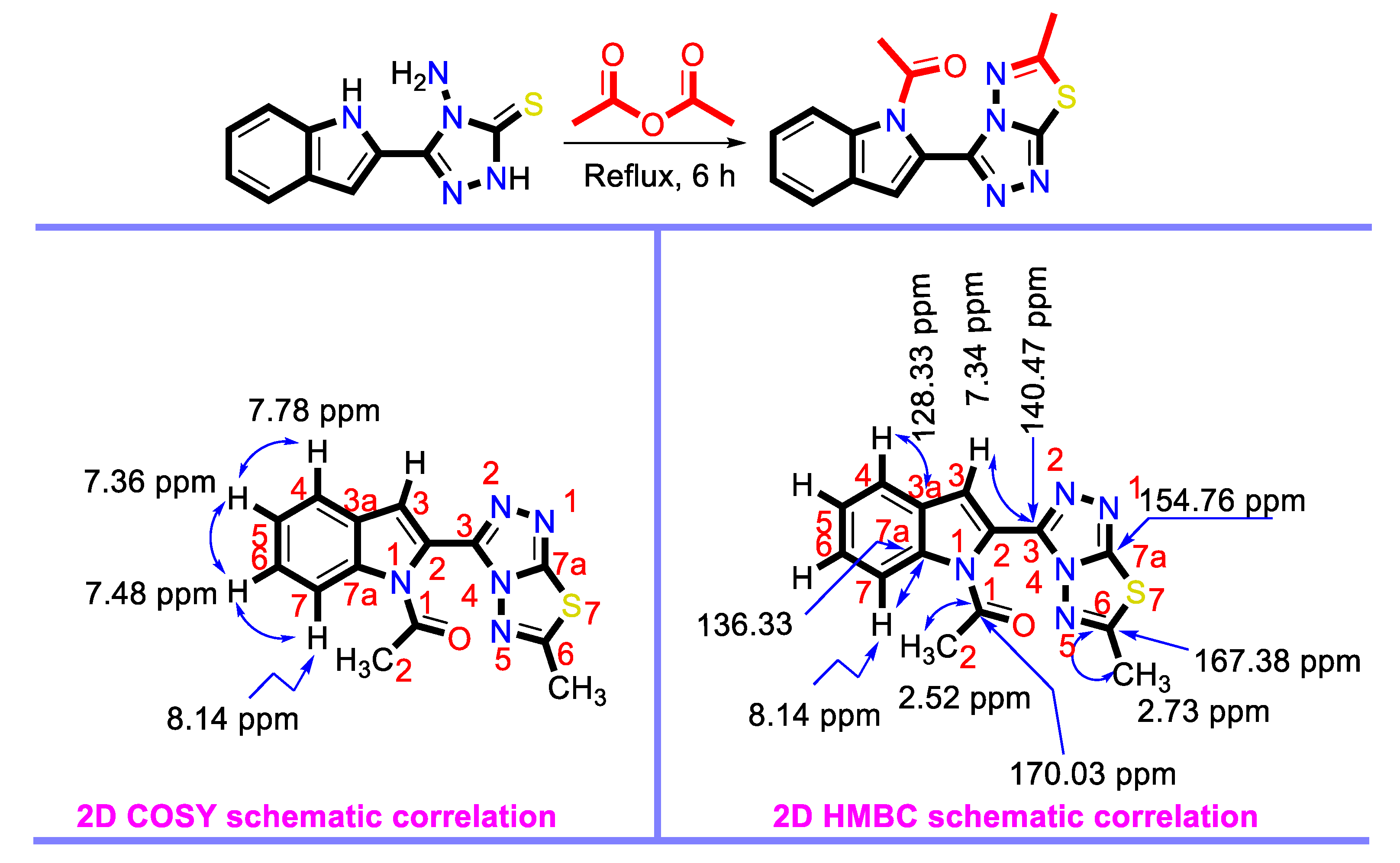
3.1. Synthesis of the Target Compound
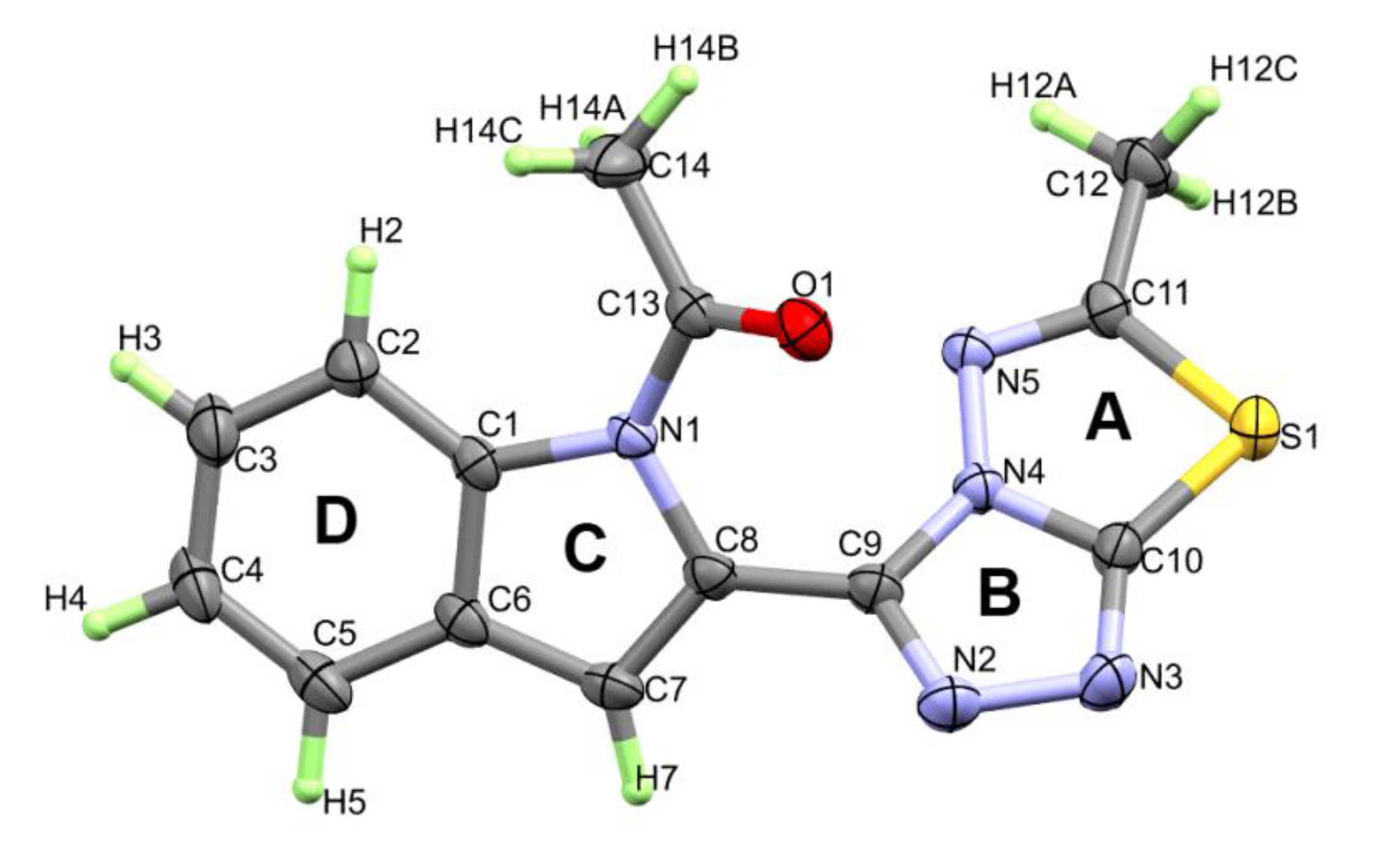
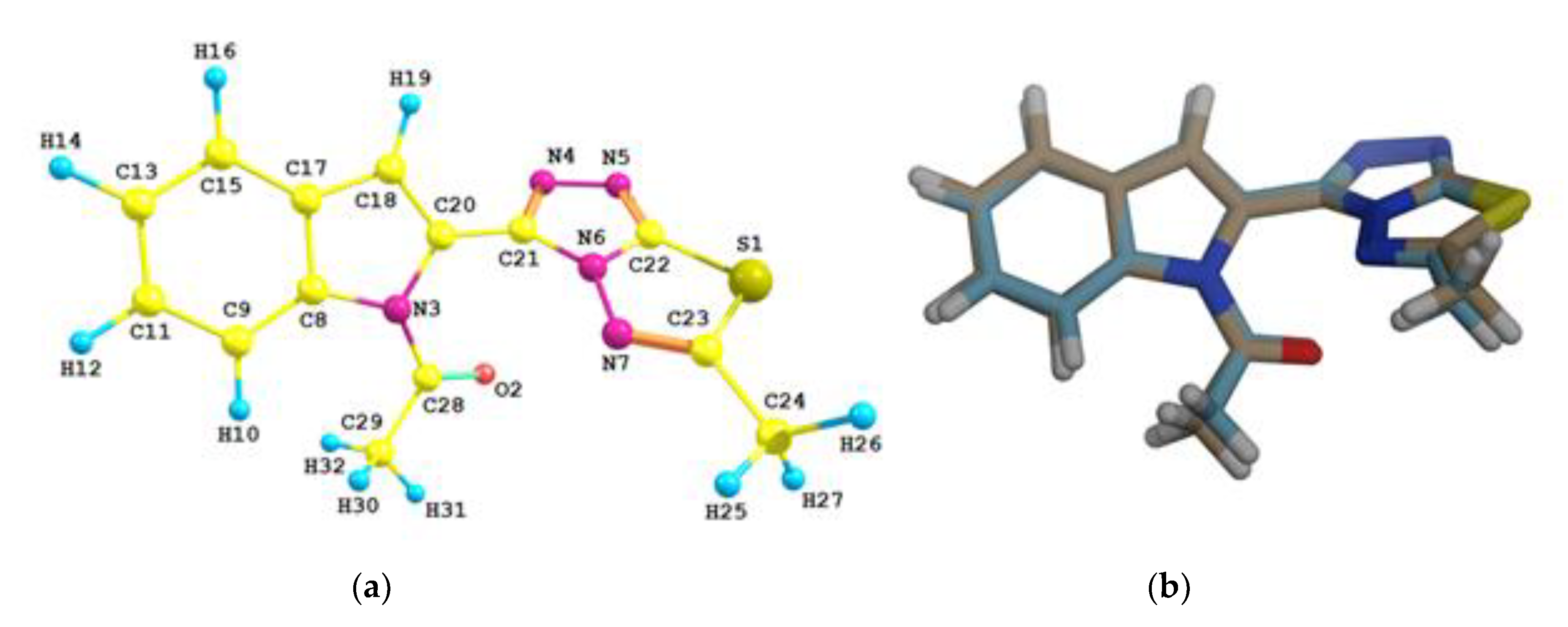
3.2. Structural Features of the Target Compound
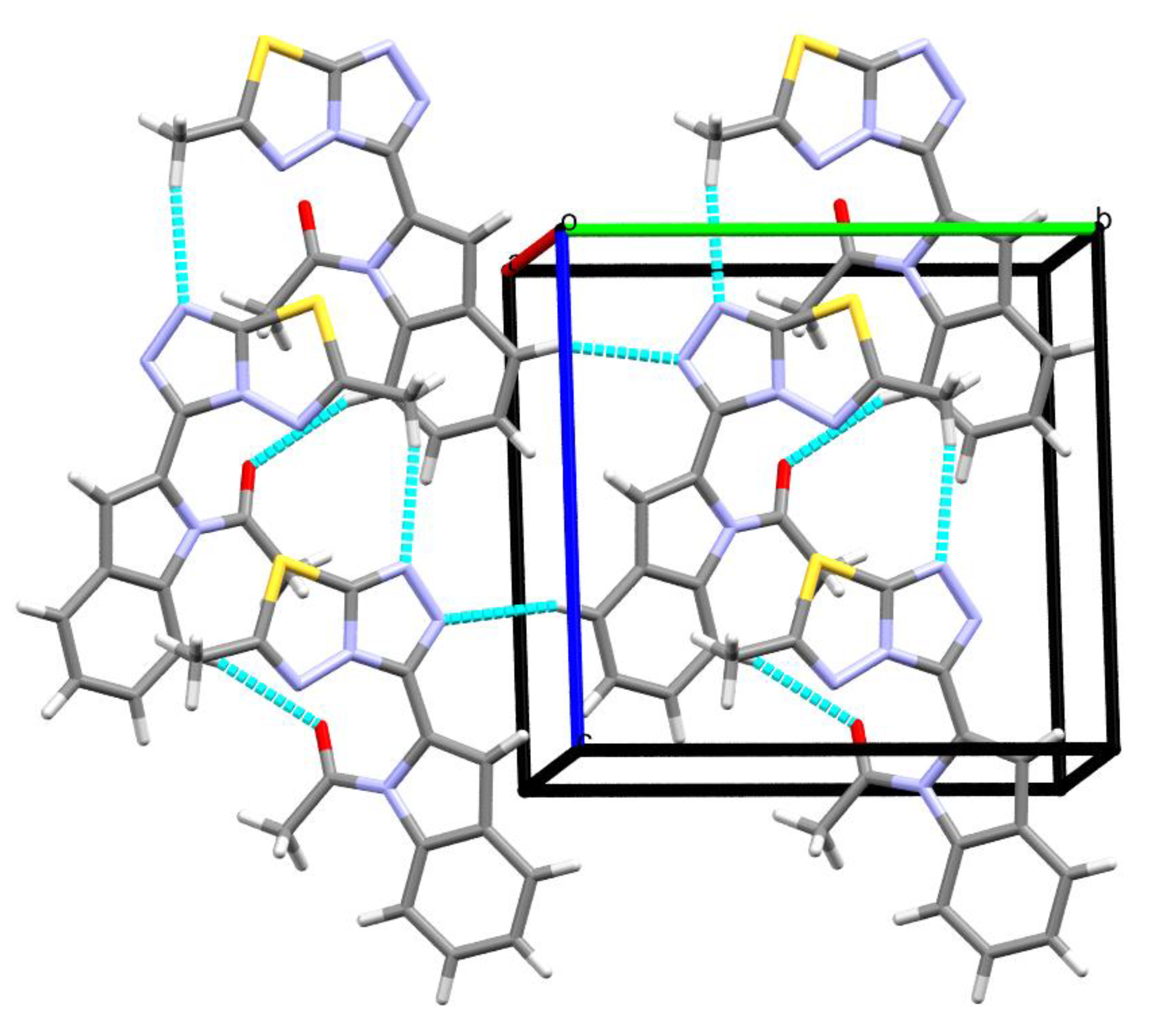
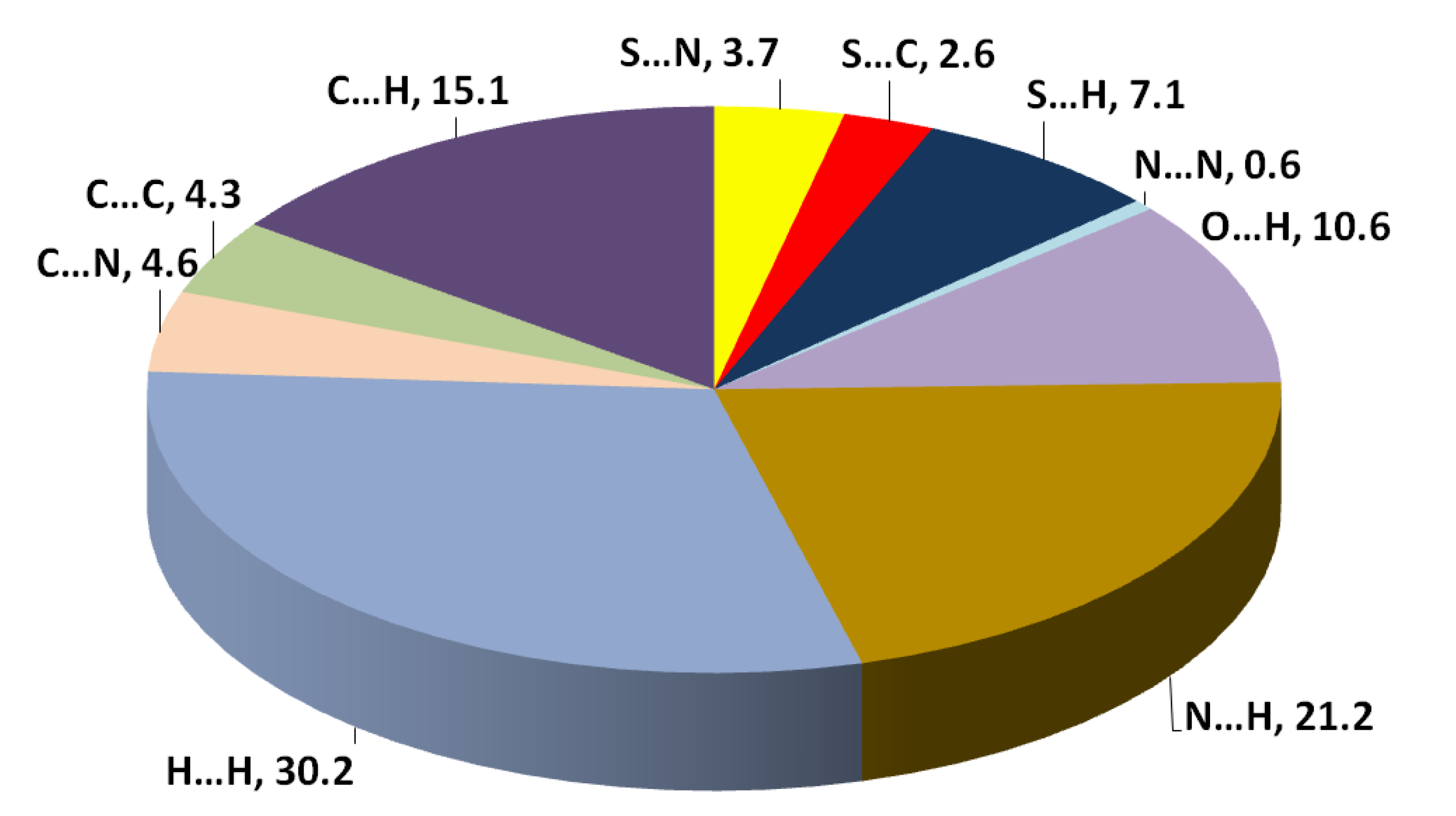
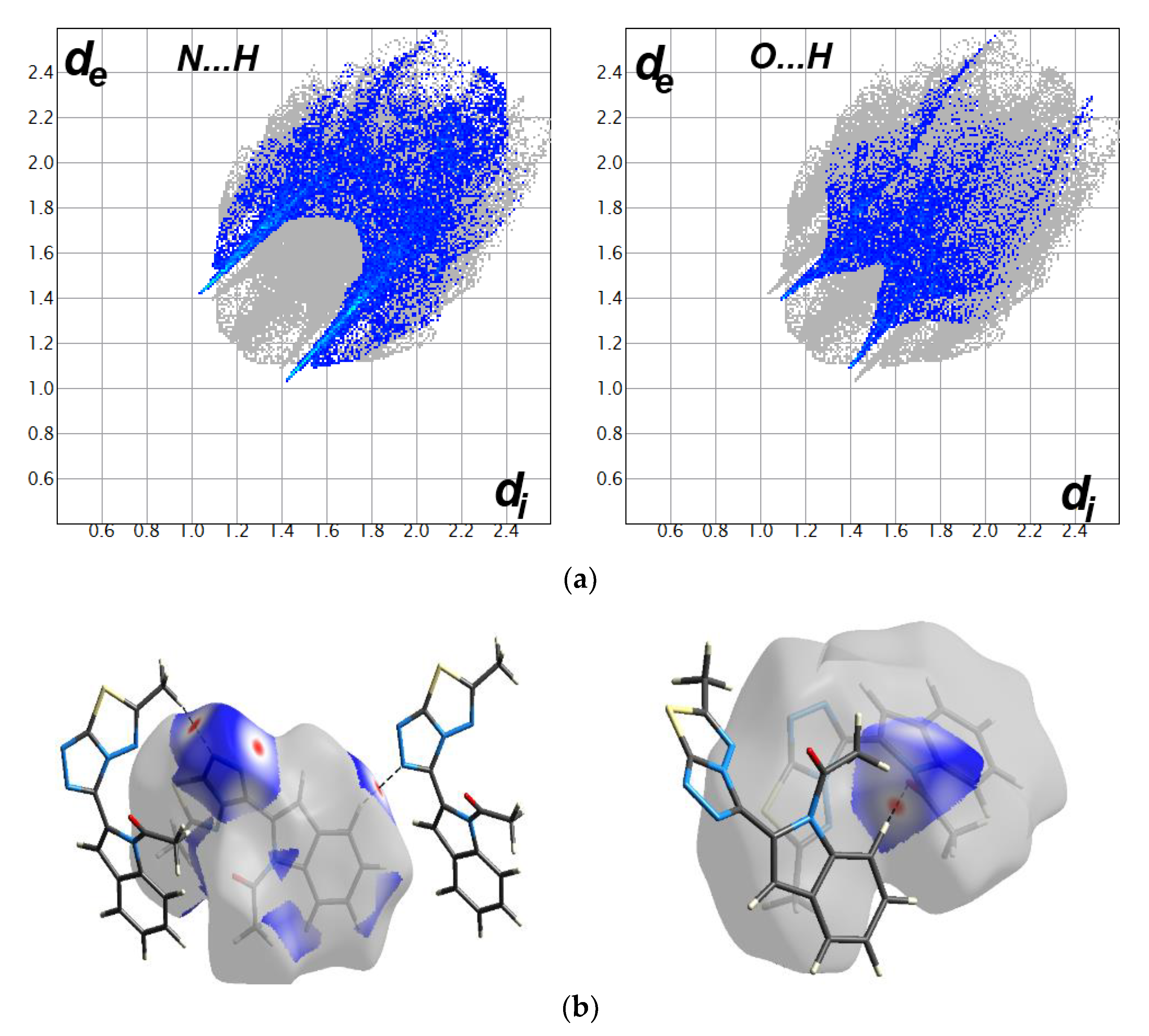
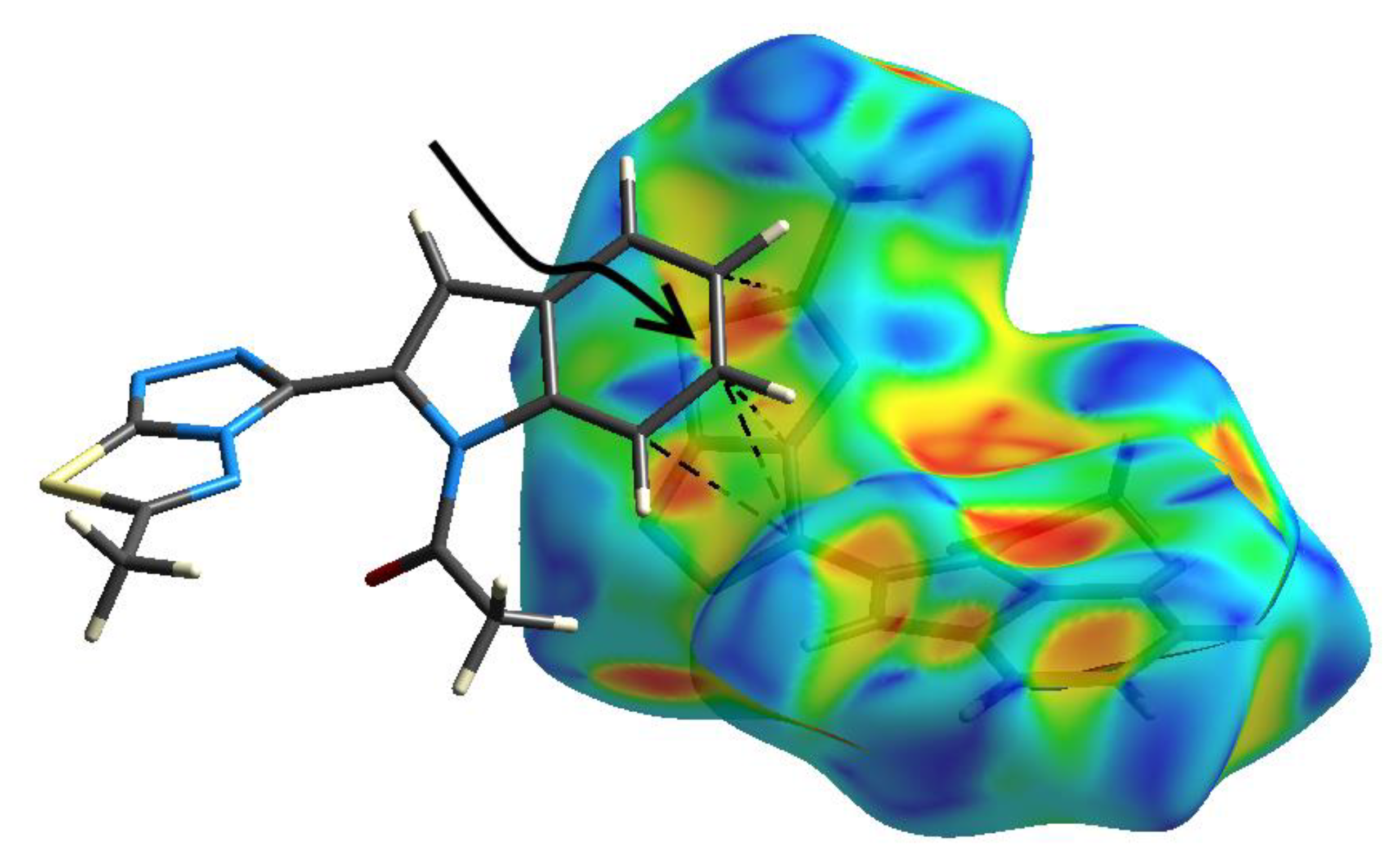
3.3. Hirshfeld Analysis of Molecular Packing
3.4. DFT Studies
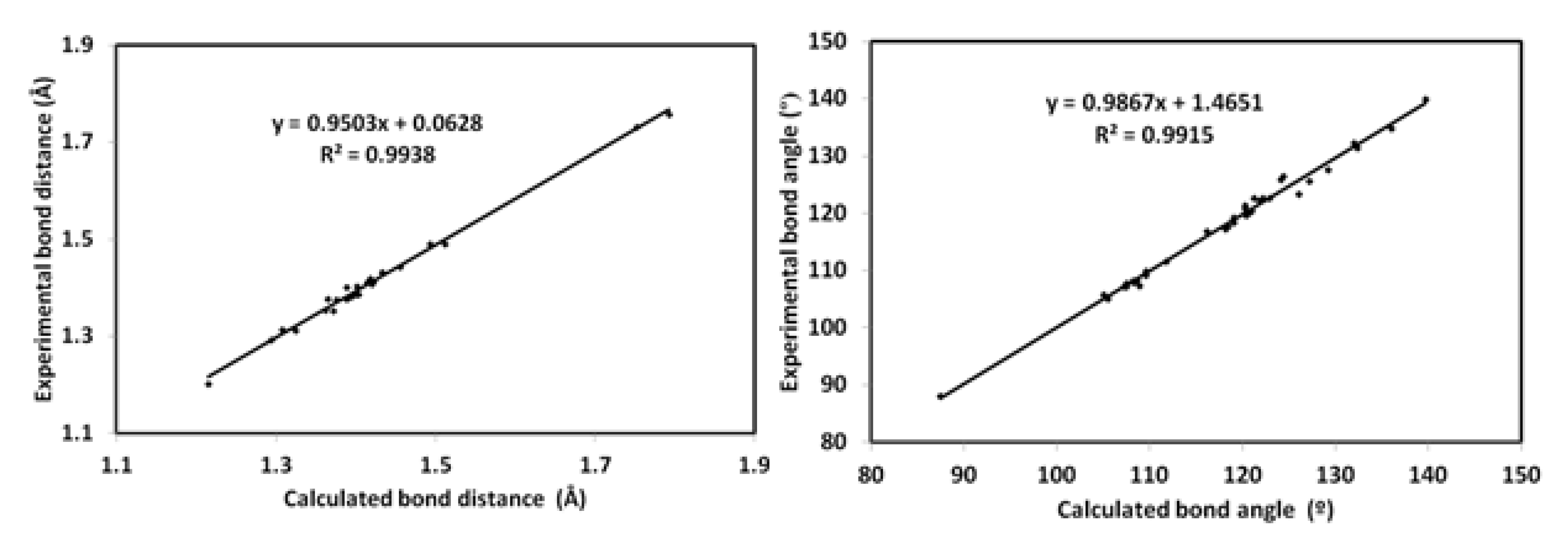
3.4.1. Optimized Geometry
3.4.2. Reactivity Studies
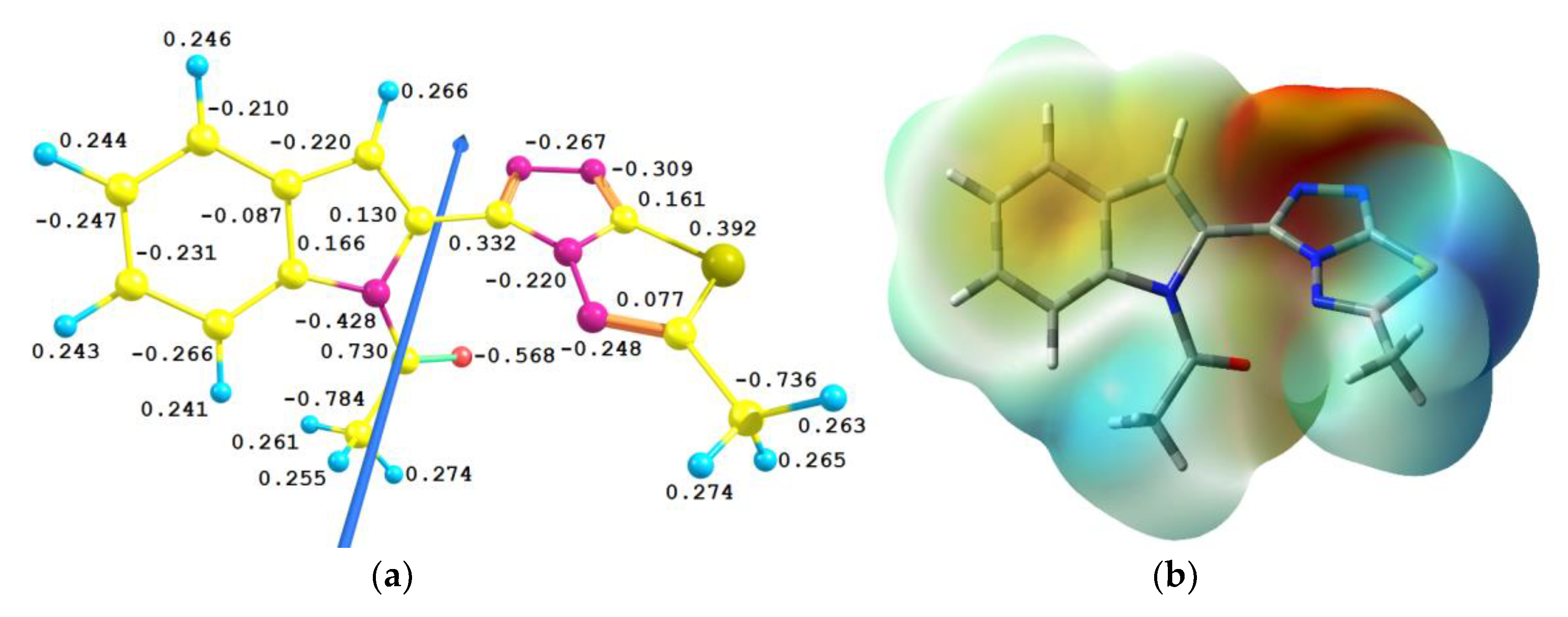
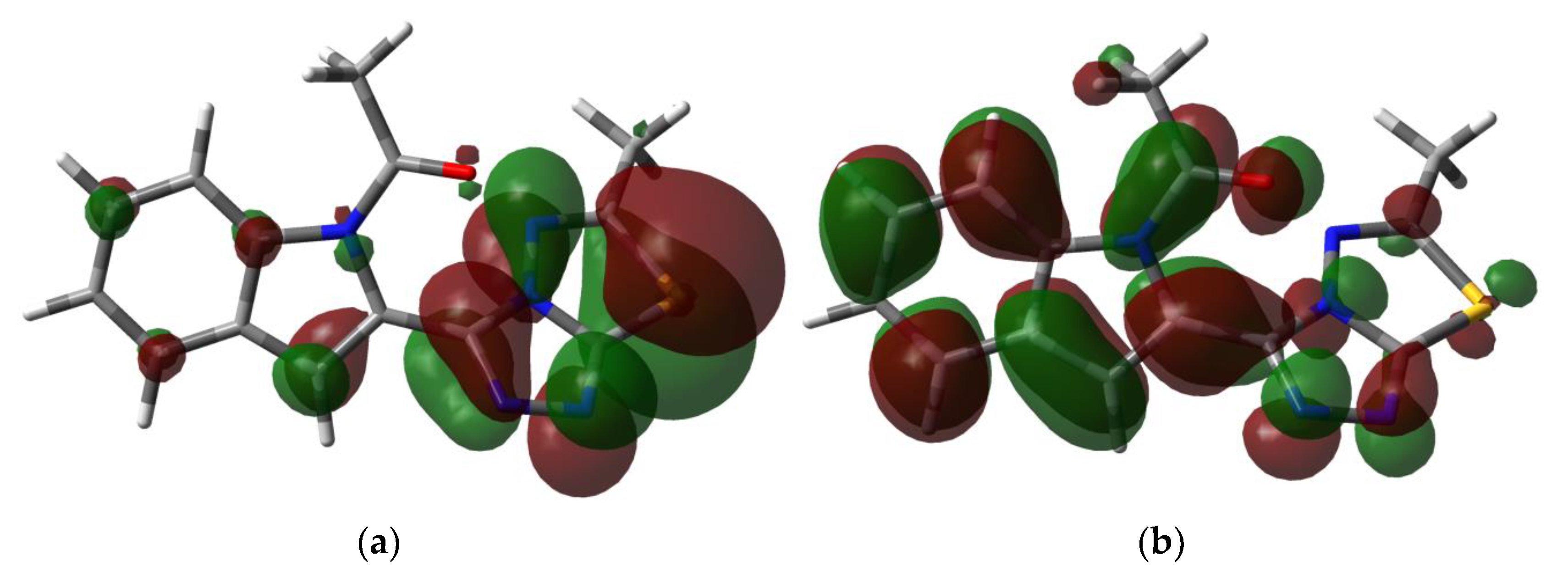
3.4.3. NBO Analysis
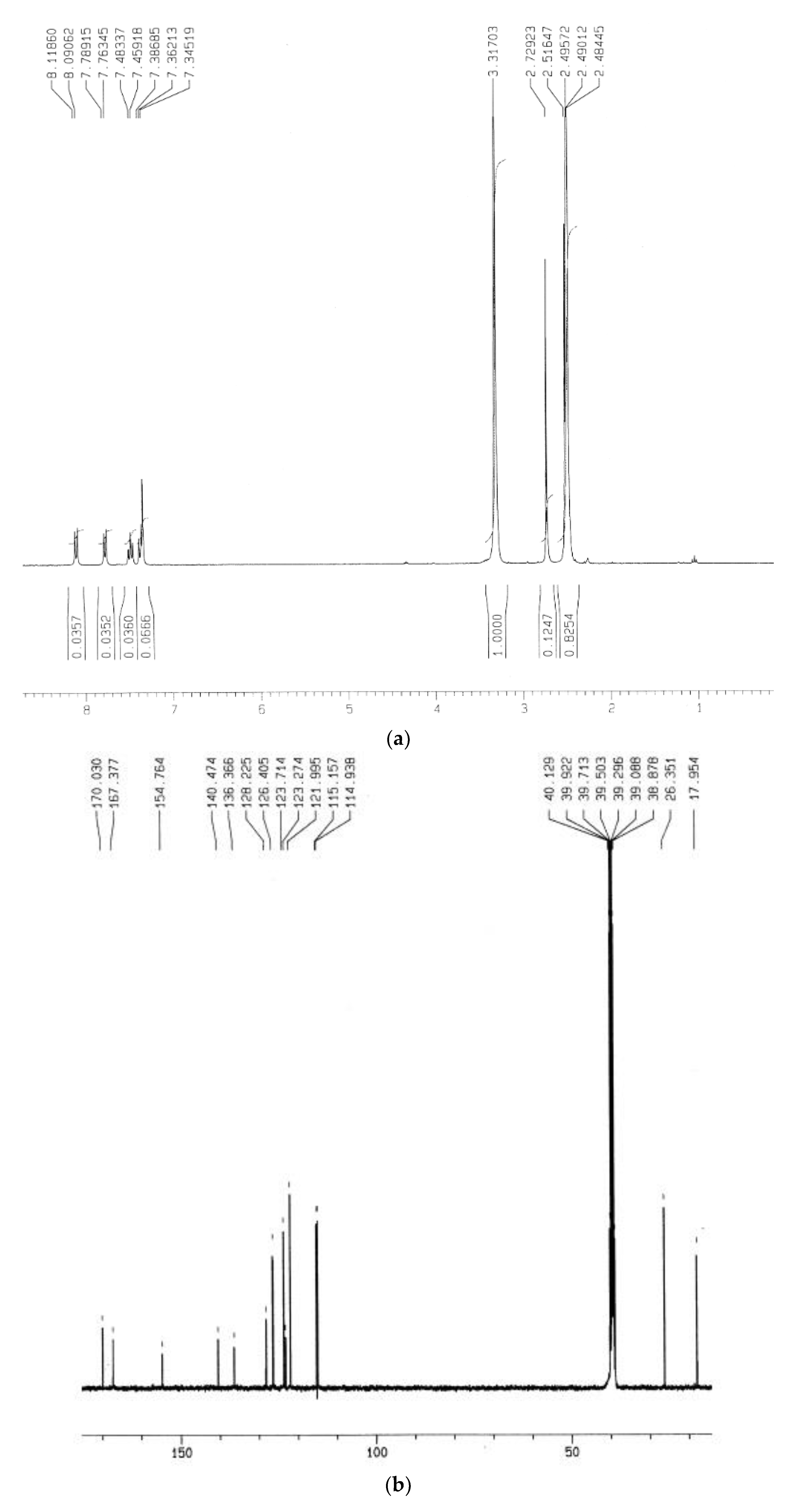
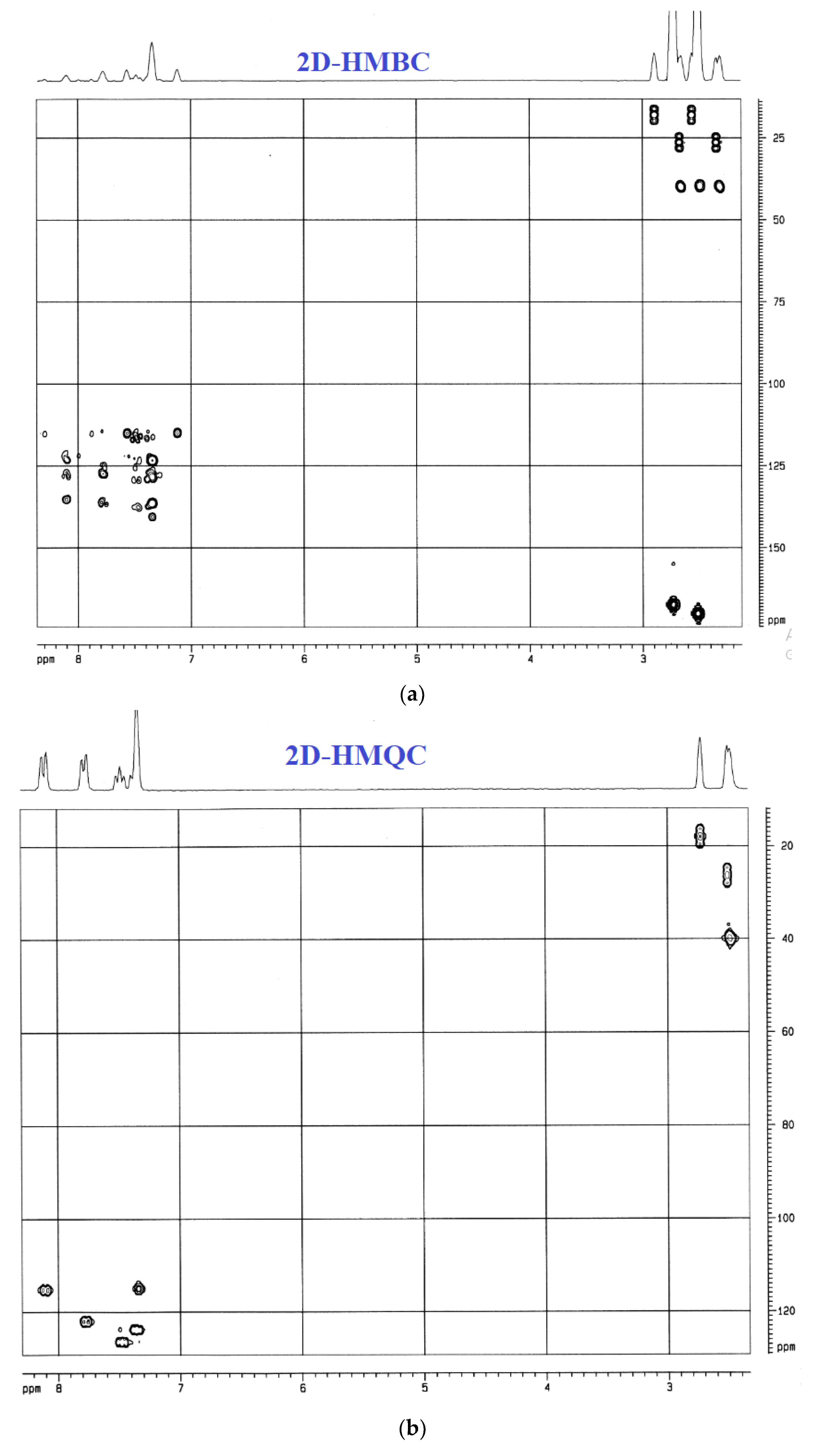
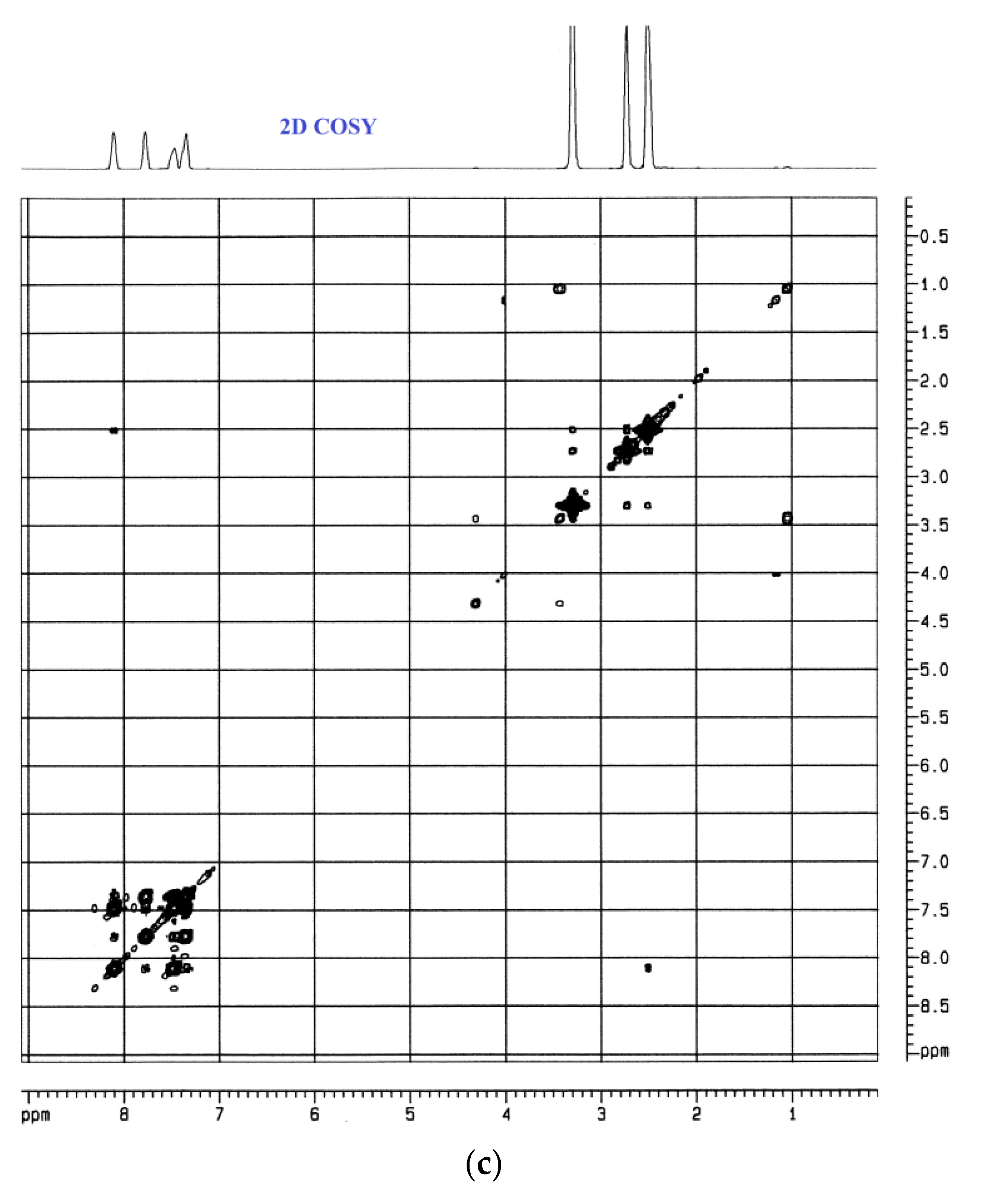
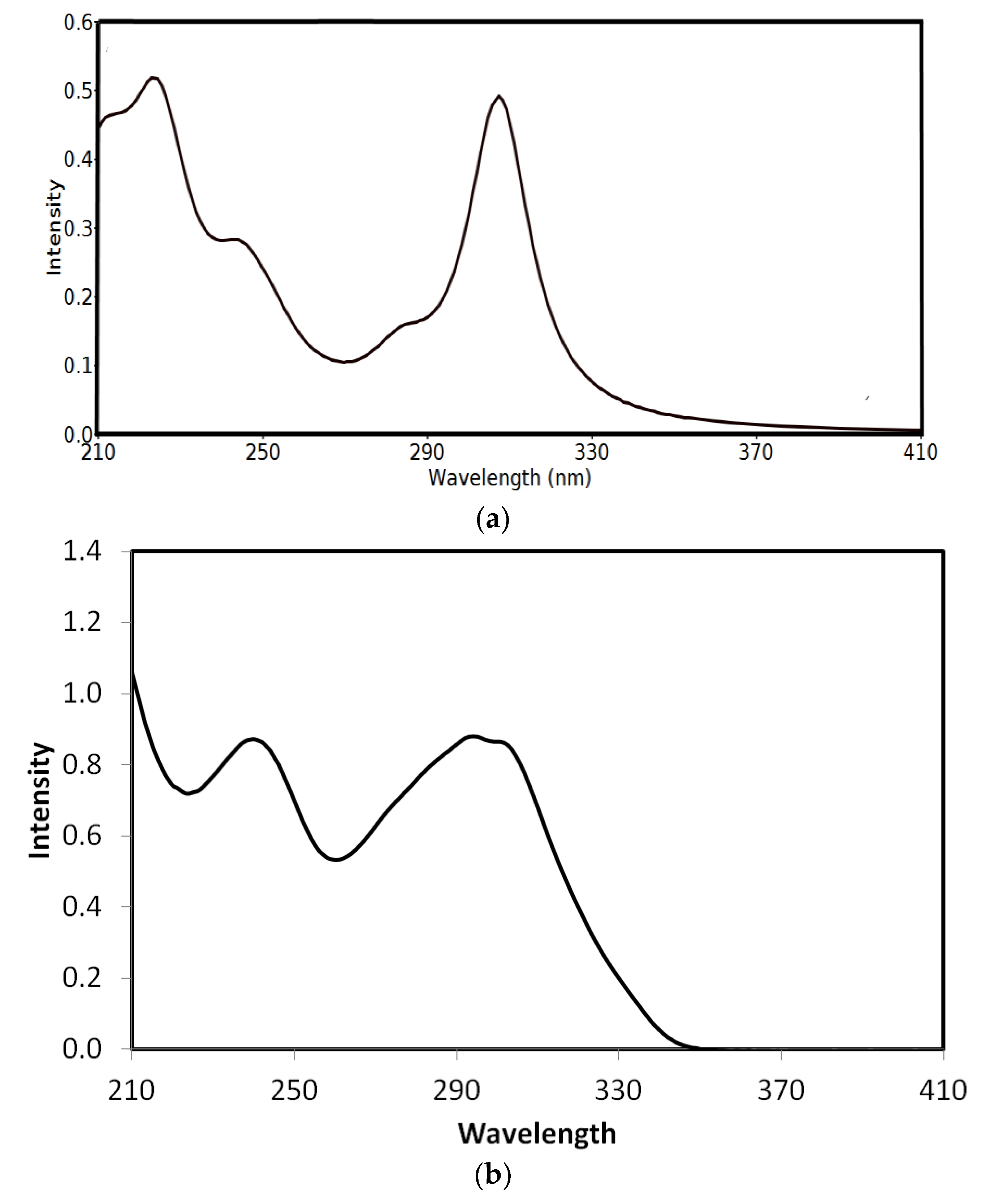
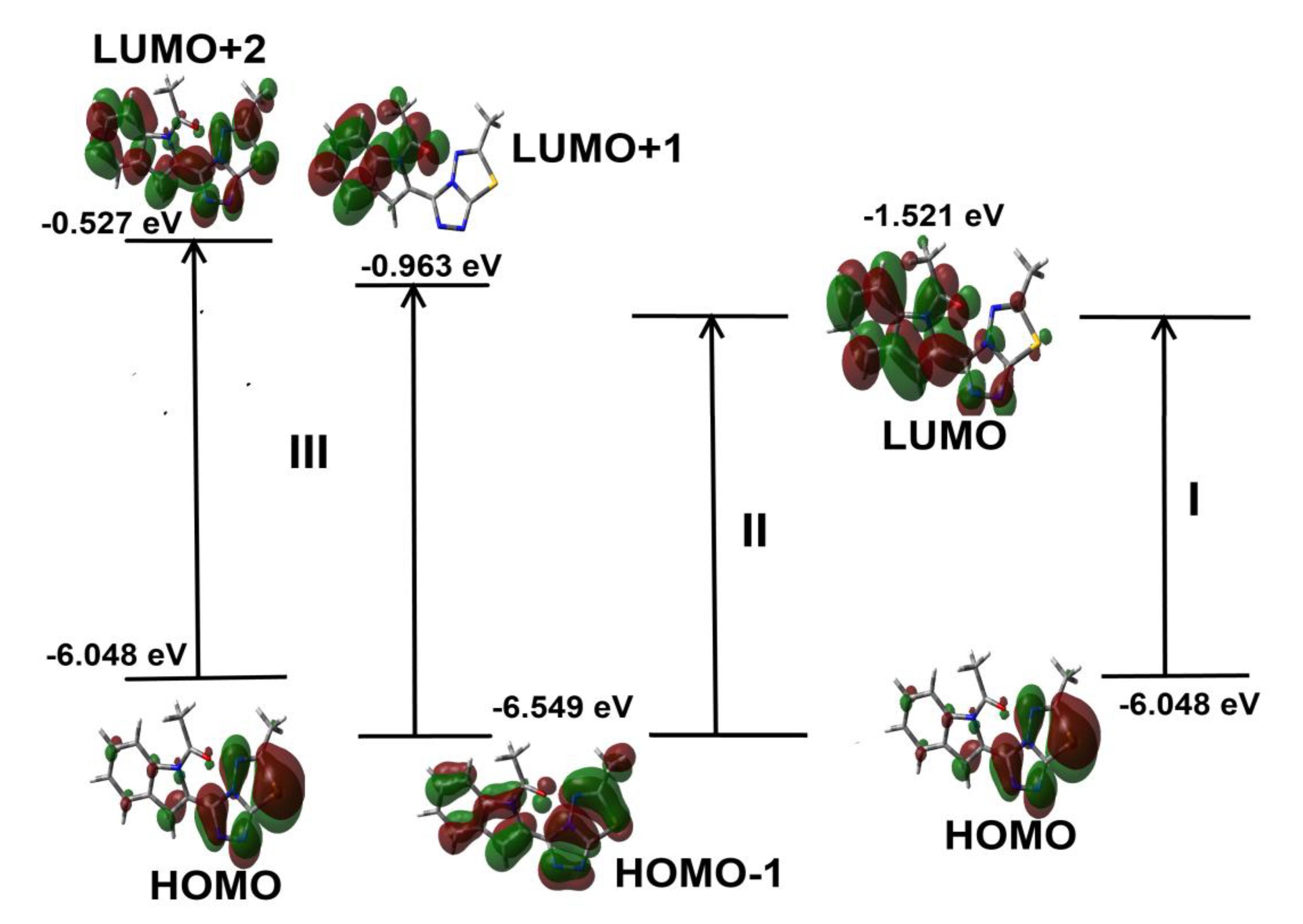
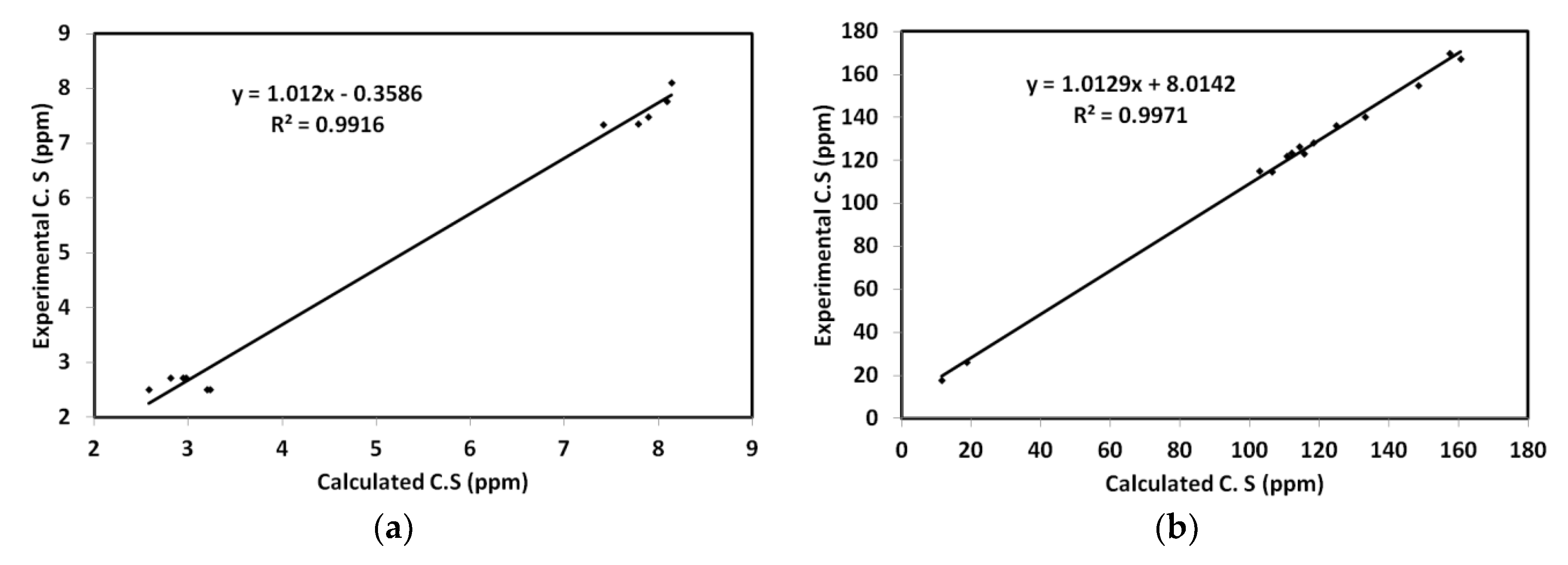
3.4.4. UV-Vis and NMR Spectra
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ji, R.V.; Sethi, A.; Nath, M.; Pratap, R. The Chemistry of Heterocycles: Nomenclature and Chemistry of Three to Five Membered Heterocycles; Elsevier: Amsterdam, The Netherlands, 2019. [Google Scholar]
- Chen, M.; Lu, S.; Yuan, G.; Yang, S.; Du, X. Synthesis and antibacterial activity of some heterocyclic β-enamino ester derivatives with 1, 2, 3-triazole. Heterocycl. Commun. 2000, 6, 421–426. [Google Scholar] [CrossRef]
- Gujjar, R.; Marwaha, A.; El Mazouni, F.; White, J.; White, K.L.; Creason, S.; Shackleford, D.M.; Baldwin, J.; Charman, W.N.; Buckner, F.S.; et al. Identification of a metabolically stable triazolopyrimidine-based dihydroorotate dehydrogenase inhibitor with antimalarial activity in mice. J. Med. Chem. 2009, 52, 1864–1872. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Deng, Z.; Tian, J.; Liu, T. Synthesis and biological evaluation of salinomycin triazole analogues as anticancer agents. Eur. J. Med. Chem. 2017, 127, 900–908. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, Y.; Fazili, K.M.; Bhat, K.A.; Ara, T. Synthesis and biological evaluation of novel 3-O-tethered triazoles of diosgenin as potent antiproliferative agents. Steroids 2017, 118, 1–8. [Google Scholar]
- Ayati, A.; Emami, S.; Foroumadi, A. The importance of triazole scaffold in the development of anticonvulsant agents. Eur. J. Med. Chem. 2016, 109, 380–392. [Google Scholar] [CrossRef]
- Akhtar, T.; Hameed, S.; Khan, K.M.; Choudhary, M.I. Syntheses, urease inhibition, and antimicrobial studies of some chiral 3-substituted-4-amino-5-thioxo-1H, 4H-1,2,4-triazoles. Med. Chem. 2008, 4, 539–543. [Google Scholar] [CrossRef]
- Timur, İ.; Kocyigit, Ü.M.; Dastan, T.; Sandal, S.; Ceribası, A.O.; Taslimi, P.; Gulcin, İ.; Koparir, M.; Karatepe, M.; Çiftçi, M. In vitro cytotoxic and in vivo antitumoral activities of some aminomethyl derivatives of 2, 4-dihydro-3H-1,2,4-triazole-3-thiones—Evaluation of their acetylcholinesterase and carbonic anhydrase enzymes inhibition profiles. J. Biochem. Mol. Toxic. 2019, 33, e22239. [Google Scholar] [CrossRef] [PubMed]
- Sevaille, L.; Gavara, L.; Bebrone, C.; De Luca, F.; Nauton, L.; Achard, M.; Mercuri, P.; Tanfoni, S.; Borgianni, L.; Guyon, C.; et al. 1,2,4-Triazole-3-thione compounds as inhibitors of dizinc metallo-β-lactamases. ChemMedChem 2017, 12, 972–985. [Google Scholar] [CrossRef] [PubMed]
- Baburajeev, C.P.; Mohan, C.D.; Ananda, H.; Rangappa, S.; Fuchs, J.E.; Jagadish, S.; Siveen5, K.S.; Chinnathambi, A.; Ali Alharbi, S.A.; Zayed, M.E.; et al. Development of novel triazolo-thiadiazoles from heterogeneous “green” catalysis as protein tyrosine phosphatase 1B inhibitors. Sci. Rep. 2015, 5, 14195. [Google Scholar] [CrossRef]
- Sechi, M.; Derudas, M.; Dallocchio, R.; Dessi, A.; Bacchi, A.; Sannia, L.; Carta, F.; Palomba, M.; Ragab, O.; Chan, C.; et al. Design and synthesis of novel indole β-diketo acid derivatives as HIV-1 integrase inhibitors. J. Med.Chem. 2014, 47, 5298–5310. [Google Scholar] [CrossRef]
- Narayana, B.; Ashalatha, B.V.; Vijayaraj, K.K.; Fernandes, J.; Sarojini, B.K. Synthesis of some new biologically active 1, 3, 4-oxadiazolyl nitroindoles and a modified Fischer indole synthesis of ethyl nitro indole-2-carboxylates. Bioorg. Med. Chem. 2005, 13, 4638–4644. [Google Scholar] [CrossRef] [PubMed]
- Mascal, M.; Modes, K.V.; Durmus, A. Concise photochemical synthesis of the antimalarial indole alkaloid decursivine. Angew. Chem. Int. Ed. 2011, 50, 4445–4446. [Google Scholar] [CrossRef] [PubMed]
- Al-Quawasmeh, R.A.; Huesca, M.; Nedunuri, V.; Peralta, R.; Wright, J.; Lee, Y.; Young, A. Potent antimicrobial activity of 3-(4, 5-diaryl-1H-imidazol-2-yl)-1H-indole derivatives against methicillin-resistant Staphylococcus aureus. Bioorg. Med. Chem. Lett. 2010, 20, 3518–3520. [Google Scholar] [CrossRef]
- Ty, N.; Dupeyre, G.; Chabot, G.G.; Seguin, J.; Tillequin, F.; Scherman, D.; Michel, S.; Cachet, X. Synthesis and biological evaluation of new disubstituted analogues of 6-methoxy-3-(3′, 4′, 5′-trimethoxybenzoyl)-1H-indole (BPR0L075), as potential antivascular agents. Bioorg. Med. Chem. 2008, 16, 7494–7503. [Google Scholar] [CrossRef] [PubMed]
- Mandour, A.H.; El-Sawy, E.R.; Shaker, K.H.; Mustafa, M.A. Synthesis, anti-inflammatory, analgesic and anticonvulsant activities of 1, 8-dihydro-1-ary1-8-alkyl pyrazolo (3, 4-b) indoles. Acta Pharm. 2010, 60, 73–88. [Google Scholar] [CrossRef]
- Islam, M.S.; Barakat, A.; Al-Majid, A.M.; Ali, M.; Yousuf, S.; Choudhary, M.I.; Khalil, R.; Ul-Haq, Z. Catalytic asymmetric synthesis of indole derivatives as novel α-glucosidase inhibitors in vitro. Bioorg. Chem. 2018, 79, 350–354. [Google Scholar] [CrossRef]
- Bi, W.; Bi, Y.; Xue, P.; Zhang, Y.; Gao, X.; Wang, Z.; Li, M.; Baudy-Floc’h, M.; Ngerebara, N.; Gibson, K.M.; et al. Synthesis and characterization of novel indole derivatives reveal improved therapeutic agents for treatment of ischemia/reperfusion (I/R) injury. J. Med. Chem. 2010, 53, 6763–6767. [Google Scholar] [CrossRef] [PubMed]
- Boraei, A.T.; Ghabbour, H.A.; Gomaa, M.S.; El Ashry, E.S.H.; Barakat, A. Synthesis and anti-proliferative assessment of triazolo-thiadiazepine and triazolo-thiadiazine scaffolds. Molecules 2019, 24, 4471. [Google Scholar] [CrossRef] [PubMed]
- Boraei, A.T.A.; Gomaa, M.S.; El Sayed, E.S.H.; Duerkop, A. Design, selective alkylation and X-ray crystal structure determination of dihydro-indolyl-1,2,4-triazole-3-thione and its 3-benzylsulfanyl analogue as potent anticancer agents. Eur. J. Med. Chem. 2017, 125, 360–371. [Google Scholar] [CrossRef]
- Boraei, A.T.; Ashour, H.K.; El Sayed, H.; Abdelmoaty, N.; El-Falouji, A.I.; Gomaa, M.S. Design and synthesis of new phthalazine-based derivatives as potential EGFR inhibitors for the treatment of hepatocellular carcinoma. Bioorg. Chem. 2019, 85, 293–307. [Google Scholar] [CrossRef] [PubMed]
- Boraei, A.T.; Singh, P.K.; Sechi, M.; Satta, S. Discovery of novel functionalized 1, 2, 4-triazoles as PARP-1 inhibitors in breast cancer: Design, synthesis and antitumor activity evaluation. Eur. J. Med. Chem. 2019, 182, 111621. [Google Scholar] [CrossRef] [PubMed]
- Boraei, A.T.A.; Sarhan, A.A.M.; Yousuf, S.; Barakat, A. Synthesis of a New Series of Nitrogen/Sulfur Heterocycles by Linking Four Rings: Indole; 1,2,4-Triazole; Pyridazine; and Quinoxaline. Molecules 2020, 25, 450. [Google Scholar] [CrossRef] [PubMed]
- Sarhan, A.A.; Boraei, A.T.; Barakat, A.; Nafie, M.S. Discovery of hydrazide-based pyridazino [4, 5-b] indole scaffold as a new phosphoinositide 3-kinase (PI3K) inhibitor for breast cancer therapy. RSC Advances 2020, 10, 19534–19541. [Google Scholar] [CrossRef]
- Badria, F.A.; Soliman, S.M.; Atef, S.; Islam, M.S.; Al-Majid, A.M.; Dege, N.; Ghabbour, H.A.; Ali, M.; El-Senduny, F.F.; Barakat, A. Anticancer Indole-Based Chalcones: A Structural and Theoretical Analysis. Molecules 2019, 24, 3728. [Google Scholar] [CrossRef]
- Boraei, A.T.; El Ashry, E.S.H.; Barakat, A.; Ghabbour, H.A. Synthesis of new functionalized indoles based on ethyl indol-2-carboxylate. Molecules 2016, 21, 333. [Google Scholar] [CrossRef]
- Bruker AXS. SAINT Software reference manual; Bruker AXS: Madison, WI, USA, 1998. [Google Scholar]
- Spek, L.A. Single-crystal structure validation with the program PLATON. J. Appl. Chem. 2002, 36, 7–13. [Google Scholar]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. C. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P.A. Mercury CSD 2.0–new features for the visualization and investigation of crystal structures. J. Appl. Cryst. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. Crystal Explorer17. 2017. University of Western Australia. Available online: http://hirshfeldsurface.net (accessed on 3 June 2020).
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. GAUSSIAN 09, Revision A02 ed; Gaussian Inc.: Wallingford, CT, USA, 2009; GaussView; Version 4.1; Dennington II, R., Keith, T., Millam, J., Eds.; Semichem Inc.: Shawnee Mission, KS, USA, 2007; Available online: https://gaussian.com (accessed on 3 June 2020).
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Marten, B.; Kim, K.; Cortis, C.; Friesner, R.A.; Murphy, R.B.; Ringnalda, M.N.; Sitkoff, D.; Honig, B. New Model for Calculation of Solvation Free Energies: Correction of Self-Consistent Reaction Field Continuum Dielectric Theory for Short-Range Hydrogen-Bonding Effects. J. Phys. Chem. 1996, 100, 11775–11788. [Google Scholar] [CrossRef]
- Tannor, D.J.; Marten, B.; Murphy, R.; Friesner, R.A.; Sitkoff, D.; Nicholls, A.; Ringnalda, M.; Goddard, W.A.; Honig, B. Accurate first principles calculation of molecular charge distributions and solvation energies from ab initio quantum mechanics and continuum dielectric theory. J. Am. Chem. Soc. 1994, 116, 11875–11882. [Google Scholar] [CrossRef]
- Cheeseman, J.R.; Trucks, G.W.; Keith, T.A.; Frisch, M.J. A Comparison of Models for Calculating Nuclear Magnetic Resonance Shielding Tensors. J. Chem. Phys. 1996, 104, 5497–5509. [Google Scholar] [CrossRef]
- Foresman, J.B.; Frisch, Æ. Exploring Chemistry with Electronic Structure Methods, 2nd ed.; Gaussian: Pittsburgh, PA, USA, 1996. [Google Scholar]
- Chang, R. Chemistry, 7th ed.; McGraw-Hill: New York, NY, USA, 2001. [Google Scholar]
- Kosar, B.; Albayrak, C. Spectroscopic investigations and quantum chemical computational study of (E)-4-methoxy-2-[(p-tolylimino) methyl] phenol. Spectrochim. Acta 2011, 78, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Koopmans, T.A. Ordering of wave functions and eigenenergies to the individual electrons of an atom. Physica 1933, 1, 104–113. [Google Scholar] [CrossRef]
- Parr, R.G.; Yang, W. Density-Functional Theory of Atoms and Molecules; Oxford University Press: New York, NY, USA, 1989. [Google Scholar]
- Parr, R.G.; Szentpaly, L.V.; Liu, S. Electrophilicity index. J. Am. Chem. Soc. 1999, 121, 1922–1924. [Google Scholar] [CrossRef]
- Singh, R.N.; Kumar, A.; Tiwari, R.K.; Rawat, P.; Gupta, V.P. A combined experimental and quantum chemical (DFT and AIM) study on molecular structure, spectroscopic properties, NBO and multiple interaction analysis in a novel ethyl 4-[2-(carbamoyl) hydrazinylidene]-3, 5-dimethyl-1H-pyrrole-2-carboxylate and its dimer. J. Mol. Strut. 2013, 1035, 427–440. [Google Scholar] [CrossRef]
- Hubert, I.J.; Kostova, I.; Ravikumar, C.; Amalanathan, M.; Pinzaru, S.C. Theoretical and vibrational spectral investigation of sodium salt of acenocoumarol. J. Raman Spectrosc. 2009, 40, 1033–1038. [Google Scholar]
- Sundaraganesan, N.; Sebastian, S. The spectroscopic (FT-IR, FT-IR gas phase, FT-Raman and UV) and NBO analysis of 4-Hydroxypiperidine by density functional method. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2010, 75, 941–952. [Google Scholar]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound Description | |
|---|---|
| Chemical formula | C14H11N5OS |
| Formula weight | 297.34 |
| Temperature | 104(2) K |
| Wavelength | 1.54184 A |
| Crystal system | Monoclinic |
| Space group | Cc |
| Unit cell dimensions | a = 10.9381(19)Å b = 11.2508(19)Å c = 11.289 (2)Å β = 101.404 (13)° |
| Volume | 1361.8(4) A3 |
| Z | 4 |
| Calculated density | 1.450 Mg/m3 |
| Absorption coefficient | 2.175 mm−1 |
| F (000) | 616 |
| Crystal size | 0.40 × 0.14 × 0.13 |
| Theta range for data collection | 7.425 to 68.432° |
| Limiting indices | −13 <= h <= 13, −13 <= k <= 13, −13 <= l <= 12 |
| Reflections collected | 15863 |
| Completeness to theta = 67.684 | 98.5% |
| Refinement method | Full-matrix least-squares on F2 |
| Independent reflections | 2410 |
| Data/restraints/parameters | 2410/2/193 |
| Goodness-of-fit on F2 | 1.019 |
| Flack parameter | 0.067(5) |
| R indices (all data) | R1 = 0.0254, wR2 = 0.0697 |
| Final R indices (I > 2sigma (I)) | R1 = 0.0256, wR2 = 0.0701 |
| Largest diff. peak and hole | 0.119 and −0.152 eA−3 |
| CCDC | 2010068 |
| D—H···A | D—H | H···A | D···A | D—H···A |
|---|---|---|---|---|
| C2—H2···O1 (i) | 0.950 | 2.590 | 3.397(3) | 142.00 |
| C5—H5···N2 (ii) | 0.950 | 2.620 | 3.522(3) | 158.00 |
| C12—H12A···N3 (i) | 0.980 | 2.560 | 3.528(4) | 168.00 |
| Donor NBO | Acceptor NBO | E(2) | Donor NBO | Acceptor NBO | E(2) |
|---|---|---|---|---|---|
| σ→σ* | n→σ* | ||||
| BD(1)S1–C23 | BD*(1)N5–C22 | 6.39 | LP(1)O2 | BD*(1)N3–C28 | 29.33 |
| BD(1)N4–N5 | BD*(1)S1–C22 | 9.84 | LP(1)O2 | BD*(1)C28–C29 | 18.88 |
| BD(1)N4–N5 | BD*(1)C20–C21 | 5.35 | LP(1)N4 | BD*(1)N5–C22 | 5.30 |
| BD(1)N4–C21 | BD*(1)N6–N7 | 5.29 | LP(1)N4 | BD*(1)N6–C21 | 8.43 |
| BD(1)N6–C22 | BD*(1)C20–C21 | 4.89 | LP(1)N5 | BD*(1)N4–C21 | 5.69 |
| BD(1)N7–C23 | BD*(1)N6–C21 | 4.93 | LP(1)N5 | BD*(1)N6–C22 | 8.73 |
| BD(1)C9–C11 | BD*(1)N3–C8 | 6.82 | LP(1)N7 | BD*(1)S1–C23 | 14.68 |
| BD(1)C17–C18 | BD*(1)C20–C21 | 5.81 | LP(1)N7 | BD*(1)N6–C22 | 7.94 |
| BD(1)C18–C20 | BD*(1)C15–C17 | 4.90 | |||
| π→π* | n→π* | ||||
| BD(2)N4–C21 | BD*(2)N5–C22 | 12.82 | LP(2)S1 | BD*(2)N5–C22 | 26.15 |
| BD(2)N4–C21 | BD*(2)C18–C20 | 5.40 | LP(2)S1 | BD*(2)N7–C23 | 26.45 |
| BD(2)N5–C22 | BD*(2)N4–C21 | 13.12 | LP(1)N3 | BD*(2)O2–C28 | 44.97 |
| BD(2)C8–C17 | BD*(2)C9–C11 | 19.38 | LP(1)N3 | BD*(2)C8–C17 | 29.26 |
| BD(2)C8–C17 | BD*(2)C13–C15 | 18.54 | LP(1)N3 | BD*(2)C18–C20 | 25.74 |
| BD(2)C8–C17 | BD*(2)C18–C20 | 16.47 | LP(1)N6 | BD*(2)N4–C21 | 40.63 |
| BD(2)C9–C11 | BD*(2)C8–C17 | 19.34 | LP(1)N6 | BD*(2)N5–C22 | 45.82 |
| BD(2)C9–C11 | BD*(2)C13–C15 | 17.83 | LP(1)N6 | BD*(2)N7–C3 | 27.30 |
| BD(2)C13–C15 | BD*(2)C8–C17 | 19.07 | |||
| BD(2)C13–C15 | BD*(2)C9–C11 | 20.65 | |||
| BD(2)C18–C20 | BD*(2)N4–C21 | 9.96 | |||
| BD(2)C18–C20 | BD*(2)C8–C17 | 15.43 |
| No. | (λmax)calc | fosc a | Assignment | (λmax)observ |
|---|---|---|---|---|
| I | 307.4 | 0.4712 | HOMO→LUMO (94%) | 301 |
| II | 283.6 | 0.0674 | H−1→LUMO (83%) | 293 |
| III | 245.1 | 0.1208 | H−1→L+1 (16%), HOMO→L+2 (65%) | 239 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boraei, A.T.A.; Soliman, S.M.; Yousuf, S.; Bibi, M.; Barakat, A. N-Acetyl Indole Linked to a Fused Triazolo/Thiadiazole Scaffold: Synthesis, Single Crystal X-Ray Structure, and Molecular Insight. Crystals 2020, 10, 600. https://doi.org/10.3390/cryst10070600
Boraei ATA, Soliman SM, Yousuf S, Bibi M, Barakat A. N-Acetyl Indole Linked to a Fused Triazolo/Thiadiazole Scaffold: Synthesis, Single Crystal X-Ray Structure, and Molecular Insight. Crystals. 2020; 10(7):600. https://doi.org/10.3390/cryst10070600
Chicago/Turabian StyleBoraei, Ahmed T. A., Saied M. Soliman, Sammer Yousuf, Memoona Bibi, and Assem Barakat. 2020. "N-Acetyl Indole Linked to a Fused Triazolo/Thiadiazole Scaffold: Synthesis, Single Crystal X-Ray Structure, and Molecular Insight" Crystals 10, no. 7: 600. https://doi.org/10.3390/cryst10070600
APA StyleBoraei, A. T. A., Soliman, S. M., Yousuf, S., Bibi, M., & Barakat, A. (2020). N-Acetyl Indole Linked to a Fused Triazolo/Thiadiazole Scaffold: Synthesis, Single Crystal X-Ray Structure, and Molecular Insight. Crystals, 10(7), 600. https://doi.org/10.3390/cryst10070600