Abstract
An efficient and convenient synthesis of benzoxazole/benzothiazole-substituted esters in a one-pot strategy is reported. In this investigation, a selective construction of C-N and C-S bonds via simple addition is performed. Thus, using substituted 2-aminophenols/2-aminobenzenethiols, TMTD (tetramethylthiuram disulfide) and α,β-unsaturated esters as starting substrates, C-N and C-S bonds can be selectively constructed by means of the Michael addition reaction. This protocol features high selectivity, high atomic economy, mild conditions, good functional tolerance and good to excellent yields, showing the potential value for the preparation of some biologically and pharmaceutically active compounds.
1. Introduction
Compounds containing C-N/C-S bonds have become a key part in organic synthesis, biomedicine, material chemistry and industrial manufacturing [1,2,3,4,5,6,7,8,9,10,11,12,13]. Many heterocyclic compounds bearing C-N/C-S bonds are indispensable in pharmaceutical chemistry due to their unique biological and pharmaceutical activities (Figure 1). These compounds can be used as an anti-inflammatory (a) [14], a heat shock protein 90 (Hsp 90) an inhibitor (b) [15], a botanical fungicide or insecticide (c) [16], a CpIMPDH inhibitor (d) [17], a CCR3 selective antagonist (e) [18], a lipoxygenase inhibitor (f) [19], an anticancer agent (g) [20] and an antinematode drug (h) [21]. Therefore, the construction of the C-N bond and the C-S bond has aroused the interest of the organic chemistry community.

Figure 1.
Representative biologically active compounds containing C-N/C-S bonds.
So far, various methods for constructing C-N/S bonds have been reported (Scheme 1). Bolm [22], Buchwald [23], Chen [24] and Biswasa [25] realized the construction of the C-N bond by using aryl halides or alcohols to react with nitrogen-containing heterocyclic compounds. The most traditional method to form the C-S bond is the coupling reaction between thiols and aryl halides [26,27,28,29,30,31]. The Sokolova [32] and Ma [33] group completed the construction of the C-S bond under base conditions. In addition, Yuan and Hajra [34,35] used phenylhydrazine and phenylthiophenol as starting materials to obtain various compounds containing a C-S bond via visible-light-mediated synthesis or aerobic-oxidative coupling. Our laboratory and other groups [36,37,38] also completed the construction of the C-S bond through the Chan-Lam coupling reaction. To the best of our knowledge, the selective construction of C-N/C-S bonds has rarely been reported [39,40]. Though, efficient, the previously reported methods for the construction of C-N/C-S bonds involved the use of toxic, foul-smelling, and expensive reagents, transition metals as catalysts, a high reaction temperature, a long reaction time, as well as low conversion, which limited the application of these methods in pharmaceutical synthesis. Thus, it is still desirable to develop efficient protocols for the selective construction of C-N/C-S bonds, especially the ones using a tandem or one-pot synthesis strategy.

Scheme 1.
Previous strategies for the synthesis of C-N/C-S bonds [22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38].
As part of our long-term interest in constructing various C-X (X = O, S, N, P) bonds and studying the synthesis of benzoheterocyclic compounds [41,42,43,44,45], we report here an effective and practical method for the selective formation of C-N/C-S bonds by Michael addition reactions, starting from substituted 2-aminophenols/2-aminobenzenethiols and TMTD (tetramethylthiuram disulfide) with α,β-unsaturated esters in a one-pot strategy (Scheme 2, the present study).
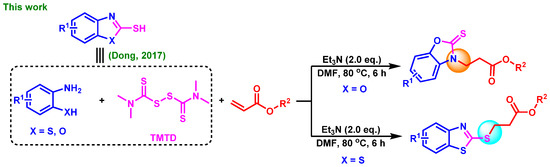
Scheme 2.
Selective formation of C-N/C-S bonds in one-pot strategy [45].
2. Results and Discussion
For our initial study, we chose 2-aminophenol (1a), tetramethylthiuram disulfide (TMTD) and ethyl acrylate (2a) as the starting materials for the model reaction to explore the optimal reaction conditions (Table 1). Our laboratory has previously made good progress in the synthesis of benzoheterocyclic compounds. The reaction conditions for the first step to produce 2-aminophenols were slightly adjusted based on our previous work [41,42,43,44,45], and our attempts were focused on the second step (the Michael addition reaction). Using CuI as the catalyst and Et3N as the base in the initial attempt, the ideal product 3aa was obtained with a 19% yield of EtOH (Entry 1, Table 1). Next we tried other catalysts and found that the catalytic effect of CuI was slightly better (Entries 1–4, Table 1). We were surprised to find that the yield was greatly improved when DMF was used as the solvent (Entry 5, Table 1), and the result was better without adding metal salt as the catalyst (entry 6, Table 1). The screening of bases and reaction temperature showed that Et3N was the best base and 80 °C was the optimal temperature (entries 7–12, Table 1). Furthermore, the loading of the base and the type of solvent were also investigated, and the results are shown in entries 13–16, Table 1. The optimal reaction conditions are summarized in Entry 10, Table 1. It is worth noting that the model reaction in Table 1 always produced the by-product 3aa’ in traces which formed the C-S bond in the additional step.

Table 1.
Optimization of the reaction conditions a.
According to the optimal conditions obtained above, a variety of related substrates are explored, and the results are presented in Table 2. First, the reaction effect of various substituted 2-aminophenols and ethyl acrylate was investigated, and the experimental results showed that 2-aminophenols with either electron-withdrawing groups (-Br, -Cl) or electron-donating groups (-CH3, -tBu) could react readily with ethyl acrylate to give the desired products with a moderate to good yield (3aa–3ah). When tert-butyl acrylate and cyclohexyl acrylate were used to react with various substituted 2-aminophenols, the reaction also worked well and provided the expected products (3ai–3av) readily. When 2-amino-3-methylphenol was used as the starting material to react with ethyl acrylate, tert-butyl acrylate and cyclohexyl acrylate, the compounds (3ah, 3aj, 3ar) could only be obtained in 28%, 29% and 36% yields, respectively, which might be due to the steric hindrance.
Table 2.
One-pot synthesis of benzoxazole-substituted ester compounds a,b.
To further determine the structure of the target products, we performed an X-ray diffraction analysis of 3ai (CCDC: 2152821, Figure 2) to show the exact C-N bond formation of the product.
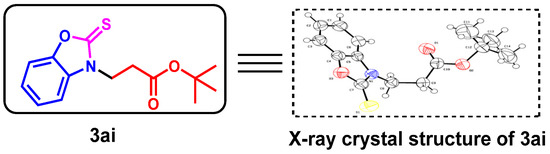
Figure 2.
X-ray crystallography of 3ai.
The above optimal reaction conditions were also applicable to the reactions between various substituted 2-aminobenzenethiols (1) and α,β-unsaturated esters (2). However, this reaction would give the C-S formation products (all oily) as major ones via Michael addition, and the C-N formation products were obtained in traces. Thus, under standard conditions, a variety of substituted 2-aminobenzenethiols (1) and TMTD could react with ethyl acrylate, tert-butyl acrylate and cyclohexyl acrylate, smoothly, giving the target products with a moderate yield (5aa–5ag, Table 3). To determine the detailed structure of the target product, the X-ray diffraction analysis of by-product 5ag’ (CCDC: 2152790, Figure 3) was performed, showing the existence of the C-N bond formation, which could further indicate the C-S bond formation of the major product 5ag.
Table 3.
One-pot synthesis of benzothiazole-substituted ester compounds a,b.
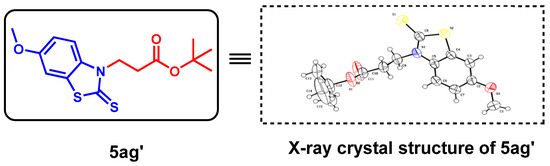
Figure 3.
X-ray crystallography of by-product 5ag’.
3. Conclusions
In summary, we developed an efficient and convenient one-pot method to selectively construct C-N and C-S bonds by a simple Michael addition. Using substituted 2-aminophenols/2-aminobenzenethiols, TMTD (tetramethylthiuram disulfide) and α,β-unsaturated esters as starting substrates, a variety of benzoxazole/benzothiazole substituted esters were obtainedwithout problem, forming C-N and C-S bonds selectively. This strategy features high selectivity, high atomic economy, mild conditions, good functional tolerance. and good yields, showing its potential value for the preparation of some biologically and pharmaceutically active compounds.
4. Experimental Section
General Information. All starting materials were commercially purchased. Yields refer to isolated compounds estimated to be >95% pure as determined by 1H NMR and capillary GC analysis. NMR spectra were recorded on a Bruker AM400 or AM 600 NMR instrument in CDCl3 using TMS as an internal standard. Chemical shifts are given in ppm, and coupling constants (J) are given in Hz. All melting points were determined on a RY-1G melting point instrument without correction. High-resolution mass spectra (HRMS) were recorded on a Angilent 6545LC/Q-TOF mass instrument (ESI). TLC was performed using aluminum plates coated with SiO2 (Merck 60, F-254) and visualized with UV light at 254 nm. Column chromatography was performed on silica gel (200–300 mesh) with PE (petroleum ether)-EA (ethyl acetate) as an eluent.
General procedure for the synthesis of desired products forming C-N bonds (3aa–3av).
A mixture of substituted 2-aminophenols (1, 0.5 mmol) and tetramethylthiuram disulfide (TMTD, 0.3 mmol) in DMF (2 mL) was stirred at 80 °C for 3 h until the substrates completely disappeared. Subsequently, the sealed tube was cooled to room temperature. 2 (ethyl acrylate, tert-butyl acrylate, or cyclohexyl acrylate, 1.0 mmol) and Et3N (2.0 eq.) were added, and the mixture was stirred at 80 °C for 6 h. After the reaction was completed, it was quenched with saturated NH4Cl, and the crude solution was separated after diluting with ethyl acetate and dried-over anhydrous Na2SO4. The solvent was removed in a vacuum to obtain the crude product, which was further separated and purified by column chromatography to give the desired products (3aa–3av).
General procedure for the synthesis of desired products forming C-S bonds (5aa–5ag).
A mixture of substituted 2-aminobenzenethiols (4, 0.5 mmol) and tetramethylthiuram disulfide (TMTD, 0.3 mmol) in DMF (2 mL) was stirred at 80 °C for 3 h until the substrates completely disappeared. Subsequently, the sealed tube was cooled to room temperature, 2 (ethyl acrylate, tert-butyl acrylate, or cyclohexyl acrylate, 1.0 mmol) and Et3N (2.0 eq.) were added, and the mixture was stirred at 80 °C for 6 h. After the reaction was completed, it was quenched with saturated NH4Cl, and the crude solution was separated after diluting with ethyl acetate and dried over anhydrous Na2SO4. The solvent was removed in a vacuum to obtain the crude product, which was further separated and purified by column chromatography to give the desired products (5aa–5ag).
ethyl 3-(2-thioxobenzo[d]oxazol-3(2H)-yl)propanoate (3aa): The target product 3aa (106.8 mg, 85%) was synthesized as a colorless oil, and purified by column chromatography (ethyl acetate/petroleum ether = 1:10). 1H NMR (400 MHz, CDCl3) δ ppm: 7.27–7.14 (m, 4H), 4.37 (t, J = 6.8 Hz, 2H), 4.02 (q, J = 7.2 Hz, 2H), 2.88 (t, J = 6.8, 2H), 1.11 (t, J = 7.2 Hz, 3H). 13C{1H} NMR (100 MHz, CDCl3) δ ppm: 179.0, 169.8, 146.1, 130.8, 123.9, 123.3, 109.3, 109.0, 60.1, 40.4, 30.3, 13.0. HRMS (ESI) m/z [M + H]+ Calcd for C12H14NO3S+ 252.0689; Found 252.0682.
ethyl 3-(5-bromo-2-thioxobenzo[d]oxazol-3(2H)-yl)propanoate (3ab): The target product 3ab (138.7 mg, 84%) was synthesized as a white solid, and purified by column chromatography (ethyl acetate/petroleum ether = 1:7). mp: 72–74 °C. 1H NMR (400 MHz, CDCl3) δ ppm: 7.36 (d, J = 2.0, 1H), 7.29–7.25 (m, 1H), 7.12 (d, J = 8.4 Hz, 1H), 4.32 (t, J = 6.8, 2H), 4.05 (q, J = 7.1 Hz, 2H), 1.14 (t, J = 7.0 Hz, 3H). 13C{1H} NMR (100 MHz, CDCl3) δ ppm: 179.3, 169.6, 145.1, 132.3, 126.1, 116.8, 112.2, 110.4, 60.2, 40.6, 30.2, 13.1. HRMS (ESI) m/z [M + H]+ Calcd for C12H13BrNO3S+ 329.9794; Found 329.9797.
ethyl 3-(6-methyl-2-thioxobenzo[d]oxazol-3(2H)-yl)propanoate (3ac): The target product 3ac (108.8 mg, 82%) was synthesized as a white solid, and purified by column chromatography (ethyl acetate/petroleum ether = 1:7). mp: 58–60 °C. 1H NMR (400 MHz, CDCl3) δ ppm: 7.10–7.01 (m, 3H), 4.35 (t, J = 6.8 Hz, 2H), 4.03 (q, J = 7.1 Hz, 2H), 2.87 (t, J = 8.6, 2H), 2.35 (s, 3H), 1.13 (t, J = 7.2 Hz, 3H). 13C{1H} NMR (100 MHz, CDCl3) δ ppm: 178.9, 169.9, 146.4, 138.7, 133.8, 128.6, 124.6, 109.8, 108.5, 60.1, 40.4, 30.3, 20.4, 13.0. HRMS (ESI) m/z [M + H]+ Calcd for C13H16NO3S+ 266.0845; Found 266.0842.
ethyl 3-(5-chloro-2-thioxobenzo[d]oxazol-3(2H)-yl)propanoate (3ad): The target product 3ad (111.4 mg, 78%) was synthesized as a white solid, and purified by column chromatography (ethyl acetate/petroleum ether = 1:7). mp: 58–60 °C. 1H NMR (400 MHz, CDCl3) δ ppm: 7.23 (s, 1H), 7.18–7.08 (m, 2H), 4.32 (t, J = 6.6, 2H), 4.04 (q, J = 7.2, 2H), 2.88 (t, J = 6.6 Hz, 2H), 1.14 (t, J = 7.0 Hz, 3H). 13C{1H} NMR (100 MHz, CDCl3) δ ppm: 179.4, 169.6, 144.6, 132.0, 129.6, 123.2, 109.9, 109.5, 60.2, 40.7, 30.2, 13.1. HRMS (ESI) m/z [M + H]+ Calcd for C12H13ClNO3S+ 286.0299; Found 286.0294.
ethyl 3-(5-(tert-butyl)-2-mercaptobenzo[d]oxazol-3(2H)-yl)propanoate (3ae): The target product 3ae (135.3 mg, 88%) was synthesized as a yellow oil, and purified by column chromatography (ethyl acetate/petroleum ether = 1:7). 1H NMR (400 MHz, CDCl3) δ ppm: 7.23–7.13 (m, 3H), 4.40 (t, J = 6.8 Hz, 2H), 4.03 (q, J = 7.1 Hz, 2H), 2.86 (t, J = 6.8, 2H), 1.28 (s, 9H), 1.10 (t, J = 7.2 Hz, 3H). 13C{1H} NMR (100 MHz, CDCl3) δ ppm: 179.3, 169.9, 147.8, 144.2, 130.5, 120.5, 108.5, 105.9, 60.0, 40.3, 34.1, 30.6, 30.5, 13.0. HRMS (ESI) m/z [M + H]+ Calcd for C16H22NO3S+ 308.1315; Found 308.1316.
ethyl 3-(6-chloro-2-thioxobenzo[d]oxazol-3(2H)-yl)propanoate (3af): The target product 3af (75.7 mg, 53%) was synthesized as a white solid, and purified by column chromatography (ethyl acetate/petroleum ether = 1:7). mp: 90–92 °C. 1H NMR (400 MHz, CDCl3) δ ppm: 7.27 (s, 1H), 7.20 (q, J = 9.9 Hz, 2H), 4.34 (t, J = 6.4 Hz, 2H), 4.03 (q, J = 7.1, 2H), 2.89 (t, J = 6.4 Hz, 2H), 1.13 (t, J = 7.1 Hz, 3H). 13C{1H} NMR (100 MHz, CDCl3) δ ppm: 179.1, 169.9, 146.2, 129.9, 129.1, 124.1, 109.9, 109.7, 60.2, 40.6, 30.2, 13.1. HRMS (ESI) m/z [M + H]+ Calcd for C12H13ClNO3S+ 286.0299; Found 286.0297.
ethyl 3-(5-methyl-2-thioxobenzo[d]oxazol-3(2H)-yl)propanoate (3ag): The target product 3ag (92.9 mg, 70%) was synthesized as a colorless oil, and purified by column chromatography (ethyl acetate/petroleum ether = 1:7). 1H NMR (400 MHz, CDCl3) δ ppm: 7.20 (d, J = 8.9 Hz, 1H), 7.04 (d, J = 7.2 Hz, 2H), 4.42 (t, J = 6.9, 2H), 4.12 (q, J = 7.2, 2H), 2.94 (t, J = 6.9 Hz, 2H), 2.44 (s, 3H), 1.20 (t, J = 7.2 Hz, 3H). 13C{1H} NMR (100 MHz, CDCl3) δ ppm: 179.2, 169.8, 144.4, 134.2, 130.8, 124.0, 109.1, 108.9, 60.1, 40.3, 30.3, 20.5, 13.0. HRMS (ESI) m/z [M + H]+ Calcd for C13H16NO3S+ 266.0845; Found 266.0851.
ethyl 3-(4-methyl-2-thioxobenzo[d]oxazol-3(2H)-yl)propanoate (3ah): The target product 3ah (37.1 mg, 28%) was synthesized as a white solid, and purified by column chromatography (ethyl acetate/petroleum ether = 1:7). mp: 70–72 °C. 1H NMR (400 MHz, CDCl3) δ ppm: 7.19 (d, J = 8.0 Hz, 1H), 7.12 (t, J = 7.8, 1H), 7.04 (d, J = 7.6, 1H), 4.68 (t, J = 7.8, 2H), 4.15 (q, J = 7.1 Hz, 2H), 2.93 (t, J = 7.8, 2H), 2.63 (s, 3H), 1.23 (t, J = 7.1, 3H). 13C{1H} NMR (100 MHz, CDCl3) δ ppm: 179.4, 169.3, 146.4, 128.7, 127.0, 123.2, 119.7, 107.5, 60.1, 41.6, 31.2, 16.7, 13.1. HRMS (ESI) m/z [M + H]+ Calcd for C13H16NO3S+ 266.0845; Found 266.0843
tert-butyl 3-(2-thioxobenzo[d]oxazol-3(2H)-yl)propanoate (3ai): The target product 3ai (120.1 mg, 86%) was synthesized as a white solid, and purified by column chromatography (ethyl acetate/petroleum ether = 1:7). mp: 88–90 °C. 1H NMR (400 MHz, CDCl3) δ ppm: 7.28–7.15 (m, 4H), 4.35 (t, J = 6.9 Hz, 2H), 2.78 (t, J = 6.9 Hz, 2H), 1.31 (s, 9H). 13C{1H} NMR (100 MHz, CDCl3) δ ppm: 179.1, 169.0, 146.1, 130.8, 123.9, 123.3, 109.3, 109.1, 80.6, 40.5, 31.5, 27.0. HRMS (ESI) m/z [M + H]+ Calcd for C14H18NO3S+ 280.1002; Found 280.1006.
tert-butyl 3-(4-methyl-2-thioxobenzo[d]oxazol-3(2H)-yl)propanoate (3aj): The target product 3aj (42.5 mg, 29%) was synthesized as a yellow oil, and purified by column chromatography (ethyl acetate/petroleum ether = 1:7). 1H NMR (400 MHz, CDCl3) δ ppm: 7.20 (d, J = 7.6 Hz, 1H), 7.13 (t, J = 7.8 Hz, 1H), 7.04 (d, J = 7.6 Hz, 1H), 4.65 (t, J = 7.9 Hz, 2H), 2.84 (t, J = 8.0 Hz, 2H), 2.63 (s, 3H), 1.43 (s, 9H). 13C{1H} NMR (100 MHz, CDCl3) δ ppm: 179.3, 168.5, 146.4, 128.7, 127.0, 123.2, 119.8, 107.5, 80.7, 41.8, 32.4, 27.3, 16.7. HRMS (ESI) m/z [M + H]+ Calcd for C15H20NO3S+ 294.1158; Found 294.1156.
tert-butyl 3-(6-methyl-2-thioxobenzo[d]oxazol-3(2H)-yl)propanoate (3ak): The target product 3ak (130.6 mg, 89%) was synthesized as a white solid, and purified by column chromatography (ethyl acetate/petroleum ether = 1:7). mp: 66–68 °C. 1H NMR (400 MHz, CDCl3) δ ppm: 7.13 (d, J = 7.8 Hz, 2H), 7.08 (d, J = 8.4 Hz, 1H), 4.37 (t, J = 6.9 Hz, 2H), 2.82 (t, J = 6.9 Hz, 2H), 2.41 (s, 3H), 1.38 (s, 9H). 13C{1H} NMR (100 MHz, CDCl3) δ ppm: 178.9, 169.1, 146.4, 133.8, 128.6, 124.5, 109.7, 108.6, 80.6, 40.5, 31.5, 27.0, 20.4. HRMS (ESI) m/z [M + H]+ Calcd for C15H20NO3S+ 294.1158; Found 294.1163.
tert-butyl 3-(5-(tert-butyl)-2-thioxobenzo[d]oxazol-3(2H)-yl)propanoate (3al): The target product 3al (122.4 mg, 73%) was synthesized as a colorless oil, and purified by column chromatography (ethyl acetate/petroleum ether = 1:7). 1H NMR (400 MHz, CDCl3) δ ppm: 7.29–7.26 (m, 2H), 7.24 (d, J = 9.0 Hz, 1H), 4.44 (t, J = 6.9 Hz, 2H), 2.84 (t, J = 6.9 Hz, 2H), 1.38 (s, 9H), 1.36 (s, 9H). 13C{1H} NMR (100 MHz, CDCl3) δ ppm: 180.4, 170.3, 148.8, 145.2, 131.6, 121.5, 109.6, 107.1, 81.6, 41.4, 35.1, 32.6, 31.6, 28.0. HRMS (ESI) m/z [M + H]+ Calcd for C18H26NO3S+ 336.1628; Found 336.1626.
tert-butyl 3-(5-bromo-2-thioxobenzo[d]oxazol-3(2H)-yl)propanoate (3am): The target product 3am (150.5 mg, 84%) was synthesized as a white solid, and purified by column chromatography (ethyl acetate/petroleum ether = 1:7). mp: 136–138 °C. 1H NMR (400 MHz, CDCl3) δ ppm: 7.43 (d, J = 1.8 Hz, 1H), 7.35 (dd, J = 8.6, 1.8 Hz, 1H), 7.19 (d, J = 8.6 Hz, 1H), 4.36 (t, J = 6.8 Hz, 2H), 2.82 (t, J = 6.8 Hz, 2H), 1.40 (s, 9H). 13C{1H} NMR (100 MHz, CDCl3) δ ppm: 179.3, 168.9, 145.1, 132.2, 126.1, 116.8, 112.2, 110.4, 80.9, 40.8, 31.5, 27.0. HRMS (ESI) m/z [M + H]+ Calcd for C14H17BrNO3S+ 358.0107; Found 358.0112.
tert-butyl 3-(6-chloro-2-thioxobenzo[d]oxazol-3(2H)-yl)propanoate (3an): The target product 3an (109.8 mg, 70%) was synthesized as a white solid, and purified by column chromatography (ethyl acetate/petroleum ether = 1:7). mp: 88–90 °C. 1H NMR (400 MHz, CDCl3) δ ppm: 7.27 (d, J = 1.7 Hz, 1H), 7.21 (dd, J = 8.5, 1.7 Hz, 1H), 7.17 (d, J = 8.5 Hz, 1H), 4.31 (t, J = 6.6 Hz, 2H), 2.78 (t, J = 6.6 Hz, 2H), 1.32 (s, 9H). 13C{1H} NMR (100 MHz, CDCl3) δ ppm: 179.1, 169.1, 146.2, 129.8, 129.0, 124.1, 109.9, 109.7, 80.7, 40.7, 31.4, 27.0. HRMS (ESI) m/z [M + H]+ Calcd for C14H17ClNO3S+ 314.0612; Found 314.0606.
cyclohexyl 3-(2-thioxobenzo[d]oxazol-3(2H)-yl)propanoate (3ao): The target product 3ao (135.9 mg, 89%) was synthesized as a colorless oil, and purified by column chromatography (ethyl acetate/petroleum ether = 1:7). 1H NMR (400 MHz, CDCl3) δ ppm: 7.26–7.13 (m, 4H), 4.68–4.60 (m, 1H), 4.37 (t, J = 6.8 Hz, 2H), 4.79–4.71 (t, J = 6.8 Hz, 2H), 3.47 (t, J = 6.8 Hz, 2H), 2.86 (t, J = 6.7 Hz, 2H), 1.71–1.64 (m, 2H), 1.61–1.54 (m, 2H), 1.46–1.14 (m, 6H). 13C{1H} NMR (100 MHz, CDCl3) δ ppm: 179.1, 169.4, 146.2, 130.9, 123.9, 123.3, 109.3, 109.1, 72.7, 40.5, 30.7, 30.4, 24.2, 22.6. HRMS (ESI) m/z [M + H]+ Calcd for C16H20NO3S+ 306.1158; Found 306.1155.
cyclohexyl 3-((6-methylbenzo[d]oxazol-2-yl)thio)propanoate (3ap): The target product 3ap (110.2 mg, 69%) was synthesized as a colorless oil, and purified by column chromatography (ethyl acetate/petroleum ether = 1:7). 1H NMR (400 MHz, CDCl3) δ ppm: 7.45 (d, J = 8.1 Hz, 1H), 7.23 (s, 1H), 7.08 (dd, J = 8.1, 0.8 Hz, 1H), 4.85–4.78 (m, 1H), 3.52 (t, J = 7.1 Hz, 2H), 2.90 (t, J = 6.9 Hz, 2H), 2.45 (s, 3H), 1.90–1.81 (m, 2H), 1.76–1.67 (m, 2H), 1.54–1.23 (m, 6H). 13C{1H} NMR (100 MHz, CDCl3) δ ppm: 169.8, 162.6, 151.2, 138.7, 133.3, 124.3, 116.7, 109.2, 72.4, 33.7, 30.6, 26.2, 24.3, 22.7, 20.6. HRMS (ESI) m/z [M + H]+ Calcd for C17H22NO3S+ 320.1315; Found 320.1316.
cyclohexyl 3-(5-(tert-butyl)-2-thioxobenzo[d]oxazol-3(2H)-yl)propanoate (3aq): The target product 3aq (126.5 mg, 70%) was synthesized as a colorless oil, and purified by column chromatography (ethyl acetate/petroleum ether = 1:7). 1H NMR (400 MHz, CDCl3) δ ppm: 7.20 (dd, J = 8.5, 1.7 Hz, 1H), 7.18–7.14 (m, 2H), 4.68–4.59 (m, 1H), 4.39 (t, J = 6.8 Hz, 2H), 2.83 (t, J = 6.8 Hz, 2H), 1.67–1.53 (m, 4H), 1.46–1.06 (m, 16H). 13C{1H} NMR (100 MHz, CDCl3) δ ppm: 179.2, 169.4, 147.8, 144.2, 130.5, 120.5, 108.5, 106.0, 72.5, 40.4, 34.1, 30.8, 30.6, 30.4, 24.2, 22.6. HRMS (ESI) m/z [M + H]+ Calcd for C20H28NO3S+ 362.1784; Found 362.1777.
cyclohexyl 3-(4-methyl-2-thioxobenzo[d]oxazol-3(2H)-yl)propanoate (3ar): The target product 3ar (57.5 mg, 36%) was synthesized as a white solid, and purified by column chromatography (ethyl acetate/petroleum ether = 1:7). mp: 78–80 °C. 1H NMR (400 MHz, CDCl3) δ ppm: 7.20 (d, J = 7.7 Hz, 1H), 7.13 (t, J = 7.8 Hz, 1H), 7.04 (d, J = 7.6 Hz, 1H), 4.82–4.75 (m, 1H), 4.68 (t, J = 7.8 Hz, 2H), 2.92 (t, J = 7.8 Hz, 2H), 2.63 (s, 3H), 1.85–1.78 (m, 2H), 1.73–1.65 (m, 2H), 1.56–1.26 (m, 6H). 13C{1H} NMR (100 MHz, CDCl3) δ ppm: 179.3, 168.7, 146.4, 128.7, 127.0, 123.2, 119.8, 107.5, 72.6, 41.7, 31.6, 30.5, 24.3, 22.6, 16.7. HRMS (ESI) m/z [M + H]+ Calcd for C17H22NO3S+ 320.1315; Found 320.1313.
cyclohexyl 3-(5-bromo-2-thioxobenzo[d]oxazol-3(2H)-yl)propanoate (3as): The target product 3as (153.7 mg, 80%) was synthesized as a white solid, and purified by column chromatography (ethyl acetate/petroleum ether = 1:7). mp: 96–98 °C. 1H NMR (400 MHz, CDCl3) δ ppm: 7.42 (d, J = 1.8 Hz, 1H), 7.34 (dd, J = 8.6, 1.8 Hz, 1H), 7.18 (d, J = 8.6 Hz, 1H), 4.77–4.68 (m, 1H), 4.37 (t, J = 6.6 Hz, 2H), 2.91 (t, J = 6.6 Hz, 2H), 1.82–1.73 (m, 2H), 1.69–1.62 (m, 2H), 1.55–1.21 (m, 6H). 13C{1H} NMR (100 MHz, CDCl3) δ ppm: 179.3, 169.2, 145.1, 132.3, 126.1, 116.8, 112.3, 110.4, 72.9, 40.7, 30.6, 30.5, 24.2, 22.7. HRMS (ESI) m/z [M + H]+ Calcd for C16H19BrNO3S+ 384.0264; Found 384.0266.
cyclohexyl 3-(6-chloro-2-thioxobenzo[d]oxazol-3(2H)-yl)propanoate (3at): The target product 3at (149.5 mg, 88%) was synthesized as a white solid, and purified by column chromatography (ethyl acetate/petroleum ether = 1:7). mp: 86–88 °C. 1H NMR (400 MHz, CDCl3) δ ppm: 7.27 (d, J = 1.6 Hz, 1H), 7.22 (dd, J = 8.5, 1.7 Hz, 1H), 7.18 (d, J = 8.5 Hz, 1H), 4.68–4.60 (m, 1H), 4.33 (t, J = 6.5 Hz, 2H), 2.87 (t, J = 6.5 Hz, 2H), 1.72–1.65 (m, 2H), 1.62–1.55 (m, 2H), 1.46–1.14 (m, 6H). 13C{1H} NMR (100 MHz, CDCl3) δ ppm: 179.1, 169.4, 146.2, 129.9, 129.0, 124.1, 109.9, 109.7, 72.8, 40.7, 30.5, 30.4, 24.2, 22.6. HRMS (ESI) m/z [M + H]+ Calcd for C16H19ClNO3S+ 340.0769; Found 340.0776.
cyclohexyl 3-(5-chloro-2-thioxobenzo[d]oxazol-3(2H)-yl)propanoate (3au): The target product 3au (95.2 mg, 56%) was synthesized as a white solid, and purified by column chromatography (ethyl acetate/petroleum ether = 1:7). mp: 110–112 °C. 1H NMR (400 MHz, CDCl3) δ ppm: 7.30 (d, J = 1.6 Hz, 1H), 7.24 (d, J = 8.6 Hz, 1H), 7.20 (dd, J = 8.6, 1.8 Hz, 1H), 4.77–4.69 (m, 1H), 4.38 (t, J = 6.6 Hz, 2H), 2.93 (t, J = 6.6 Hz, 2H), 1.82–1.73 (m, 2H), 1.72–1.63 (m, 2H), 1.52–1.20 (m, 6H). 13C{1H} NMR (100 MHz, CDCl3) δ ppm: 179.5, 169.2, 144.6, 132.0, 129.8, 123.2, 109.9, 109.5, 72.9, 40.7, 30.6, 30.5, 24.2, 22.7. HRMS (ESI) m/z [M + H]+ Calcd for C16H19ClNO3S+ 340.0769; Found 340.0766.
cyclohexyl 3-(5-methyl-2-thioxobenzo[d]oxazol-3(2H)-yl)propanoate (3av): The target product 3av (138.9 mg, 87%) was synthesized as a white solid, and purified by column chromatography (ethyl acetate/petroleum ether = 1:7). mp: 98–100 °C. 1H NMR (400 MHz, CDCl3) δ ppm: 7.19 (d, J = 8.2 Hz, 1H), 7.06–7.00 (m, 2H), 4.76–4.69 (m, 1H), 4.41 (t, J = 6.9 Hz, 2H), 2.91 (t, J = 6.9 Hz, 2H), 2.43 (s, 3H), 1.80–1.72 (m, 2H), 1.69–1.63 (m, 2H), 1.54–1.19 (m, 6H). 13C{1H} NMR (100 MHz, CDCl3) δ ppm: 179.2, 169.3, 144.4, 134.1, 130.8, 124.0, 109.2, 108.8, 72.7, 40.4, 30.7, 30.5, 24.2, 22.6, 20.5. HRMS (ESI) m/z [M + H]+ Calcd for C17H22NO3S+ 320.1315; Found 320.1321.
ethyl 3-(benzo[d]thiazol-2-ylthio)propanoate (5aa): The target product 5aa (88.2 mg, 66%) was synthesized as a colorless oil, and purified by column chromatography (ethyl acetate/petroleum ether = 1:10). 1H NMR (400 MHz, CDCl3) δ ppm: 7.48 (dd, J = 8.0, 4.0 Hz, 1H), 7.44–7.39 (m, 1H), 7.34–7.27 (m, 2H), 5.00 (t, J = 4.0, 2H), 4.12 (q, J = 8.0 Hz, 2H), 2,87 (t, J = 8.0 Hz, 2H), 1.21 (t, J = 8.0 Hz, 3H). 13C{1H} NMR (100 MHz, CDCl3) δ ppm: 186.6, 169.8, 156.5, 134.2, 127.9, 113.4, 112.0, 104.7, 60.1, 54.9, 41.0, 30.3, 13.1. HRMS (ESI) m/z [M + H]+ Calcd for C12H13NO2S2+ 268.0460; Found 268.0463.
tert-butyl 3-(benzo[d]thiazol-2-ylthio)propanoate (5ab): The target product 5ab (104.9 mg, 71%) was synthesized as a colorless oil, and purified by column chromatography (ethyl acetate/petroleum ether = 1:7). 1H NMR (400 MHz, CDCl3) δ ppm: 7.30 (d, J = 8.0 Hz, 1H), 7.23 (d, J = 7.6, 1H), 7.18–7.09 (m, 2H), 4.49 (t, J = 7.6, 2H), 2.60 (t, J = 7.6 Hz, 2H), 1.25 (s, 9H). 13C{1H} NMR (100 MHz, CDCl3) δ ppm: 188.1, 169.0, 140.2, 126.7, 126.0, 123.8, 120.4, 111.5, 80.5, 41.0, 31.4, 27.0. HRMS (ESI) m/z [M + H]+ Calcd for C14H18NO2S2+ 296.0773; Found 296.0771.
cyclohexyl 3-(benzo[d]thiazol-2-ylthio)propanoate (5ac): The target product 5ac (99.7 mg, 62%) was synthesized as a colorless oil, and purified by column chromatography (ethyl acetate/petroleum ether = 1:7). 1H NMR (400 MHz, CDCl3) δ ppm: 7.40 (d, J = 7.8 Hz, 1H), 7.34 (t, J = 7.2 Hz, 1H), 7.28–7.18 (m, 2H), 4.71–4.64 (m, 1H), 4.61 (t, J = 7.4 Hz, 2H), 2.78 (t, J = 7.4 Hz, 2H), 1.73–1.57 (m, 4H), 1.46–1.1.13 (m, 6H). 13C{1H} NMR (100 MHz, CDCl3) δ ppm: 188.1, 169.2, 140.2, 126.7, 126.0, 123.8, 120.4, 111.5, 72.5, 41.0, 30.6, 30.4, 24.3, 22.6. HRMS (ESI) m/z [M + H]+ Calcd for C16H20NO2S2+ 322.0930; Found 322.0935.
ethyl 3-((5-chlorobenzo[d]thiazol-2-yl)thio)propanoate (5ad): The target product 5ad (66.4 mg, 44%) was synthesized as a colorless oil, and purified by column chromatography (ethyl acetate/petroleum ether = 1:15). 1H NMR (400 MHz, CDCl3) δ ppm: 7.31 (d, J = 8.4 Hz, 1H), 7.27 (d, J = 2.0 Hz, 1H), 7.21 (dd, J = 8.4, 1.8 Hz, 1H), 4.58 (t, J = 7.4, 2H), 4.09 (q, J = 7.2 Hz, 2H), 2.81 (t, J = 7.2 Hz, 2H), 1.18 (t, J = 7.2 Hz, 3H). 13C{1H} NMR (100 MHz, CDCl3) δ ppm: 188.9, 169.6, 141.1, 132.4, 124.9, 124.0, 121.0, 111.7, 60.2, 401.1, 30.2, 13.1. HRMS (ESI) m/z [M + H]+ Calcd for C12H13ClNO2S2+ 302.0071; Found 302.0068.
tert-butyl 3-((5-chlorobenzo[d]thiazol-2-yl)thio)propanoate (5ae): The target product 5ae (100.6 mg, 61%) was synthesized as a colorless oil, and purified by column chromatography (ethyl acetate/petroleum ether = 1:7). 1H NMR (400 MHz, CDCl3) δ ppm: 7.35 (d, J = 8.4 Hz, 1H), 7.31 (d, J = 1.6 Hz, 1H), 7.26–7.23 (m, 1H), 4.59 (t, J = 7.4 Hz, 2H), 2.74 (t, J = 7.4 Hz, 2H), 1.41 (s, 9H). 13C{1H} NMR (100 MHz, CDCl3) δ ppm: 188.1, 169.8, 140.2, 126.7, 126.0, 123.8, 120.4, 111.4, 60.1, 40.9, 30.3, 13.1. HRMS (ESI) m/z [M + H]+ Calcd for C14H17ClNO2S2+ 330.0384; Found 330.0386.
cyclohexyl 3-((5-chlorobenzo[d]thiazol-2-yl)thio)propanoate (5af): The target product 5af (99.7 mg, 62%) was synthesized as a colorless oil, and purified by column chromatography (ethyl acetate/petroleum ether = 1:7). 1H NMR (400 MHz, CDCl3) δ ppm: 7.39 (d, J = 8.4 Hz, 1H), 7.35 (s, 1H), 7.22–7.17 (m, 1H), 4.74–4.66 (m, 1H), 4.64 (t, J = 7.2 Hz, 2H), 2.86 (t, J = 7.2 Hz, 2H), 1.87–1.77 (m, 2H), 1.74–1.65 (m, 2H), 1.52–1.18 (m, 6H). 13C{1H} NMR (100 MHz, CDCl3) δ ppm: 188.8, 169.1, 141.1, 132.4, 124.9, 124.0, 121.0, 111.7, 72.7, 41.2, 30.6, 30.5, 24.3, 22.7. HRMS (ESI) m/z [M + H]+ Calcd for C16H19ClNO2S2+ 356.0540; Found 356.0536.
tert-butyl 3-((6-methoxybenzo[d]thiazol-2-yl)thio)propanoate (5ag): The target product 5ag (113.9 mg, 70%) was synthesized as a colorless oil, and purified by column chromatography (ethyl acetate/petroleum ether = 1:7). 1H NMR (400 MHz, CDCl3) δ ppm: 7.16 (d, J = 8.8 Hz, 1H), 6.93–6.87 (m, 2H), 4.53 (t, J = 7.4 Hz, 2H), 3.75 (s, 3H), 2.69 (t, J = 7.4 Hz, 2H), 1.34 (s, 9H). 13C NMR (100 MHz, CDCl3) δ ppm: 186.1, 168.5, 156.0, 133.8, 127.4, 112.9, 111.6, 104.1, 80.0, 54.4, 40.6, 30.9, 26.5. HRMS (ESI) m/z [M + H]+ Calcd for C15H20NO3S2+ 326.0879; Found 326.0871.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/catal13040658/s1, Characterization data for all products, X-ray diffraction analysis of compound 3ai and 5ag’, 1H-NMR and 13C-NMR spectra of all products. Deposition Numbers 2152790 (for 5ag’), 2152821 (for 3ai) contain the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe “http://www.ccdc.cam.ac.uk/structures”.
Author Contributions
Methodology, Z.-Y.G.; writing—review and editing, Z.-Y.G. and C.-L.Y.; methodology, D.W.; review and editing, L.H.; supervision, Z.-B.D.; project administration, Z.-B.D. All authors have read and agreed to the published version of the manuscript.
Funding
The financial support from the Open Research Fund of School of Chemistry and Chemical Engineering, Henan Normal University (2020ZD02), the Natural Science Foundation of Henan Province (232300421126) is greatly appreciated.
Data Availability Statement
Data supporting reported results can be found in Supplementary Materails.
Acknowledgments
Z.-Y.G. is thankful for the support of Postgraduate Innovation Foundation from Wuhan Institute of Technology.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Huang, W.; Yang, G.F. Microwave-Assisted, One-Pot Syntheses and Fungicidal Activity of Polyfluorinated 2-Benzylthiobenzothiazoles. Bioorg. Med. Chem. 2006, 14, 8280–8285. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.P.; Segal, S. Study of Fungicidal Activities of Some Benzothiazoles. Indian J. Chem. 1988, 27B, 941–943. [Google Scholar]
- Akhtar, T.; Hameed, S.; Al-Masoudi, N.A.; Loddo, R.; Colla, P. In Vitro Antitumor and Antiviral Activities of New Benzothiazole and 1, 3, 4-Oxadiazole-2-Thione Derivatives. Acta Pharm. 2008, 58, 135–149. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.T.; Hsei, I.J.; Chen, C. Synthesis and Anticancer Evaluation of Bis (Benzimidazoles), Bis (Benzoxazoles), and Benzothiazoles. Bioorg. Med. Chem. 2006, 14, 6106–6119. [Google Scholar]
- Palmer, P.J.; Trigg, R.B.; Warrington, J.V. Benzothiazolines as Antituberculous Agents. J. Med. Chem. 1971, 14, 248–251. [Google Scholar] [CrossRef]
- Rao, A.J.; Rao, P.V.; Rao, V.K.; Mohan, C.; Raju, C.N.; Reddy, C.S. Microwave Assisted One-Pot Synthesis of Novel a-Aminophosphonates and Their Biological Activity. Bull. Korean Chem. Soc. 2010, 31, 1863–1868. [Google Scholar] [CrossRef]
- Singh, M.; Singh, S.K.; Gangwar, M.; Nath, G.; Singh, S.K. Design, Synthesis and Mode of Action of Some Benzothiazole Derivatives Bearing an Amide Moiety as Antibacterial Agents. RSC Adv. 2014, 4, 19013–19023. [Google Scholar] [CrossRef]
- Suresh, C.H.; Rao, J.V.; Jayaveera, K.N.; Subudhi, S.K. Synthesis and Anthelminitc Activity of 3 (2-Hydrazino Benzothiazoles)-Substituted Indole-2-One. Int. Res. J. Pharm. 2011, 2, 257–261. [Google Scholar]
- Reddy, P.; Lin, Y.; Chang, H. Synthesis of Novel Benzothiazole Compounds with an Extended Conjugated System. Arkivoc 2007, 16, 113–122. [Google Scholar] [CrossRef]
- Heo, Y.; Song, Y.S.; Kim, B.T.; Heo, J.N. A Highly Regioselective Synthesis of 2-Aryl-6-Chlorobenzothiazoles Employing Microwave-Promoted Suzuki-Miyaura Coupling Reaction. Tetrahedron Lett. 2006, 47, 3091–3094. [Google Scholar] [CrossRef]
- Azam, M.A.; Suresh, B. Biological Activities of 2-Mercaptobenzothiazole Derivatives: A Review. Sci. Pharm. 2012, 80, 789–824. [Google Scholar] [CrossRef]
- Dumas, J.; Brittelli, D.; Chen, J.; Dixon, B.; Hatoum-Mokdad, H.; Konig, G.; Sibley, R.; Witowsky, J.; Wong, S. Synthesis and Structure Activity Relationships of Novel Small Molecule Cathepsin D Inhibitors. Bioorg. Med. Chem. Lett. 1999, 9, 2531–2536. [Google Scholar] [CrossRef]
- Pejin, B.; Iodice, C.; Tommonaro, G.; Rosa, S.D. Synthesis and Biological Activities of Thio-Avarol Derivatives. J. Nat. Prod. 2008, 71, 1850–1853. [Google Scholar] [CrossRef]
- Han, Y.; Dong, W.; Guo, Q.Q.; Li, X.F.; Huang, L.J. The Importance of Indole and Azaindole Scaffold in the Development of Antitumor Agents. Eur. J. Med. Chem. 2020, 203, 112506. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Fan, J.; Vu, K.; Hong, K.; Le Brazidec, J.Y.; Shi, J.; Biamonte, M.; Busch, D.J.; Lough, R.E.; Grecko, R.; et al. 7′-Substituted Benzothiazolothio-and Pyridinothiazolothio-Purines as Potent Heat Shock Protein 90 Inhibitors. J. Med. Chem. 2006, 49, 5352–5362. [Google Scholar] [CrossRef] [PubMed]
- Zhi, X.Y.; Jiang, L.Y.; Li, T.; Song, L.L.; Wu, L.J.; Cao, H.; Yang, C. Natural Product-Based Semisynthesis and Biological Evaluation of Thiol/Amino-Michael Adduct of Xanthatin Derived from Xanthium Strumarium as Potential Pesticidal Agents. Bioorg. Chem. 2020, 97, 103696. [Google Scholar] [CrossRef]
- Gorla, S.K.; Kavitha, M.; Zhang, M.; Chin, J.E.; Liu, X.; Striepen, B.; Makowska-Grzyska, M.; Kim, Y.; Joachimiak, A.; Hedstrom, L.; et al. Optimization of Benzoxazole-Based Inhibitors of Cryptosporidium Parvum Inosine 5′-Monophosphate Dehydrogenase. J. Med. Chem. 2013, 56, 4028–4043. [Google Scholar] [CrossRef]
- Naya, A.; Kobayashi, K.; Ishikawa, M.; Ohwaki, K.; Saeki, T.; Noguchi, K.; Ohtake, N. Discovery of a Novel CCR3 Selective Antagonist. Bioorg. Med. Chem. Lett. 2001, 11, 1219–1223. [Google Scholar] [CrossRef]
- Choudhary, A.N.; Kumar, A.; Juyal, V. Quantitative Structure Activity Relationship (QSAR) Analysis of Substituted 4-Oxothiazolidines and 5-Arylidines as Lipoxygenase Inhibitors. Mini-Rev. Med. Chem. 2010, 10, 705–714. [Google Scholar] [CrossRef] [PubMed]
- Chekal, B.P.; Guinness, S.M.; Lillie, B.M.; McLaughlin, R.W.; Palmer, C.W.; Post, R.J.; Sieser, J.E.; Singer, R.A.; Sluggett, G.W.; Vaidyanathan, R.; et al. Development of an Efficient Pd-Catalyzed Coupling Process for Axitinib. Org. Process Res. Dev. 2013, 18, 266–274. [Google Scholar] [CrossRef]
- Klimešová, V.; Kočí, J.; Pour, M.; Stachel, J.; Waisser, K.; Kaustová, J. Synthesis and Preliminary Evaluation of Benzimidazole Derivatives as Antimicrobial Agents. Eur. J. Med. Chem. 2002, 37, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Larsson, P.F.; Correa, A.; Carril, M.; Norrby, P.O.; Bolm, C. Copper-Catalyzed Cross-Couplings with Part-per-Million Catalyst Loadings. Angew. Chem. Int. Ed. 2009, 48, 5691. [Google Scholar] [CrossRef] [PubMed]
- Ueda, S.; Buchwald, S.L. Catalyst-Controlled Chemoselective Arylation of 2-Aminobenzimidazoles. Angew. Chem. Int. Ed. 2012, 51, 10364–10367. [Google Scholar] [CrossRef]
- Li, X.; Yuan, T.; Yang, Y.; Chen, J. Novel Copper/PEG-400 Catalyst Systems for Chemoselective S- and N-arylation of 2-Mercaptobenzothiazole. Tetrahedron 2014, 70, 9652–9660. [Google Scholar] [CrossRef]
- Duari, S.; Biswas, S.; Roy, A.; Maity, S.; Mishra, A.K.; Souza, A.R.; Elsharif, A.M.; Morgon, N.H.; Biswas, S. Regioselective N-Functionalization of Tautomerizable Heterocycles through Methyl Trifluoromethanesulfonate-Catalyzed Substitution of Alcohols and Alkyl Group Migrations. Adv. Synth. Catal. 2022, 364, 865–872. [Google Scholar] [CrossRef]
- Herrera Cano, N.; Ballari, M.S.; Lopez, A.G.; Santiago, A.N. New Synthesis and Biological Evaluation of Benzothiazole Derivates as Antifungal Agents. J. Agric. Food. Chem. 2015, 63, 3681–3686. [Google Scholar] [CrossRef] [PubMed]
- Ghaderi-Shekhi Abadi, P.; Rafiee, E.; Joshaghani, M. Pd-PVP-Fe (Palladium-Poly(N-vinylpyrrolidone)-iron) Catalyzed S-arylation of Thiols with Aryl Halides in Aqueous Media. Inorg. Chim. Acta. 2016, 451, 162–170. [Google Scholar] [CrossRef]
- Sikari, R.; Sinha, S.; Das, S.; Saha, A.; Chakraborty, G.; Mondal, R.; Paul, N.D. Achieving Nickel Catalyzed C-S Cross-Coupling under Mild Conditions Using Metal-Ligand Cooperativity. J. Org. Chem. 2019, 84, 4072–4085. [Google Scholar] [CrossRef]
- Feng, Y.-S.; Li, Y.-Y.; Tang, L.; Wu, W.; Xu, H.-J. Efficient Ligand-Free Copper-Catalyzed C-S Cross-Coupling of Thiols with Aryl Iodides Using KF/Al2O3 as Base. Tetrahedron Lett. 2010, 51, 2489–2492. [Google Scholar] [CrossRef]
- Wu, W.-Y.; Wang, J.-C.; Tsai, F.-Y. A Reusable FeCl3-6H2O/Cationic 2,2′-Bipyridyl Catalytic System for the Coupling of Aryl Iodides with Thiols in Water under Aerobic Conditionst. Green Chem. 2009, 11, 326–329. [Google Scholar] [CrossRef]
- Correa, A.; Carril, M.; Bolm, C. Iron-Catalyzed S-Arylation of Thiols with Aryl Iodides. Angew. Chem. Int. Ed. 2008, 47, 2880–2883. [Google Scholar] [CrossRef] [PubMed]
- Sokolova, A.S.; Yarovaya, O.I.; Shtro, A.A.; Borisova, M.S.; Morozova, E.A.; Tolstikova, T.G.; Zarubaev, V.V.; Salakhutdinov, N.F. Synthesis and Biological Activity of Heterocyclic Borneol Derivatives. Chem. Heterocycl. Compd. 2017, 53, 371–377. [Google Scholar] [CrossRef]
- Shi, L.; Liu, X.; Zhang, H.; Jiang, Y.; Ma, D. Synthesis of 2-Thio-Substituted Benzothiazoles via a Domino Condensation/S-Arylation/Heterocyclization Process. J. Org. Chem. 2011, 76, 4200–4204. [Google Scholar] [CrossRef] [PubMed]
- Kibriya, G.; Mondal, S.; Hajra, A. Visible-Light-Mediated Synthesis of Unsymmetrical Diaryl Sulfides via Oxidative Coupling of Arylhydrazine with Thiol. Org. Lett. 2018, 20, 7740–7743. [Google Scholar] [CrossRef]
- Ren, X.; Tang, S.; Li, L.; Li, J.; Liang, H.; Li, G.; Yang, G.; Li, H.; Yuan, B. Surfactant-Type Catalyst for Aerobic Oxidative Coupling of Hydrazine with Thiol in Water. J. Org. Chem. 2019, 84, 8683–8690. [Google Scholar] [CrossRef]
- Xu, H.J.; Zhao, Y.Q.; Feng, T.; Feng, Y.S. Chan-Lam-Type S-Arylation of Thiols with Boronic Acids at Room Temperature. J. Org. Chem. 2012, 77, 2878–2884. [Google Scholar] [CrossRef]
- Liu, X.; Dong, Z.B. Chemoselective Chan-Lam Coupling Reactions between Benzimidazoline-2-thiones and Arylboronic Acids. J. Org. Chem. 2019, 84, 11524–11532. [Google Scholar] [CrossRef]
- Bhowmik, A.; Yadav, M.; Fernandes, R.A. Room Temperature Nickel-Catalyzed Cross-Coupling of Aryl-Boronic Acids with Thiophenols: Synthesis of Diarylsulfides. Org. Biomol. Chem. 2020, 18, 2447–2458. [Google Scholar] [CrossRef]
- Quan, Z.J.; Ren, R.G.; Da, Y.X.; Zhang, Z.; Wang, X.C. Alkylation of SH-Heterocycles with Diethyl Phosphite Using Tetrachloroethylene as an Efficient Solvent. Heteroat. Chem. 2011, 22, 653–658. [Google Scholar] [CrossRef]
- Anan’Eva, K.V.; Rozhkova, N.K. Benzazolin-2-Thiones in the Michael Reaction. 2. Reaction of Benzothiazolin-and Benzoxazolin-2-Thiones with Acrylonitrile, Acrylamide, and Methyl Acrylate in the Presence of Basic Catalysts. Chem. Heterocycl. Compd. 1986, 22, 564–567. [Google Scholar] [CrossRef]
- Dong, Z.B.; Liu, X.; Bolm, C. Copper-Catalyzed C(sp2)-S Coupling Reactions for the Synthesis of Aryl Dithiocarbamates with Thiuram Disulfide Reagents. Org. Lett. 2017, 19, 5916–5919. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.B.; Balkenhohl, M.; Tan, E.; Knochel, P. Synthesis of Functionalized Diaryl Sulfides by Cobalt-Catalyzed Coupling between Arylzinc Pivalates and Diaryl Disulfides. Org. Lett. 2018, 20, 7581–7584. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.Y.; Li, J.H.; Zhang, S.B.; Chen, L.J.; Li, Y.S.; Dong, Z.B. A Mild Synthesis of 2-Substituted Benzothiazoles via Nickel-Catalyzed Intramolecular Oxidative C-H Functionalization. J. Org. Chem. 2020, 85, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Peng, H.Y.; Yang, M.M.; Hao, E.J.; Li, Y.S.; Dong, Z.B. Cs2CO3-Promoted Hydrothiolation of Alkynes with Aryl Thioureas: Stereoselective Synthesis of (Z)-Vinyl Sulfides. J. Org. Chem. 2021, 86, 8457–8464. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Liu, M.; Xu, W.; Zeng, M.T.; Zhu, H.; Chang, C.Z.; Dong, Z.B. An Environmentally Benign and Efficient Synthesis of Substituted Benzothiazole-2-thiols, Benzoxazole-2-thiols, and Benzimidazoline-2-thiones in Water. Green Chem. 2017, 19, 5591–5598. [Google Scholar] [CrossRef]





































Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).