Mechanistic Investigation on Hydrocyanation of Butadiene: A DFT Study

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
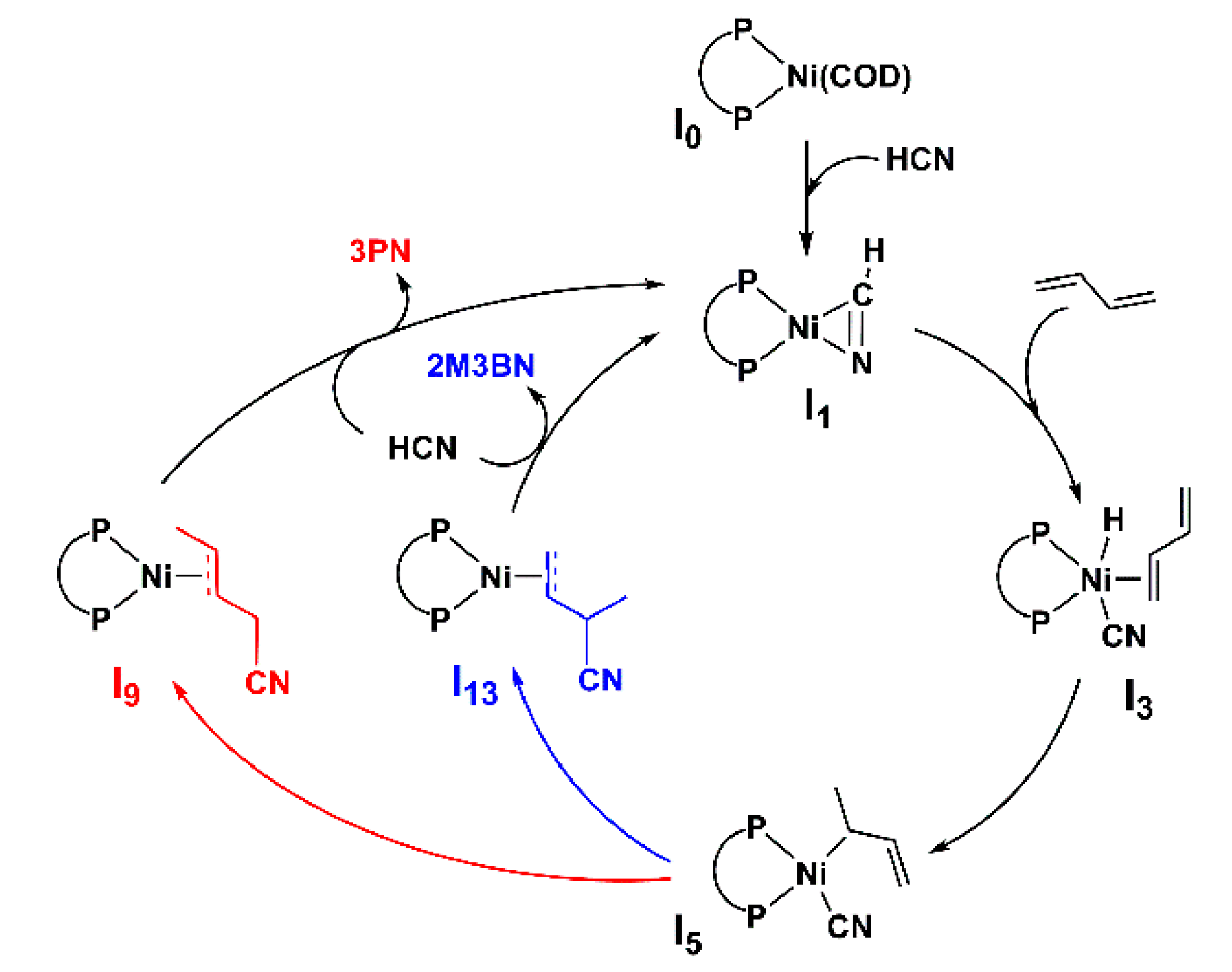
2.1. HCN Addition and Butadiene Insertion
2.2. Mechanism of the Formation of 3PN
2.3. Mechanism of the Formation of 2M3BN
2.4. Isomerization of 2M3BN to 3PN
2.5. Key Intermediate I5
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A
Appendix B

References
- Gillespie, J.A.; Zuidema, E.; van Leeuwen, P.W.N.M.; Kamer, P.C.J. Phosphorus(III) Ligands in Homogeneous Catalysis: Design and Synthesis, 1st ed.; Wiley: John Wiley & Sons, Ltd: Chichester, UK, 2012; pp. 1–26. [Google Scholar] [CrossRef]
- Vogt, D.; Wilting, J. Reduction: Hydrocyanation of C=C. In Comprehensive Chirality; Carreira, E.M., Yamamoto, H., Eds.; Elsevier: Amsterdam, The Netherlands, 2012; pp. 343–354. [Google Scholar] [CrossRef]
- RajanBabu, T.V. Hydrocyanation in Organic Synthesis, in Comprehensive Organic Synthesis II, 2nd ed.; Knochel, P., Ed.; Elsevier: Amsterdam, The Netherlands, 2014; pp. 1772–1793. [Google Scholar] [CrossRef]
- Seidel, W.C.; Tolman, C.A. Homogeneous nickel-catalyzed olefin hydrocyanation. Annal. N. Y. Acad. Sci. 1983, 415, 201–221. [Google Scholar] [CrossRef]
- Tolman, C.A.; Seidel, W.C.; Gosser, L.W. Formation of three-coordinate nickel(0) complexes by phosphorus ligand dissociation from NiL4. J. Am. Chem. Soc. 1974, 96, 53–60. [Google Scholar] [CrossRef]
- Tolman, C.A.; McKinney, R.J.; Seidel, W.C.; Druliner, J.D.; Stevens, W.R. Homogeneous Nickel-Catalyzed Olefin Hydrocyanation. Adv. Catal. 1985, 33, 1–46. [Google Scholar] [CrossRef]
- Baker, M.J.; Pringle, P.G. Chiral aryl diphosphites: A new class of ligands for hydrocyanation catalysis. J. Chem. Soc. Chem. Commun. 1991, 18, 1292–1293. [Google Scholar] [CrossRef]
- Tolman, C.A. Electron donor-acceptor properties of phosphorus ligands. Substituent additivity. J. Am. Chem. Soc. 1970, 92, 2953–2956. [Google Scholar] [CrossRef]
- Tolman, C.A. Phosphorus ligand exchange equilibriums on zerovalent nickel. Dominant role for steric effects. J. Am. Chem. Soc. 1970, 92, 2956–2965. [Google Scholar] [CrossRef]
- Tolman, C.A. The 16 and 18 electron rule in organometallic chemistry and homogeneous catalysis. Chem. Soc. Rev. 1972, 1, 337–353. [Google Scholar] [CrossRef]
- Tolman, C.A. Steric effects of phosphorus ligands in organometallic chemistry and homogeneous catalysis. Chem. Rev. 1977, 77, 313–348. [Google Scholar] [CrossRef]
- Van der Vlugt, J.I.; Hewat, A.C.; Neto, S.; Sablong, R.; Mills, A.M.; Lutz, M.; Spek, A.L.; Müller, C.; Vogt, D. Sterically Demanding Diphosphonite Ligands-Synthesis and Application in Nickel-Catalyzed Isomerization of 2-Methyl-3-Butenenitrile. Adv. Synth. Catal. 2004, 346, 993–1003. [Google Scholar] [CrossRef]
- Wilting, J.; Müller, C.; Hewat, A.C.; Ellis, D.D.; Tooke, D.M.; Spek, A.L.; Vogt, D. Nickel-Catalyzed Isomerization of 2-Methyl-3-butenenitrile. Organometallics 2005, 24, 13–15. [Google Scholar] [CrossRef][Green Version]
- Bini, L.; Müller, C.; Vogt, D. Mechanistic Studies on Hydrocyanation Reactions. ChemCatChem 2010, 2, 590–608. [Google Scholar] [CrossRef]
- Bini, L.; Müller, C.; Vogt, D. Ligand development in the Ni-catalyzed hydrocyanation of alkenes. Chem. Commun. 2010, 46, 8325–8334. [Google Scholar] [CrossRef] [PubMed]
- García, J.J.; Arévalo, A.; Brunkan, N.M.; Jones, W.D. Cleavage of Carbon-Carbon Bonds in Alkyl Cyanides Using Nickel(0). Organometallics 2004, 23, 3997–4002. [Google Scholar] [CrossRef]
- Acosta-Ramírez, A.; Flores-Gaspar, A.; Muñoz-Hernández, M.; Arévalo, A.; Jones, W.D.; García, J.J. Nickel Complexes Involved in the Isomerization of 2-Methyl-3-butenenitrile. Organometallics 2007, 26, 1712–1720. [Google Scholar] [CrossRef]
- Swartz, B.D.; Reinartz, N.M.; Brennessel, W.W.; García, J.J.; Jones, W.D. Solvent Effects and Activation Parameters in the Competitive Cleavage of C-CN and C-H Bonds in 2-Methyl-3-Butenenitrile Using [(dippe)NiH]2. J. Am. Chem. Soc. 2008, 130, 8548–8554. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Jones, W.D. DFT Calculations of the Isomerization of 2-Methyl-3-butenenitrile by [Ni(bisphosphine)] in Relation to the DuPont Adiponitrile Process. Organometallics 2011, 30, 547–555. [Google Scholar] [CrossRef]
- Tolman, C.A.; Seidel, W.C.; Druliner, J.D.; Domaille, P.J. Catalytic hydrocyanation of olefins by nickel(0) phosphite complexes-effects of Lewis acids. Organometallics 1984, 3, 33–38. [Google Scholar] [CrossRef]
- Chaumonnot, A.; Lamy, F.; Sabo-Etienne, S.; Donnadieu, B.; Chaudret, B.; Barthelat, J.-C.; Galland, J.-C. Catalytic Isomerization of Cyanoolefins Involved in the Adiponitrile Process. C-CN Bond Cleavage and Structure of the Nickel π-Allyl Cyanide Complex Ni(η3-1-Me-C3H4)(CN)(dppb). Organometallics 2004, 23, 3363–3365. [Google Scholar] [CrossRef]
- Liu, K.; Liu, K.-K.; Cheng, M.-J.; Han, M.-H. Theoretical and experimental study of the nickel-catalyzed isomerization of 2-Methyl-3-butenenitrile and the effect of a Lewis acid. J. Organomet. Chem. 2016, 822, 29–38. [Google Scholar] [CrossRef]
- Liu, K.; Xin, H.; Han, M. Elucidation of key factors in nickel-diphosphines catalyzed isomerization of 2-methyl-3-butenenitrile. J. Catal. 2019, 377, 13–19. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.; et al. Gaussian 09, Revision E.01; Gaussian, Inc.: Wallingford, CT, USA, 2015. [Google Scholar]
- Peverati, R.; Truhlar, D.G. M11-L: A Local Density Functional That Provides Improved Accuracy for Electronic Structure Calculations in Chemistry and Physics. J. Phys. Chem. Let. 2012, 3, 117–124. [Google Scholar] [CrossRef]
- Hariharan, P.C.; Pople, J.A.J.T.C.A. The influence of polarization functions on molecular orbital hydrogenation energies. Theor. Chim. Acta 1973, 28, 213–222. [Google Scholar] [CrossRef]
- Francl, M.M.; Pietro, W.J.; Hehre, W.J.; Binkley, J.S.; Gordon, M.S.; Defrees, D.J.; Pople, J.A. Self-consistent Molecular-orbital Methods. XXIII. A Polarization-type Basis Set For 2nd-row Elements. J. Chem. Phys. 1982, 77, 3654–3665. [Google Scholar] [CrossRef]
- Dolg, M.; Wedig, U.; Stoll, H.; Preuss, H. Energy adjusted ab initio pseudopotentials for the first row transition elements. J. Chem. Phys. 1987, 86, 866–872. [Google Scholar] [CrossRef]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- McLean, A.D.; Chandler, G.S. Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z=11-18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar] [CrossRef]
- Blaudeau, J.-P.; McGrath, M.P.; Curtiss, L.A.; Radom, L. Extension of Gaussian-2 (G2) theory to molecules containing third-row atoms K and Ca. J. Chem. Phys. 1997, 107, 5016–5021. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Performance of SM6, SM8, and SMD on the SAMPL1 Test Set for the Prediction of Small-Molecule Solvation Free Energies. J. Phys. Chem. B. 2009, 113, 4538–4543. [Google Scholar] [CrossRef]
- Liu, K.; Han, M. Solvent-Controlled the Isomerization of 2-Methyl-3-Butenenitrile. Molecules. under review.
- Legault, C.Y. CYLview. 2009, Université de Sherbrooke. Available online: http://www.cylview.org (accessed on 11 February 2018).








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, K.; Zhang, S.; Han, M. Mechanistic Investigation on Hydrocyanation of Butadiene: A DFT Study. Catalysts 2020, 10, 818. https://doi.org/10.3390/catal10080818
Liu K, Zhang S, Han M. Mechanistic Investigation on Hydrocyanation of Butadiene: A DFT Study. Catalysts. 2020; 10(8):818. https://doi.org/10.3390/catal10080818
Chicago/Turabian StyleLiu, Kaikai, Shuai Zhang, and Minghan Han. 2020. "Mechanistic Investigation on Hydrocyanation of Butadiene: A DFT Study" Catalysts 10, no. 8: 818. https://doi.org/10.3390/catal10080818
APA StyleLiu, K., Zhang, S., & Han, M. (2020). Mechanistic Investigation on Hydrocyanation of Butadiene: A DFT Study. Catalysts, 10(8), 818. https://doi.org/10.3390/catal10080818