Abstract
Pt monolayers (PtML) supported on nanoparticles with different compositions (i.e., Ru, Rh, Pd, Ir, and Au) were synthesized by the surface–limited redox replacement of underpotentially deposited Cu monolayers on nanoparticle supports. Nanoparticle supports with different compositions were directly deposited on the conducting substrate by a clean and one-step electrodeposition method with controlled deposition potential and time. The whole synthesis process of the electrode was free of surfactants, binders, capping agents and reductants, and without an additional coating process of electrocatalysts. The results show that the specific activity (SA) of PtML electrocatalysts depended strongly on the composition of the nanoparticle support. For example, the PtML supported on the Au nanoparticle exhibited 8.3 times higher SA than that supported on the Ru and Pd nanoparticles. The change in the SA of the PtML supported on different nanoparticles was related to the substrate–induced strain in the PtML resulting from the lattice mismatch between the PtML and the nanoparticle support. As the strain in the PtML changed from the tensile strain to the compressive strain, the SA of the PtML electrocatalysts decreased remarkably.
1. Introduction
Growing concern in energy and environmental issues has stimulated considerable research on the electro-oxidation of ammonia, since it addresses both clean energy supply and removal of pollutants [1,2,3,4,5,6]. Ammonia is a carbon–free chemical energy carrier, which has a high capacity of hydrogen storage (17.7 wt.%) and high energy density (3000 Wh kg−1) [1,7]. On the other hand, ammonia is an environmental pollutant and a toxic gas. Therefore, the electro-oxidation of ammonia has important applications in direct fuel cells [8,9,10], hydrogen production [1,11,12], ammonia decomposition in wastewaters [8,13] and electrochemical sensors for detecting ammonia [14]. Since ammonia electro-oxidation is a sluggish reaction, electrocatalysts are required to catalyze the oxidation of ammonia to N2. Among various investigated electrocatalysts, Pt has been acknowledged as the most effective single–component electrocatalyst for ammonia electro-oxidation [15,16]. However, it is a limited resource and has high cost which been the main obstacles for wide usage of Pt [17,18,19,20], strongly limiting the large-scale applications of ammonia electro-oxidation technologies. Therefore, great efforts have been paid to the development of Pt-based electrocatalysts with a high activity for ammonia electro-oxidation and simultaneously low Pt loading.
Developing PtML catalysts is one of the most promising strategies because of its unique advantages, such as an extremely high utilization degree of Pt atoms and tunable activity through the electronic and structural effects between the PtML and the substrate [15,16,17,18]. Adzic’s group successfully developed a class of electrocatalysts consisting of a PtML on different metals (e.g., Au, Pd, Rh, Ir, and Ru) [15,19,20], alloys (e.g., PdCo [21], PdFe [22], PdAu [23], IrNi [24], and AuNiFe [25]). PtML electrocatalysts can be synthesized by deposition of a Cu monolayer on the substrate through underpotential deposition (UPD), followed by a surface-limited replacement reaction between Pt ions and Cu monolayer [26,27]. It has been well demonstrated that a Cu monolayer can be replaced by a PtML [26]. Due to the high Pt utilization, the mass activity of these PtML electrocatalysts could be more than an order of magnitude higher than traditional all-Pt electrocatalysts [15,18,27]. Since the catalytic activity of the PtML electrocatalysts is strongly dependent on the interaction between the PtML and the supporting substrate [18,19,28,29], it is of great interest to investigate the effect of the supporting substrate on the catalytic activity of electrocatalysts. Such studies are important for further enhancing the electrocatalytic activity of PtML electrocatalysts by rationally selecting or designing substrate materials. To date, a number of characteristics of substrate materials, such as composition [19,30,31], crystalline orientation [20,21,28,31] and particle size [17,21], have been studied as PtML electrocatalysts for oxygen reduction reactions and methanol electro-oxidation. However, there has so far been very limited work on PtML electrocatalysts for ammonia electro-oxidation [18,32]. Previous work from our group synthesized the PtML on an Au substrate (bulk Au electrode [32] or Au particles [18]) for ammonia electro-oxidation. These PtML electrocatalysts exhibited several times, to a more than 10-fold increase, in the mass activity compared with the all-Pt electrocatalysts for ammonia electro-oxidation. It has also been found that the surface morphology of substrate material greatly influences the SA of PtML electrocatalysts [18]. Nevertheless, the effect of the composition of the substrate on the catalytic activity of PtML nanoparticles for ammonia electro-oxidation has remained unclear.
The purpose of the present work is to illustrate whether and how the composition of the substrate material would have an influence on the activity of the surface PtML nanoparticles. For practical electrocatalytic applications, it is required to support PtML on high surface area nanoparticles. Therefore, we investigated the activity of the PtML on nanoparticles with various compositions including Ru, Rh, Pd, Ir and Au. A clean, facile and one-step electrodeposition method was proposed to synthesize nanoparticles with different compositions as the supporting core, synthesized directly on the surface of conducting substrate. PtML was synthesized on the surface of these nanoparticles by a well-established method, consisting of Cu UPD on nanoparticles and subsequently the galvanic replacement of Pt [15,33]. The entire synthesis process was free of any surfactants, binders, capping agents and reductants, ensuring an extremely clean surface of electrocatalysts. In addition, the electrocatalysts were synthesized directly on the conducting substrate surface, avoiding the transfer process of the electrocatalysts. This greatly decreases the uncertainty resulting from the synthesis procedures, allowing the truly comparison of the impact of the composition of the supporting core on the electrocatalytic activity of the supported PtML. Furthermore, the relationship between the composition of the nanoparticle core of PtML electrocatalysts and the electrocatalytic activity for the ammonia oxidation was investigated by cyclic voltammetry (CV). To the best of our knowledge, this is the first time to report the synthesis of PtML electrocatalysts with nanoparticle core of different compositions by the electrochemical method as well as the compositional effect of the core materials on the activity of PtML electrocatalysts for electro-oxidation of ammonia. Interestingly, this study shows that the composition of the nanoparticle core had a great influence on the intrinsic activity of PtML electrocatalysts.
2. Results and Discussion
Figure 1a–e shows the SEM images of electrodeposited Ru, Rh, Pd, Ir, and Au nanoparticles, respectively. It can be seen that all these deposited nanoparticles were randomly distributed over the surface of the glassy carbon electrode (GCE). Due to the different nature of these five metals, it is very difficult to obtain different kinds of nanoparticles with exactly the same particle size. As mentioned in the experimental part, preliminary work has tried various electrodeposition parameters including potentials and times, and finally found the optimal conditions to synthesize these five kinds of nanoparticles with a similar particle size. The particle size distribution histograms in insets of Figure 1 show that the average particle sizes for electrodeposited Ru, Rh, Pd, Ir, and Au nanoparticles were 34.2 ± 7.9 nm, 15.2 ± 3.2 nm, 18.8 ± 1.7 nm, 24.2 ± 2.9 nm, 21.4 ± 5.1 nm, respectively. It was also noticed that much larger overpotential (i.e., more negative deposition potential) was required for the electrodeposition of Ru and Ir compared to other metals. Similar results have also been reported by previous studies for the electrodeposition of Ru [34] and Ir [35], due to the slow kinetics for their deposition. For example, Le Vot et al. [35] reported that the deposition of Ir on GCE required a large overpotential for the reduction of Ir3+ ions in 1.0 mM IrCl3 + 0.5 M H2SO4 solution. In their work, the lowest deposition potential was −0.6 V (vs. Ag/AgCl). The Ir deposits electrodeposited at this potential were characterized by large aggregates at several micrometers in size, indicating that the deposition process was very slow, and Ir tended to be deposited on previously formed Ir deposits than on the carbon substrate [35]. However, in the present work, Ir nanoparticles with good dispersion and small particle size of 24.2 ± 2.9 nm could be obtained at a further decreasing deposition potential of −0.7 V (vs. reversible hydrogen electrode (RHE)) in 5 mM IrCl3 + 0.05 M H2SO4 solution (Figure 1d). Such well-dispersed Ir nanoparticles are beneficial for increasing the effective surface area.
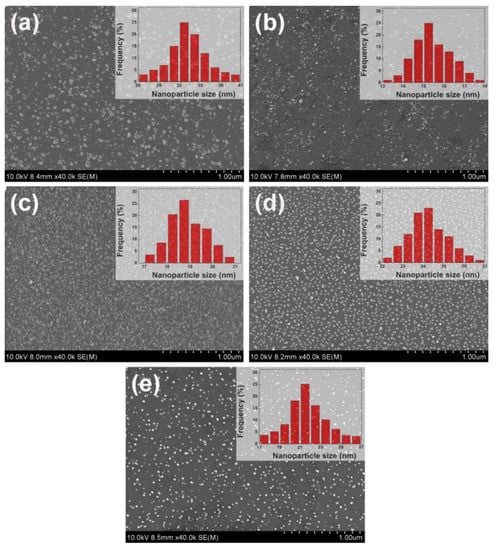
Figure 1.
Scanning electron microscopy (SEM) images of (a) Ru, (b) Rh, (c) Pd, (d) Ir, and (e) Au nanoparticles electrodeposited on the GCE, respectively. Insets of SEM images show the corresponding particle size distribution histogram.
Figure 2a–e shows the cyclic voltammetry (CV) curves of pure Ru, Rh, Pd, Ir, and Au nanoparticles electrodeposited on the GCE in 0.5 M H2SO4 solution at a scan rate of 0.05 V s−1. Rh, Ir and Pd nanoparticles showed characteristic hydrogen adsorption and desorption (0.05 to 0.2 V (vs. RHE)) peaks while no such hydrogen adsorption/desorption peaks could be observed for Au and Ru nanoparticles. All these CV curves exhibited very similar features compared to previously reported nanoparticles synthesized by a water-in-oil microemulsion method [36].
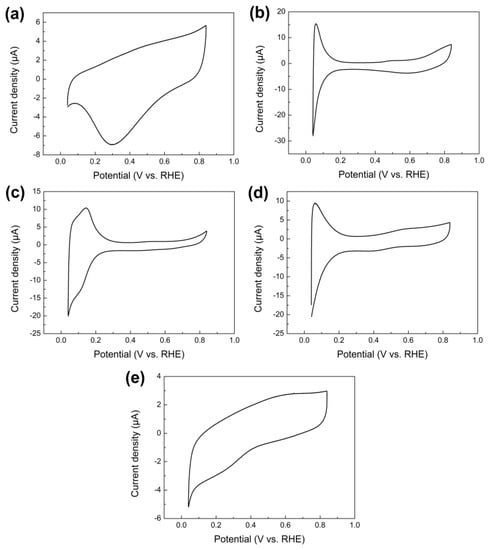
Figure 2.
CVs measured on pure (a) Ru, (b) Rh, (c) Pd, (d) Ir, and (e) Au nanoparticles electrodeposited on the GCE in 0.5 M H2SO4 solution at 0.05 V s−1, respectively.
Figure 3a–e shows CVs measured on pure Ru, Rh, Pd, Ir, and Au nanoparticles electrodeposited on the GCE in 0.1 M ammonia + 1 M KOH aqueous solution at 0.05 V s−1. Only for Ir nanoparticles did the CV curve exhibited an anodic current peak at 0.59 V (vs. RHE) (Figure 3d). Such an oxidation current peak has been extensively reported on pure Ir and also Pt electrodes and is ascribed to the electro-oxidation of ammonia to N2 [12,13,37]. Previous work performed CV measurements in the absence of ammonia and found that a such characteristic current peak did not appear [13,37,38]. For 4d transition metals (i.e., Ru, Rh and Pd), all CV plots showed no anodic oxidation current peak related with the ammonia electro-oxidation from 0.4 to 0.7 V (vs. RHE). This is in good agreement with previous results reported by de Vooys et al. [13] and Vidal-Iglesias et al. [36]. Based on differential electrochemical mass spectroscopy (DEMS) measurements, de Vooys et al. [13] found the dehydrogenation of ammonia occurred at significantly lower potentials on Ru, Rh and Pd than that on Pt and Ir, resulting in the surface poisoning by adsorbed nitrogen atoms (Nads) at much lower potentials on Ru, Rh and Pd. As a result, Ru, Rh and Pd were inactive towards the ammonia electro-oxidation to N2 [13]. It was also observed that the peak height at 0.2 V (vs. RHE) for Rh nanoparticles (Figure 3b) is remarkably higher compared to other metals. This agrees well with previous studies reported by Cooper et al. [39], which attributed such peak to the OH− adsorption by Rh.
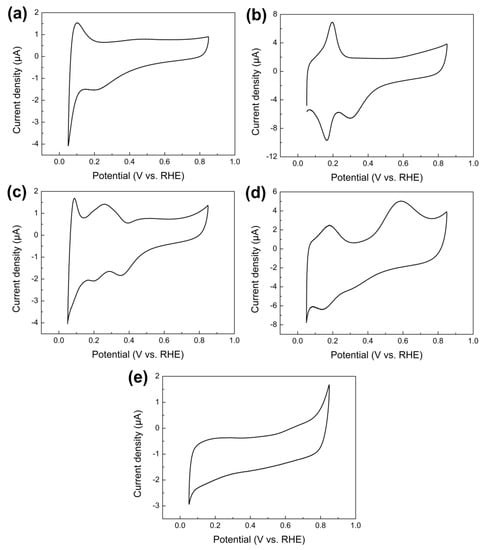
Figure 3.
CVs of the ammonia electro-oxidation on pure (a) Ru, (b) Rh, (c) Pd, (d) Ir, and (e) Au nanoparticles electrodeposited on the GCE in aqueous solution containing 1 M KOH + 0.1 M ammonia at 0.05 V s−1, respectively.
Figure 4a–e shows anodic stripping voltammograms of Cu UPD adlayer on synthesized Ru, Rh, Pd, Ir and Au nanoparticles on the GCE, respectively. All these curves showed typical Cu UPD stripping peaks. The amount and the corresponding surface area of the UPD Cu layer could be estimated from the stripping charge of the UPD Cu, as calculated by integrating the area covered by the Cu stripping current peak after subtracting the background [33,40,41]. To achieve the formation of PtML on those metal nanoparticles, the GCE was immersed into 5 mM K2PtCl4 and 0.05 M H2SO4 solution for the galvanic replacement of the surface Cu monolayer by Pt ions. Assuming that the Faraday efficiency of the replacement reaction between the UPD Cu and Pt ions is 100%, the amount and the corresponding surface area of the formed Pt can be calculated based on the amount of UPD Cu monolayer. The corresponding surface area of Pt can also be measured by the stripping charge of Cu UPD monolayer, assuming 480 μC cm−2 for PtML [19,42]. The calculated data of the Cu stripping charge, Cu amount, Pt amount and Pt surface area are summarized in Table 1.
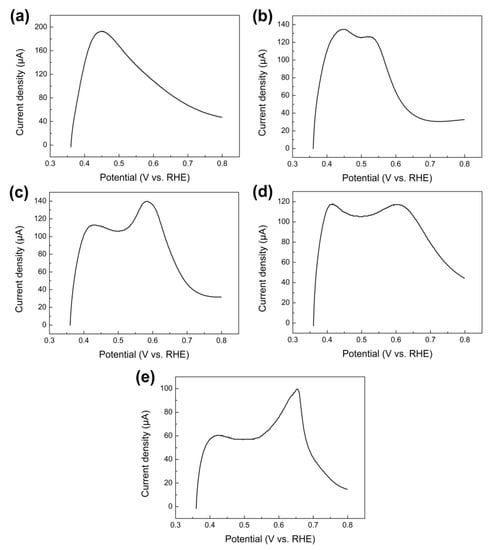
Figure 4.
Anodic stripping voltammograms after Cu UPD on electrodeposited (a) Ru, (b) Rh, (c) Pd, (d) Ir and (e) Au nanoparticles on the GCE, respectively.

Table 1.
Calculated values of the Cu stripping charge, Cu amount, Pt amount and Pt surface area.
Figure 5a–e shows the transmission electron microscopy (TEM) images of PtML nanoparticles with Ru, Rh, Pd, Ir, and Au cores, respectively. As a result of TEM sample synthesis, which is required to strip nanoparticles from GCE and then to transfer them to the Cu grid, it is unavoidable to result in the agglomeration of nanoparticles. Therefore, it could be seen that PtML nanoparticles with different composition cores were not dispersed uniformly, showing some extent of agglomeration (Figure 5) compared to freshly electrodeposited core nanoparticles (Figure 1). Nevertheless, it is clear that the overall structure of a single PtML nanoparticle with different composition cores consisted of the smaller, interconnected nanoparticles, suggesting a large surface area. Obviously, the PtML nanoparticles with different composition cores observed in SEM images (Figure 1) were composed of interconnected smaller nanoparticles. The insets of Figure 5 show the corresponding small nanoparticle size distribution histograms. It could be seen that the average nanoparticle sizes of the small PtML nanoparticles with Ru, Rh, Pd, Ir, and Au cores were approximately 4.1 nm, 5.0 nm, 2.6 nm, 4.9 nm and 4.4 nm respectively (the insets of Figure 5), and all of which had narrow nanoparticle size distribution.
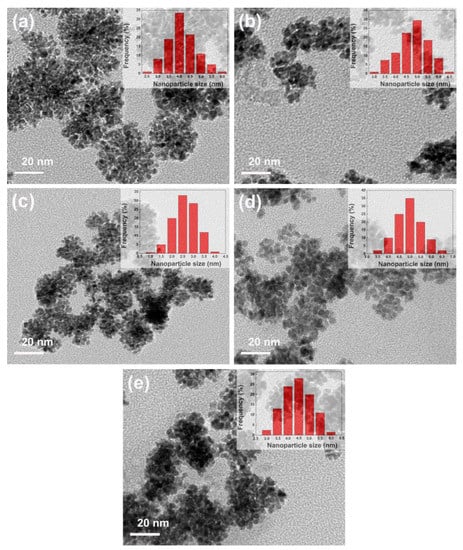
Figure 5.
TEM images of PtML particles with (a) Ru, (b) Rh, (c) Pd, (d) Ir, and (e) Au cores electrodeposited on the GCE, respectively. Insets of TEM images show the corresponding particle size distribution histogram.
Figure 6a–e shows CVs measured on PtML nanoparticles with different core compositions in 0.5 M H2SO4 solution at a scan rate of 0.05 V s−1. Compared to Figure 2, a significant change in the electrochemical behavior could be observed after applying a PtML on nanoparticles. All CV profiles had typical voltammetric characteristics of Pt in H2SO4 aqueous solution, showing characteristic potential regions including the hydrogen adsorption/desorption region from 0.05 to 0.3 V(vs. RHE) and the double-layer region from 0.3 to 0.85 V(vs. RHE) [43]. It has been extensively reported that the hydrogen desorption profile on pure polycrystalline Pt is characterized by two well-separated anodic peaks [5,36,44,45]. However, PtML nanoparticles showed only one relatively broad anodic peak in the hydrogen desorption region. Similar results have also been widely reported by previous studies, which is attributed to the interaction between the PtML and the substrate [32,46,47,48,49]. The above results provide clear evidence of the formation of the PtML on these nanoparticles. Generally, the electrochemically active surface area of pure Pt can be estimated from the CV based on the charge related with the hydrogen desorption [50,51,52,53]. However, in the case of PtML nanoparticles, the hydrogen desorption profile would be affected due to the interaction between underlying core particles and the PtML, therefore it is possibly not accurate to calculate the effective surface area of Pt. Therefore, previous studies, including the present work, used the amount of Cu monolayer to estimate the Pt surface area assuming the complete replacement of Cu by Pt (Table 1).
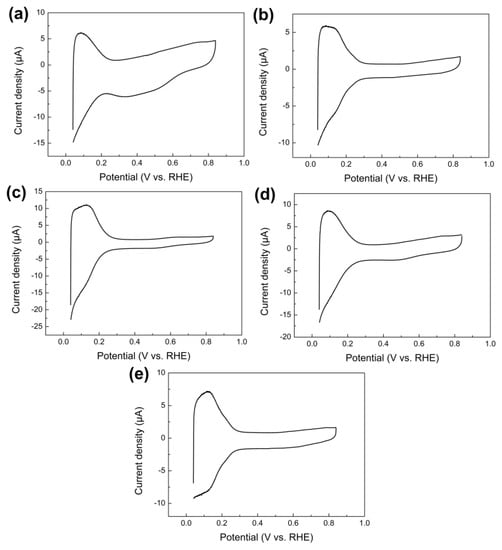
Figure 6.
CVs measured on (a) PtML/Ru nanoparticles, (b) PtML/Rh nanoparticles, (c) PtML/Pd nanoparticles, (d) PtML/Ir nanoparticles, and (e) PtML/Au nanoparticles on the GCE in 0.5 M H2SO4 solution, respectively.
To investigate the electrocatalytic activity for the electro-oxidation of ammonia, Figure 7a shows the CVs measured on five different PtML covered nanoparticles in 0.1 M ammonia + 1 M KOH. In order to reveal the SA, the current was normalized by the effective electrochemical surface area. Compared to observed CVs on bare nanoparticles (Figure 3), applying a PtML on these nanoparticles resulted in an obvious change of CV curves. All five CV curves showed a current peak around 0.7 V (vs. RHE) on the positive sweep, which is a typical feature of ammonia electro-oxidation on Pt as previously discussed. This again confirms the successful deposition of Pt on various nanoparticles by the replacement of UPD Cu. It is worth noticing that the SA of PtML covered nanoparticles strongly depends on the composition of core nanoparticles, which can be clearly seen in Figure 7b. The SA for electro-oxidation of ammonia decreases in the following order: PtML/Au nanoparticles > PtML/Ir nanoparticles ≈ PtML/Rh nanoparticles > PtML/Ru Nanoparticles ≈ PtML/Pd nanoparticles. PtML supported on Au nanoparticles showed the highest SA, which was about 8.3 times higher than that on Ru and Pd nanoparticles. In addition, compared to other Pt-based catalysts reported in previous work, the obtained PtML/Au nanoparticles exhibited relatively high SA for electrooxidation ammonia and Pt surface area among the various catalysts (Table 2). As a typical representative, as-prepared PtML/Au nanoparticles are provided in Figure 7c since it exhibited highest SA among the obtained catalysts. The high-resolution transmission electron microscopy (HRTEM) image showed the clear lattice fringes indicating a high crystallinity of the PtML/Au nanoparticles, and it also revealed that the PtML/Au nanoparticles were composed of multiple crystalline domains, suggesting a polycrystalline structure. The widely accepted mechanism of ammonia electro-oxidation on Pt was proposed by Gerischer and Mauerer [54], involving dehydrogenation of NH3,ads to NHx,ads (x = 1 or 2) intermediates and Nads. The partially dehydrogenated NH2,ads species are key precursors to promote the formation of N2H4 that is then dehydrogenated quickly to N2 [54]. This mechanism has been later confirmed by experimental methods [55,56] and density functional theory calculations [57], supporting that NH2 species are active intermediates, and therefore a Pt surface with a higher binding energy of NH2 intermediates has higher activity for ammonia electro-oxidation. Previous studies found that due to the lattice mismatch between the PtML and the substrate, different level of tensile or compressive strain is exerted on the PtML depending on the substrate material [19]. For example, Au exerts a tensile strain on PtML (over 4%) while other metals such as Pd, Ru and Rh exert on it a comprehensive strain (about −2 ~ −4%) [19]. This affects the binding abilities of the PtML and thus changes the surface activity. In consistence with theoretical calculations [58,59], Li et al. [19] demonstrated that the tensile surface strain in the PtML (e.g., PtML supported on Au) tends to upshift the weighted center of the d-band (εd) in energy and thus increases the binding of CO and OH. This contributes to a significant enhancement in the activity of the PtML on Au towards electrocatalytic oxidation of methanol and ethanol [19]. On the contrary, PtML on other substrates (i.e., Ru, Rh, Ir and Pd) is under compressive strain, resulting in decreased activity for the electro-oxidation of methanol and ethanol [19]. It is interesting to note that the substrate–induced surface strain effect on activity was also been found in the present work. For example, the PtML supported on electrodeposited Au nanoparticles showed significantly higher SA compared to that supported on electrodeposited Pd and Ru nanoparticles. Similarly, the tensile surface strain in the PtML induced by the substrate Au is expected to increase the binding of the PtML for active intermediates such as NH2 to activate ammonia electro-oxidation and thus improves the SA. On the contrary, Pd and Ru nanoparticles exert a compressive strain on the PtML [19] and therefore they have an opposite effect compared to PtML on Au, exhibiting low specific activities among all investigated PtML nanoparticles. However, it was noticed that PtML on electrodeposited Rh and Ir nanoparticles had remarkably higher activity than those on Pd and Ru nanoparticles despite of the compressive strain exerted by Rh or Ir on Pt, which is different with that reported by previous studies [19]. This was due to the positive synergistic effect between Pt and Rh or Ir. Cooper et al. [39] pointed out that the presence of Rh along with Pt will increase active sites on the Pt surface for ammonia electro-oxidation. The synergic effect of Ir to Pt has also been reported by previous studies, which was attributed to the ability of Ir to dehydrogenate ammonia molecules at lower potentials [36,60,61]. Our results agree well with these studies [36,39,60].
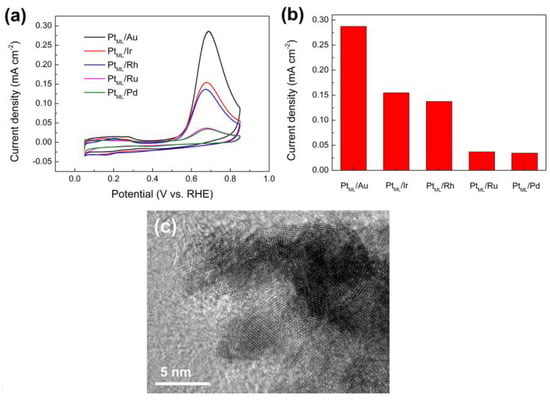
Figure 7.
(a) CVs of the ammonia electro-oxidation on PtML supported on Ru nanoparticles, Rh nanoparticles, Pd nanoparticles, Ir nanoparticles, and Au nanoparticles on the GCE in aqueous solution containing 1 M KOH + 0.1 M ammonia at 0.05 V s−1, respectively. (b) Comparison of the SA of PtML supported on Ru nanoparticles, Rh nanoparticles, Pd nanoparticles, Ir nanoparticles, and Au nanoparticles on the GCE. (c) HRTEM image of as-prepared PtML/Au nanoparticles.
Table 2.
Comparison of SA for ammonia electro-oxidation and Pt surface area of as-prepared electrocatalysts with previous work.
3. Experimental Section
3.1. Reagents
RuCl3, RhCl3, PdCl2, IrCl3, HAuCl4·3H2O, K2PtCl4, CuSO4, H2SO4, and (NH4)2SO4 were obtained from Beijing Chemicals (Beijing, China). All reagents employed were of analytical grade and used directly without further purification. All the aqueous solutions used in this work were prepared with ultrapure water (Milli-Q Millipore, > 18.2 MΩ cm).
3.2. Electrode Synthesis and Characterization
All electrochemical syntheses and characterizations were performed on a three-electrode setup. A glassy carbon electrode (GCE, 5 mm in diameter) served as the working electrode, a Pt plate (1 × 1 cm2) electrode and a mercury sulfate electrode (MSE) served as counter and reference electrodes respectively. For the convenience of comparison, the potential measured by MSE was referred to reversible hydrogen electrode (vs. RHE). Prior to the electrodeposition, the GCE was first polished and then ultrasonically cleaned in ultrapure water for 3 min. Through controlling the deposition potential under potentiostatic conditions, nanoparticles with different compositions were synthesized on the surface of GCE. Some preliminary experiments had been carried out to find out the optimal electrodeposition potentials and times, and the following conditions were employed to synthesize nanoparticles. Ru nanoparticles were synthesized on the GCE at −0.8 V (vs. RHE) for 8 s in 5 mM RuCl3 + 0.05 M H2SO4 solution. Rh nanoparticles were synthesized on the GCE at −0.05 V (vs. RHE) for 0.2 s in 5 mM RhCl3 + 0.05 M H2SO4 solution. Pd nanoparticles were synthesized on the GCE at 0 V (vs. RHE) for 0.2 s in 5 mM PdCl2 + 0.05 M H2SO4 solution. Ir nanoparticles were synthesized on the GCE at −0.7 V (vs. RHE) for 800 s in 5 mM IrCl3 + 0.05 M H2SO4 solution. Au nanoparticles were synthesized on the GCE at 0 V (vs. RHE) for 0.2 s in 5 mM HAuCl4 + 0.05 M H2SO4 solution. The synthesis of PtML on electrodeposited nanoparticles was achieved by a well-established method, as described as follows. After the synthesis of core nanoparticles on the GCE, the modified GCE, rinsed thoroughly with ultrapure water, was transferred into 0.05 M H2SO4 + 0.05 M CuSO4 aqueous solution for the subsequent Cu UPD. The modified GCE was initially kept at 0.8 V (vs. RHE) for 20 s to make sure there is no Cu on the surface [18,33]. The electrode potential was subsequently swept at 0.4 V s−1 to the Cu UPD potential of 0.36 V (vs. RHE) and held at this potential for 60 s [18,33]. When the Cu UPD was finished, the corresponding GCE, rinsed thoroughly with ultrapure water, was immediately transferred into solution containing 5 mM K2PtCl4 and 0.05 M H2SO4 under the protection of high-purity Ar (99.999%) atmosphere to achieve the replacement of Cu with Pt2+ ions for 20 min. After the replacement reaction, the PtML covered nanoparticles on the GCE were rinsed thoroughly with ultrapure water and dried in a nitrogen stream. The amount of the deposited Cu monolayer by UPD could be measured by the anodic stripping technique. For anodic stripping tests, the electrode potential was kept at 0.36 V (vs. RHE) for 60 s to complete the Cu UPD, and then swept back to 0.8 V (vs. RHE) at 0.4 V s−1 to strip off the Cu layer [33].
The particle size and dispersion degree of the PtML decorated nanoparticles were determined by scanning electron microscopy (SEM, S–4800, Hitachi, Tokyo, Japan) and transmission scanning electron microscopy (TEM, JEM–2100F, JEOL, Tokyo, Japan). For the TEM measurements, the electrodeposited nanoparticles were gently scraped and dispersed in ethanol on surface of GCE, and then the nanoparticle dispersions were transferred onto a Cu grid, which was directly conducted on surface of GCE.
3.3. Electrochemical Tests
Electrochemical tests were also carried out on the electrochemical workstation (IviumStat, Eindhoven, Netherland). Electrodeposited pure nanoparticles, or PtML covered nanoparticles, on the GCE served as the working electrode. A Pt plate (1 × 1 cm2) served as the counter electrode. A CV technique was used to investigate the electrochemical performance of nanoparticles with different compositions before and after the coverage of the PtML. For the CVs tested in 0.5 M H2SO4 aqueous solution, an MSE was used as the reference electrode with a scan rate of 0.05 V s−1. The electro-oxidation of ammonia was conducted in aqueous solution containing 1 M KOH + 0.1 M ammonia by CV at 0.01 V s−1, and a Hg/HgO (filled with 1 M KOH) served as the reference electrode in this case. All the electrochemical measurements were performed in testing solutions saturated with argon gas (99.999%). All the measured potentials by the reference electrode were referred to as the RHE for the convenience of comparison. All experiments were carried out at controlled temperature of 25 ± 1 °C.
4. Conclusions
The effect of the composition of nanoparticle supports on the electrocatalytic activity of PtML electrocatalysts for ammonia oxidation was investigated. A clean electrochemical approach free of binders, surfactants, capping agents and reductants was proposed to synthesis nanoparticle supports with various compositions including Ru, Rh, Pd, Ir and Au. PtML supported on different nanoparticles were obtained by the Cu UPD and subsequent replacement of Pt ions. The SA of the PtML electrocatalysts was significantly dependent on the composition of nanoparticle supports, which decreased in the following order: PtML/Au nanoparticles > PtML/Ir nanoparticles ≈ PtML/Rh nanoparticles > PtML/Ru Nanoparticles ≈ PtML/Pd nanoparticles. The SA of the PtML supported on Au nanoparticles was about 8.3 times higher than that of the PtML supported on Ru Nanoparticles and Pd Nanoparticles. This trend was generally related to the change of the surface strain in the PtML exerted by the nanoparticle support. As the surface strain in the PtML exerted by the nanoparticle support changed from a tensile strain to a compressive strain, the SA of PtML electrocatalysts decreased. Therefore, the highest SA was observed for the PtML supported on the Au nanoparticles, where the PtML was under a tensile strain. On the contrary, for Ru, Rh and Pd nanoparticle supports that exerted on the PtML a tensile strain had much lower specific activities.
Author Contributions
J.L. and B.L. contributed equally to this work, that is, they performed the synthesis, electrochemical characterizations of the catalysts and wrote the draft. X.C. and J.Z. carried out structural characterizations with SEM and TEM. Y.W. and Y.D. contributed to the data analysis. W.H. supervised the project. C.Z. supervised the project, designed the experiments and revised manuscript.
Funding
This work was supported by the National Science Foundation for Excellent Young Scholar (No. 51722403), the National Natural Science Foundation of China (Nos. 51771134, 51571151, 51801134 and 51701139), the National Natural Science Foundation of China and Guangdong Province (No. U1601216), the Tianjin Natural Science Foundation (Nos. 16JCYBJC17600 and 18JCJQJC46500), and the National Youth Talent Support Program.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Li, G.; Kanezashi, M.; Tsuru, T. Catalytic Ammonia Decomposition over High-Performance Ru/Graphene Nanocomposites for Efficient COx-Free Hydrogen Production. Catalysts 2017, 7, 23. [Google Scholar] [CrossRef]
- Liu, J.; Chen, B.; Kou, Y.; Liu, Z.; Chen, X.; Li, Y.B.; Deng, Y.D.; Han, X.P.; Hu, W.B.; Zhong, C. Pt-Decorated highly porous flower-like Ni particles with high mass activity for ammonia electro-oxidation. J. Mater. Chem. A 2016, 4, 11060–11068. [Google Scholar] [CrossRef]
- Liao, C.H.; Huang, C.W.; Wu, J.C.S. Hydrogen Production from Semiconductor-based Photocatalysis via Water Splitting. Catalysts 2012, 2, 490–516. [Google Scholar] [CrossRef]
- Liu, J.; Liu, B.; Ni, Z.Y.; Deng, Y.D.; Zhong, C.; Hu, W.B. Improved Catalytic Performance of Pt/TiO2 Nanotubes Electrode for Ammonia Oxidation under UV-light Illumination. Electrochim. Acta 2014, 150, 146–150. [Google Scholar] [CrossRef]
- Liu, J.; Zhong, C.; Du, X.T.; Wu, Y.T.; Xu, P.Z.; Liu, J.B.; Hu, W.B. Pulsed Electrodeposition of Pt Particles on Indium Tin Oxide Substrates and Their Electrocatalytic Properties for Methanol Oxidation. Electrochim. Acta 2013, 100, 164–170. [Google Scholar] [CrossRef]
- Sala, R.; Dzida, J.; Krasowski, J. Ammonia concentration distribution measurements on selective catalytic reduction catalysts. Catalysts 2018, 8, 231. [Google Scholar] [CrossRef]
- Strickland, G. Hydrogen Derived from Ammonia: Small-scale Costs. Int. J. Hydrogen Energy 1984, 9, 759–766. [Google Scholar] [CrossRef]
- Jiang, D.; Zhang, S.; Zeng, Y.; Wang, P.; Zhong, Q. Active site of O2 and its improvement mechanism over Ce-Ti catalyst for NH3-SCR reaction. Catalysts 2018, 8, 336. [Google Scholar] [CrossRef]
- Yao, K.; Cheng, Y.F. Fabrication by electrolytic deposition of Pt-Ni electrocatalyst for oxidation of ammonia in alkaline solution. Int. J. Hydrogen Energy 2008, 33, 6681–6686. [Google Scholar] [CrossRef]
- Wang, R.; Wu, X.; Zou, C.; Li, X.; Du, Y. NOx removal by selective catalytic reduction with ammonia over a hydrotalcite-derived NiFe mixed oxide. Catalysts 2018, 8, 384. [Google Scholar] [CrossRef]
- Boggs, B.K.; Botte, G.G. On-board Hydrogen Storage and Production: An Application of Ammonia Electrolysis. J. Power Sources 2009, 192, 573–581. [Google Scholar] [CrossRef]
- Galipaud, J.; Roy, C.; Martin, M.H.; Garbarino, S.; Roué, L.; Guay, D. Electrooxidation of Ammonia at Tuned (100)Pt Surfaces by Using Epitaxial Thin Films. ChemElectroChem 2015, 2, 1187–1198. [Google Scholar] [CrossRef]
- De Vooys, A.C.A.; Koper, M.T.M.; van Santen, R.A.; van Veen, J.A.R. The Role of Adsorbates in the Electrochemical Oxidation of Ammonia on Noble and Transition Metal Electrodes. J. Electroanal. Chem. 2001, 506, 127–137. [Google Scholar] [CrossRef]
- Liu, J.; Fan, X.Y.; Liu, X.R.; Song, Z.S.; Deng, Y.D.; Han, X.P.; Hu, W.B.; Zhong, C. Synthesis of Cubic-Shaped Pt Particles with (100) Preferential Orientation by a Quick, One-step and Clean Electrochemical Method. ACS Appl. Mater. Interfaces 2017, 9, 18856–18864. [Google Scholar] [CrossRef] [PubMed]
- Adzic, R.R.; Zhang, J.; Sasaki, K.; Vukmirovic, M.B.; Shao, M.; Wang, J.X.; Nilekar, A.U.; Mavrikakis, M.; Valerio, J.A.; Uribe, F. Platinum Monolayer Fuel Cell Electrocatalysts. Top. Catal. 2007, 46, 249–262. [Google Scholar] [CrossRef]
- Alayoglu, S.; Nilekar, A.U.; Mavrikakis, M.; Eichhorn, B. Ru–Pt Core–Shell Nanoparticles for Preferential Oxidation of Carbon Monoxide in Hydrogen. Nat. Mater. 2008, 7, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Shao, M.H.; Peles, A.; Shoemaker, K.; Gummalla, M.; Njoki, P.N.; Luo, J.; Zhong, C.J. Enhanced Oxygen Reduction Activity of Platinum Monolayer on Gold Nanoparticles. J. Phys. Chem. Lett. 2011, 2, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chen, B.; Ni, Z.; Deng, Y.; Han, X.; Hu, W.; Zhong, C. Improving the Electrocatalytic Activity of Pt Monolayer Catalysts for Electro-oxidation of Methanol, Ethanol and Ammonia by Tailoring the Surface Morphology of the Supporting Core. ChemElectroChem 2016, 3, 537–551. [Google Scholar] [CrossRef]
- Li, M.; Liu, P.; Adzic, R.R. Platinum Monolayer Electrocatalysts for Anodic Oxidation of Alcohols. J. Phys. Chem. Lett. 2012, 3, 3480–3485. [Google Scholar] [CrossRef] [PubMed]
- Gong, K.; Vukmirovic, M.B.; Ma, C.; Zhu, Y.M.; Adzic, R.R. Synthesis and Catalytic Activity of Pt Monolayer on Pd Tetrahedral Nanocrystals with CO-adsorption-induced Removal of Surfactants. J. Electroanal. Chem. 2011, 662, 213–218. [Google Scholar] [CrossRef]
- Wang, J.X.; Inada, H.; Wu, L.J.; Zhu, Y.M.; Choi, Y.M.; Liu, P.; Zhou, W.P.; Adzic, R.R. Oxygen Reduction on Well-Defined Core-Shell Nanocatalysts: Particle Size, Facet, and Pt Shell Thickness Effects. J. Am. Chem. Soc. 2009, 131, 17298–17302. [Google Scholar] [CrossRef] [PubMed]
- Shao, M.H.; Liu, P.; Zhang, J.L.; Adzic, R. Origin of Enhanced Activity in Palladium Alloy Electrocatalysts for Oxygen Reduction Reaction. J. Phys. Chem. B 2007, 111, 6772–6775. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, K.; Naohara, H.; Choi, Y.; Cai, Y.; Chen, W.F.; Liu, P.; Adzic, R.R. Highly Stable Pt Monolayer on PdAu Nanoparticle Electrocatalysts for the Oxygen Reduction Reaction. Nat. Commun. 2012, 3, 1115. [Google Scholar] [CrossRef] [PubMed]
- Kuttiyiel, K.A.; Sasaki, K.; Choi, Y.; Su, D.; Liu, P.; Adzic, R.R. Bimetallic IrNi Core Platinum Monolayer Shell Electrocatalysts for the Oxygen Reduction Reaction. Energy Environ. Sci. 2012, 5, 5297–5304. [Google Scholar] [CrossRef]
- Gong, K.; Su, D.; Adzic, R.R. Platinum-monolayer Shell on AuNi0.5Fe Nanoparticle Core Electrocatalyst With High Activity and Stability for the Oxygen Reduction Reaction. J. Am. Chem. Soc. 2010, 132, 14364–14366. [Google Scholar] [CrossRef] [PubMed]
- Brankovic, S.R.; Wang, J.X.; Adžić, R.R. Metal Monolayer Deposition by Replacement of Metal Adlayers on Electrode Surfaces. Surf. Sci. 2001, 474, L173–L179. [Google Scholar] [CrossRef]
- Sasaki, K.; Wang, J.X.; Naohara, H.; Marinkovic, N.; More, K.; Inada, H.; Adzic, R.R. Recent Advances in Platinum Monolayer Electrocatalysts for Oxygen Reduction Reaction: Scale-up Synthesis, Structure and Activity of Pt Shells on Pd Cores. Electrochim. Acta 2010, 55, 2645–2652. [Google Scholar] [CrossRef]
- Shao, M.H.; He, G.N.; Peles, A.; Odell, J.H.; Zeng, J.; Su, D.; Tao, J.; Yu, T.; Zhu, Y.; Xia, Y.N. Manipulating the Oxygen Reduction Activity of Platinum Shells with Shape-controlled Palladium Nanocrystal Cores. Chem. Commun. 2013, 49, 9030–9032. [Google Scholar] [CrossRef]
- Wang, X.M.; Orikasa, Y.; Takesue, Y.; Inoue, H.; Nakamura, M.; Minato, T.; Hoshi, N.; Uchimoto, Y. Quantitating the Lattice Strain Dependence of Monolayer Pt Shell Activity Toward Oxygen Reduction. J. Am. Chem. Soc. 2013, 135, 5938–5941. [Google Scholar] [CrossRef]
- Zhang, J.; Lima, F.H.B.; Shao, M.H.; Sasaki, K.; Wang, J.X.; Hanson, J.; Adzic, R.R. Platinum Monolayer on Nonnoble Metal-noble Metal Core-Shell Nanoparticle Electrocatalysts for O2 Reduction. J. Phys. Chem. B 2005, 109, 22701–22704. [Google Scholar] [CrossRef]
- Zhang, J.L.; Vukmirovic, M.B.; Xu, Y.; Mavrikakis, M.; Adzic, R.R. Controlling the Catalytic Activity of Platinum-monolayer Electrocatalysts for Oxygen Reduction with Different Substrates. Angew. Chem. Int. Ed. 2005, 117, 2170–2173. [Google Scholar] [CrossRef]
- Ni, Z.Y.; Liu, J.; Wu, Y.T.; Liu, B.; Zhao, C.K.; Deng, Y.D.; Hu, W.B.; Zhong, C. Fabrication of Platinum Submonolayer Electrodes and Their High Electrocatalytic Activities for Ammonia Oxidation. Electrochim. Acta 2015, 177, 30–35. [Google Scholar] [CrossRef]
- Yu, Y.L.; Hu, Y.P.; Liu, X.W.; Deng, W.Q.; Wang, X. The Study of Pt@Au Electrocatalyst Based on Cu Underpotential Deposition and Pt Redox Replacement. Electrochim. Acta 2009, 54, 3092–3097. [Google Scholar] [CrossRef]
- Oppedisano, D.K.; Jones, L.A.; Junk, T.; Bhargava, S.K. Ruthenium Electrodeposition from Aqueous Solution at High Cathodic Overpotential. J. Electrochem. Soc. 2014, 161, D489–D494. [Google Scholar] [CrossRef]
- Le Vot, S.; Roué, L.; Bélanger, D. Electrodeposition of Iridium onto Glassy Carbon and Platinum Electrodes. Electrochim. Acta 2012, 59, 49–56. [Google Scholar] [CrossRef]
- Vidal-Iglesias, F.J.; Solla-Gullón, J.; Montiel, V.; Feliu, J.M.; Aldaz, A. Screening of Electrocatalysts for Direct Ammonia Fuel Cell: Ammonia Oxidation on PtMe (Me: Ir, Rh, Pd, Ru) and Preferentially Oriented Pt(100) Nanoparticles. J. Power Sources 2007, 171, 448–456. [Google Scholar] [CrossRef]
- Yao, K.; Cheng, Y.F. Electrodeposited Ni–Pt Binary Alloys as Electrocatalysts for Oxidation of Ammonia. J. Power Sources 2007, 173, 96–101. [Google Scholar] [CrossRef]
- Deng, X.H.; Wu, Y.T.; He, M.F.; Dan, C.Y.; Chen, Y.J.; Deng, Y.D.; Jiang, D.H.; Zhong, C. Electrochemical Deposition of Pt Particles on Indium Tin Oxide Electrode and Their Electrocatalytic Applications in Ammonia Oxidation. Acta Chim. Sin. 2011, 69, 1041–1046. (In Chinese) [Google Scholar]
- Cooper, M.; Botte, G.G. Hydrogen Production from the Electro-oxidation of Ammonia Catalyzed by Platinum and Rhodium on Raney Nickel Substrate. J. Electrochem. Soc. 2006, 153, A1894. [Google Scholar] [CrossRef]
- Kazemi, R.; Kiani, A. Deposition of Palladium Submonolayer on Nanoporous Gold Film and Investigation of Its Performance for the Methanol Electrooxidation Reaction. Int. J. Hydrogen Energy 2012, 37, 4098–4106. [Google Scholar] [CrossRef]
- Santos, M.C.; Mascaro, L.H.; Machado, S.A.S. Voltammetric and Rotating Ring-disk Studies of Underpotential Deposition of Ag and Cu on Polycrystalline Au Electrodes in Aqueous H2SO4. Electrochim. Acta 1998, 43, 2263–2272. [Google Scholar] [CrossRef]
- Markovic, N.M.; Gasteiger, H.A.; Ross, P.N.J. Copper Electrodeposition on Pt (111) in the Presence of Chloride and (bi) Sulfate: Rotating Ring-Pt (111) Disk Electrode Studies. Langmuir 1995, 11, 4098–4108. [Google Scholar] [CrossRef]
- Markovíc, N.M.; Ross, P.N., Jr. Surface Science Studies of Model Fuel Cell Electrocatalysts. Surf. Sci. Rep. 2002, 45, 117–229. [Google Scholar] [CrossRef]
- Zhong, C.; Hu, W.B.; Cheng, Y.F. On the Essential Role of Current Density in Electrocatalytic Activity of The Electrodeposited Platinum for Oxidation of Ammonia. J. Power Sources 2011, 196, 8064–8072. [Google Scholar] [CrossRef]
- Liu, J.; Hu, W.B.; Zhong, C.; Cheng, Y.F. Surfactant-free Electrochemical Synthesis of Hierarchical Platinum Particle Electrocatalysts for Oxidation of Ammonia. J. Power Sources 2013, 223, 165–174. [Google Scholar] [CrossRef]
- Liu, P.; Ge, X.; Wang, R.; Ma, H.; Ding, Y. Facile Fabrication of Ultrathin Pt Overlayers onto Nanoporous Metal Membranes via Repeated Cu UPD and In situ Redox Replacement Reaction. Langmuir 2009, 25, 561–567. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Kuttiyiel, K.A.; Sasaki, K.; Su, D.; Yang, T.H.; Park, G.G.; Zhang, C.; Chen, G.; Adzic, R.R. Pt Monolayer Shell on Nitrided Alloy Core—A Path to Highly Stable Oxygen Reduction Catalyst. Catalysts 2015, 5, 1321–1332. [Google Scholar] [CrossRef]
- Kuttiyiel, K.A.; Sasaki, K.; Su, D.; Vukmirovic, M.B.; Marinkovic, N.S.; Adzic, R.R. Pt Monolayer on Au-stabilized PdNi Core-Shell Nanoparticles for Oxygen Reduction Reaction. Electrochim. Acta 2013, 110, 267–272. [Google Scholar] [CrossRef]
- Choi, Y.; Kuttiyiel, K.A.; Labis, J.P.; Sasaki, K.; Park, G.G.; Yang, T.H.; Adzic, R.R. Enhanced Oxygen Reduction Activity of IrCu Core Platinum Monolayer Shell Nano-Electrocatalysts. Top. Catal. 2013, 56, 1059–1064. [Google Scholar] [CrossRef]
- Lee, E.P.; Peng, Z.; Cate, D.M.; Yang, H.; Campbell, C.T.; Xia, Y. Growing Pt Nanowires as a Densely Packed Array on Metal Gauze. J. Am. Chem. Soc. 2007, 129, 10634–10635. [Google Scholar] [CrossRef]
- Lim, B.; Jiang, M.; Camargo, P.H.; Cho, E.C.; Tao, J.; Lu, X.; Zhu, Y.; Xia, Y. Pd-Pt Bimetallic Nanodendrites with High Activity for Oxygen Reduction. Science 2009, 324, 1302–1305. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Du, X.T.; Yang, Y.; Deng, Y.D.; Hu, W.B.; Zhong, C. A One-step, Clean, Capping-agent-free Electrochemical Approach to Prepare Pt nanoparticles with Preferential (100) Orientation and Their High Electrocatalytic Activities. Electrochem. Commun. 2015, 58, 6–10. [Google Scholar] [CrossRef]
- Wu, S.; Liu, J.; Tian, Z.; Cai, Y.; Ye, Y.; Yuan, Q.; Liang, C. Highly Dispersed Ultrafine Pt Nanoparticles on Reduced Graphene Oxide Nanosheets: In Situ Sacrificial Template Synthesis and Superior Electrocatalytic Performance for Methanol Oxidation. ACS Appl. Mater. Interfaces 2015, 7, 22935–22940. [Google Scholar] [CrossRef] [PubMed]
- Gerischer, H.; Mauerer, A. Untersuchungen Zur Anodischen Oxidation Von Ammoniak an Platin-elektroden. J. Electroanal. Chem. 1970, 25, 421–433. [Google Scholar] [CrossRef]
- Vidaliglesias, F.; Sollagullon, J.; Perez, J.; Aldaz, A. Evidence by SERS of Azide Anion Participation in Ammonia Electrooxidation in Alkaline Medium on Nanostructured Pt Electrodes. Electrochem. Commun. 2006, 8, 102–106. [Google Scholar] [CrossRef]
- Rosca, V.; Koper, M.T. Electrocatalytic Oxidation of Ammonia on Pt(111) and Pt(100) Surfaces. Phys. Chem. Chem. Phys. 2006, 8, 2513–2524. [Google Scholar] [CrossRef] [PubMed]
- Novell-Leruth, G.; Valcárcel, A.; Clotet, A.; Ricart, J.M.; Pérez-Ramírez, J. DFT Characterization of Adsorbed NHx Species on Pt(100) and Pt(111) Surfaces. J. Phys. Chem. B 2005, 109, 18061–18069. [Google Scholar] [CrossRef] [PubMed]
- Hammer, B.; Nørskov, J.K. Theoretical Surface Science and Catalysis—Calculations and Concepts. Adv. Catal. 2000, 45, 71–129. [Google Scholar]
- Greeley, J.; Nørskov, J.K.; Mavrikakis, M. Electronic Structure and Catalysis on Metal Surfaces. Annu. Rev. Phys. Chem. 2002, 53, 319–348. [Google Scholar] [CrossRef]
- Endo, K.; Katayama, Y.; Miura, T. Pt–Ir and Pt–Cu Binary Alloys as the Electrocatalyst for Ammonia Oxidation. Electrochim. Acta 2004, 49, 1635–1638. [Google Scholar] [CrossRef]
- Zhong, C.; Liu, J.; Ni, Z.Y.; Deng, Y.D.; Chen, B.; Hu, W.B. Shape-controlled Synthesis of Pt-Ir Nanocubes with Preferential (100) Orientation and Their Unusual Enhanced Electrocatalytic Activities. Sci. China Mater. 2014, 57, 13–25. [Google Scholar] [CrossRef]
- Endo, K.; Nakamura, K.; Katayama, Y.; Miura, T. Pt–Me (Me = Ir, Ru, Ni) binary alloys as an ammonia oxidation anode. Electrochim. Acta 2004, 49, 2503–2509. [Google Scholar] [CrossRef]
- Vidal-Iglesias, F.J.; Solla-Gullón, J.; Rodríguez, P.; Herrero, E.; Montiel, V.; Feliu, J.M.; Aldaz, A. Shape-dependent electrocatalysis: Ammonia oxidation on platinum nanoparticles with preferential (100) surfaces. Electrochem. Commun. 2004, 6, 1080–1084. [Google Scholar] [CrossRef]
- Vidal-Iglesias, F.J.; García-Aráez, N.; Montiel, V.; Feliu, J.M.; Aldaz, A. Selective electrocatalysis of ammonia oxidation on Pt(100) sites in alkaline medium. Electrochem. Commun. 2003, 5, 22–26. [Google Scholar] [CrossRef]
- Bertin, E.; Garbarino, S.; Guay, D.; Solla-Gullón, J.; Vidal-Iglesias, F.J.; Feliu, J.M. Electrodeposited Platinum Thin Films with Preferential (100) Orientation: Characterization and Electrocatalytic Properties for Ammonia and Formic Acid Oxidation. J. Power Sources 2013, 225, 323–329. [Google Scholar] [CrossRef]
- Ojani, R.; Hasheminejad, E.; Raoof, J.B. Direct growth of 3D flower-like Pt nanostructures by a template-free electrochemical route as an efficient electrocatalyst for methanol oxidation reaction. Energy 2015, 90, 1122–1131. [Google Scholar] [CrossRef]
- Li, C.A.; Han, K.N.; Bui, M.P.N.; Pham, X.H.; Hong, M.H.; Irfan, M.; Kim, Y.S.; Seong, G.H. Morphology-controlled synthesis and electrocatalytic characteristics of platinum structures on micro-patterned carbon nanotube platforms. J. Appl. Electrochem. 2011, 41, 1425–1431. [Google Scholar] [CrossRef]
- El-Nagar, G.A.; Mohammad, A.M.; El-Deab, M.S.; El-Anadouli, B.E. Electrocatalysis by design: Enhanced electrooxidation of formic acid at platinum nanoparticles–nickel oxide nanoparticles binary catalysts. Electrochim. Acta 2013, 94, 62–71. [Google Scholar] [CrossRef]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).