Enhanced Direct Dimethyl Ether Synthesis from CO2-Rich Syngas with Cu/ZnO/ZrO2 Catalysts Prepared by Continuous Co-Precipitation

 , , , , ,
, , , , ,  , , and
, , and
Abstract

1. Introduction
| CO + 2 H2 ⇌ CH3OH | ∆rH298 = −90.6 kJ mol−1 | (1) |
| CO2 + 3 H2 ⇌ CH3OH + H2O | ∆rH298 = −49.1 kJ mol−1 | (2) |
| CO2 + H2 ⇌ CO + H2O | ∆rH298 = 41.0 kJ mol−1 | (3) |
| 2 CH3OH ⇌ C2H6O + H2O | ∆rH298 = −23.4 kJ mol−1 | (4) |
2. Results and Discussion
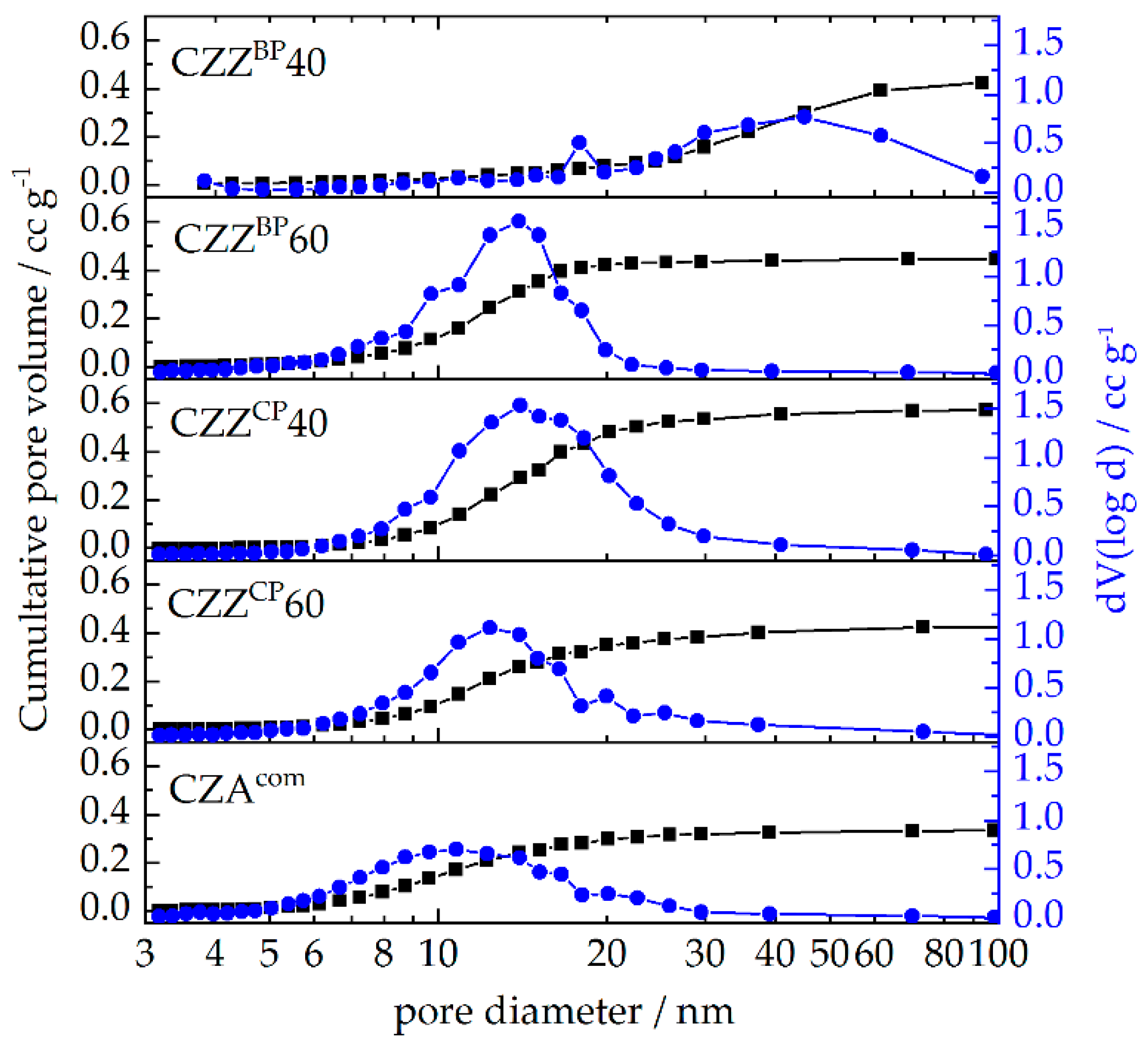
2.1. Textural Properties: Metal Composition and N2 Physisorption
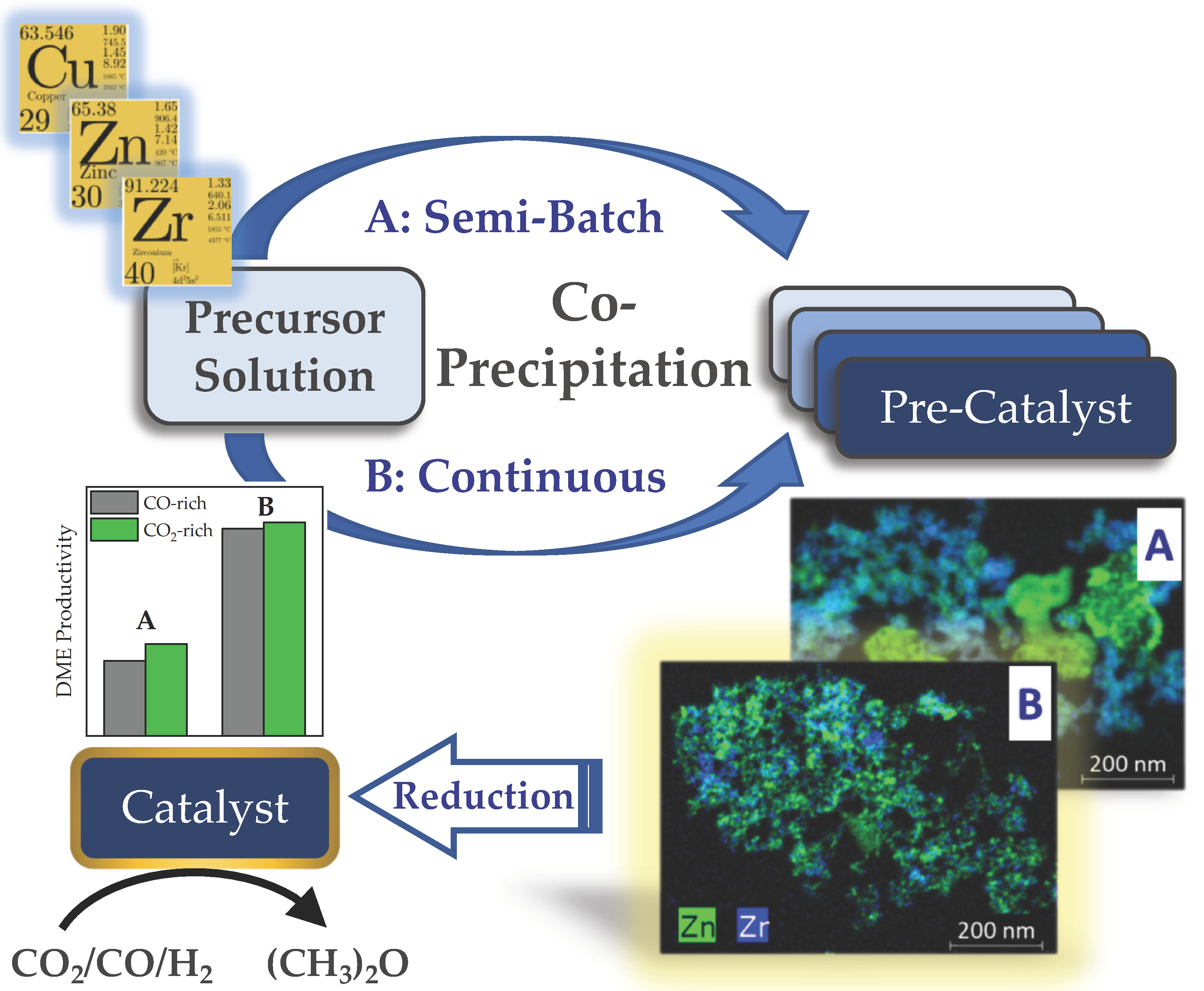
- The different mixing intensities and time-dependencies of semi-batch and continuous co-precipitation results in different rates of phase-specific solid formation in semi-batch and continuous co-precipitation;
- Therefore, the continuous precipitation variant yields a more homogeneous material in terms of phase composition and phase distribution than precipitation in the semi-batch variant;
- Furthermore, the phase composition of the aged product is governed by thermodynamic equilibrium.
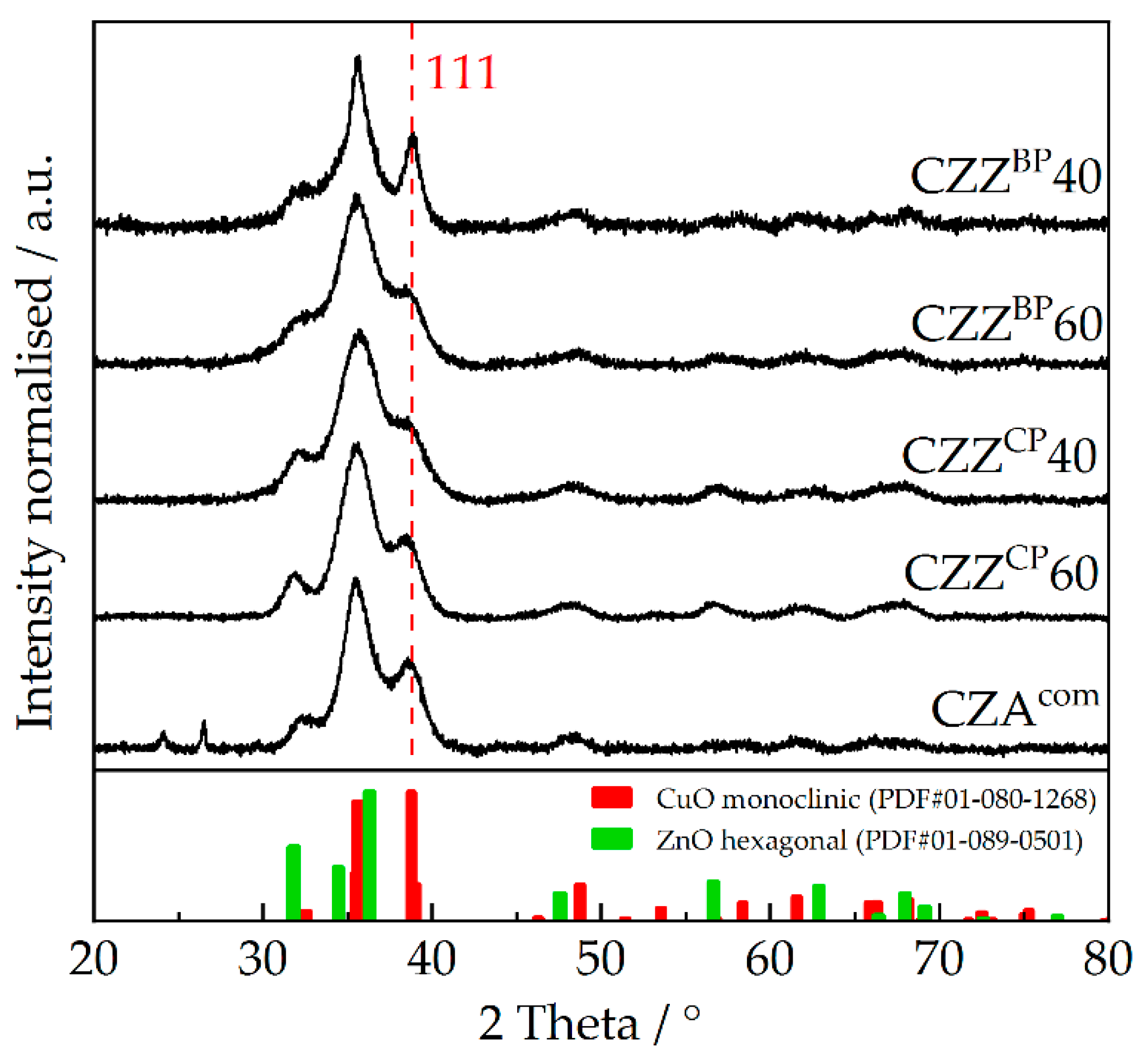
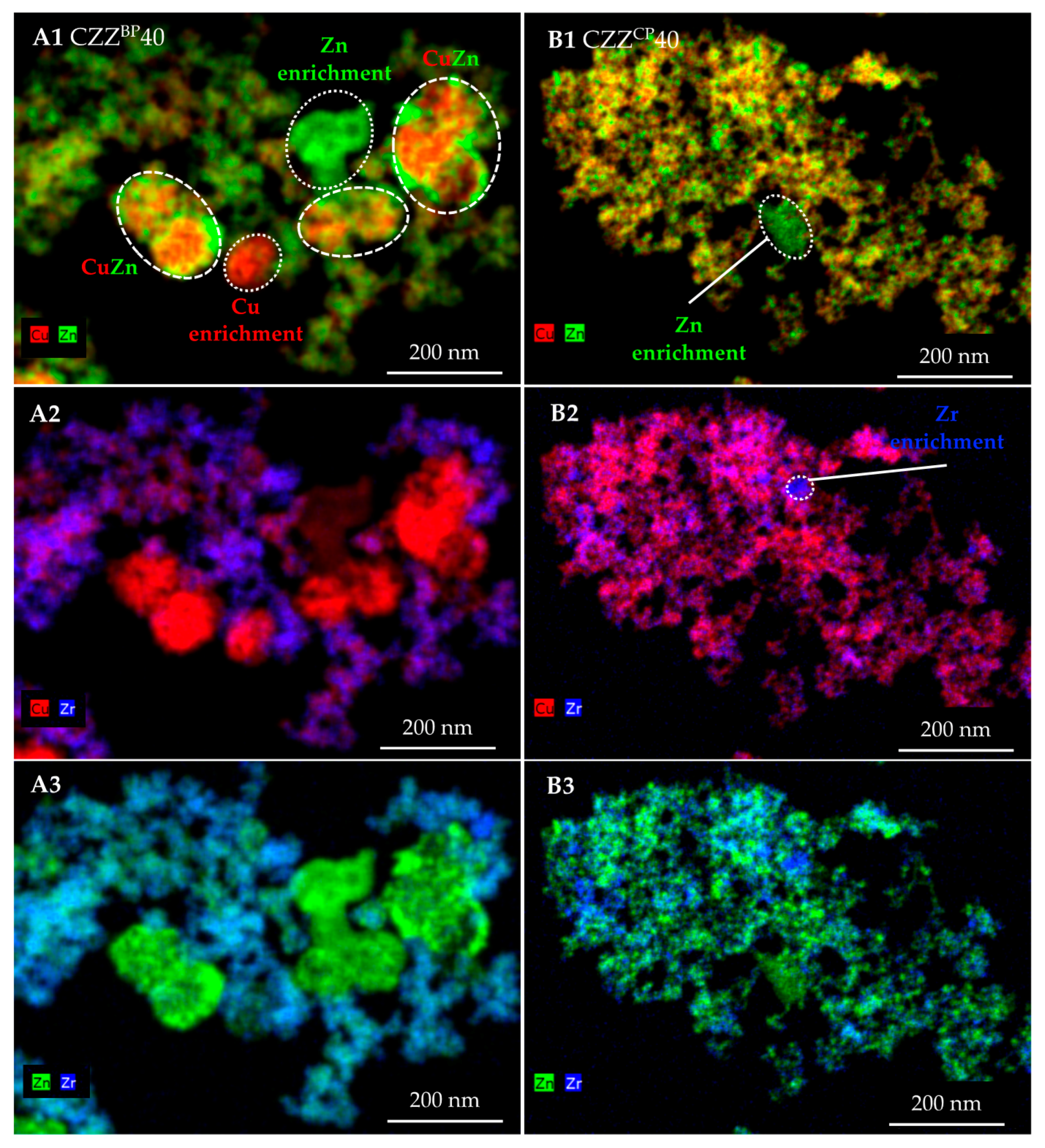
2.2. Structural Properties: XRD, TEM and EDXS Analyses
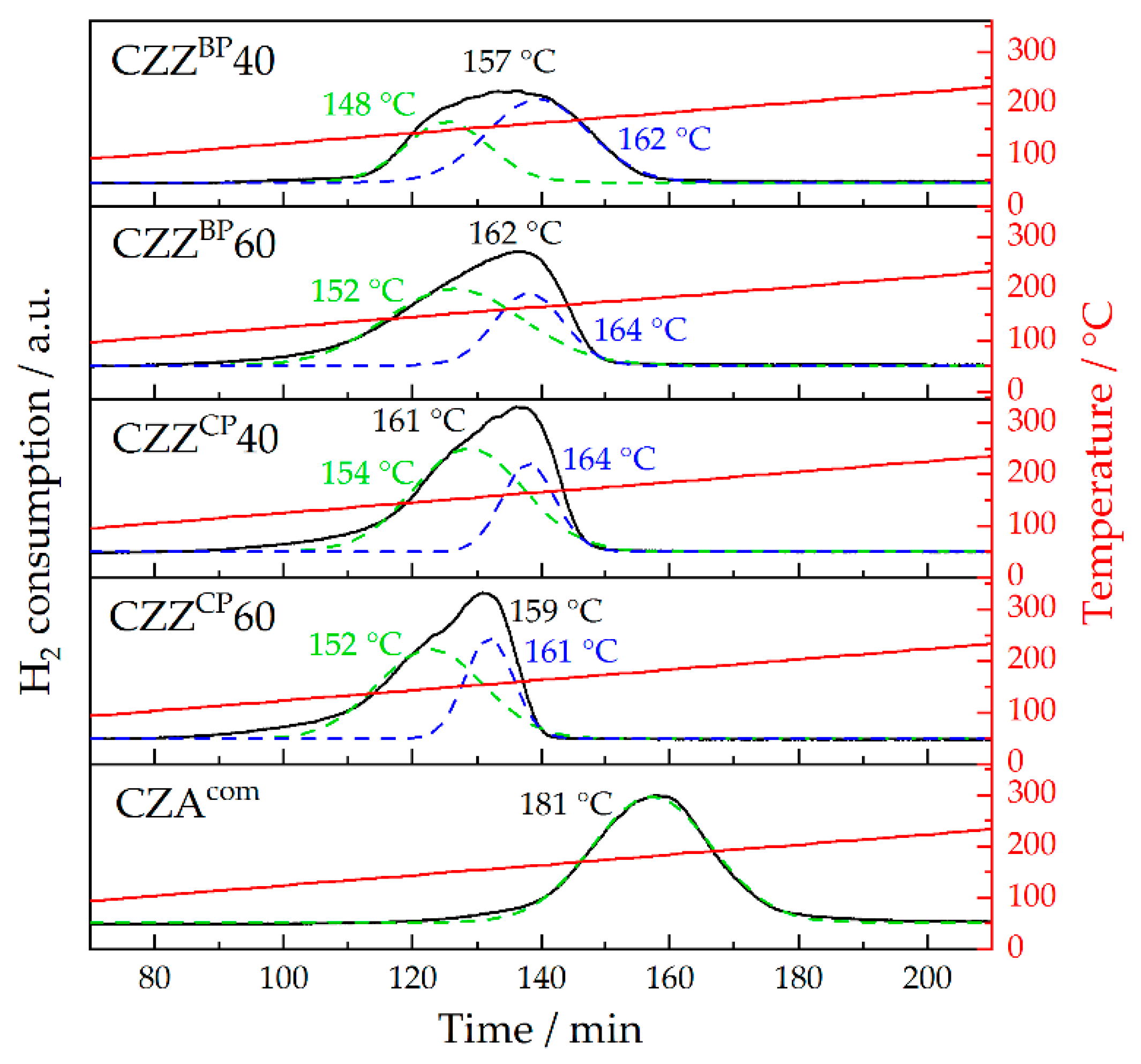
2.3. Chemisorption Measurements: H2-TPR and N2O-RFC
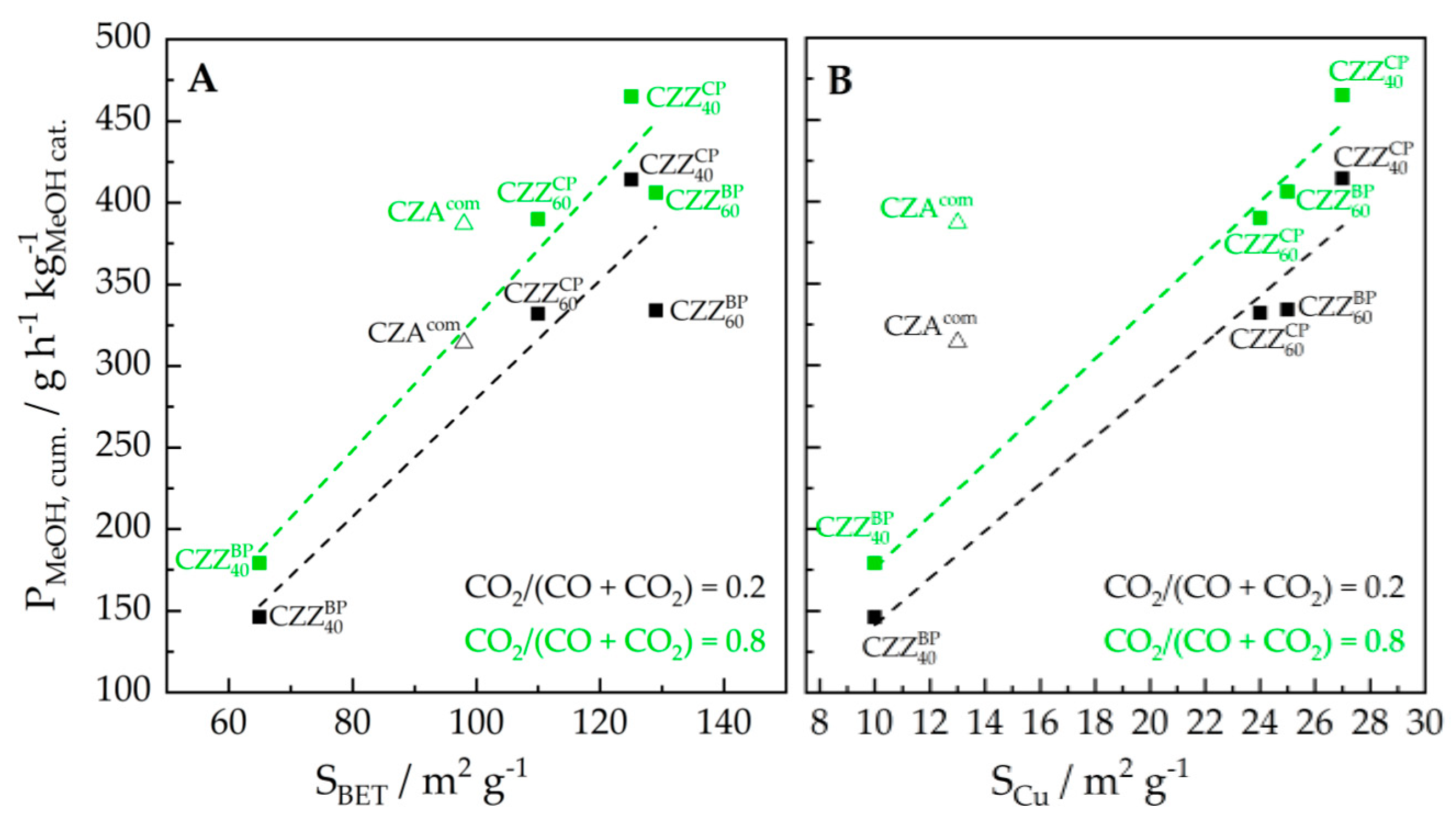
2.4. Evaluation of the Catalytic Activity and Correlation with Material Properties
3. Materials and Methods
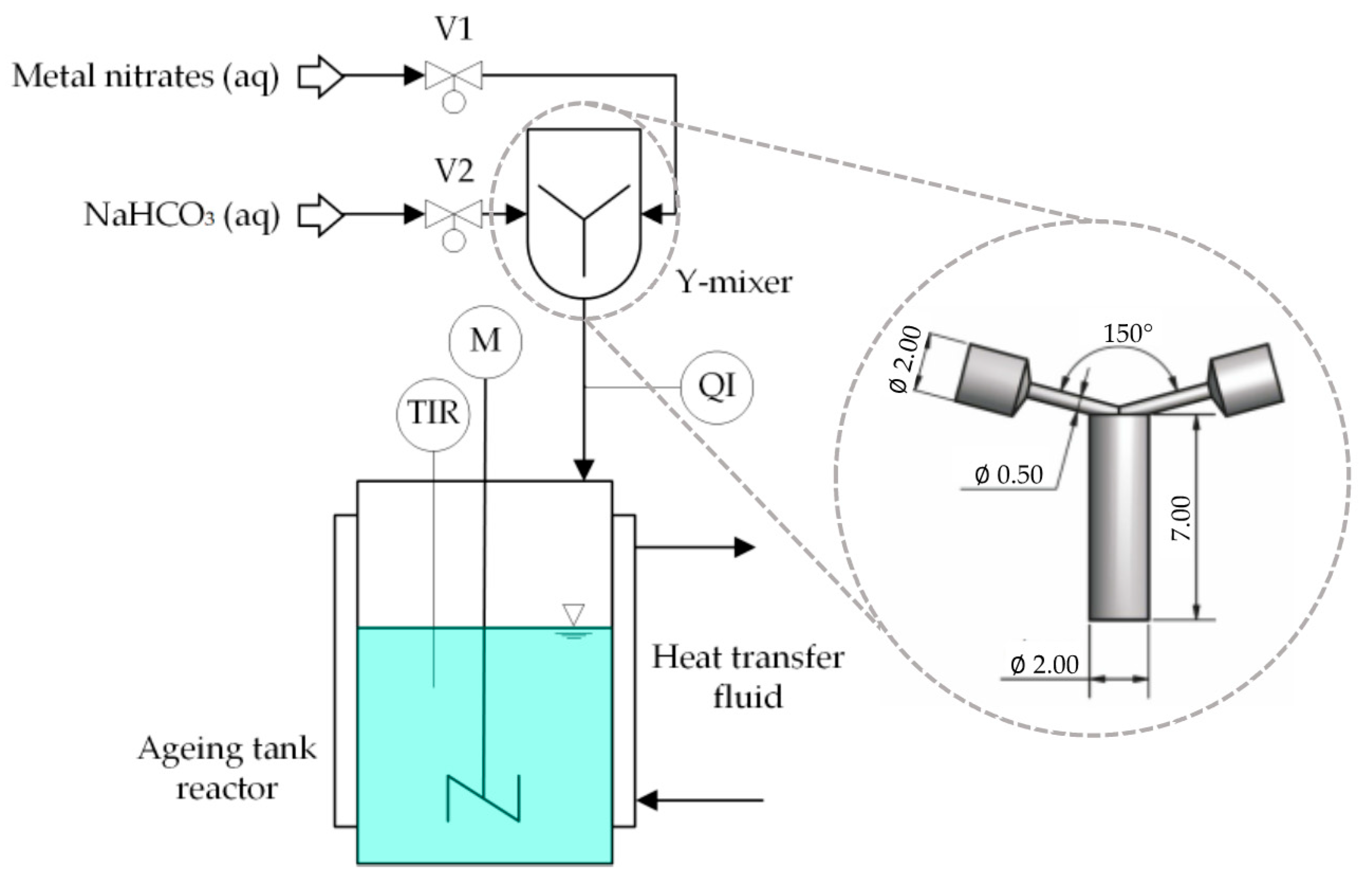
3.1. Catalyst Preparation
3.1.1. Semi-Batch Co-Precipitation
3.1.2. Continuous Co-Precipitation
3.2. Cataylst Characterisation
3.2.1. X-Ray Fluorescence (XRF)
3.2.2. X-Ray Diffraction (XRD)
3.2.3. Transmission Electron Microscopy (TEM) and Energy Dispersive X-Ray Spectroscopy (EDXS)
3.2.4. N2 Physisorption
3.2.5. H2 Temperature Programmed Reduction (H2-TPR)
3.2.6. N2O Reactive Frontal Chromatography (N2O-RFC)
3.3. Catalyst Performance in Direct DME Synthesis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Henrich, E.; Dahmen, N.; Dinjus, E.; Sauer, J. The role of biomass in a future world without fossil fuels. Chem. Ing. Tech. 2015, 87, 1667–1685. [Google Scholar] [CrossRef]
- Arshadi, M.; Sellstedt, A. Biomass-based energy production. In Introduction to Chemicals from Biomass, 2nd ed.; Clark, J., Deswarte, F., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2015; pp. 249–284. [Google Scholar]
- Sternberg, A.; Bardow, A. Life cycle assessment of Power-to-Gas: Syngas vs methane. ACS Sustain. Chem. Eng. 2016, 4, 4156–4165. [Google Scholar] [CrossRef]
- Olah, G.A.; Goeppert, A.; Prakash, G.K.S. Chemical recycling of carbon dioxide to methanol and dimethyl ether: From greenhouse gas to renewable, environmentally carbon neutral fuels and synthetic hydrocarbons. J. Org. Chem. 2009, 74, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Niethammer, B.; Wodarz, S.; Betz, M.; Haltenort, P.; Oestreich, D.; Hackbarth, K.; Arnold, U.; Otto, T.; Sauer, J. Alternative liquid fuels from renewable resources. Chem. Ing. Tech. 2018, 90, 99–112. [Google Scholar] [CrossRef]
- Zimmermann, M.C.; Otto, T.N.; Wodarz, S.; Zevaco, T.A.; Pitter, S. Mesoporous H-ZSM-5 for the conversion of dimethyl ether to hydrocarbons. Chem. Ing. Tech. 2019, 91, 1302–1313. [Google Scholar] [CrossRef]
- Saravanan, K.; Ham, H.; Tsubaki, N.; Bae, J.W. Recent progress for direct synthesis of dimethyl ether from syngas on the heterogeneous bifunctional hybrid catalysts. Appl. Catal. B 2017, 217, 494–522. [Google Scholar] [CrossRef]
- Peral, E.; Martín, M. Optimal production of dimethyl ether from Switchgrass-Based syngas via direct synthesis. Ind. Eng. Chem. Res. 2015, 54, 7465–7475. [Google Scholar] [CrossRef]
- Arena, F.; Barbera, K.; Italiano, G.; Bonura, G.; Spadaro, L.; Frusteri, F. Synthesis, characterization and activity pattern of Cu–ZnO/ZrO2 catalysts in the hydrogenation of carbon dioxide to methanol. J. Catal. 2007, 249, 185–194. [Google Scholar] [CrossRef]
- Arena, F.; Italiano, G.; Barbera, K.; Bonura, G.; Spadaro, L.; Frusteri, F. Basic evidences for methanol-synthesis catalyst design. Catal. Today 2009, 143, 80–85. [Google Scholar] [CrossRef]
- Arena, F.; Italiano, G.; Barbera, K.; Bordiga, S.; Bonura, G.; Spadaro, L.; Frusteri, F. Solid-state interactions, adsorption sites and functionality of Cu-ZnO/ZrO2 catalysts in the CO2 hydrogenation to CH3OH. Appl. Catal. A 2008, 350, 16–23. [Google Scholar] [CrossRef]
- Bonura, G.; Cordaro, M.; Cannilla, C.; Arena, F.; Frusteri, F. The changing nature of the active site of Cu-Zn-Zr catalysts for the CO2 hydrogenation reaction to methanol. Appl. Catal. B 2014, 152–153, 152–161. [Google Scholar] [CrossRef]
- Chen, S.; Zhang, J.; Wang, P.; Wang, X.; Song, F.; Bai, Y.; Zhang, M.; Wu, Y.; Xie, H.; Tan, Y. Effect of Vapor-phase-treatment to CuZnZr Catalyst on the reaction behaviors in CO2 hydrogenation into methanol. ChemCatChem 2019, 11, 1448–1457. [Google Scholar] [CrossRef]
- Larmier, K.; Liao, W.C.; Tada, S.; Lam, E.; Verel, R.; Bansode, A.; Urakawa, A.; Comas-Vives, A.; Copéret, C. CO2-to-Methanol hydrogenation on zirconia-supported copper nanoparticles: Reaction intermediates and the role of the Metal-Support interface. Angew. Chem. Int. Ed. 2017, 56, 2318–2323. [Google Scholar] [CrossRef]
- Samson, K.; Śliwa, M.; Socha, R.P.; Góra-Marek, K.; Mucha, D.; Rutkowska-Zbik, D.; Paul, J.F.; Ruggiero-Mikołajczyk, M.; Grabowski, R.; Słoczyński, J. Influence of ZrO2 structure and copper electronic state on activity of Cu/ZrO2 catalysts in methanol synthesis from CO2. ACS Catal. 2014, 4, 3730–3741. [Google Scholar] [CrossRef]
- Tada, S.; Katagiri, A.; Kiyota, K.; Honma, T.; Kamei, H.; Nariyuki, A.; Uchida, S.; Satokawa, S. Cu species incorporated into amorphous ZrO2 with high activity and selectivity in CO2-to-Methanol hydrogenation. J. Phys. Chem. C 2018, 122, 5430–5442. [Google Scholar] [CrossRef]
- Tada, S.; Kayamori, S.; Honma, T.; Kamei, H.; Nariyuki, A.; Kon, K.; Toyao, T.; Shimizu, K.-i.; Satokawa, S. Design of interfacial sites between Cu and amorphous ZrO2 dedicated to CO2-to-Methanol hydrogenation. ACS Catal. 2018, 8, 7809–7819. [Google Scholar] [CrossRef]
- Tada, S.; Larmier, K.; Büchel, R.; Copéret, C. Methanol synthesis via CO2 hydrogenation over CuO–ZrO2 prepared by two-nozzle flame spray pyrolysis. Catal. Sci. Technol. 2018, 8, 2056–2060. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhong, L.; Wang, H.; Gao, P.; Li, X.; Xiao, S.; Ding, G.; Wei, W.; Sun, Y. Catalytic performance of spray-dried Cu/ZnO/Al2O3/ZrO2 catalysts for slurry methanol synthesis from CO2 hydrogenation. J. CO2 Util. 2016, 15, 72–82. [Google Scholar] [CrossRef]
- Scotti, N.; Bossola, F.; Zaccheria, F.; Ravasio, N. Copper–Zirconia catalysts: Powerful multifunctional catalytic tools to approach sustainable processes. Catalysts 2020, 10, 168. [Google Scholar] [CrossRef]
- Hong, Q.-J.; Liu, Z.-P. Mechanism of CO2 hydrogenation over Cu/ZrO2(2̅12) interface from first-principles kinetics Monte Carlo simulations. Surf. Sci. 2010, 604, 1869–1876. [Google Scholar] [CrossRef]
- Lam, E.; Larmier, K.; Wolf, P.; Tada, S.; Safonova, O.V.; Copéret, C. Isolated Zr surface sites on silica promote hydrogenation of CO2 to CH3OH in supported Cu catalysts. J. Am. Chem. Soc. 2018, 140, 10530–10535. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Qu, Z.; Song, L.; Fu, Q. CO2 hydrogenation to methanol over Cu/CeO2 and Cu/ZrO2 catalysts: Tuning methanol selectivity via metal-support interaction. J. Energy Chem. 2020, 40, 22–30. [Google Scholar] [CrossRef]
- Tang, Q.-L.; Hong, Q.-J.; Liu, Z.-P. CO2 fixation into methanol at Cu/ZrO2 interface from first principles kinetic Monte Carlo. J. Catal. 2009, 263, 114–122. [Google Scholar] [CrossRef]
- Polierer, S.; Jelic, J.; Pitter, S.; Studt, F. On the reactivity of the Cu/ZrO2 system for the hydrogenation of CO2 to methanol: A density functional theory study. J. Phys. Chem. C 2019, 123, 26904–26911. [Google Scholar] [CrossRef]
- Behrens, M.; Brennecke, D.; Girgsdies, F.; Kißner, S.; Trunschke, A.; Nasrudin, N.; Zakaria, S.; Idris, N.F.; Hamid, S.B.A.; Kniep, B.; et al. Understanding the complexity of a catalyst synthesis: Co-precipitation of mixed Cu,Zn,Al hydroxycarbonate precursors for Cu/ZnO/Al2O3 catalysts investigated by titration experiments. Appl. Catal. A 2011, 392, 93–102. [Google Scholar] [CrossRef]
- Bems, B.; Schur, M.; Dassenoy, A.; Junkes, H.; Herein, D.; Schlögl, R. Relations between synthesis and microstructural properties of copper/zinc hydroxycarbonates. Chem. Eur. J. 2003, 9, 2039–2052. [Google Scholar] [CrossRef]
- Raudaskoski, R.; Niemelä, M.V.; Keiski, R.L. The effect of ageing time on co-precipitated Cu/ZnO/ZrO2 catalysts used in methanol synthesis from CO2 and H2. Top. Catal. 2007, 45, 57–60. [Google Scholar] [CrossRef]
- Jung, K.T.; Bell, A.T. Effects of zirconia phase on the synthesis of methanol over zirconia-supported copper. Catal. Lett. 2002, 80, 63–68. [Google Scholar] [CrossRef]
- Frusteri, F.; Cordaro, M.; Cannilla, C.; Bonura, G. Multifunctionality of Cu–ZnO–ZrO2/H-ZSM5 catalysts for the one-step CO2-to-DME hydrogenation reaction. Appl. Catal. B 2015, 162, 57–65. [Google Scholar] [CrossRef]
- Ahmad, R.; Hellinger, M.; Buchholz, M.; Sezen, H.; Gharnati, L.; Wöll, C.; Sauer, J.; Döring, M.; Grunwaldt, J.-D.; Arnold, U. Flame-made Cu/ZnO/Al2O3 catalyst for dimethyl ether production. Catal. Commun. 2014, 43, 52–56. [Google Scholar] [CrossRef]
- Jiang, X.; Qin, X.; Ling, C.; Wang, Z.; Lu, J. The effect of mixing on Co-precipitation and evolution of microstructure of Cu-ZnO catalyst. AIChE J. 2018, 64, 2647–2654. [Google Scholar] [CrossRef]
- Jiang, X.; Zheng, L.; Wang, Z.; Lu, J. Microstructure characters of Cu/ZnO catalyst precipitated inside microchannel reactor. J. Mol. Catal. A Chem. 2016, 423, 457–462. [Google Scholar] [CrossRef]
- Kaluza, S.; Behrens, M.; Schiefenhövel, N.; Kniep, B.; Fischer, R.; Schlögl, R.; Muhler, M. A Novel synthesis route for Cu/ZnO/Al2O3 catalysts used in methanol synthesis: Combining continuous consecutive precipitation with continuous aging of the precipitate. ChemCatChem 2011, 3, 189–199. [Google Scholar] [CrossRef]
- Simson, G.; Prasetyo, E.; Reiner, S.; Hinrichsen, O. Continuous precipitation of Cu/ZnO/Al2O3 catalysts for methanol synthesis in microstructured reactors with alternative precipitating agents. Appl. Catal. A 2013, 450, 1–12. [Google Scholar] [CrossRef]
- Zhang, Q.-C.; Cheng, K.-P.; Wen, L.-X.; Guo, K.; Chen, J.-F. A study on the precipitating and aging processes of CuO/ZnO/Al2O3 catalysts synthesized in micro-impinging stream reactors. RSC Adv. 2016, 6, 33611–33621. [Google Scholar] [CrossRef]
- Schur, M.; Bems, B.; Dassenoy, A.; Kassatkine, I.; Urban, J.; Wilmes, H.; Hinrichsen, O.; Muhler, M.; Schlögl, R. Continuous coprecipitation of catalysts in a micromixer: Nanostructured Cu/ZnO composite for the synthesis of methanol. Angew. Chem. Int. Ed. 2003, 42, 3815–3817. [Google Scholar] [CrossRef]
- Angelo, L.; Girleanu, M.; Ersen, O.; Serra, C.; Parkhomenko, K.; Roger, A.-C. Catalyst synthesis by continuous coprecipitation under micro-fluidic conditions: Application to the preparation of catalysts for methanol synthesis from CO2/H2. Catal. Today 2016, 270, 59–67. [Google Scholar] [CrossRef]
- Huang, C.; Mao, D.; Guo, X.; Yu, J. Microwave-assisted hydrothermal synthesis of CuO-ZnO-ZrO2 as catalyst for direct synthesis of methanol by carbon dioxide hydrogenation. Energy Technol. 2017, 5, 2100–2107. [Google Scholar] [CrossRef]
- Wolf, A.; Michele, V.; Schlüter, O.F.K.; Herbstritt, F.; Heck, J.; Mleczko, L. Precipitation in a Micromixer—From laboratory to industrial scale. Chem. Eng. Technol. 2015, 38, 2017–2024. [Google Scholar] [CrossRef]
- Hartig, M.A.J.; Jacobsen, N.; Peukert, W. Multi-component and multi-phase population balance model: The case of Georgeite formation as methanol catalyst precursor phase. Chem. Eng. Sci. 2014, 109, 158–170. [Google Scholar] [CrossRef]
- Behrens, M.; Schlögl, R. How to prepare a good Cu/ZnO catalyst or the role of solid state chemistry for the synthesis of nanostructured catalysts. Z. Anorg. Allg. Chem. 2013, 639, 2683–2695. [Google Scholar] [CrossRef]
- Muhr, H.; David, R.; Villermaux, J. Crystallization and precipitation engineering -V. Simulation of the precipitation of silver bromide octahedral crystals in a double-jet semi-batch reactor. Chem. Eng. Sci. 1995, 50, 345–355. [Google Scholar] [CrossRef]
- Rehage, H.; Scherer, S.; Kind, M. A steady-state precipitation model for flowsheet simulation and its application. Comput. Chem. Eng. 2019, 128, 524–537. [Google Scholar] [CrossRef]
- Vicum, L.; Mazzotti, M. Multi-scale modeling of a mixing-precipitation process in a semibatch stirred tank. Chem. Eng. Sci. 2007, 62, 3513–3527. [Google Scholar] [CrossRef]
- Ahmadi, F.; Haghighi, M.; Ajamein, H. Sonochemically coprecipitation synthesis of CuO/ZnO/ZrO2/Al2O3 nanocatalyst for fuel cell grade hydrogen production via steam methanol reforming. J. Mol. Catal. A Chem. 2016, 421, 196–208. [Google Scholar] [CrossRef]
- Allahyari, S.; Haghighi, M.; Ebadi, A.; Hosseinzadeh, S. Effect of irradiation power and time on ultrasound assisted co-precipitation of nanostructured CuO–ZnO–Al2O3 over HZSM-5 used for direct conversion of syngas to DME as a green fuel. Energy Convers. Manag. 2014, 83, 212–222. [Google Scholar] [CrossRef]
- Baldyga, J.; Bourne, J.R. Turbulent Mixing and Chemical Reactions; Wiley: Chichester, UK, 1999. [Google Scholar]
- Metzger, L.; Kind, M. On the mixing in confined impinging jet mixers—Time scale analysis and scale-up using CFD coarse-graining methods. Chem. Eng. Res. Des. 2016, 109, 464–476. [Google Scholar] [CrossRef]
- Rehage, H.; Nikq, F.; Kind, M. Experimental investigation of a two-zone model for semi-batch precipitation in stirred-tank reactors. Chem. Eng. Sci. 2019, 207, 258–270. [Google Scholar] [CrossRef]
- Wei, H.; Zhou, W.; Garside, J. Computational fluid dynamics modeling of the precipitation process in a semibatch crystallizer. Ind. Eng. Chem. Res. 2001, 40, 5255–5261. [Google Scholar] [CrossRef]
- Kügler, R.T.; Beißert, K.; Kind, M. On heterogeneous nucleation during the precipitation of barium sulfate. Chem. Eng. Res. Des. 2016, 114, 30–38. [Google Scholar] [CrossRef]
- Kügler, R.T.; Kind, M. Experimental study about plugging in confined impinging jet mixers during the precipitation of strontium sulfate. Chem. Eng. Process. 2016, 101, 25–32. [Google Scholar] [CrossRef]
- Schwarzer, H.-C.; Peukert, W. Combined experimental/numerical study on the precipitation of nanoparticles. AIChE J. 2004, 50, 3234–3247. [Google Scholar] [CrossRef]
- Catizzone, E.; Migliori, M.; Purita, A.; Giordano, G. Ferrierite vs. γ-Al2O3: The superiority of zeolites in terms of water-resistance in vapour-phase dehydration of methanol to dimethyl ether. J. Energy Chem. 2019, 30, 162–169. [Google Scholar] [CrossRef]
- Frusteri, F.; Migliori, M.; Cannilla, C.; Frusteri, L.; Catizzone, E.; Aloise, A.; Giordano, G.; Bonura, G. Direct CO2-to-DME hydrogenation reaction: New evidences of a superior behaviour of FER-based hybrid systems to obtain high DME yield. J. CO2 Util. 2017, 18, 353–361. [Google Scholar] [CrossRef]
- Migliori, M.; Aloise, A.; Catizzone, E.; Giordano, G. Kinetic analysis of methanol to dimethyl ether reaction over H-MFI catalyst. Ind. Eng. Chem. Res. 2014, 53, 14885–14891. [Google Scholar] [CrossRef]
- Stiefel, M.; Ahmad, R.; Arnold, U.; Döring, M. Direct synthesis of dimethyl ether from carbon-monoxide-rich synthesis gas: Influence of dehydration catalysts and operating conditions. Fuel Process. Technol. 2011, 92, 1466–1474. [Google Scholar] [CrossRef]
- Tokay, K.C.; Dogu, T.; Dogu, G. Dimethyl ether synthesis over alumina based catalysts. Chem. Eng. J. 2012, 184, 278–285. [Google Scholar] [CrossRef]
- Bae, J.W.; Kang, S.-H.; Lee, Y.-J.; Jun, K.-W. Synthesis of DME from syngas on the bifunctional Cu–ZnO–Al2O3/Zr-modified ferrierite: Effect of Zr content. Appl. Catal. B 2009, 90, 426–435. [Google Scholar] [CrossRef]
- Bae, J.W.; Kang, S.-H.; Lee, Y.-J.; Jun, K.-W. Effect of precipitants during the preparation of Cu-ZnO-Al2O3/Zr-ferrierite catalyst on the DME synthesis from syngas. J. Ind. Eng. Chem. 2009, 15, 566–572. [Google Scholar] [CrossRef]
- Bonura, G.; Cannilla, C.; Frusteri, L.; Frusteri, F. The influence of different promoter oxides on the functionality of hybrid CuZn-ferrierite systems for the production of DME from CO2-H2 mixtures. Appl. Catal. A 2017, 544, 21–29. [Google Scholar] [CrossRef]
- Bonura, G.; Cannilla, C.; Frusteri, L.; Mezzapica, A.; Frusteri, F. DME production by CO2 hydrogenation: Key factors affecting the behaviour of CuZnZr/ferrierite catalysts. Catal. Today 2017, 281, 337–344. [Google Scholar] [CrossRef]
- Catizzone, E.; Daele, S.V.; Bianco, M.; Di Michele, A.; Aloise, A.; Migliori, M.; Valtchev, V.; Giordano, G. Catalytic application of ferrierite nanocrystals in vapour-phase dehydration of methanol to dimethyl ether. Appl. Catal. B 2019, 243, 273–282. [Google Scholar] [CrossRef]
- Lee, Y.J.; Jung, M.H.; Lee, J.-B.; Jeong, K.-E.; Roh, H.-S.; Suh, Y.-W.; Bae, J.W. Single-step synthesis of dimethyl ether from syngas on Al2O3-modified CuO–ZnO–Al2O3/ferrierite catalysts: Effects of Al2O3 content. Catal. Today 2014, 228, 175–182. [Google Scholar] [CrossRef]
- Roman-Leshkov, Y.; Moliner, M.; Davis, M.E. Impact of controlling the site distribution of Al atoms on catalytic properties in Ferrierite-Type zeolites. J. Phys. Chem. C 2011, 115, 1096–1102. [Google Scholar] [CrossRef]
- Sai Prasad, P.S.; Bae, J.W.; Kang, S.-H.; Lee, Y.-J.; Jun, K.-W. Single-step synthesis of DME from syngas on Cu–ZnO–Al2O3/zeolite bifunctional catalysts: The superiority of ferrierite over the other zeolites. Fuel Process. Technol. 2008, 89, 1281–1286. [Google Scholar] [CrossRef]
- Wu, J.; Luo, S.; Toyir, J.; Saito, M.; Takeuchi, M.; Watanabe, T. Optimization of preparation conditions and improvement of stability of Cu/ZnO-based multicomponent catalysts for methanol synthesis from CO2 and H2. Catal. Today 1998, 45, 215–220. [Google Scholar] [CrossRef]
- Frei, E.; Schaadt, A.; Ludwig, T.; Hillebrecht, H.; Krossing, I. The influence of the precipitation/ageing temperature on a Cu/ZnO/ZrO2 catalyst for methanol synthesis from H2 and CO2. ChemCatChem 2014, 6, 1721–1730. [Google Scholar] [CrossRef]
- Ruland, H.; Song, H.; Laudenschleger, D.; Stürmer, S.; Schmidt, S.; He, J.; Kähler, K.; Muhler, M.; Schlögl, R. CO2 hydrogenation with Cu/ZnO/Al2O3: A benchmark study. ChemCatChem 2020, 12, 1–8. [Google Scholar] [CrossRef]
- Dadgar, F.; Myrstad, R.; Pfeifer, P.; Holmen, A.; Venvik, H.J. Direct dimethyl ether synthesis from synthesis gas: The influence of methanol dehydration on methanol synthesis reaction. Catal. Today 2016, 270, 76–84. [Google Scholar] [CrossRef]
- Chanchlani, K.G.; Hudgins, R.R.; Silveston, P.L. Methanol synthesis from H2, CO, and CO2 over Cu/ZnO catalysts. J. Catal. 1992, 136, 59–75. [Google Scholar] [CrossRef]
- Liu, G.; Willcox, G.; Garland, M.; Kung, H.H. The rate of methanol production on a Copper-Zinc oxide catalyst: The dependence on the feed composition. J. Catal. 1984, 90, 139–146. [Google Scholar] [CrossRef]
- Studt, F.; Behrens, M.; Kunkes, E.L.; Thomas, N.; Zander, S.; Tarasov, A.; Schumann, J.; Frei, E.; Varley, J.B.; Abild-Pedersen, F.; et al. The mechanism of CO and CO2 hydrogenation to methanol over Cu-Based catalysts. ChemCatChem 2015, 7, 1105–1111. [Google Scholar] [CrossRef]
- Jones, A.J.; Carr, R.T.; Zones, S.I.; Iglesia, E. Acid strength and solvation in catalysis by MFI zeolites and effects of the identity, concentration and location of framework heteroatoms. J. Catal. 2014, 312, 58–68. [Google Scholar] [CrossRef]
- Baltes, C.; Vukojevic, S.; Schüth, F. Correlations between synthesis, precursor, and catalyst structure and activity of a large set of CuO/ZnO/Al2O3 catalysts for methanol synthesis. J. Catal. 2008, 258, 334–344. [Google Scholar] [CrossRef]
- Behrens, M. Coprecipitation: An excellent tool for the synthesis of supported metal catalysts—From the understanding of the well known recipes to new materials. Catal. Today 2015, 246, 46–54. [Google Scholar] [CrossRef]
- Behrens, M.; Studt, F.; Kasatkin, I.; Kühl, S.; Hävecker, M.; Abild-Pedersen, F.; Zander, S.; Girgsdies, F.; Kurr, P.; Kniep, B.-L.; et al. The active site of methanol synthesis over Cu/ZnO/Al2O3 industrial catalysts. Science 2012, 336, 893–897. [Google Scholar] [CrossRef]
- Kurtz, M.; Bauer, N.; Büscher, C.; Wilmer, H.; Hinrichsen, O.; Becker, R.; Rabe, S.; Merz, K.; Driess, M.; Fischer, R.A.; et al. New synthetic routes to more active Cu/ZnO catalysts used for methanol synthesis. Catal. Lett. 2004, 92, 49–52. [Google Scholar] [CrossRef]
- Kügler, R.T.; Doyle, S.; Kind, M. Fundamental insights into barium sulfate precipitation by time-resolved in situ synchrotron radiation wide-angle X-ray scattering (WAXS). Chem. Eng. Sci. 2015, 133, 140–147. [Google Scholar] [CrossRef]
- Kügler, R.T.; Kind, M. On precipitation of sparingly soluble fluoride salts. Cryst. Growth Des. 2018, 18, 728–733. [Google Scholar] [CrossRef]
- Spencer, M.S. Precursors of copper/zinc oxide catalysts. Catal. Lett. 2000, 66, 255–257. [Google Scholar] [CrossRef]
- Thommes, M.; Kaneko, K.; Neimark, A.V.; Olivier, J.P.; Rodriguez-Reinoso, F.; Rouquerol, J.; Sing, K.S.W. Physisorption of gases, with special reference to the evaluation of surface area and pore size distribution (IUPAC Technical Report). Pure Appl. Chem. 2015, 87, 1051–1069. [Google Scholar] [CrossRef]
- Brunauer, S.; Emmet, P.H.; Teller, E. Adsorption of gases in multimolecular layers. J. Am. Chem. Soc. 1938, 60, 309–319. [Google Scholar] [CrossRef]
- Barrett, E.P.; Joyner, L.G.; Halenda, P.P. The determination of pore volume and area distributions in porous substances. i.computations from nitrogen isotherms. J. Am. Chem. Soc. 1951, 73, 373–380. [Google Scholar] [CrossRef]
- Hinrichsen, O.; Genger, T.; Muhler, M. Chemisorption of N2O and H2 for the surface determination of copper catalysts. Chem. Eng. Technol. 2000, 23, 956–959. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pre-Catalyst | Metal Composition/wt% | SBET/m2 g−1 | PV/cm3 g−1 | dpore, max. 1/nm | dpore,range/nm | |||
|---|---|---|---|---|---|---|---|---|
| Cu | Zn | Zr | Al | |||||
| CZZBP40 | 57 | 29 | 14 | - | 65 | 0.424 | 18, 45 | 20–94 |
| CZZBP60 | 57 | 30 | 11 | - | 129 | 0.448 | 14 | 6–22 |
| CZZCP40 | 61 | 31 | 8 | - | 125 | 0.571 | 14 | 7–41 |
| CZZCP60 | 61 | 33 | 5 | - | 110 | 0.423 | 12 | 6–37 |
| CZAcom | 64 | 29 | - | 6 | 98 | 0.332 | 11 | 5–26 |
| Catalyst | Fresh Calcined Pre-Catalyst | Spent Catalyst | ||
|---|---|---|---|---|
| 2θ (CuO)/° | dCuO/nm | 2θ (CuO)/° | dCuO/nm | |
| CZZBP40 | 38.8 | 8 | 38.7 | 10 |
| CZZBP60 | 38.4 | 3 | 38.6 | 10 |
| CZZCP40 | 38.6 | 4 | 38.6 | 10 |
| CZZCP60 | 38.5 | 4 | 38.7 | 10 |
| CZAcom | 38.5 | 4 | 38.8 | 8 |
| Pre-Catalyst | Tred., range/°C | Tred., max./°C | SCu/m2 g−1 |
|---|---|---|---|
| CZZBP40 | 122–183 | 157 | 10 |
| CZZBP60 | 113–177 | 162 | 25 |
| CZZCP40 | 114–174 | 161 | 27 |
| CZZCP60 | 111–171 | 159 | 24 |
| CZAcom | 145–218 | 181 | 13 |
| Catalyst | PDME/g h−1 kg−1 (0.2/0.8) | PMeOH/g h−1 kg−1 (0.2/0.8) | PMeOH, cum./g h−1 kg−1 (0.2/0.8) |
|---|---|---|---|
| CZZBP40/FER | 105/129 | 0/0 | 146/179 |
| CZZBP60/FER | 240/261 | 0/42 | 334/406 |
| CZZCP40/FER | 291/300 | 9/48 | 414/465 |
| CZZCP60/FER | 231/247 | 11/46 | 332/390 |
| CZAcom/FER | 226/261 | 0/24 | 314/387 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Polierer, S.; Guse, D.; Wild, S.; Herrera Delgado, K.; Otto, T.N.; Zevaco, T.A.; Kind, M.; Sauer, J.; Studt, F.; Pitter, S. Enhanced Direct Dimethyl Ether Synthesis from CO2-Rich Syngas with Cu/ZnO/ZrO2 Catalysts Prepared by Continuous Co-Precipitation. Catalysts 2020, 10, 816. https://doi.org/10.3390/catal10080816
Polierer S, Guse D, Wild S, Herrera Delgado K, Otto TN, Zevaco TA, Kind M, Sauer J, Studt F, Pitter S. Enhanced Direct Dimethyl Ether Synthesis from CO2-Rich Syngas with Cu/ZnO/ZrO2 Catalysts Prepared by Continuous Co-Precipitation. Catalysts. 2020; 10(8):816. https://doi.org/10.3390/catal10080816
Chicago/Turabian StylePolierer, Sabrina, David Guse, Stefan Wild, Karla Herrera Delgado, Thomas N. Otto, Thomas A. Zevaco, Matthias Kind, Jörg Sauer, Felix Studt, and Stephan Pitter. 2020. "Enhanced Direct Dimethyl Ether Synthesis from CO2-Rich Syngas with Cu/ZnO/ZrO2 Catalysts Prepared by Continuous Co-Precipitation" Catalysts 10, no. 8: 816. https://doi.org/10.3390/catal10080816
APA StylePolierer, S., Guse, D., Wild, S., Herrera Delgado, K., Otto, T. N., Zevaco, T. A., Kind, M., Sauer, J., Studt, F., & Pitter, S. (2020). Enhanced Direct Dimethyl Ether Synthesis from CO2-Rich Syngas with Cu/ZnO/ZrO2 Catalysts Prepared by Continuous Co-Precipitation. Catalysts, 10(8), 816. https://doi.org/10.3390/catal10080816