Abstract
This short review is aimed at giving an overview of catalytic carbonylative double cyclization reactions, which are processes in which suitable organic substrates and carbon monoxide are sequentially activated by a promoting a catalyst to form two new cycles with the concomitant incorporation of carbon monoxide as a carbonyl function in the final product. Paradigmatic examples of this powerful synthetic methodology, which allows the one-step synthesis of complex molecular architectures from simple building blocks using the simplest and readily available C-1 unit (CO), are illustrated and discussed. The review is divided into five sections: (1) Introduction, (2) Functionalized Olefinic Substrates, (3) Functionalized Acetylenic Substrates, (4) Functionalized Halides, (5) Conclusions and Future Perspectives.
1. Introduction
The importance of carbon monoxide as a C-1 unit in organic synthesis can hardly be overemphasized [1]. It is a readily available feedstock that can be easily obtained by steam reforming of light hydrocarbons (including natural gas), partial oxidation of petroleum hydrocarbons, or gasification of coal to give syngas (CO and H2) [2]. It can be installed into an organic substrate, usually under catalytic conditions, leading to the direct formation of high value-added carbonylated compounds with 100% atom economy (carbonylation reactions) [1]. It should also be noted that recent progress in the chemical utilization of carbon dioxide has led to the implementation of efficient methods for reducing CO2 to CO [3]. Therefore, carbonylation reactions may also represent a very important indirect method for the conversion of carbon dioxide (the main waste currently produced by human activities, and principal responsible for the greenhouse effect [4]) into useful chemicals and materials.
Since their discovery at the beginning of the 19th century, carbonylation reactions have acquired steadily increasing importance both at the industrial and academic level. A huge number of examples of these important processes have been reported in the scientific as well as the patent literature [1]. In particular, the development of more efficient and selective catalytic systems associated with the use of suitably functionalized starting materials has opened the way to achieving sophisticated synthetic processes, with the formation of complex carbonylated molecular architectures that have potential applications in many fields of science (including drug discovery and material science) in one step. Among these processes, carbonylative double cyclization represents a particularly important methodology as it makes possible the construction of two new cycles in one synthetic procedure with formation of carbonylated polycyclic structures starting from readily available and suitably functionalized substrates.
The present short review is aimed at offering a description of paradigmatic synthetic methodologies based on catalytic carbonylative double cyclization reactions.
2. Functionalized Olefinic Substrates
It is well-know that palladium(II)-based catalysts activate unsaturated carbon–carbon bonds towards the attack of a variety of nucleophilic groups (mainly oxygen- or nitrogen-based). The intramolecular version of this reactivity is of particular importance as it allows the construction of heterocyclic derivatives in a straightforward manner and under mild reaction conditions (Pd(II)-catalyzed heterocyclization reactions) [5]. On the other hand, it is also very well known that Pd(II) catalysts promote many important kinds of carbonylation processes, particularly under oxidative conditions, including cyclization processes in which carbon monoxide is inserted as a carbonyl function inside the newly formed ring (cyclocarbonylation reactions) [6]. It is therefore not surprising that several important methods have been developed in which a single Pd(II)-based catalytic system promotes, in one synthetic step, the sequential heterocyclization−cyclocarbonylation of suitably functionalized olefinic substrates that carry two nucleophilic moieties placed in appropriate positions to undergo double cyclization.
Pioneering studies on this kind of reactivity were conducted by the Semmelhack and Yoshida research groups during the 1980s. In 1984, Semmelhack et al. reported the Pd(II)-promoted stereoselective carbonylative double cyclization of 1-(2-(hydroxymethyl)phenyl)prop-2-en-1-ols to give 3,3a,5,9b-tetrahydro-2H-furo[3,2-c]isochromen-2-ones with a cis junction between the newly formed rings using a stoichiometric amount of Pd(OAc)2 [7]. The process started with the intramolecular 6-exo-trig nucleophilic attack of the benzylic hydroxyl group to the double bond, activated by coordination to the Pd(II) center. This led to the formation of a cis-type alkylpalladium intermediate stabilized by chelation of the second hydroxyl group. The final bicyclic product was then formed through CO migratory insertion followed by intramolecular nucleophilic displacement by the hydroxyl, possibly via the formation of a palladacycle followed by reductive elimination (Scheme 1).
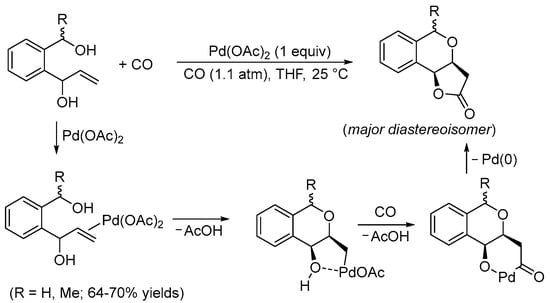
Scheme 1.
Synthesis of 3,3a,5,9b-tetrahydro-2H-furo[3,2-c]isochromen-2-ones from 1-(2-(hydroxymethyl)phenyl)prop-2-en-1-ols [7].
Interestingly, when an oxidant for Pd(0) such as CuCl2 was employed to make the process catalytic, the reaction led to the formation of (E)-(2-(3-chloroprop-1-en-1-yl)phenyl)methanol from allylic chlorination (74% yield) [7]. Later on, however, suitable conditions were elaborated by the Yoshida group for performing the carbonylative double cyclization of 3-hydroxy-4-pentenoic acids to stereoselectively give tetrahydrofuro[3,2-b]furan-2,5-diones with a cis junction between the rings under Pd(II) catalysis (10 mol% PdCl2 in the presence of 3 equiv of CuCl2 and 3 equiv of AcONa, in glacial acetic acid as the solvent, at room temperature and under 1 atm of CO) (Scheme 2) [8]. The process took place through 5-exo-trig cyclization by the intramolecular nucleophilic attack of the carboxylic group to the double bond coordinated to the metal center, stabilized by hydroxyl chelation, to give a cis-type alkylpalladium complex followed by CO insertion and intramolecular nucleophilic displacement, possibly via the formation of a palladacycle followed by reductive elimination (Scheme 2) [8].
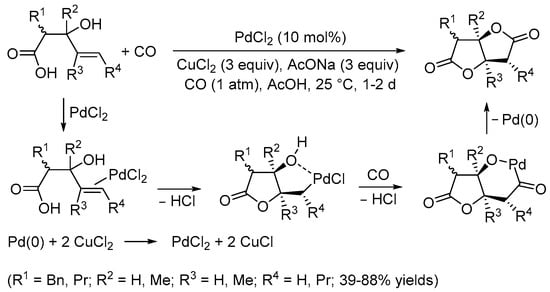
Scheme 2.
Synthesis of tetrahydrofuro[3,2-b]furan-2,5-diones from 3-hydroxy-4-pentenoic acids [8].
The same research group then published the carbonylative double cyclization of 4-ene-1,3-diols under similar reaction conditions to obtain tetrahydrofuro[3,2-b]furan-2(3H)-ones (Scheme 3) [9].
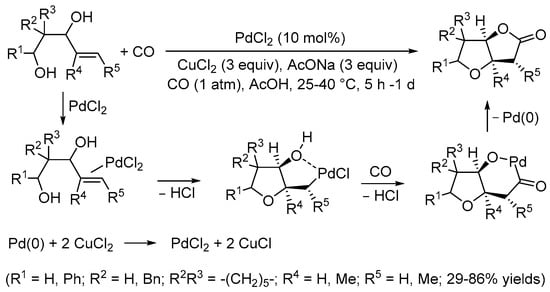
Scheme 3.
Synthesis of tetrahydrofuro[3,2-b]furan-2(3H)-ones from 4-ene-1,3-diols [9].
Considering that the bicyclic tetrahydrofuro[3,2-b]furan-2(3H)-one substructure is largely found in natural and biologically active molecules, the methods disclosed by Semmelhack and Yoshida for constructing this important core by the carbonylative double cyclization of enediol derivatives have been largely employed as the key step in the semi- or total synthesis of natural products and bioactive compounds. Representative examples are shown in Table 1.

Table 1.
Representative examples of the Pd(II)-promoted carbonylative double cyclization of enediol derivatives in the synthesis of natural and bioactive products.
Interestingly, using the appropriate enantiopure ligand, a kinetic resolution of (±)-pent-4-ene-1,3-diols was possible with the formation of the corresponding bicyclic lactone in noracemic form. This was exemplified by the Pd(OAc)2-catalyzed carbonylation of (±)-pent-4-ene-1,3-diol performed in the presence of an enantiopure bis(oxazoline) ligand and p-benzoquinone as an external oxidant to give (3aR,6aR)-tetrahydrofuro[3,2-b]furan-2(3H)-one in 29% yield and 62% ee (Scheme 4) [32].
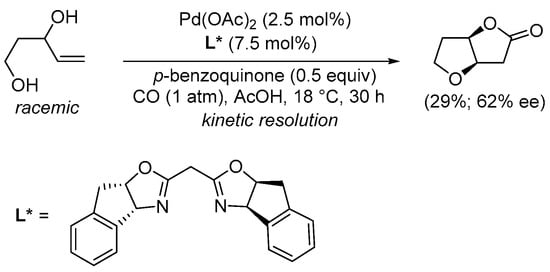
Scheme 4.
Kinetic resolution of (±)-pent-4-ene-1,3-diol leading to enantioenriched (3aR,6aR)-tetrahydrofuro[3,2-b]furan-2(3H)-one [32].
More recently, the kinetic resolution of (±)-pent-4-ene-1,3-diols to give nonracemic tetrahydrofuro[3,2-b]furan-2(3H)-ones [2-(S,S) up to 80% ee, 2-(R,R) up to 57% ee] has been realized under similar conditions [4 mol% of Pd(OAc)2, 12 mol% of 2,6-bis[(4R)-4-phenyl-2-oxazolinyl]pyridine as enantiopure ligand, 0.5 equiv of p-benzoquinone, and 10 equiv AcOH] using an ionic liquid as the solvent (such as 1-ethyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide, [Bmim][NTf2], 10 equiv), as shown in Scheme 5 [33].
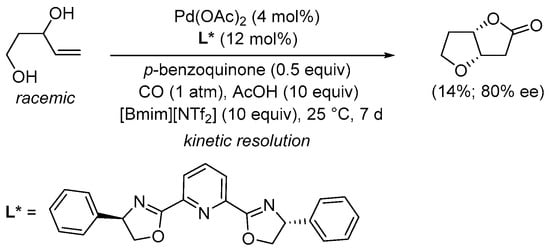
Scheme 5.
Kinetic resolution of (±)-pent-4-ene-1,3-diol in [bmim][NTf2] leading to enantioenriched (3aS,6aS)-tetrahydrofuro[3,2-b]furan-2(3H)-one [33].
Interestingly, the Gracza group reported that the use of iron pentacarbonyl as an in situ liquid CO source may lead to improved results (significantly shorter reaction times, in particular) in the Pd(II)-catalyzed carbonylative double cyclization of enediols with a terminal double bond, as shown in Scheme 6 [29,34,35].

Scheme 6.
Carbonylative double cyclization of pent-4-ene-1,3-diol using [Fe(CO)5] as in situ CO source [34].
The same research group recently reported their reaction under flow conditions using a continuous microflow system, as shown in Scheme 7 [31,36].
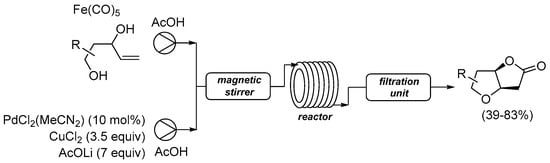
Scheme 7.
Carbonylative double cyclization of 4-ene-1,3-diols using [Fe(CO)5] as in situ CO source under flow conditions [36].
The carbonylative double cyclization process of enediols has also been reported to occur with 4-ene-1,2-diol derivatives. In this case, after the initial 5-exo-trig O-cyclization, in the cyclocarbonylation it is the free hydroxyl at C-2 that acts as internal nucleophile, with the formation of a 6-membered ring. This is illustrated by the formation of 8-((tert-butyldimethylsilyl)oxy)-2,6-dioxabicyclo[3.2.1]octan-3-one from 3-((tert-butyldimethylsilyl)oxy)pent-4-ene-1,2-diol, as shown in Scheme 8 [37].
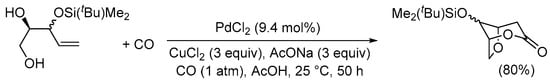
Scheme 8.
5-exo-trig O-cyclization followed by cyclocarbonylation with 6-membered ring closure [37].
The nucleophilic group undergoing initial heterocyclization can also be nitrogen-based. Thus, as early as 1985 the Tamaru and Yoshida group found that the Pd(II)-catalyzed carbonylative double cyclization of the N-protected 5-aminopent-1-en-3-ols yielded N-protected 6-hydroxyhexahydro-2H-furo[3,2-b]pyrrol-2-ones, using the same conditions employed for 4-penten-1,3-diols [38]. Among the protective groups tested, the –CO2Me group turned out as the most suitable, as exemplified in Scheme 9 [39].
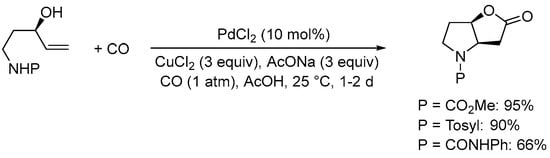
Scheme 9.
Formation of 6-hydroxyhexahydro-2H-furo[3,2-b]pyrrol-2-one derivatives from N-protected 5-aminopent-1-en-3-ols [39].
As predicted, owing to the higher degrees of freedom of the alkyl chain, N-protected 6-aminohex-1-en-3-ols were significantly less reactive, and relatively good results were usually observed with P = CONHPh, as shown in Scheme 10 [39].
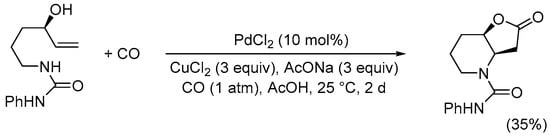
Scheme 10.
Synthesis of 2-oxo-N-phenylhexahydrofuro[3,2-b]pyridine-4(2H)-carboxamide from 1-(4-hydroxyhex-5-en-1-yl)-3-phenylurea [39].
Later on, Jäger et al. reported the carbonylation of benzyl ((2R,3S)-2,3-dihydroxypent-4-en-1-yl)carbamate (Scheme 11a) as a key step in the synthesis of novel 1,4-iminoglycitol derivatives as potential glycosidase inhibitors [40]. On the other hand, the PdCl2-catalyzed carbonylation of benzyl ((2S,3S)-2,3-dihydroxypent-4-en-1-yl)carbamate gave benzyl (3aR,6S,6aS)-6-hydroxy-2-oxohexahydro-4H-furo[3,2-b]pyrrole-4-carboxylate in a 14% yield (Scheme 11b) [41].

Scheme 11.
Synthesis of (a) benzyl (3aR,6R,6aS)-6-hydroxy-2-oxohexahydro-4H-furo[3,2-b]pyrrole-4-carboxylate (a precursor for the formation of glycosidase inhibitor derivatives) [40] and (b) benzyl (3aR,6S,6aS)-6-hydroxy-2-oxohexahydro-4H-furo[3,2-b]pyrrole-4-carboxylate [41].
Kinetic resolution of N-protected 5-aminopent-1-en-3-ols in the Pd(II)-catalyzed carbonylative double cyclization has been reported by Gracza et al. Thus, using enantiopure bisoxazoline ligands, nonracemic hexahydro-2H-furo[3,2-b]pyrrol-2-ones could be obtained as in Scheme 12 [42].
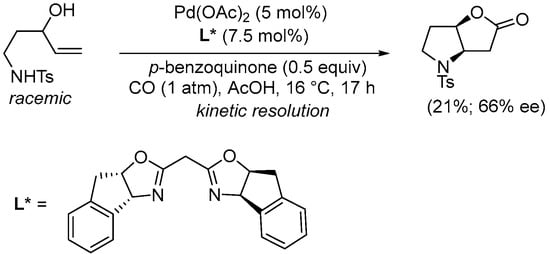
Scheme 12.
Kinetic resolution of (±)-N-(3-hydroxypent-4-en-1-yl)-4-methylbenzenesulfonamide leading to enantioenriched (3aR,6aR)-4-tosylhexahydro-2H-furo[3,2-b]pyrrol-2-one [42].
The gaseous CO-free conditions elaborated by the Gracza group for the carbonylation of 4-ene-1,3-diols, involving the use of liquid [Fe(CO)5] as an in situ CO source (Scheme 6), have also been successfully employed by the same research team in the Pd(II)-catalyzed carboylative double cyclization of N-protected 5-aminopent-1-en-3-ols, as in Scheme 13 [34].

Scheme 13.
Carbonylative double cyclization of tert-butyl (3-hydroxypent-4-en-1-yl)carbamate using [Fe(CO)5] as in situ CO source [34].
An interesting carbonylative double cyclization process has recently been developed by the Li group [43]. It involved the reaction of N-(2-aminoethyl)pent-4-enamide or N-(2-hydroxyethyl)pent-4-enamide derivatives with CO (1 atm) in the presence of PdCl2 as a catalyst (1 mol%) and p-benzoquinone as an oxidant (1.2 equiv) (Scheme 14). As shown in Scheme 14, the initial 5-exo-trig N-cyclization was followed by CO insertion and intramolecular nucleophilic displacement via the formation of an 8-membered ring palladacycle followed by reduction elimination [43].
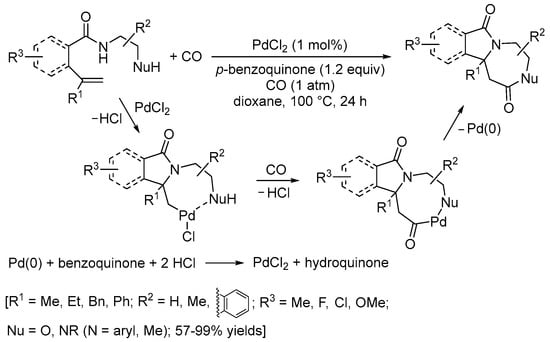
Scheme 14.
Carbonylative double cyclization of N-(2-aminoethyl)pent-4-enamide and N-(2-hydroxyethyl)pent-4-enamide derivatives [43].
A general approach leading to carbonylative double cyclization is the intramolecular Pauson–Khand reaction starting from suitable diene or enyne substrates. Since several excellent reviews have been published on this reaction [44,45,46], even in the most recent literature [47,48,49], this process will not be treated here; however, a particularly striking example in Scheme 15 gives the reader an idea of the powerfulness of this synthetic method for constructing complex carbonylated polycyclic compounds [50].
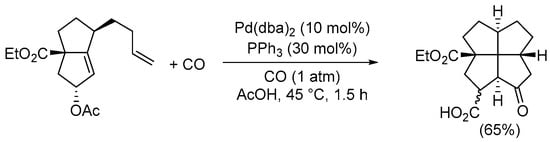
Scheme 15.
Synthesis of 2a-(ethoxycarbonyl)-8-oxododecahydropentaleno[1,6-cd]pentalene-1-carboxylic acid from ethyl 5-acetoxy-1-(but-3-en-1-yl)-2,3,4,5-tetrahydropentalene-3a(1H)-carboxylate by Pauson–Khand-type intramolecular reaction [50].
3. Functionalized Acetylenic Substrates
Starting from suitably functionalized acetylenic substrates bearing a nucleophilic group in the appropriate position, Kato et al. reported an interesting Pd(II)-catalyzed carbonylative double cyclization process without incorporating CO into the rings, which leads to di(hetero)cyclic ketones [51]. According to Scheme 16, the process, which the authors call the “cyclization–carbonylation–cyclization coupling reaction” or ”CCC reaction”, starts with the electrophilic activation of the triple bond by a Pd(II) species followed by an intramolecular nucleophilic attack on the activated triple bond with the formation of the first ring. (Only the endo cyclization mode is shown in Scheme 16 for simplicity.) The ensuing cyclic vinylpalladium intermediate then undergoes carbon monoxide insertion followed by the coordination of another molecule of the acetylenic substrate. This opens the way to a second cyclization, which is followed by reductive elimination to give the final product and Pd(0). The latter is reoxidized to give catalytically active Pd(II) by the use of an external oxidant, usually p-benzoquinone. The system CuCl2/O2 has also been used occasionally [52] although it should be noted that in this case the CO–O2 mixture fell within the explosion limits. In fact, these mixtures are potentially explosive over a large range of composition: the flammability range for CO in O2 is 16.7–93.5% at room temperature and becomes even larger at higher temperatures [53].
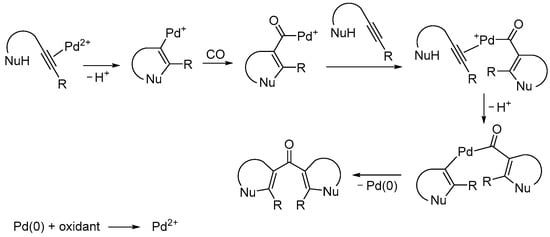
Scheme 16.
The “cyclization–carbonylation–cyclization coupling” concept leading to di(hetero)cyclic ketones [51].
This method has been successfully employed by the Kato group to synthesize a variety of di(heterocyclic)ketones; representative examples are shown in Table 2, entries 1–8. Entry 9 shows a recent extension of the concept to allenic substrates (2-methyl-1-phenyl-2,3-dien-1-ones, in particular) for the synthesis of bis(3-furanyl)methanones.
Table 2.
Examples of the “cyclization–carbonylation–cyclization coupling” concept leading to di(hetero)cyclic ketones.
The Pd(II)-catalyzed carbonylative cyclization of functionalized acetylenic derivatives with CO incorporation into the cycle was disclosed by our research group a few years ago [62,63,64,65] using the PdI2/KI catalytic system, which we had already successfully used to promote a plethora of carbonylation reactions [66,67,68,69,70]. Thus, starting from readily available 4-yne-1,3-diols under oxidative conditions (using oxygen from the air as a benign oxidative agent), novel dihydrofurofuranone derivatives with antitumor activity were synthesized (Scheme 17). In particular, 5,5-dimethyl-6a-phenyl-3-(trimethylsilyl)-6,6a-dihydrofuro[3,2-b]furan-2(5H)-one showed a significant antiproliferative activity in vitro on human breast cancer cell lines, including the most aggressive triple-negative breast cancer cells (MDA-MB-231 and MDAMB-468) while being practically non-toxic to normal cells (human mammary epithelia cells, MCF-10A, as well as murine fibroblasts 3T3-L1) [62,63,64].
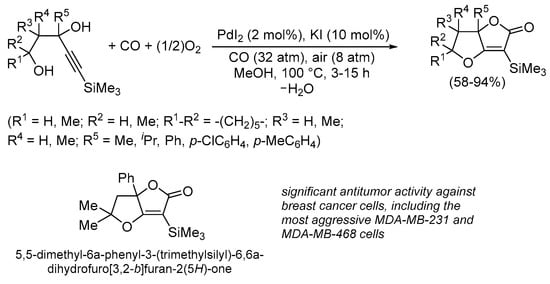
Scheme 17.
Synthesis of 6,6a-dihydrofuro[3,2-b]furan-2(5H)ones by PdI2/KI-catalyzed carbonylative double cyclization of 4-yne-1,3-diols [62,63,64].
Mechanistically, the process involved an initial 5-exo-dig heterocyclization by an intramolecular nucleophilic attack of the terminal hydroxyl group to the triple bond coordinated to Pd(II) followed by carbon monoxide insertion. Intramolecular nucleophilic displacement then took place, probably by forming a palladacycle followed by a reductive elimination, to give the product and Pd(0). The latter was then reoxidized to PdI2 according to the mechanism we demonstrated several years ago in the PdI2/KI-catalyzed oxidative dialkoxycarbonylation of alkynes [71], which involves oxidation of 2 mol of HI (formed during the process) to give I2 followed by the oxidative addition of I2 to Pd(0) (Scheme 18; anionic iodide ligands are omitted for clarity) [64].
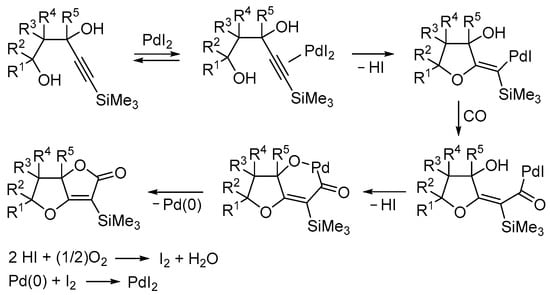
Scheme 18.
Proposed mechanism for the PdI2/KI-catalyzed carbonylative double cyclization of 4-yne-1,3-diols leading to 6,6a-dihydrofuro[3,2-b]furan-2(5H)ones [64].
The method was then extended to using 2-(3-hydroxy-1-yn-1-yl)phenols as substrates to give furo[3,4-b]benzofuran-1(3H)ones in the ionic liquid BmimBF4 (1-butyl-3-methylimidazolium tetrafluoroborate) as an unconventional solvent (Scheme 19) [72]. The catalyst–solvent system could be conveniently recycled several times without appreciable loss of activity. Interestingly, this process turned out to be unselective when carried out in a classical solvent (such as DME or MeCN), in forming mixtures of the desired furobenzofuranone derivative and the simple benzofuran product from non-carbonylative heterocyclization.
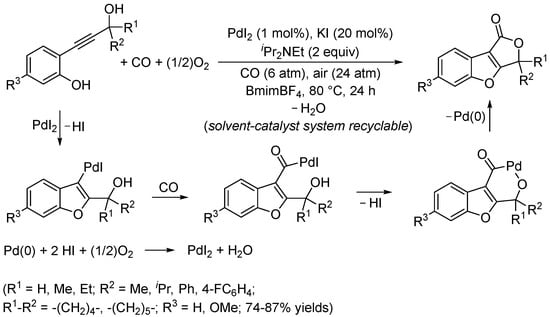
Scheme 19.
Synthesis of furo[3,4-b]benzofuran-1(3H)ones by PdI2/KI-catalyzed carbonylative double cyclization of 2-(3-hydroxy-1-yn-1-yl)phenols in ionic liquid BmimBF4 [72].
In a similar way, starting from 2-(hydroxyprop-1-ynyl)anilines as the substrates, 3,4-dihydrofuro[3,4-b]indol-1-ones were synthesized in one step with yields up to 98% from an initial 5-endo-dig N-heterocyclization followed by cyclocarbonylation (Scheme 20) [65].
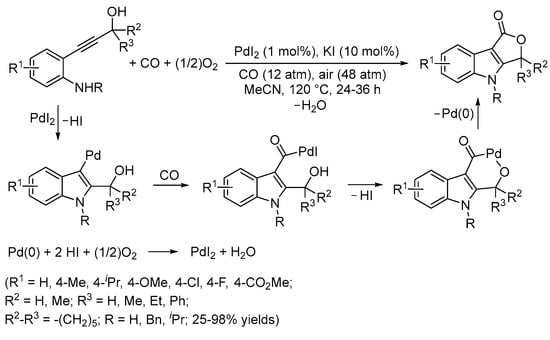
Scheme 20.
Synthesis of 3,4-dihydrofuro[3,4-b]indol-1-ones by PdI2/KI-catalyzed carbonylative double cyclization of 2-(hydroxyprop-1-ynyl)anilines [65].
Interestingly, the use of the analogue substrates having a secondary propargylaminic moiety rather than the propargylacoholic group [2-(3-(alkylamino)prop-1-yn-1-yl)anilines] led to a complex reaction mixture when allowed to react under conditions similar to those shown in Scheme 20. However, a selective and novel double cyclization process was observed with the N-acyl derivatives, i.e., in the case of N-(3-(2-aminophenyl)prop-2-yn-1-yl)acetamides, with formation of 4,6-dihydro-5H-[1,3]oxazino[5,6-c]quinolin-5-ones. (Scheme 21) [73]. In this case, the reaction began with an intramolecular nucleophilic attack by the amide carbonyl oxygen on the coordinated triple bond leading to 6-endo-dig ring closure and the formation of a vinylpalladium intermediate stabilized by coordination of the aniline amino group. Carbon monoxide insertion followed by intramolecular nucleophilic displacement, possibly through the formation of a palladacycle, then delivers the product (Scheme 21) [73].
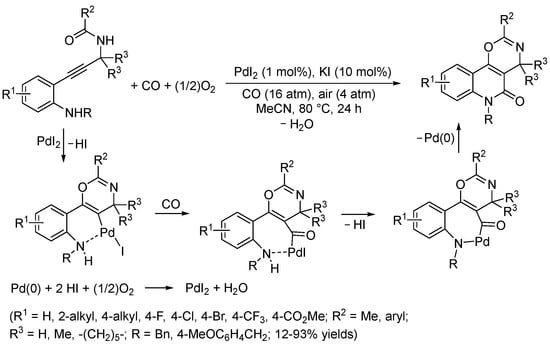
Scheme 21.
Synthesis of 4,6-dihydro-5H-[1,3]oxazino[5,6-c]quinolin-5-ones by PdI2/KI-catalyzed carbonylative double cyclization of N-(3-(2-aminophenyl)prop-2-yn-1-yl)acetamides [73].
A striking carbonylative tetracyclization process was observed in the case of 2-(3-amino-3-methylbut-1-yn-1-yl)anilines having a primary propargylaminic moiety, which led to 7,7,16,16-tetramethyl-5H,14H-benzopyrido[3’’,4’’:5’,6’]pyrimido[2’,1’:2,3][1,3]oxazino[5,6-c]quinoline-6,15-diones in one step (Scheme 22) [73].
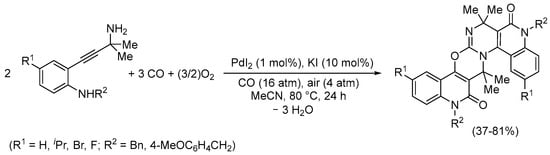
Scheme 22.
Synthesis of 7,7,16,16-tetramethyl-5H,14H-benzopyrido[3’’,4’’:5’,6’]pyrimido[2’,1’:2,3][1,3]oxazino[5,6-c]quinoline-6,15-diones by PdI2/KI-catalyzed carbonylative tetracyclization of 2-(3-amino-3-methylbut-1-yn-1-yl)anilines [73].
In fact, these substrates first underwent PdI2-catalyzed oxidative carbonylation of the primary amino group to give the corresponding urea [74,75], which then reacted through O-6-endo-dig cyclization from the ureidic carbonyl group followed by two successive cyclocarbonylations to yield the final product (Scheme 23) [73].
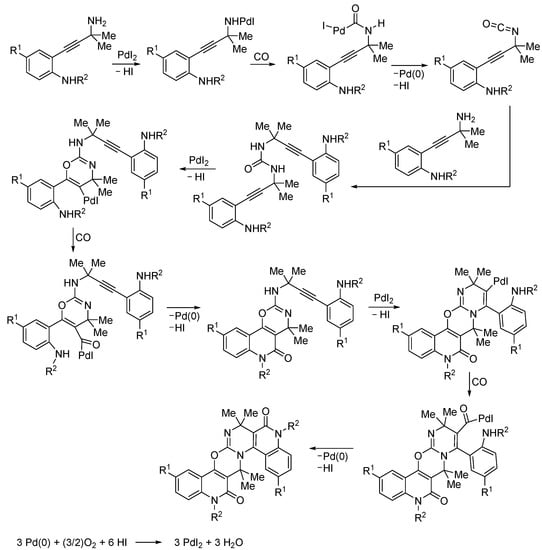
Scheme 23.
Plausible mechanism for the PdI2/KI-catalyzed carbonylative tetracyclization of 2-(3-amino-3-methylbut-1-yn-1-yl)anilines to give 7,7,16,16-tetramethyl-5H,14H-benzopyrido[3’’,4’’:5’,6’]pyrimido[2’,1’:2,3][1,3]oxazino[5,6-c]quinoline-6,15-diones [73].
More recently, we studied the reactivity of thiophenecarboxylic acids having an ω-hydroxyalkynyl substituent in vicinal position under PdI2/KI-catalyzed oxidative carbonylation conditions and found that also these substrates underwent carbonylative double cyclization to give previously unknown 1H-furo[3,4-b]thieno[3,2-d]pyran-1,5(3H)-dione (Scheme 24a), 4H-furo[3,4-b]thieno[2,3-d]pyran-4,8(6H)-dione (Scheme 24b), 3,4-dihydro-1H,6H-pyrano[4,3-b]thieno[3,2-d]pyran-1,6-dione (Scheme 24c), and 6,7-dihydro-4H,9H-pyrano[4,3-b]thieno[2,3-d]pyran-4,9-dione (Scheme 24d) derivatives [76]. The process begins with 6-endo-dig cyclization from the carboxylic group followed by cyclocarbonylation, as exemplified in Scheme 24a to synthesize 1H-furo[3,4-b]thieno[3,2-d]pyran-1,5(3H)-diones from 3-(3-hydroxyprop-1-yn-1-yl)thiophene-2-carboxylic acids [76].
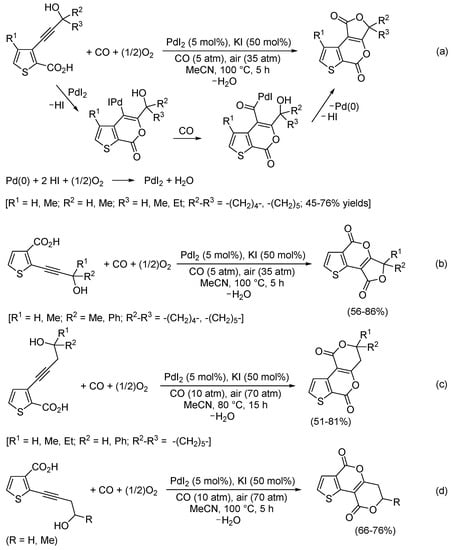
Scheme 24.
Synthesis of (a) 1H-furo[3,4-b]thieno[3,2-d]pyran-1,5(3H)-diones, (b) 4H-furo[3,4-b]thieno[2,3-d]pyran-4,8(6H)-diones, (c) 3,4-dihydro-1H,6H-pyrano[4,3-b]thieno[3,2-d]pyran-1,6-diones, and (d) 6,7-dihydro-4H,9H-pyrano[4,3-b]thieno[2,3-d]pyran-4,9-diones by PdI2/KI-catalyzed carbonylative double cyclization of thiophenecarboxylic acids bearing an ω-hydroxyalkynyl substituent in vicinal position [76].
We also found that even sulfurated acetylenic substrates, under the right oxidative conditions, can undergo PdI2/KI-catalyzed carbonylative double cyclization. However, considering the instability of the free thiol group to oxygen [77,78], it was necessary to protect the sulfur atom with an easily removable methyl group. This group, in fact, could be removed after cyclization because of the presence of excess iodide anions. Accordingly, starting from 5-(methylthio)-1-yn-3-ols, we were able to synthesize 6,6a-dihydrothieno[3,2-b]furan-2(5H)-ones as a new class of S,O-bicyclic heterocycles as shown in Scheme 25 [79].
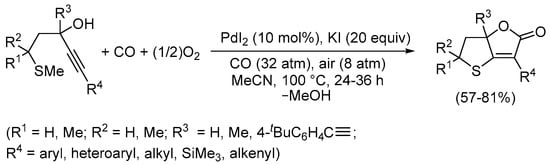
Scheme 25.
Synthesis of 6,6a-dihydrothieno[3,2-b]furan-2(5H)-ones by PdI2/KI-catalyzed carbonylative double cyclization of 5-(methylthio)-1-yn-3-ols [79].
Mechanistically, the process began with 5-exo-dig S-cyclization by the intramolecular nucleophilic attack by the sulfur in the thiomethyl group on the triple bond coordinated to PdI2. This was followed by demethylation of the ensuing sulfonium cation by the iodide anion, with the formation of the corresponding vinylpalladium intermediate and methyl iodide. The latter readily reacted with water that was initially present as an impurity and then also formed in the final Pd(0) reoxidation step to give MeOH and one mol of HI. On the other hand, the vinylpalladium intermediate underwent carbon monoxide insertion followed by nucleophilic displacement to give the final product together with Pd(0) and a second mol of HI. Lastly, Pd(0) was, as usual, oxidized back to PdI2 by reacting with 2 mol of HI and 0.5 mol of O2 (Scheme 26) [79].
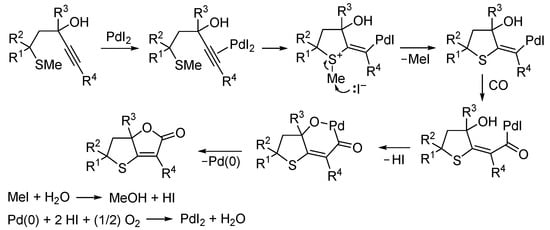
Scheme 26.
Proposed mechanism for the PdI2/KI-catalyzed carbonylative double cyclization of 5-(methylthio)-1-yn-3-ols to give 6,6a-dihydrothieno[3,2-b]furan-2(5H)-ones [79].
4. Functionalized Halides
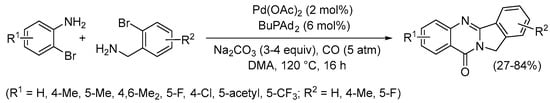
Under non-oxidative conditions, suitably functionalized halides may undergo Pd(0)-catalyzed double cyclization leading to high value-added polycyclic heterocyclic compounds. Thus, 1,2-dibromoarenes have been reported by Beller and Wu to undergo carbonylative double cyclization when allowed to react with 2-aminobenzyl amine using Pd(OAc)2 in the presence of the catalyst precursor BuPAd2 (Ad = 1-adamantyl) in the solvent N,N-dimethylacetamide (DMA) with Et3N as the base and under 10 atm of CO. In this manner, several isoindolo[1,2-b]quinazolin-12(10H)-one derivatives (analogues of the anticancer agent batracylin) were prepared in a 36–84% yield (Scheme 27) [80].
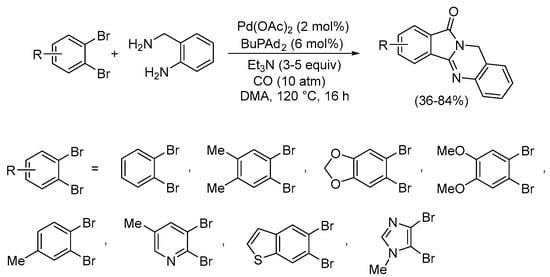
Scheme 27.
Carbonylative double cyclization of 1,2-dibromoarenes with 2-aminobenzyl amine to yield isoindolo[1,2-b]quinazolin-12(10H)-ones [80].
The process started with the oxidative addition of a C–Br bond to Pd(0) followed by CO insertion. Then the more nucleophilic benzylic amino group of the diamine caused nucleophilic displacement, which was followed by a further oxidative addition and CO insertion from the second C–Br bond, intramolecular nucleophilic displacement by the second amino group, and intramolecular condensation (Scheme 28) [80].
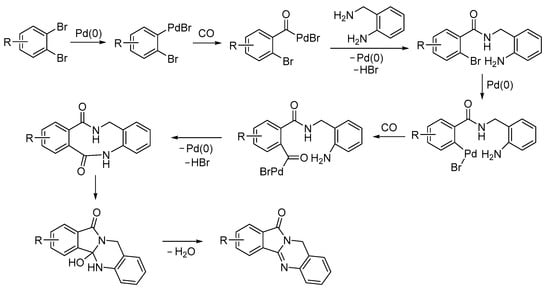
Scheme 28.
Proposed mechanism for the carbonylative double cyclization of 1,2-dibromoarenes with 2-aminobenzyl amine leading to isoindolo[1,2-b]quinazolin-12(10H)-ones [80].
In a similar way, and under similar conditions, isoindolo[1,2-b]quinazolin-10(12H)-ones were synthesized by the Wu group starting from 2-bromoanilines and 2-bromobenzyl amines, as shown in Scheme 29 [81].
Scheme 29.
Carbonylative double cyclization of 2-bromoanilines with 2-bromobenzyl amines to yield isoindolo[1,2-b]quinazolin-10(12H)-ones [81].
In this case, it was the 2-bromoaniline derivative that underwent initial oxidative addition to Pd(0), followed by CO insertion (Scheme 30). This was followed by nucleophilic displacement by the 2-bromobenzyl amine, the oxidative addition to Pd(0) of the second C–Br bond, CO insertion, intramolecular nucleophilic displacement, and intramolecular condensation (Scheme 30) [81].
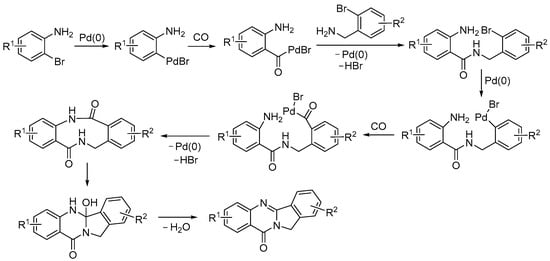
Scheme 30.
Proposed mechanism for the carbonylative double cyclization of 2-bromoanilines with 2-bromobenzyl amines to yield isoindolo[1,2-b]quinazolin-10(12H)-ones [81].
The Beller and Wu group also reported the Pd(0)-catalyzed reaction of 2-bromoanilines with 2-bromobenzaldehyde and CO, which resulted in a carbonylative double cyclization leading to 5H-benzo[4,5][1,3]oxazino[2,3-a]isoindole-5,11(6aH)-diones (Scheme 31a) [82]. 2-Bromobenzonitriles also underwent carbonylative double cyclization when allowed to react with 2-bromoanilines to give isoindolo[1,2-b]quinazoline-10,12-diones (Scheme 31b) [83].
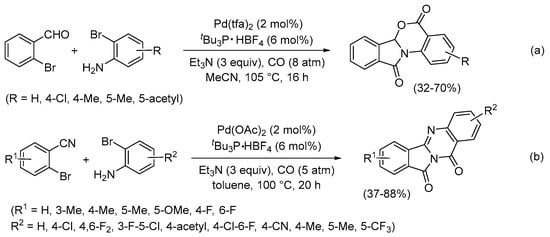
Scheme 31.
Carbonylative double cyclization of 2-bromoanilines with (a) 2-bromobenzaldehyde to give 5H-benzo[4,5][1,3]oxazino[2,3-a]isoindole-5,11(6aH)-diones [82] or (b) 2-bromobenzonitriles to give isoindolo[1,2-b]quinazoline-10,12-diones [83].
As shown in Scheme 32, the process leading to 5H-benzo[4,5][1,3]oxazino[2,3-a]isoindole-5,11(6aH)-diones began with the oxidative addition of 2-bromobenzaldehyde to Pd(0), followed by CO insertion and nucleophilic displacement by the amino group of the 2-bromoaniline derivative. Then, there was an intramolecular nucleophilic attack of the nitrogen of the newly formed amido group on the formyl group, followed by the oxidative addition of the second C–Br group to Pd(0), CO insertion, and intramolecular nucleophilic displacement by the hydroxyl group. 2-Bromobenzoic acid could also be employed as a substrate in this reaction in place of 2-bromobenzaldehyde [82].
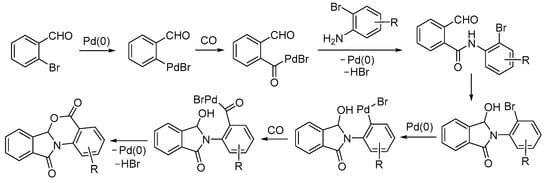
Scheme 32.
Proposed mechanism for the carbonylative double cyclization of 2-bromoanilines with 2-bromobenzaldehyde to give 5H-benzo[4,5][1,3]oxazino[2,3-a]isoindole-5,11(6aH)-diones [82].
The first steps of the mechanistic pathway leading to isoindolo[1,2-b]quinazoline-10,12-diones are similar to those seen above for the reaction of 2-bromoanilines with 2-bromobenzaldehyde. Thus, the oxidative addition of the 2-bromobenzonitrile derivative was followed by CO insertion, nucleophilic displacement by the 2-bromoaniline, and an intramolecular nucleophilic attack by the nitrogen of the newly formed amide group on the cyano group, which formed the corresponding 2-(2-bromophenyl)-3-iminoisoindolin-1-one derivative (Scheme 33). Then, an unexpected isomerization of this intermediate took place, probably due to steric effects, to give a (Z)-3-((2-bromophenyl)imino)isoindolin-1-one intermediate. The oxidative addition of the C–Br bond of the latter to Pd(0) followed by CO insertion and intramolecular nucleophilic displacement delivered the final product (Scheme 33) [83].
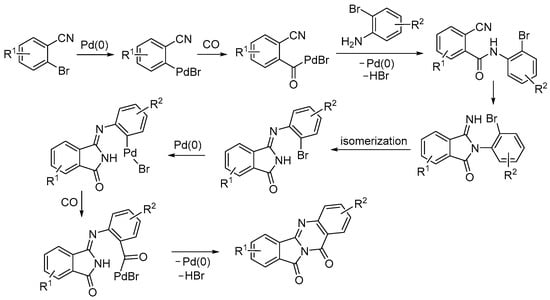
Scheme 33.
Proposed mechanism for the carbonylative double cyclization of 2-bromoanilines with 2-bromobenzonitriles to give isoindolo[1,2-b]quinazoline-10,12-diones [83].
The carbonylative double cyclization of substrates having two aryl halide bonds and a suitable nucleophile, such as an enolate formed in situ under basic conditions, in an appropriate position was also reported, as shown by the synthesis of 5H-isochromeno[3,4-b]quinoline-5,12(7H)-diones starting from N-(2-bromophenyl)-2-(2-iodophenyl)acetamides (Scheme 34) [84].
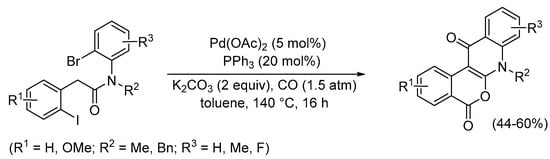
Scheme 34.
Carbonylative double cyclization of N-(2-bromophenyl)-2-(2-iodophenyl)acetamides to give 5H-isochromeno[3,4-b]quinoline-5,12(7H)-diones [84].
In this case, the more reactive C–I bond gave the initial oxidative addition to Pd(0), followed by CO insertion (Scheme 35). Then, intramolecular nucleophilic displacement by the enolate oxygen occurred, which led to the first cyclization. The oxidative addition of the C–Br bond to Pd(0), followed by Csp2–H activation, CO insertion, and reductive elimination, eventually gave the final product [84].
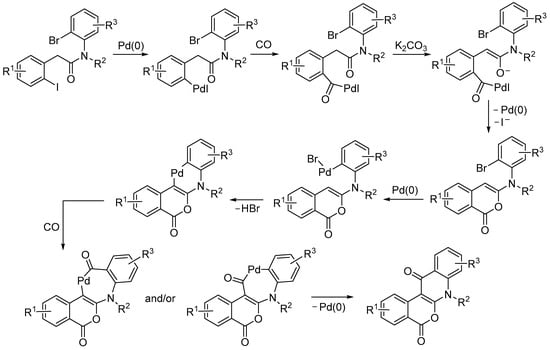
Scheme 35.
Proposed mechanism for the carbonylative double cyclization of N-(2-bromophenyl)-2-(2-iodophenyl)acetamides to give 5H-isochromeno[3,4-b]quinoline-5,12(7H)-diones [84].
5. Conclusions and Future Perspectives
Catalytic carbonylative double cyclization is a powerful technique for constructing two novel rings in sequential order with the simultaneous incorporation of carbon monoxide in the final product. Starting from simple building blocks, it allows the direct synthesis of high value-added, complex molecular architectures.
So far, several important examples have been reported in the literature, in particular starting from suitably functionalized olefinic, acetylenic, or halide substrates and under the catalysis of either Pd(II) or Pd(0) species. These reactions led to the formation of important polycyclic heterocyclic derivatives, which have shown important biological activity (including anticancer activity) or have been used as precursors to the synthesis of bioactive natural products.
Progress in catalysis is expected to give further impetus to this very attractive field of synthetic chemistry through the discovery of novel and more efficient one-step catalytic processes that produce polycyclic heterocycles with potential applications in fields such as material science and pharmaceutical chemistry.
Author Contributions
Conceptualization, B.G.; Validation, B.G., R.M., N.D.C., L.V. and I.Z.; Investigation, all authors; writing—original draft preparation, B.G.; writing—review and editing, all authors; supervision, B.G. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
No new data are reported with respect to those published in the original manuscripts cited in this review paper.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Gabriele, B. (Ed.) Carbon Monoxide in Organic Synthesis—Carbonylation Chemistry; Wiley-VCH: Weinheim, Germany, 2022. [Google Scholar]
- Reimert, R.; Marschner, F.; Renner, H.-J.; Boll, W.; Supp, E.; Brejc, M.; Liebner, W.; Schaub, G. Gas production, 2. In Ullmann’s Encyclopedia of Industrial Chemistry; Baltes, H., Göpel, W., Hesse, J., Eds.; Wiley-VCH: Weinheim, Germany, 2011; pp. 423–479. [Google Scholar]
- Karl, J.; Pröll, T. Steam Gasification of Biomass in Dual Fluidized Bed Gasifiers: A Review. Renew. Sustain. Energy Rev. 2018, 98, 64–78. [Google Scholar] [CrossRef]
- Figueres, C.; Le Quéré, C.; Mahindra, A.; Bäte, O.; Whiteman, G.; Peters, G.; Guan, D. Emissions Are Still Rising: Ramp up the Cuts. Nature 2018, 564, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Li, J.J.; Gribble, G. (Eds.) Palladium in Heterocyclic Chemistry—A Guide for the Synthetic Chemist, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2006. [Google Scholar]
- Gabriele, B.; Della Ca’, N.; Mancuso, R.; Veltri, L.; Ziccarelli, I. Palladium(II)-Catalyzed Carbonylations. In Carbon Monoxide in Organic Synthesis—Carbonylation Chemistry; Gabriele, B., Ed.; Wiley-VCH: Weinheim, Germany, 2022; Chapter 7. [Google Scholar]
- Semmelhack, M.F.; Bodurow, C.; Baum, M. Direct Synthesis of Pyran-Lactones Related to Nafhthoquinone Antibiotics. Tetrahadron Lett. 1984, 25, 3171–3174. [Google Scholar] [CrossRef]
- Tamaru, H.; Higashimura, K.; Hojo, M.; Yoshida, Z. PdII-Catalyzed Stereoselective Bis-Lactonization. Angew. Chem. Int. Ed. 1985, 24, 1045–1046. [Google Scholar] [CrossRef]
- Tamaru, H.; Kobayashi, T.; Kawamura, S.; Hojo, M.; Yoshida, Z. Palladium Catalyzed Oxycarbonylation of 4-Penten-1,3-diols: Efficient Stereoselective Synthesis of cis 3-Hydroxytetrahydrofuran 2-Acetic Acid Lactones. Tetrahedron Lett. 1985, 26, 3207–3210. [Google Scholar] [CrossRef]
- Gracza, T.; Hasenöhrl, T.; Stahl, T.; Jäger, V. Synthesis of 3,5-Anhydro-2-deoxy-1,4-glyconolactones by Palladium(II)-Catalyzed, Regioselective Oxycarbonylation of C5- and C6-enitols. ω-Homologation of Aldoses to Produce Intermediates for C-Glycoside/C-Nucleoside Synthesis. Synthesis 1991, 1991, 1108–1118. [Google Scholar] [CrossRef]
- Kraus, G.A.; Li, J. Regiocontrol by Remote Substituents. An Enantioselective Total Synthesis of Frenolicin B via a Highly Regioselective Diels-Alder Reaction. J. Am. Chem. Soc. 1993, 115, 5859–5860. [Google Scholar] [CrossRef]
- Gracza, T.; Jäger, V. Synthesis of Natural and Unnatural Enantiomers of Goniofufurone and Its 7-Epimers from D-Glucose. Application of Palladium(II)—Catalyzed Oxycarbonylation of Unsaturated Polyols. Synthesis 1994, 1994, 1359–1368. [Google Scholar] [CrossRef]
- Boukouvalas, J.; Fortier, G.; Radu, I.-I. Efficient Synthesis of (−)-trans-Kumausyne via Tandem Intramolecular Alkoxycarbonylation-Lactonization. J. Org. Chem. 1998, 63, 916–917. [Google Scholar] [CrossRef]
- Dixon, D.J.; Ley, S.V.; Gracza, T.; Szolcsanyi, P. Total Synthesis of the Polyenoyltetramic acid Mycotoxin Erythroskyrine. J. Chem. Soc. Perkin Trans. 1 1999, 1999, 831–841. [Google Scholar] [CrossRef]
- Paddon-Jones, G.C.; Hungerford, N.L.; Haynes, P.; Kitching, W. Efficient Palladium(II)-Mediated Construction of Functionalized Plakortone Cores. Org. Lett. 1999, 1, 1905–1908. [Google Scholar] [CrossRef]
- Semmelhack, M.F.; Shanmugam, P. Development of an Approach to the Synthesis of the Plakortones. Tetrahedron Lett. 2000, 41, 3567–3571. [Google Scholar] [CrossRef]
- Paddon-Jones, G.C.; McErlean, C.S.P.; Haynes, P.; Moore, C.J.; Konig, W.A.; Kitching, W. Synthesis and Stereochemistry of Some Bicyclic γ-Lactones from Parasitic Wasps (Hymenoptera: Braconidae). Utility of Hydrolytic Kinetic Resolution of Epoxides and Palladium(II)-Catalyzed Hydroxycyclization-Carbonylation-Lactonization of Ene-diols. J. Org. Chem. 2001, 66, 7487–7495. [Google Scholar] [CrossRef] [PubMed]
- Haynes, P.Y.; Kitching, W. Total Synthesis and Absolute Stereochemistry of Plakortone D. J. Am. Chem. Soc. 2002, 124, 9718–9719. [Google Scholar]
- Haynes, P.Y.; Kitching, W. Synthesis of the Plakortone Series: Plakortone E. Heterocycles 2004, 62, 173–177. [Google Scholar]
- Babjak, M.; Kapitán, P.; Gracza, T. Synthesis of (+)-Goniothalesdiol and (+)-7-epi-Goniothalesdiol. Tetrahedron 2005, 61, 2471–2479. [Google Scholar] [CrossRef]
- Semmelhack, M.F.; Hooley, R.J.; Kraml, C.M. Synthesis of Plakortone B and Analogs. Org. Lett. 2006, 8, 5203–5206. [Google Scholar] [CrossRef]
- Boukouvalas, J.; Pouliot, M.; Robichaud, J.; MacNeil, S.; Snieckus, V. Asymmetric Total Synthesis of (−)-Panacene and Correction of Its Relative Configuration. Org. Lett. 2006, 8, 3597–3599. [Google Scholar] [CrossRef]
- Kapitán, P.; Gracza, T. Stereocontrolled Oxycarbonylation of 4-Benzyloxyhepta-1,6-diene-3,5-diols Promoted by Chiral Palladium(II) Complexes. Tetrahedron Asymm. 2008, 19, 38–44. [Google Scholar] [CrossRef]
- Nesbitt, C.L.; McErlean, C.S.P. An Expedient Synthesis of 2,5-Disubstituted-3-oxygenated Tetrahydrofurans. Tetrahedron Lett. 2009, 50, 6318–6320. [Google Scholar] [CrossRef]
- Nesbitt, C.L.; McErlean, C.S.P. Total Synthesis of C19 Lipid Diols Containing a 2,5-Disubstituted-3-Oxygenated Tetrahydrofuran. Org. Biomol. Chem. 2011, 9, 2198–2208. [Google Scholar] [CrossRef] [PubMed]
- Haynes, P.Y.; Chow, S.; Rahm, F.; Bernhardt, P.V.; De Voss, J.J.; Kitching, W. Synthesis of the Sponge-Derived Plakortone Series of Bioactive Compounds. J. Org. Chem. 2010, 75, 6489–6501. [Google Scholar]
- Werness, J.B.; Tang, W. Stereoselective Total Synthesis of (−)-Kumausallene. Org. Lett. 2011, 13, 3664–3666. [Google Scholar] [CrossRef] [PubMed]
- Markovič, M.; Ďuranová, M.; Koóš, P.; Szolcsányi, P.; Gracza, T. Synthesis of bis-Tetrahydrofuran Subunit of (−)-Neopallavicinin. Tetrahedron 2013, 69, 4185–4189. [Google Scholar] [CrossRef]
- Markovič, M.; Koóš, P.; Čarný, T.; Sokoliová, S.; Bohačiková, N.; Moncol’, J.; Gracza, T. Total Synthesis, Configuration Assignment, and Cytotoxic Activity Evaluation of Protulactone A. J. Nat. Prod. 2017, 80, 1631–1638. [Google Scholar] [CrossRef]
- Markovič, M.; Koóš, P.; Gracza, T. A Short Asymmetric Synthesis of Sauropunols A–D. Synthesis 2017, 49, 2939–2942. [Google Scholar]
- Lopatka, P.; Gavenda, M.; Markovič, M.; Koóš, P.; Gracza, T. Flow Pd(II)-Catalyzed Cyclisation in the Total Synthesis of Jaspine B. Catalysts 2021, 11, 1513. [Google Scholar] [CrossRef]
- Kapitán, P.; Gracza, T. Asymmetric Intramolecular Pd(II)-catalyzed Oxycarbonylation of Alkene-1,3-diols. Arkivoc 2008, viii, 8–17. [Google Scholar]
- Doháňošová, J.; Lásikivá, A.; Toffano, M.; Gracza, T.; Vo-Thanh, G. Kinetic Resolution of Pent-4-ene-1,3-diol by Pd(II)-Catalysed Oxycarbonylation in Ionic Liquids. New J. Chem. 2012, 36, 1744–1750. [Google Scholar] [CrossRef]
- Babjak, M.; Markovič, K.; Kandríkova, B.; Gracza, T. Homogeneous Cyclocarbonylation of Alkenols with Iron Pentacarbonyl. Synthesis 2014, 46, 809–816. [Google Scholar] [CrossRef]
- Markovič, K.; Lopatka, P.; Koóš, P.; Gracza, T. Asymmetric Formal Synthesis of (+)-Pyrenolide D. Synthesis 2014, 46, 817–821. [Google Scholar]
- Lopatka, P.; Markovič, K.; Koóš, P.; Ley, S.V.; Gracza, T. Continuous Pd-Catalyzed Carbonylative Cyclization Using Iron Pentacarbonyl as a CO Source. J. Org. Chem. 2019, 84, 14394–14406. [Google Scholar] [CrossRef] [PubMed]
- Babjak, M.; Zálupský, P.; Gracza, T. Regiocontrol in the Palladium(II)-Catalysed Oxycarbonylation of Unsaturated Polyols. Arkivoc 2005, 45, 57. [Google Scholar] [CrossRef]
- Tamaru, Y.; Kobayashi, T.; Kawamura, S.; Ochiai, H.; Yoshida, Z. Stereoselective Intramolecular Aminocarbonylation of 3-Hydroxypent-4-enylamides Catalyzed by Palladium. Tetrahedron Lett. 1985, 26, 4479–4482. [Google Scholar] [CrossRef]
- Tamaru, Y.; Hojo, M.; Yoshida, Z. Palladium(2+)-Catalyzed Intramolecular Aminocarbonylation of 3-Hydroxy-4-pentenylamines and 4-Hydroxy-5-hexenylamines. J. Org. Chem. 1988, 53, 5731–5741. [Google Scholar] [CrossRef]
- Hümmer, W.; Dubois, E.; Gracza, T.; Jäger, V. Halocyclization and Palladium(II)-Catalyzed Amidocarbonylation of Unsaturated Aminopolyols. Synthesis of 1,4-Iminoglycitols as Potential Glycosidase Inhibitors. Synthesis 1997, 1997, 634–642. [Google Scholar] [CrossRef]
- Caletková, O.; Ďurišová, N.; Gracza, T. Aminohydroxylation of Divinylcarbinol and its Application to the Synthesis of Bicyclic hydroxypyrrolidine and Aminotetrahydrofuran Building Blocks. Chem. Pap. 2013, 67, 66–75. [Google Scholar] [CrossRef]
- Koóš, P.; Špánik, I.; Gracza, T. Asymmetric Intramolecular Pd(II)-Catalysed Amidocarbonylation of Unsaturated Amino Alcohols. Tetrahedron Asymm. 2009, 20, 2720–2723. [Google Scholar] [CrossRef]
- Shi, L.; Weng, M.; Li, F. Palladium-Catalyzed Tandem Carbonylative Aza-Wacker-Type Cyclization of Nucleophile Tethered Alkene to Access Fused N-Heterocycles. Chin. J. Chem. 2021, 39, 317–322. [Google Scholar] [CrossRef]
- Lee, H.-W.; Kwong, F.-Y. A Decade of Advancements in Pauson-Khand-Type Reactions. Eur. J. Org. Chem. 2010, 2010, 789–811. [Google Scholar] [CrossRef]
- Shibata, T. Recent Advances in the Catalytic Pauson-Khand-type Reaction. Adv. Synth. Catal. 2006, 348, 2328–2336. [Google Scholar] [CrossRef]
- Blanco-Urgoiti, J.; Añorbe, L.; Pérez-Serrano, L.; Domínguez, G.; Pérez-Castells, J. The Pauson–Khand Reaction, a Powerful Synthetic Tool for the Synthesis of Complex Molecules. Chem. Soc. Rev. 2004, 33, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Heravi, M.M.; Mohammadi, L. Application of Pauson-Khand Reaction in the Total Synthesis of Terpenes. RSC Adv. 2021, 11, 38325–38373. [Google Scholar] [CrossRef]
- Yang, Z. Navigating the Pauson-Khand Reaction in Total Syntheses of Complex Natural Products. Acc. Chem. Res. 2021, 54, 556–568. [Google Scholar] [CrossRef]
- Chen, S.; Jiang, C.; Zheng, N.; Yang, Z.; Shi, L. Evolution of Pauson-Khand Reaction: Strategic Applications in Total Syntheses of Architecturally Complex Natural Products (2016–2020). Catalysts 2020, 10, 1199. [Google Scholar] [CrossRef]
- Keese, F.; Guidetti-Grept, R.; Herzog, B. Synthesis of [5.5.5.5]Fenestranes by Pd-Catalyzed Carbonylation-Cyclisation. Tetrahedron Lett. 1992, 33, 1207–1210. [Google Scholar] [CrossRef]
- Yasuhara, S.; Sasa, M.; Kusakabe, T.; Takayama, H.; Kimura, M.; Mochida, T.; Kato, K. Cyclization–Carbonylation–Cyclization Coupling Reactions of Propargyl Acetates and Amides with Palladium(II)–Bisoxazoline Catalysts. Angew. Chem. Int. Ed. 2011, 50, 3912–3915. [Google Scholar] [CrossRef] [PubMed]
- Shen, R.; Kusakabe, T.; Yatsu, T.; Kanno, Y.; Takahashi, K.; Nemoto, K.; Kato, K. Palladium(II) Catalyzed Cyclization-Carbonylation-Cyclization Coupling Reaction of (ortho-Alkynyl Phenyl) (Methoxymethyl) Sulfides Using Molecular Oxygen as the Terminal Oxidant. Molecules 2016, 21, 1177. [Google Scholar] [CrossRef]
- Bartish, C.M.; Drissel, G.M. Kirk-Othmer Encyclopedia of Chemical Technology, 3rd ed.; Grayson, M., Eckroth, D., Bushey, G.J., Campbell, L., Klingsberg, A., van Nes, L., Eds.; John Wiley & Sons: New York, NY, USA, 1978; Volume 4, p. 774. [Google Scholar]
- Kusakabe, T.; Kawaguchi, K.; Kawamura, M.; Niimura, N.; Shen, R.; Takayama, H.; Kato, K. Cyclization-Carbonylation-Cyclization Coupling Reaction of Propargyl Ureas with Palladium(II)-bisoxazoline Catalyst. Molecules 2012, 17, 9220–9230. [Google Scholar] [CrossRef]
- Kusakabe, T.; Kawai, Y.; Shen, R.; Mochida, T.; Kato, K. Cyclization–Carbonylation–Cyclization Coupling Reaction of γ-Propynyl-1,3-diketones with Palladium(II)-bisoxazoline Catalyst. Org. Biomol. Chem. 2012, 10, 3192–3194. [Google Scholar] [CrossRef]
- Kusakabe, T.; Sekiyama, E.; Ishino, Y.; Motodate, S.; Kato, S.; Mochida, T.; Kato, K. Cyclization–Carbonylation–Cyclization Coupling Reactions of N-Propargylanilines and o-Alkynylphenols with Palladium(II)–bisoxazoline Catalysts. Synthesis 2012, 44, 1825–1832. [Google Scholar] [CrossRef]
- Kusakabe, T.; Sagae, H.; Kato, K. Cyclization–Carbonylation–Cyclization Coupling reaction of α,β-Alkynic Hydrazones with Palladium(II)-bisoxazoline Catalyst. Org. Biomol. Chem. 2013, 11, 4943–4948. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Kusakabe, T.; Takahashi, K.; Kato, K. A Cyclization–Carbonylation–Cyclization Coupling Reaction of (ortho-Alkynyl phenyl) (Methoxymethyl) Sulfides with the Palladium(II)-bisoxazoline Catalyst. Org. Biomol. Chem. 2014, 12, 3380–3385. [Google Scholar] [CrossRef] [PubMed]
- Shen, R.; Kusakabe, T.; Takahashi, K.; Kato, K. Pd(II)-Catalyzed Ligand Controlled Synthesis of Methyl 1-benzyl-1H-indole-3-carboxylates and Bis(1-benzyl-1H-indol-3-yl)methanones. Org. Biomol. Chem. 2014, 12, 4602–4609. [Google Scholar] [CrossRef]
- Ariyama, T.; Kusakabe, T.; Sato, K.; Funatogawa, M.; Lee, D.; Takahashi, K.; Kato, K. Pd(II)-Catalyzed Ligand-Controlled Synthesis of 2,3-Dihydroisoxazole-4-carboxylates and Bis(2,3-dihydroisoxazol-4-yl)methanones. Heterocycles 2016, 93, 512–518. [Google Scholar]
- Kubasabe, T.; Mochida, T.; Ariyama, T.; Lee, D.; Ohkubo, S.; Takahashi, K.; Kato, K. PdII Catalyzed Ligand Controlled Synthesis of Bis(3-furanyl)methanones and Methyl 3-furancarboxylates. Org. Biomol. Chem. 2019, 17, 6860–6865. [Google Scholar]
- Gabriele, B.; Chimento, A.; Mancuso, R.; Pezzi, V.; Ziccarelli, I.; Sirianni, R. Derivati 6,6a-diidrofuro[3,2-b]furan-2-(5H)onici, loro Preparazione e Uso nel Trattamento dei Tumori. Italian Patent 102017000078586, 8 October 2019. [Google Scholar]
- Gabriele, B.; Chimento, A.; Mancuso, R.; Pezzi, V.; Ziccarelli, I.; Sirianni, R. 6,6a-Dihydrofuro[3,2-b]furan-2-(5H)one Derivatives, their Preparation and Use for Treating Tumors. European Patent EP3428169, 20 January 2021. [Google Scholar]
- Mancuso, R.; Ziccarelli, I.; Chimento, A.; Marino, N.; Della Ca’, N.; Sirianni, R.; Pezzi, V.; Gabriele, B. Catalytic Double Cyclization Process for Antitumor Agents against Breast Cancer Cell Lines. iScience 2018, 3, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Acerbi, A.; Carfagna, C.; Costa, M.; Mancuso, R.; Gabriele, B.; Della Ca’, N. An Unprecedented Pd-Catyalyzed Carbonylative Route to Fused Furo[3,4-b]indol-1-ones. Chem. Eur. J. 2018, 24, 4835–4840. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, R.; Della Ca’, N.; Veltri, L.; Ziccarelli, I.; Gabriele, B. PdI2–Based Catalysis for Carbonylation Reactions: A Personal Account. Catalysts 2019, 9, 610. [Google Scholar] [CrossRef]
- Gabriele, B. Recent Advances in the PdI2-Catalyzed Carbonylative Synthesis of Heterocycles from Acetylenic Substrates: A Personal Account. Targets Heterocycl. Syst. 2018, 22, 41–55. [Google Scholar]
- Gabriele, B.; Salerno, G. PdI2. In e-EROS (Electronic Encyclopedia of Reagents for Organic Synthesis); Crich, D., Ed.; Wiley–Interscience: New York, NY, USA, 2006. [Google Scholar]
- Gabriele, B.; Salerno, G.; Costa, M. PdI2-Catalyzed Synthesis of Heterocycles. Synlett 2004, 2004, 2468–2483. [Google Scholar] [CrossRef]
- Gabriele, B.; Salerno, G.; Costa, M.; Chiusoli, G.P. Recent Developments in the Synthesis of Heterocyclic Derivatives by PdI2-Catalyzed Oxidative Carbonylation Reactions. J. Organomet. Chem. 2003, 687, 219–228. [Google Scholar] [CrossRef]
- Gabriele, B.; Costa, M.; Salerno, G.; Chiusoli, G.P. An Efficient and Selective Palladium-Catalysed Oxidative Sicarbonylation of Alkynes to Alkyl- or Aryl-Maleic Esters. J. Chem. Soc. Perkin Trans. 1 1994, 1994, 83–87. [Google Scholar] [CrossRef]
- Mancuso, R.; Miliè, R.; Palumbo Piccionello, A.; Olivieri, D.; Della Ca’, N.; Carfagna, C.; Gabriele, B. Catalytic Carbonylative Double Cyclization of 2-(3-Hydroxy-1-yn-1-yl)phenols in Ionic Liquids Leading to Furobenzofuranone Derivatives. J. Org. Chem. 2019, 84, 7303–7311. [Google Scholar] [CrossRef]
- Pancrazzi, F.; Sarti, N.; Mazzeo, P.P.; Bacchi, A.; Carfagna, C.; Mancuso, R.; Gabriele, B.; Stirling, A.; Della Ca’, N. Site-Selective Double and Tetracyclization Routes to Fused Polyheterocyclic Structures by Pd-Catalyzed Carbonylation Reactions. Org. Lett. 2020, 22, 1569–1574. [Google Scholar] [CrossRef]
- Gabriele, B.; Salerno, G.; Mancuso, R.; Costa, M. Efficient Synthesis of Ureas by Direct Palladium-Catalyzed Oxidative Carbonylation of Amines. J. Org. Chem. 2004, 69, 4741–4750. [Google Scholar] [CrossRef]
- Della Ca’, N.; Bottarelli, P.; Dibenedetto, A.; Aresta, M.; Gabriele, B.; Salerno, G.; Costa, M. Palladium-Catalyzed Synthesis of Symmetrical Urea Derivatives by Oxidative Carbonylation of Primary Amines in Carbon Dioxide Medium. J. Catal. 2011, 282, 120–127. [Google Scholar]
- Mancuso, R.; Russo, P.; Lettieri, M.; Santandrea, M.; Cuocci, C.; Gabriele, B. Disclosing Polycyclic Heterocycles: Synthesis of Furothienopyran and Pyranothienopyran Derivatives by Palladium Iodide Catalyzed Carbonylative Double Cyclization. Adv. Synth. Catal. 2022, 364, 3917–3926. [Google Scholar] [CrossRef]
- Mancuso, R.; Strangis, R.; Ziccarelli, I.; Della Ca’, N.; Gabriele, B. Palladium Catalysis with Sulfurated Substrates under Aerobic Conditions: A Direct Oxidative Carbonylation Approach to Thiophene-3-carboxylic Esters. J. Catal. 2021, 393, 335–343. [Google Scholar] [CrossRef]
- Mancuso, R.; Cuglietta, S.; Strangis, R.; Gabriele, B. Synthesis of Benzothiophene-3-carboxylic Esters by Palladium Iodide-Catalyzed Oxidative Cyclization−Deprotection−Alkoxycarbonylation Sequence under Aerobic Conditions. J. Org. Chem. 2023, 88, 5180–5186. [Google Scholar] [CrossRef]
- Mancuso, R.; Russo, P.; Miliè, R.; Dell’Aera, M.; Grande, F.; Della Ca’, N.; Gabriele, B. Palladium Iodide Catalyzed Carbonylative Double Cyclization to a New Class of S,O-Bicyclic Heterocycles. Catal. Today 2022, 397–399, 631–638. [Google Scholar] [CrossRef]
- Chen, J.; Neumann, H.; Beller, M.; Wu, X.-F. Palladium-Catalyzed Synthesis of Isoindoloquinazolinones via Dicarbonylation of 1,2-Dibromoarenes. Org. Biomol. Chem. 2014, 12, 5835–5838. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Man, N.Y.T.; Stewart, S.; Wu, X.-F. Palladium-Catalyzed Dicarbonylative Synthesis of Tetracycle Quinazolinones. Org. Biomol. Chem. 2015, 13, 4422–4425. [Google Scholar] [CrossRef] [PubMed]
- Natte, K.; Chen, J.; Li, H.; Neumann, H.; Beller, M.; Wu, X.-F. Palladium-Catalyzed Carbonylation of 2-Bromoanilines with 2-Formylbenzoic Acid and 2-Halobenzaldehydes: Efficient Synthesis of Functionalized Isoindolinones. Chem. Eur. J. 2014, 20, 14184–14188. [Google Scholar] [CrossRef]
- Li, H.; Li, W.; Spannenberg, A.; Baumann, W.; Neumann, H.; Beller, M.; Wu, X.-F. A Novel Domino Synthesis of Quinazolinediones by Palladium-Catalyzed Double Carbonylation. Chem. Eur. J. 2014, 20, 8541–8544. [Google Scholar] [CrossRef]
- Frutos-Pedreño, R.; García-López, J.-A. 2-Arylacetamides as Versatile Precursors for 3-Aminoisocoumarin and Homophthalimide Derivatives: Palladium-Catalyzed Cascade Double Carbonylation Reactions. Adv. Synth. Catal. 2016, 358, 2692–2700. [Google Scholar] [CrossRef]








































































































Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).