
Current State of Melanoma Therapy and Next Steps: Battling Therapeutic Resistance


Abstract
Simple Summary
Abstract
1. Introduction to Melanoma
2. Treatment Options Available for Cutaneous Melanoma and Their Efficacy
2.1. Radiation Therapy, Topical Therapeutics and Chemotherapy
2.2. Targeted Therapy
2.3. Immunotherapy
3. Available Treatment for Acral, Mucosal and Uveal Melanoma
4. mGluR1-Driven Melanoma
4.1. Glutamate in Cancer Development and Progression
4.2. mGluR1-Driven Melanoma
5. Therapeutic Potential to Treat mGluR1-Driven Melanoma and Other Cancers with Riluzole
5.1. Use of Riluzole in Melanoma
5.2. Use of Riluzole in Cancers Other Than Melanoma
6. Resistance to Different Treatment Options in Melanoma
7. Resistance to Targeted Therapies
7.1. MAPK Proteins
7.2. Receptor Tyrosine Kinases
7.3. MITF and Phenotypic Switching
7.4. NF-κB Signaling and Tumor Microenvironment
7.5. Parallel Signaling Cascades
7.6. Autophagy
7.7. MicroRNAs
8. Resistance to Immunotherapies
8.1. Tumor Mutational Burden
8.2. Antigen Presentation: LAG-3 and B2M
8.3. IFN Signaling and Other Cellular Regulators
8.4. Regulatory T-Cells
8.5. Phenotypic Switching
8.6. Epigenetic Regulation
9. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Giaquinto, A.N.; Jemal, A. Cancer statistics, 2024. CA Cancer J. Clin. 2024, 74, 12–49. [Google Scholar] [CrossRef]
- American Cancer Society. Cancer Facts & Figures 2023; American Cancer Society: Atlanta, GA, USA, 2023. [Google Scholar]
- American Society of Clinical Oncology Cancer. Net Melanoma Statistics. Available online: https://www.cancer.net/cancer-types/melanoma/statistics (accessed on 15 May 2023).
- Jung, S.; Johnson, D.B. Management of Acral and Mucosal Melanoma: Medical Oncology Perspective. Oncologist 2022, 27, 703–710. [Google Scholar] [CrossRef]
- American Cancer Society Risk Factors for Melanoma Skin Cancer. Available online: https://www.cancer.org/cancer/types/melanoma-skin-cancer/causes-risks-prevention/risk-factors.html (accessed on 15 May 2023).
- American Cancer Society Skin Cancer (Non-Melanoma): Risk Factors and Prevention. Available online: https://www.cancer.net/cancer-types/skin-cancer-non-melanoma/risk-factors-and-prevention (accessed on 15 May 2023).
- Massand, S.; Neves, R.I. Emerging Therapies in the Treatment of Advanced Melanoma. Clin. Plast. Surg. 2021, 48, 713–733. [Google Scholar] [CrossRef]
- Tétu, P.; Baroudjian, B.; Lebbe, C. Targeting BRAF and MEK inhibitors in melanoma in the metastatic, neoadjuvant and adjuvant setting. Curr. Opin. Oncol. 2020, 32, 85–90. [Google Scholar] [CrossRef]
- Curtin, J.A.; Fridlyand, J.; Kageshita, T.; Patel, H.N.; Busam, K.J.; Kutzner, H.; Cho, K.H.; Aiba, S.; Bröcker, E.B.; LeBoit, P.E.; et al. Distinct sets of genetic alterations in melanoma. N. Engl. J. Med. 2005, 353, 2135–2147. [Google Scholar] [CrossRef]
- Ascierto, P.A.; Kirkwood, J.M.; Grob, J.J.; Simeone, E.; Grimaldi, A.M.; Maio, M.; Palmieri, G.; Testori, A.; Marincola, F.M.; Mozzillo, N. The role of BRAF V600 mutation in melanoma. J. Transl. Med. 2012, 10, 85. [Google Scholar] [CrossRef]
- Ito, T.; Tanaka, Y.; Murata, M.; Kaku-Ito, Y.; Furue, K.; Furue, M. BRAF Heterogeneity in Melanoma. Curr. Treat. Options Oncol. 2021, 22, 20. [Google Scholar] [CrossRef]
- Dankort, D.; Curley, D.P.; Cartlidge, R.A.; Nelson, B.; Karnezis, A.N.; Damsky, W.E., Jr.; You, M.J.; DePinho, R.A.; McMahon, M.; Bosenberg, M. Braf(V600E) cooperates with Pten loss to induce metastatic melanoma. Nat. Genet. 2009, 41, 544–552. [Google Scholar] [CrossRef]
- Milagre, C.; Dhomen, N.; Geyer, F.C.; Hayward, R.; Lambros, M.; Reis-Filho, J.S.; Marais, R. A mouse model of melanoma driven by oncogenic KRAS. Cancer Res. 2010, 70, 5549–5557. [Google Scholar] [CrossRef]
- Shi, W. Role for radiation therapy in melanoma. Surg. Oncol. Clin. N. Am. 2015, 24, 323–335. [Google Scholar] [CrossRef]
- Powell, A.M.; Russell-Jones, R.; Barlow, R.J. Topical imiquimod immunotherapy in the management of lentigo maligna. Clin. Exp. Dermatol. 2004, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Teimouri, F.; Nikfar, S.; Abdollahi, M. Efficacy and side effects of dacarbazine in comparison with temozolomide in the treatment of malignant melanoma: A meta-analysis consisting of 1314 patients. Melanoma Res. 2013, 23, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Eggermont, A.M.; Kirkwood, J.M. Re-evaluating the role of dacarbazine in metastatic melanoma: What have we learned in 30 years? Eur. J. Cancer 2004, 40, 1825–1836. [Google Scholar] [CrossRef] [PubMed]
- National Cancer Institute. Drugs Approved for Melanoma. Available online: https://www.cancer.gov/about-cancer/treatment/drugs/melanoma (accessed on 27 March 2023).
- Velho, T.R. Metastatic melanoma-a review of current and future drugs. Drugs Context 2012, 2012, 212242. [Google Scholar] [CrossRef]
- Ascierto, P.A.; Kirkwood, J.M. Adjuvant therapy of melanoma with interferon: Lessons of the past decade. J. Transl. Med. 2008, 6, 62. [Google Scholar] [CrossRef]
- Cook, J.; Zitelli, J.A. Treating patients with melanoma with interferon. Arch. Dermatol. 1997, 133, 387–389. [Google Scholar] [CrossRef] [PubMed]
- Atkins, M.B.; Lotze, M.T.; Dutcher, J.P.; Fisher, R.I.; Weiss, G.; Margolin, K.; Abrams, J.; Sznol, M.; Parkinson, D.; Hawkins, M.; et al. High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: Analysis of 270 patients treated between 1985 and 1993. J. Clin. Oncol. 1999, 17, 2105–2116. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food & Drug Administration. Search Orphan Drug Designations and Approvals. Available online: https://www.accessdata.fda.gov/scripts/opdlisting/oopd/detailedIndex.cfm?cfgridkey=100696 (accessed on 27 March 2023).
- Tsai, J.; Lee, J.T.; Wang, W.; Zhang, J.; Cho, H.; Mamo, S.; Bremer, R.; Gillette, S.; Kong, J.; Haass, N.K.; et al. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc. Natl. Acad. Sci. USA 2008, 105, 3041–3046. [Google Scholar] [CrossRef] [PubMed]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef]
- Chapman, P.B.; Robert, C.; Larkin, J.; Haanen, J.B.; Ribas, A.; Hogg, D.; Hamid, O.; Ascierto, P.A.; Testori, A.; Lorigan, P.C.; et al. Vemurafenib in patients with BRAFV600 mutation-positive metastatic melanoma: Final overall survival results of the randomized BRIM-3 study. Ann. Oncol. 2017, 28, 2581–2587. [Google Scholar] [CrossRef]
- Huang, A.C.; Zappasodi, R. A decade of checkpoint blockade immunotherapy in melanoma: Understanding the molecular basis for immune sensitivity and resistance. Nat. Immunol. 2022, 23, 660–670. [Google Scholar] [CrossRef]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Robert, C.; Long, G.V.; Brady, B.; Dutriaux, C.; Maio, M.; Mortier, L.; Hassel, J.C.; Rutkowski, P.; McNeil, C.; Kalinka-Warzocha, E.; et al. Nivolumab in previously untreated melanoma without BRAF mutation. N. Engl. J. Med. 2015, 372, 320–330. [Google Scholar] [CrossRef]
- Weber, J.S.; Minor, D.R.; D’Angelo, S.P.; Hodi, F.S.; Gutzmer, R.; Neyns, B.; Hoeller, C.; Khushalani, N.I.; Miller, W.H.; Grob, J.J.; et al. A phase 3 randomized, open-label study of nivolumab (anti-PD-1; BMS-936558; ONO-4538) versus investigator’s choice chemotherapy (ICC) in patients with advanced melanoma with prior anti-CTLA-4 therapy. In Proceedings of the Presented at the European Society for Medical Oncology 2014 Congress, Madrid, Spain, 26–30 September 2014. [Google Scholar]
- Robert, C.; Schachter, J.; Long, G.V.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2015, 372, 2521–2532. [Google Scholar] [CrossRef]
- National Cancer Institute. FDA Approves Cobimetinib as Part of Drug Combination for Advanced Melanoma. 2015. Available online: https://www.cancer.gov/news-events/cancer-currents-blog/2015/cobimetinib-melanoma (accessed on 27 March 2023).
- Wang, C.; Lu, N.; Yan, L.; Li, Y. The efficacy and safety assessment of oncolytic virotherapies in the treatment of advanced melanoma: A systematic review and meta-analysis. Virol. J. 2023, 20, 252. [Google Scholar] [CrossRef]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Long-Term Outcomes With Nivolumab Plus Ipilimumab or Nivolumab Alone Versus Ipilimumab in Patients With Advanced Melanoma. J. Clin. Oncol. 2022, 40, 127–137. [Google Scholar] [CrossRef]
- Long, G.V.; Hauschild, A.; Santinami, M.; Atkinson, V.; Mandalà, M.; Chiarion-Sileni, V.; Larkin, J.; Nyakas, M.; Dutriaux, C.; Haydon, A.; et al. Adjuvant Dabrafenib plus Trametinib in Stage III BRAF-Mutated Melanoma. N. Engl. J. Med. 2017, 377, 1813–1823. [Google Scholar] [CrossRef]
- U.S. Food & Drug Administration. FDA Approves Dabrafenib Plus Trametinib for Adjuvant Treatment of Melanoma with BRAF V600E or V600K Mutations. 2018. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-dabrafenib-plus-trametinib-adjuvant-treatment-melanoma-braf-v600e-or-v600k-mutations (accessed on 27 March 2023).
- U.S. Food & Drug Administration. FDA Approves Atezolizumab for BRAF V600 Unresectable or Metastatic Melanoma. 2020. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-atezolizumab-braf-v600-unresectable-or-metastatic-melanoma (accessed on 27 March 2023).
- Song, S.; Han, M.; Zhang, H.; Wang, Y.; Jiang, H. Full screening and accurate subtyping of HLA-A*02 alleles through group-specific amplification and mono-allelic sequencing. Cell Mol. Immunol. 2013, 10, 490–496. [Google Scholar] [CrossRef]
- Tawbi, H.A.; Schadendorf, D.; Lipson, E.J.; Ascierto, P.A.; Matamala, L.; Castillo Gutiérrez, E.; Rutkowski, P.; Gogas, H.J.; Lao, C.D.; De Menezes, J.J.; et al. Relatlimab and Nivolumab versus Nivolumab in Untreated Advanced Melanoma. N. Engl. J. Med. 2022, 386, 24–34. [Google Scholar] [CrossRef]
- Au, L.; Larkin, J.; Turajlic, S. Relatlimab and nivolumab in the treatment of melanoma. Cell 2022, 185, 4866–4869. [Google Scholar] [CrossRef]
- U.S. Food & Drug Administration. FDA Approves Melphalan as a Liver-Directed Treatment for Uveal Melanoma. 2023. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-melphalan-liver-directed-treatment-uveal-melanoma (accessed on 27 March 2023).
- FDA Press Announcement. FDA Approves First Cellular Therapy to Treat Patients with Unresectable or Metastatic Melanoma; U.S. Food and Drug Administration: Washington, DC, USA, 2024. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-first-cellular-therapy-treat-patients-unresectable-or-metastatic-melanoma (accessed on 22 February 2024).
- Subbiah, V.; Baik, C.; Kirkwood, J.M. Clinical Development of BRAF plus MEK Inhibitor Combinations. Trends Cancer 2020, 6, 797–810. [Google Scholar] [CrossRef] [PubMed]
- Eisen, T.; Ahmad, T.; Flaherty, K.T.; Gore, M.; Kaye, S.; Marais, R.; Gibbens, I.; Hackett, S.; James, M.; Schuchter, L.M.; et al. Sorafenib in advanced melanoma: A Phase II randomised discontinuation trial analysis. Br. J. Cancer 2006, 95, 581–586. [Google Scholar] [CrossRef] [PubMed]
- Hauschild, A.; Agarwala, S.S.; Trefzer, U.; Hogg, D.; Robert, C.; Hersey, P.; Eggermont, A.; Grabbe, S.; Gonzalez, R.; Gille, J.; et al. Results of a phase III, randomized, placebo-controlled study of sorafenib in combination with carboplatin and paclitaxel as second-line treatment in patients with unresectable stage III or stage IV melanoma. J. Clin. Oncol. 2009, 27, 2823–2830. [Google Scholar] [CrossRef] [PubMed]
- Koelblinger, P.; Thuerigen, O.; Dummer, R. Development of encorafenib for BRAF-mutated advanced melanoma. Curr. Opin. Oncol. 2018, 30, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Rheault, T.R.; Stellwagen, J.C.; Adjabeng, G.M.; Hornberger, K.R.; Petrov, K.G.; Waterson, A.G.; Dickerson, S.H.; Mook, R.A., Jr.; Laquerre, S.G.; King, A.J.; et al. Discovery of Dabrafenib: A Selective Inhibitor of Raf Kinases with Antitumor Activity against B-Raf-Driven Tumors. ACS Med. Chem. Lett. 2013, 4, 358–362. [Google Scholar] [CrossRef] [PubMed]
- Delord, J.P.; Robert, C.; Nyakas, M.; McArthur, G.A.; Kudchakar, R.; Mahipal, A.; Yamada, Y.; Sullivan, R.; Arance, A.; Kefford, R.F.; et al. Phase I Dose-Escalation and -Expansion Study of the BRAF Inhibitor Encorafenib (LGX818) in Metastatic BRAF-Mutant Melanoma. Clin. Cancer Res. 2017, 23, 5339–5348. [Google Scholar] [CrossRef]
- Yang, H.; Higgins, B.; Kolinsky, K.; Packman, K.; Go, Z.; Iyer, R.; Kolis, S.; Zhao, S.; Lee, R.; Grippo, J.F.; et al. RG7204 (PLX4032), a selective BRAFV600E inhibitor, displays potent antitumor activity in preclinical melanoma models. Cancer Res. 2010, 70, 5518–5527. [Google Scholar] [CrossRef] [PubMed]
- Hauschild, A.; Grob, J.J.; Demidov, L.V.; Jouary, T.; Gutzmer, R.; Millward, M.; Rutkowski, P.; Blank, C.U.; Miller, W.H., Jr.; Kaempgen, E.; et al. Dabrafenib in BRAF-mutated metastatic melanoma: A multicentre, open-label, phase 3 randomised controlled trial. Lancet 2012, 380, 358–365. [Google Scholar] [CrossRef]
- Davis, L.E.; Shalin, S.C.; Tackett, A.J. Current state of melanoma diagnosis and treatment. Cancer Biol. Ther. 2019, 20, 1366–1379. [Google Scholar] [CrossRef]
- Flaherty, K.T.; Robert, C.; Hersey, P.; Nathan, P.; Garbe, C.; Milhem, M.; Demidov, L.V.; Hassel, J.C.; Rutkowski, P.; Mohr, P.; et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N. Engl. J. Med. 2012, 367, 107–114. [Google Scholar] [CrossRef]
- Ugurel, S.; Röhmel, J.; Ascierto, P.A.; Flaherty, K.T.; Grob, J.J.; Hauschild, A.; Larkin, J.; Long, G.V.; Lorigan, P.; McArthur, G.A.; et al. Survival of patients with advanced metastatic melanoma: The impact of novel therapies-update 2017. Eur. J. Cancer 2017, 83, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Karaszewska, B.; Schachter, J.; Rutkowski, P.; Mackiewicz, A.; Stroiakovski, D.; Lichinitser, M.; Dummer, R.; Grange, F.; Mortier, L.; et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N. Engl. J. Med. 2015, 372, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Long, G.V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.J.; et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N. Engl. J. Med. 2014, 371, 1877–1888. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Ascierto, P.A.; Dréno, B.; Atkinson, V.; Liszkay, G.; Maio, M.; Mandalà, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N. Engl. J. Med. 2014, 371, 1867–1876. [Google Scholar] [CrossRef] [PubMed]
- Dummer, R.; Flaherty, K.T.; Robert, C.; Arance, A.; de Groot, J.W.B.; Garbe, C.; Gogas, H.J.; Gutzmer, R.; Krajsová, I.; Liszkay, G.; et al. COLUMBUS 5-Year Update: A Randomized, Open-Label, Phase III Trial of Encorafenib Plus Binimetinib Versus Vemurafenib or Encorafenib in Patients With BRAF V600-Mutant Melanoma. J. Clin. Oncol. 2022, 40, 4178–4188. [Google Scholar] [CrossRef] [PubMed]
- Schadendorf, D.; Long, G.V.; Stroiakovski, D.; Karaszewska, B.; Hauschild, A.; Levchenko, E.; Chiarion-Sileni, V.; Schachter, J.; Garbe, C.; Dutriaux, C.; et al. Three-year pooled analysis of factors associated with clinical outcomes across dabrafenib and trametinib combination therapy phase 3 randomised trials. Eur. J. Cancer 2017, 82, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Ascierto, P.A.; McArthur, G.A.; Dréno, B.; Atkinson, V.; Liszkay, G.; Di Giacomo, A.M.; Mandalà, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. Cobimetinib combined with vemurafenib in advanced BRAF(V600)-mutant melanoma (coBRIM): Updated efficacy results from a randomised, double-blind, phase 3 trial. Lancet Oncol. 2016, 17, 1248–1260. [Google Scholar] [CrossRef] [PubMed]
- Dummer, R.; Ascierto, P.A.; Gogas, H.J.; Arance, A.; Mandala, M.; Liszkay, G.; Garbe, C.; Schadendorf, D.; Krajsova, I.; Gutzmer, R.; et al. Overall survival in patients with BRAF-mutant melanoma receiving encorafenib plus binimetinib versus vemurafenib or encorafenib (COLUMBUS): A multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2018, 19, 1315–1327. [Google Scholar] [CrossRef]
- Heinzerling, L.; Eigentler, T.K.; Fluck, M.; Hassel, J.C.; Heller-Schenck, D.; Leipe, J.; Pauschinger, M.; Vogel, A.; Zimmer, L.; Gutzmer, R. Tolerability of BRAF/MEK inhibitor combinations: Adverse event evaluation and management. ESMO Open 2019, 4, e000491. [Google Scholar] [CrossRef]
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996, 271, 1734–1736. [Google Scholar] [CrossRef]
- Robert, C.; Thomas, L.; Bondarenko, I.; O’Day, S.; Weber, J.; Garbe, C.; Lebbe, C.; Baurain, J.F.; Testori, A.; Grob, J.J.; et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N. Engl. J. Med. 2011, 364, 2517–2526. [Google Scholar] [CrossRef]
- Syn, N.L.; Teng, M.W.L.; Mok, T.S.K.; Soo, R.A. De-novo and acquired resistance to immune checkpoint targeting. Lancet Oncol. 2017, 18, e731–e741. [Google Scholar] [CrossRef]
- Wang, X.; Teng, F.; Kong, L.; Yu, J. PD-L1 expression in human cancers and its association with clinical outcomes. OncoTargets Ther. 2016, 9, 5023–5039. [Google Scholar] [CrossRef]
- Gandini, S.; Massi, D.; Mandalà, M. PD-L1 expression in cancer patients receiving anti PD-1/PD-L1 antibodies: A systematic review and meta-analysis. Crit. Rev. Oncol. Hematol. 2016, 100, 88–98. [Google Scholar] [CrossRef]
- Strome, S.E.; Dong, H.; Tamura, H.; Voss, S.G.; Flies, D.B.; Tamada, K.; Salomao, D.; Cheville, J.; Hirano, F.; Lin, W.; et al. B7-H1 blockade augments adoptive T-cell immunotherapy for squamous cell carcinoma. Cancer Res. 2003, 63, 6501–6505. [Google Scholar]
- Iwai, Y.; Ishida, M.; Tanaka, Y.; Okazaki, T.; Honjo, T.; Minato, N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc. Natl. Acad. Sci. USA 2002, 99, 12293–12297. [Google Scholar] [CrossRef]
- Robert, C.; Ribas, A.; Schachter, J.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.M.; Lotem, M.; et al. Pembrolizumab versus ipilimumab in advanced melanoma (KEYNOTE-006): Post-hoc 5-year results from an open-label, multicentre, randomised, controlled, phase 3 study. Lancet Oncol. 2019, 20, 1239–1251. [Google Scholar] [CrossRef]
- Ascierto, P.A.; Long, G.V.; Robert, C.; Brady, B.; Dutriaux, C.; Di Giacomo, A.M.; Mortier, L.; Hassel, J.C.; Rutkowski, P.; McNeil, C.; et al. Survival Outcomes in Patients With Previously Untreated BRAF Wild-Type Advanced Melanoma Treated With Nivolumab Therapy: Three-Year Follow-up of a Randomized Phase 3 Trial. JAMA Oncol. 2019, 5, 187–194. [Google Scholar] [CrossRef]
- Brahmer, J.R.; Drake, C.G.; Wollner, I.; Powderly, J.D.; Picus, J.; Sharfman, W.H.; Stankevich, E.; Pons, A.; Salay, T.M.; McMiller, T.L.; et al. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: Safety, clinical activity, pharmacodynamics, and immunologic correlates. J. Clin. Oncol. 2010, 28, 3167–3175. [Google Scholar] [CrossRef]
- Curran, M.A.; Montalvo, W.; Yagita, H.; Allison, J.P. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc. Natl. Acad. Sci. USA 2010, 107, 4275–4280. [Google Scholar] [CrossRef]
- Carlino, M.S.; Larkin, J.; Long, G.V. Immune checkpoint inhibitors in melanoma. Lancet 2021, 398, 1002–1014. [Google Scholar] [CrossRef] [PubMed]
- De Sousa Linhares, A.; Battin, C.; Jutz, S.; Leitner, J.; Hafner, C.; Tobias, J.; Wiedermann, U.; Kundi, M.; Zlabinger, G.J.; Grabmeier-Pfistershammer, K.; et al. Therapeutic PD-L1 antibodies are more effective than PD-1 antibodies in blocking PD-1/PD-L1 signaling. Sci. Rep. 2019, 9, 11472. [Google Scholar] [CrossRef] [PubMed]
- Gutzmer, R.; Stroyakovskiy, D.; Gogas, H.; Robert, C.; Lewis, K.; Protsenko, S.; Pereira, R.P.; Eigentler, T.; Rutkowski, P.; Demidov, L.; et al. Atezolizumab, vemurafenib, and cobimetinib as first-line treatment for unresectable advanced BRAF(V600) mutation-positive melanoma (IMspire150): Primary analysis of the randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2020, 395, 1835–1844. [Google Scholar] [CrossRef]
- Ascierto, P.A.; Ferrucci, P.F.; Fisher, R.; Del Vecchio, M.; Atkinson, V.; Schmidt, H.; Schachter, J.; Queirolo, P.; Long, G.V.; Di Giacomo, A.M.; et al. Dabrafenib, trametinib and pembrolizumab or placebo in BRAF-mutant melanoma. Nat. Med. 2019, 25, 941–946. [Google Scholar] [CrossRef]
- Ferrucci, P.F.; Ascierto, P.A.; Maio, M. Updated survival in patients (pts) with BRAF mutant melanoma administered pembrolizumab (pembro), dabrafenib (D), and trametinib. In Proceedings of the International Congress of the Society for Melanoma Research, Salt Lake City, UT, USA, 20–23 November 2019. [Google Scholar]
- Melchiorri, D.; Cappuccio, I.; Ciceroni, C.; Spinsanti, P.; Mosillo, P.; Sarichelou, I.; Sale, P.; Nicoletti, F. Metabotropic glutamate receptors in stem/progenitor cells. Neuropharmacology 2007, 53, 473–480. [Google Scholar] [CrossRef]
- Tong, Q.; Ouedraogo, R.; Kirchgessner, A.L. Localization and function of group III metabotropic glutamate receptors in rat pancreatic islets. Am. J. Physiol. Endocrinol. Metab. 2002, 282, E1324–E1333. [Google Scholar] [CrossRef]
- Amaria, R.N.; Postow, M.; Burton, E.M.; Tetzlaff, M.T.; Ross, M.I.; Torres-Cabala, C.; Glitza, I.C.; Duan, F.; Milton, D.R.; Busam, K.; et al. Neoadjuvant relatlimab and nivolumab in resectable melanoma. Nature 2022, 611, 155–160. [Google Scholar] [CrossRef]
- Pol, J.; Kroemer, G.; Galluzzi, L. First oncolytic virus approved for melanoma immunotherapy. Oncoimmunology 2016, 5, e1115641. [Google Scholar] [CrossRef] [PubMed]
- Bommareddy, P.K.; Shettigar, M.; Kaufman, H.L. Integrating oncolytic viruses in combination cancer immunotherapy. Nat. Rev. Immunol. 2018, 18, 498–513. [Google Scholar] [CrossRef]
- Achard, C.; Surendran, A.; Wedge, M.E.; Ungerechts, G.; Bell, J.; Ilkow, C.S. Lighting a Fire in the Tumor Microenvironment Using Oncolytic Immunotherapy. eBioMedicine 2018, 31, 17–24. [Google Scholar] [CrossRef]
- Eddy, K.; Chen, S. Overcoming Immune Evasion in Melanoma. Int. J. Mol. Sci. 2020, 21, 8984. [Google Scholar] [CrossRef]
- Chesney, J.; Lewis, K.D.; Kluger, H.; Hamid, O.; Whitman, E.; Thomas, S.; Wermke, M.; Cusnir, M.; Domingo-Musibay, E.; Phan, G.Q.; et al. Efficacy and safety of lifileucel, a one-time autologous tumor-infiltrating lymphocyte (TIL) cell therapy, in patients with advanced melanoma after progression on immune checkpoint inhibitors and targeted therapies: Pooled analysis of consecutive cohorts of the C-144-01 study. J. Immunother. Cancer 2022, 10, e005755. [Google Scholar] [CrossRef]
- Lu, Y.G.; Wang, Y.Y.; Yang, Y.D.; Zhang, X.C.; Gao, Y.; Yang, Y.; Zhang, J.B.; Li, G.L. Efficacy of topical ALA-PDT combined with excision in the treatment of skin malignant tumor. Photodiagnosis Photodyn. Ther. 2014, 11, 122–126. [Google Scholar] [CrossRef] [PubMed]
- Yano, T.; Muto, M.; Minashi, K.; Iwasaki, J.; Kojima, T.; Fuse, N.; Doi, T.; Kaneko, K.; Ohtsu, A. Photodynamic therapy as salvage treatment for local failure after chemoradiotherapy in patients with esophageal squamous cell carcinoma: A phase II study. Int. J. Cancer 2012, 131, 1228–1234. [Google Scholar] [CrossRef]
- Lou, P.J.; Jäger, H.R.; Jones, L.; Theodossy, T.; Bown, S.G.; Hopper, C. Interstitial photodynamic therapy as salvage treatment for recurrent head and neck cancer. Br. J. Cancer 2004, 91, 441–446. [Google Scholar] [CrossRef] [PubMed]
- Isola, V.; Pece, A.; Pierro, L. Photodynamic therapy with verteporfin of choroidal malignancy from breast cancer. Am. J. Ophthalmol. 2006, 142, 885–887. [Google Scholar] [CrossRef] [PubMed]
- Simone, C.B., 2nd; Cengel, K.A. Photodynamic therapy for lung cancer and malignant pleural mesothelioma. Semin. Oncol. 2014, 41, 820–830. [Google Scholar] [CrossRef]
- Salvio, A.G.; Stringasci, M.D.; Requena, M.B.; Fregolenti, B.A.; Medeiro, M.; Santos, R.G.; Bagnato, V.S. Long-term follow-up results of a pilot study for nodular basal cell carcinoma with PDT using partial home treatment protocol. Photodiagnosis Photodyn. Ther. 2024, 45, 103930. [Google Scholar] [CrossRef] [PubMed]
- Li, X.Y.; Tan, L.C.; Dong, L.W.; Zhang, W.Q.; Shen, X.X.; Lu, X.; Zheng, H.; Lu, Y.G. Susceptibility and Resistance Mechanisms During Photodynamic Therapy of Melanoma. Front. Oncol. 2020, 10, 597. [Google Scholar] [CrossRef]
- Kim, M.M.; Ghogare, A.A.; Greer, A.; Zhu, T.C. On the in vivo photochemical rate parameters for PDT reactive oxygen species modeling. Phys. Med. Biol. 2017, 62, R1–R48. [Google Scholar] [CrossRef]
- Lim, M.E.; Lee, Y.L.; Zhang, Y.; Chu, J.J. Photodynamic inactivation of viruses using upconversion nanoparticles. Biomaterials 2012, 33, 1912–1920. [Google Scholar] [CrossRef] [PubMed]
- Saji, H.; Song, W.; Furumoto, K.; Kato, H.; Engleman, E.G. Systemic antitumor effect of intratumoral injection of dendritic cells in combination with local photodynamic therapy. Clin. Cancer Res. 2006, 12, 2568–2574. [Google Scholar] [CrossRef] [PubMed]
- Naylor, M.F.; Chen, W.R.; Teague, T.K.; Perry, L.A.; Nordquist, R.E. In situ photoimmunotherapy: A tumour-directed treatment for melanoma. Br. J. Dermatol. 2006, 155, 1287–1292. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Naylor, M.F.; Le, H.; Nordquist, R.E.; Teague, T.K.; Howard, C.A.; Murray, C.; Chen, W.R. Clinical effects of in situ photoimmunotherapy on late-stage melanoma patients: A preliminary study. Cancer Biol. Ther. 2010, 10, 1081–1087. [Google Scholar] [CrossRef] [PubMed]
- Soliman, A.H.; Youness, R.A.; Sebak, A.A.; Handoussa, H. Phytochemical-derived tumor-associated macrophage remodeling strategy using Phoenix dactylifera L. boosted photodynamic therapy in melanoma via H19/iNOS/PD-L1 axis. Photodiagnosis Photodyn. Ther. 2023, 44, 103792. [Google Scholar] [CrossRef] [PubMed]
- Naidoo, C.; Kruger, C.A.; Abrahamse, H. Photodynamic Therapy for Metastatic Melanoma Treatment: A Review. Technol. Cancer Res. Treat. 2018, 17, 1533033818791795. [Google Scholar] [CrossRef]
- Carbó-Bagué, A.; Rubió-Casadevall, J.; Puigdemont, M.; Sanvisens, A.; Oliveras, G.; Coll, M.; Del Olmo, B.; Perez-Bueno, F.; Marcos-Gragera, R. Epidemiology and Molecular Profile of Mucosal Melanoma: A Population-Based Study in Southern Europe. Cancers 2022, 14, 780. [Google Scholar] [CrossRef] [PubMed]
- Kolla, A.M.; Vitiello, G.A.; Friedman, E.B.; Sun, J.; Potdar, A.; Daou, H.; Farrow, N.E.; Farley, C.R.; Vetto, J.T.; Han, D.; et al. Acral Lentiginous Melanoma: A United States Multi-Center Substage Survival Analysis. Cancer Control 2021, 28, 10732748211053567. [Google Scholar] [CrossRef] [PubMed]
- Sureda, N.; Phan, A.; Poulalhon, N.; Balme, B.; Dalle, S.; Thomas, L. Conservative surgical management of subungual (matrix derived) melanoma: Report of seven cases and literature review. Br. J. Dermatol. 2011, 165, 852–858. [Google Scholar] [CrossRef] [PubMed]
- Walker, M.E.; Han, G.; Gabrick, K.; Kanouzi, J.; Lo, Y.C.; Galan, A.; Clune, J.; Narayan, D.; Ariyan, S.; Han, D. Acral Lentiginous Melanoma: Do Surgical Approach and Sentinel Lymph Node Biopsy Matter? Plast. Reconstr. Surg. Glob. Open 2020, 8, e2698. [Google Scholar] [CrossRef]
- Kirkwood, J.M.; Strawderman, M.H.; Ernstoff, M.S.; Smith, T.J.; Borden, E.C.; Blum, R.H. Interferon alfa-2b adjuvant therapy of high-risk resected cutaneous melanoma: The Eastern Cooperative Oncology Group Trial EST 1684. J. Clin. Oncol. 1996, 14, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Eggermont, A.M.; Suciu, S.; Testori, A.; Santinami, M.; Kruit, W.H.; Marsden, J.; Punt, C.J.; Salès, F.; Dummer, R.; Robert, C.; et al. Long-term results of the randomized phase III trial EORTC 18991 of adjuvant therapy with pegylated interferon alfa-2b versus observation in resected stage III melanoma. J. Clin. Oncol. 2012, 30, 3810–3818. [Google Scholar] [CrossRef] [PubMed]
- Mao, L.; Si, L.; Chi, Z.; Cui, C.; Sheng, X.; Li, S.; Tang, B.; Guo, J. A randomised phase II trial of 1 month versus 1 year of adjuvant high-dose interferon α-2b in high-risk acral melanoma patients. Eur. J. Cancer 2011, 47, 1498–1503. [Google Scholar] [CrossRef] [PubMed]
- Lerner, B.A.; Stewart, L.A.; Horowitz, D.P.; Carvajal, R.D. Mucosal Melanoma: New Insights and Therapeutic Options for a Unique and Aggressive Disease. Oncology 2017, 31, e23–e32. [Google Scholar]
- Bai, X.; Mao, L.L.; Chi, Z.H.; Sheng, X.N.; Cui, C.L.; Kong, Y.; Dai, J.; Wang, X.; Li, S.M.; Tang, B.X.; et al. BRAF inhibitors: Efficacious and tolerable in BRAF-mutant acral and mucosal melanoma. Neoplasma 2017, 64, 626–632. [Google Scholar] [CrossRef] [PubMed]
- Fujisawa, Y.; Ito, T.; Kato, H.; Irie, H.; Kaji, T.; Maekawa, T.; Asai, J.; Yamamoto, Y.; Fujimura, T.; Nakai, Y.; et al. Outcome of combination therapy using BRAF and MEK inhibitors among Asian patients with advanced melanoma: An analysis of 112 cases. Eur. J. Cancer 2021, 145, 210–220. [Google Scholar] [CrossRef] [PubMed]
- Curtin, J.A.; Busam, K.; Pinkel, D.; Bastian, B.C. Somatic activation of KIT in distinct subtypes of melanoma. J. Clin. Oncol. 2006, 24, 4340–4346. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; Corless, C.L.; Giobbie-Hurder, A.; Fletcher, J.A.; Zhu, M.; Marino-Enriquez, A.; Friedlander, P.; Gonzalez, R.; Weber, J.S.; Gajewski, T.F.; et al. Imatinib for melanomas harboring mutationally activated or amplified KIT arising on mucosal, acral, and chronically sun-damaged skin. J. Clin. Oncol. 2013, 31, 3182–3190. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Si, L.; Kong, Y.; Flaherty, K.T.; Xu, X.; Zhu, Y.; Corless, C.L.; Li, L.; Li, H.; Sheng, X.; et al. Phase II, open-label, single-arm trial of imatinib mesylate in patients with metastatic melanoma harboring c-Kit mutation or amplification. J. Clin. Oncol. 2011, 29, 2904–2909. [Google Scholar] [CrossRef]
- Carvajal, R.D.; Antonescu, C.R.; Wolchok, J.D.; Chapman, P.B.; Roman, R.A.; Teitcher, J.; Panageas, K.S.; Busam, K.J.; Chmielowski, B.; Lutzky, J.; et al. KIT as a therapeutic target in metastatic melanoma. JAMA 2011, 305, 2327–2334. [Google Scholar] [CrossRef]
- Montone, K.T.; van Belle, P.; Elenitsas, R.; Elder, D.E. Proto-oncogene c-kit expression in malignant melanoma: Protein loss with tumor progression. Mod. Pathol. 1997, 10, 939–944. [Google Scholar] [PubMed]
- Isabel Zhu, Y.; Fitzpatrick, J.E. Expression of c-kit (CD117) in Spitz nevus and malignant melanoma. J. Cutan. Pathol. 2006, 33, 33–37. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Luca, M.; Gutman, M.; McConkey, D.J.; Langley, K.E.; Lyman, S.D.; Bar-Eli, M. Enforced c-KIT expression renders highly metastatic human melanoma cells susceptible to stem cell factor-induced apoptosis and inhibits their tumorigenic and metastatic potential. Oncogene 1996, 13, 2339–2347. [Google Scholar] [PubMed]
- Slipicevic, A.; Herlyn, M. KIT in melanoma: Many shades of gray. J. Investig. Dermatol. 2015, 135, 337–338. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.B.; Eton, O.; Davis, D.W.; Frazier, M.L.; McConkey, D.J.; Diwan, A.H.; Papadopoulos, N.E.; Bedikian, A.Y.; Camacho, L.H.; Ross, M.I.; et al. Phase II trial of imatinib mesylate in patients with metastatic melanoma. Br. J. Cancer 2008, 99, 734–740. [Google Scholar] [CrossRef] [PubMed]
- Carvajal, R.D.; Lawrence, D.P.; Weber, J.S.; Gajewski, T.F.; Gonzalez, R.; Lutzky, J.; O’Day, S.J.; Hamid, O.; Wolchok, J.D.; Chapman, P.B.; et al. Phase II Study of Nilotinib in Melanoma Harboring KIT Alterations Following Progression to Prior KIT Inhibition. Clin. Cancer Res. 2015, 21, 2289–2296. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Carvajal, R.D.; Dummer, R.; Hauschild, A.; Daud, A.; Bastian, B.C.; Markovic, S.N.; Queirolo, P.; Arance, A.; Berking, C.; et al. Efficacy and safety of nilotinib in patients with KIT-mutated metastatic or inoperable melanoma: Final results from the global, single-arm, phase II TEAM trial. Ann. Oncol. 2017, 28, 1380–1387. [Google Scholar] [CrossRef] [PubMed]
- Ascierto, P.A.; Del Vecchio, M.; Mandalá, M.; Gogas, H.; Arance, A.M.; Dalle, S.; Cowey, C.L.; Schenker, M.; Grob, J.J.; Chiarion-Sileni, V.; et al. Adjuvant nivolumab versus ipilimumab in resected stage IIIB-C and stage IV melanoma (CheckMate 238): 4-year results from a multicentre, double-blind, randomised, controlled, phase 3 trial. Lancet Oncol. 2020, 21, 1465–1477. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, J.; Li, D.; Wen, X.; Ding, Y.; Liu, X.; Jiang, H.; Huang, F.; Zhang, X. Adjuvant PD-1 inhibitor versus high-dose interferon α-2b for Chinese patients with cutaneous and acral melanoma: A retrospective cohort analysis. Dermatol. Ther. 2021, 34, e15067. [Google Scholar] [CrossRef]
- Valero, C.; Lee, M.; Hoen, D.; Wang, J.; Nadeem, Z.; Patel, N.; Postow, M.A.; Shoushtari, A.N.; Plitas, G.; Balachandran, V.P.; et al. The association between tumor mutational burden and prognosis is dependent on treatment context. Nat. Genet. 2021, 53, 11–15. [Google Scholar] [CrossRef]
- Shoushtari, A.N.; Munhoz, R.R.; Kuk, D.; Ott, P.A.; Johnson, D.B.; Tsai, K.K.; Rapisuwon, S.; Eroglu, Z.; Sullivan, R.J.; Luke, J.J.; et al. The efficacy of anti-PD-1 agents in acral and mucosal melanoma. Cancer 2016, 122, 3354–3362. [Google Scholar] [CrossRef] [PubMed]
- Klemen, N.D.; Wang, M.; Rubinstein, J.C.; Olino, K.; Clune, J.; Ariyan, S.; Cha, C.; Weiss, S.A.; Kluger, H.M.; Sznol, M. Survival after checkpoint inhibitors for metastatic acral, mucosal and uveal melanoma. J. Immunother. Cancer 2020, 8, e000341. [Google Scholar] [CrossRef] [PubMed]
- Ogata, D.; Haydu, L.E.; Glitza, I.C.; Patel, S.P.; Tawbi, H.A.; McQuade, J.L.; Diab, A.; Ekmekcioglu, S.; Wong, M.K.; Davies, M.A.; et al. The efficacy of anti-programmed cell death protein 1 therapy among patients with metastatic acral and metastatic mucosal melanoma. Cancer Med. 2021, 10, 2293–2299. [Google Scholar] [CrossRef] [PubMed]
- Namikawa, K.; Kiyohara, Y.; Takenouchi, T.; Uhara, H.; Uchi, H.; Yoshikawa, S.; Takatsuka, S.; Koga, H.; Wada, N.; Minami, H.; et al. Efficacy and safety of nivolumab in combination with ipilimumab in Japanese patients with advanced melanoma: An open-label, single-arm, multicentre phase II study. Eur. J. Cancer 2018, 105, 114–126. [Google Scholar] [CrossRef] [PubMed]
- Nathan, P.; Ascierto, P.A.; Haanen, J.; Espinosa, E.; Demidov, L.; Garbe, C.; Guida, M.; Lorigan, P.; Chiarion-Sileni, V.; Gogas, H.; et al. Safety and efficacy of nivolumab in patients with rare melanoma subtypes who progressed on or after ipilimumab treatment: A single-arm, open-label, phase II study (CheckMate 172). Eur. J. Cancer 2019, 119, 168–178. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, S.P.; Larkin, J.; Sosman, J.A.; Lebbé, C.; Brady, B.; Neyns, B.; Schmidt, H.; Hassel, J.C.; Hodi, F.S.; Lorigan, P.; et al. Efficacy and Safety of Nivolumab Alone or in Combination With Ipilimumab in Patients With Mucosal Melanoma: A Pooled Analysis. J. Clin. Oncol. 2017, 35, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Si, L.; Zhang, X.; Shu, Y.; Pan, H.; Wu, D.; Liu, J.; Lou, F.; Mao, L.; Wang, X.; Wen, X.; et al. A Phase Ib Study of Pembrolizumab as Second-Line Therapy for Chinese Patients With Advanced or Metastatic Melanoma (KEYNOTE-151). Transl. Oncol. 2019, 12, 828–835. [Google Scholar] [CrossRef]
- Singh, A.D.; Turell, M.E.; Topham, A.K. Uveal melanoma: Trends in incidence, treatment, and survival. Ophthalmology 2011, 118, 1881–1885. [Google Scholar] [CrossRef] [PubMed]
- Carvajal, R.D.; Schwartz, G.K.; Tezel, T.; Marr, B.; Francis, J.H.; Nathan, P.D. Metastatic disease from uveal melanoma: Treatment options and future prospects. Br. J. Ophthalmol. 2017, 101, 38–44. [Google Scholar] [CrossRef]
- Shoushtari, A.N.; Carvajal, R.D. Treatment of Uveal Melanoma. Cancer Treat. Res. 2016, 167, 281–293. [Google Scholar] [CrossRef]
- Chattopadhyay, C.; Kim, D.W.; Gombos, D.S.; Oba, J.; Qin, Y.; Williams, M.D.; Esmaeli, B.; Grimm, E.A.; Wargo, J.A.; Woodman, S.E.; et al. Uveal melanoma: From diagnosis to treatment and the science in between. Cancer 2016, 122, 2299–2312. [Google Scholar] [CrossRef] [PubMed]
- Carvajal, R.D.; Sacco, J.J.; Jager, M.J.; Eschelman, D.J.; Olofsson Bagge, R.; Harbour, J.W.; Chieng, N.D.; Patel, S.P.; Joshua, A.M.; Piperno-Neumann, S. Advances in the clinical management of uveal melanoma. Nat. Rev. Clin. Oncol. 2023, 20, 99–115. [Google Scholar] [CrossRef] [PubMed]
- Hassel, J.C.; Piperno-Neumann, S.; Rutkowski, P.; Baurain, J.F.; Schlaak, M.; Butler, M.O.; Sullivan, R.J.; Dummer, R.; Kirkwood, J.M.; Orloff, M.; et al. Three-Year Overall Survival with Tebentafusp in Metastatic Uveal Melanoma. N. Engl. J. Med. 2023, 389, 2256–2266. [Google Scholar] [CrossRef] [PubMed]
- Nicoletti, F.; Arcella, A.; Iacovelli, L.; Battaglia, G.; Giangaspero, F.; Melchiorri, D. Metabotropic glutamate receptors: New targets for the control of tumor growth? Trends Pharmacol. Sci. 2007, 28, 206–213. [Google Scholar] [CrossRef]
- Storto, M.; Capobianco, L.; Battaglia, G.; Molinaro, G.; Gradini, R.; Riozzi, B.; Di Mambro, A.; Mitchell, K.J.; Bruno, V.; Vairetti, M.P.; et al. Insulin secretion is controlled by mGlu5 metabotropic glutamate receptors. Mol. Pharmacol. 2006, 69, 1234–1241. [Google Scholar] [CrossRef]
- Morimoto, R.; Uehara, S.; Yatsushiro, S.; Juge, N.; Hua, Z.; Senoh, S.; Echigo, N.; Hayashi, M.; Mizoguchi, T.; Ninomiya, T.; et al. Secretion of L-glutamate from osteoclasts through transcytosis. EMBO J. 2006, 25, 4175–4186. [Google Scholar] [CrossRef]
- Willard, S.S.; Koochekpour, S. Glutamate signaling in benign and malignant disorders: Current status, future perspectives, and therapeutic implications. Int. J. Biol. Sci. 2013, 9, 728–742. [Google Scholar] [CrossRef]
- Fazzari, J.; Linher-Melville, K.; Singh, G. Tumour-Derived Glutamate: Linking Aberrant Cancer Cell Metabolism to Peripheral Sensory Pain Pathways. Curr. Neuropharmacol. 2017, 15, 620–636. [Google Scholar] [CrossRef]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar] [CrossRef]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Wise, D.R.; Thompson, C.B. Glutamine addiction: A new therapeutic target in cancer. Trends Biochem. Sci. 2010, 35, 427–433. [Google Scholar] [CrossRef]
- Dang, C.V. MYC, microRNAs and glutamine addiction in cancers. Cell Cycle 2009, 8, 3243–3245. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.B.; Erickson, J.W.; Fuji, R.; Ramachandran, S.; Gao, P.; Dinavahi, R.; Wilson, K.F.; Ambrosio, A.L.; Dias, S.M.; Dang, C.V.; et al. Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell 2010, 18, 207–219. [Google Scholar] [CrossRef] [PubMed]
- van den Heuvel, A.P.; Jing, J.; Wooster, R.F.; Bachman, K.E. Analysis of glutamine dependency in non-small cell lung cancer: GLS1 splice variant GAC is essential for cancer cell growth. Cancer Biol. Ther. 2012, 13, 1185–1194. [Google Scholar] [CrossRef] [PubMed]
- Seltzer, M.J.; Bennett, B.D.; Joshi, A.D.; Gao, P.; Thomas, A.G.; Ferraris, D.V.; Tsukamoto, T.; Rojas, C.J.; Slusher, B.S.; Rabinowitz, J.D.; et al. Inhibition of glutaminase preferentially slows growth of glioma cells with mutant IDH1. Cancer Res. 2010, 70, 8981–8987. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Mao, S.; Guo, Y.; Wu, Y.; Yao, X.; Huang, Y. Inhibition of GLS suppresses proliferation and promotes apoptosis in prostate cancer. Biosci. Rep. 2019, 39, BSR20181826. [Google Scholar] [CrossRef] [PubMed]
- Eddy, K.; Eddin, M.N.; Fateeva, A.; Pompili, S.V.B.; Shah, R.; Doshi, S.; Chen, S. Implications of a Neuronal Receptor Family, Metabotropic Glutamate Receptors, in Cancer Development and Progression. Cells 2022, 11, 2857. [Google Scholar] [CrossRef] [PubMed]
- Pollock, P.M.; Cohen-Solal, K.; Sood, R.; Namkoong, J.; Martino, J.J.; Koganti, A.; Zhu, H.; Robbins, C.; Makalowska, I.; Shin, S.S.; et al. Melanoma mouse model implicates metabotropic glutamate signaling in melanocytic neoplasia. Nat. Genet. 2003, 34, 108–112. [Google Scholar] [CrossRef]
- Chen, S.; Zhu, H.; Wetzel, W.J.; Philbert, M.A. Spontaneous melanocytosis in transgenic mice. J. Investig. Dermatol. 1996, 106, 1145–1151. [Google Scholar] [CrossRef]
- Zhu, H.; Reuhl, K.; Zhang, X.; Botha, R.; Ryan, K.; Wei, J.; Chen, S. Development of heritable melanoma in transgenic mice. J. Investig. Dermatol. 1998, 110, 247–252. [Google Scholar] [CrossRef]
- Chen, S.; Teicher, L.C.; Kazim, D.; Pollack, R.E.; Wise, L.S. Commitment of mouse fibroblasts to adipocyte differentiation by DNA transfection. Science 1989, 244, 582–585. [Google Scholar] [CrossRef] [PubMed]
- Colón-Teicher, L.; Wise, L.S.; Martino, J.J.; Baskin, L.; Sakoulas, G.; Pollack, R.E.; Chen, S. Genomic sequences capable of committing mouse and rat fibroblasts to adipogenesis. Nucleic Acids Res. 1993, 21, 2223–2228. [Google Scholar] [CrossRef] [PubMed]
- Funusaka, Y.H.; Harada, T.; Aiba, A.; Nishigori, C. Expression of metabotropic glutamate receptor 1 and phosphorylated extracellular signal-regulated kinase 1/2 proteins in human melanocytic lesions. Pigm. Cell Res. 2006, 19, 256. [Google Scholar]
- Wangari-Talbot, J.; Wall, B.A.; Goydos, J.S.; Chen, S. Functional effects of GRM1 suppression in human melanoma cells. Mol. Cancer Res. 2012, 10, 1440–1450. [Google Scholar] [CrossRef] [PubMed]
- Namkoong, J.; Shin, S.S.; Lee, H.J.; Marín, Y.E.; Wall, B.A.; Goydos, J.S.; Chen, S. Metabotropic glutamate receptor 1 and glutamate signaling in human melanoma. Cancer Res. 2007, 67, 2298–2305. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.S.; Namkoong, J.; Wall, B.A.; Gleason, R.; Lee, H.J.; Chen, S. Oncogenic activities of metabotropic glutamate receptor 1 (Grm1) in melanocyte transformation. Pigment Cell Melanoma Res. 2008, 21, 368–378. [Google Scholar] [CrossRef]
- Ohtani, Y.; Harada, T.; Funasaka, Y.; Nakao, K.; Takahara, C.; Abdel-Daim, M.; Sakai, N.; Saito, N.; Nishigori, C.; Aiba, A. Metabotropic glutamate receptor subtype-1 is essential for in vivo growth of melanoma. Oncogene 2008, 27, 7162–7170. [Google Scholar] [CrossRef]
- Eddy, K.; Chen, S. Glutamatergic Signaling a Therapeutic Vulnerability in Melanoma. Cancers 2021, 13, 3874. [Google Scholar] [CrossRef]
- Shin, S.S.; Wall, B.A.; Goydos, J.S.; Chen, S. AKT2 is a downstream target of metabotropic glutamate receptor 1 (Grm1). Pigment Cell Melanoma Res. 2010, 23, 103–111. [Google Scholar] [CrossRef]
- Marín, Y.E.; Namkoong, J.; Cohen-Solal, K.; Shin, S.S.; Martino, J.J.; Oka, M.; Chen, S. Stimulation of oncogenic metabotropic glutamate receptor 1 in melanoma cells activates ERK1/2 via PKCepsilon. Cell Signal. 2006, 18, 1279–1286. [Google Scholar] [CrossRef]
- Wen, Y.; Li, J.; Koo, J.; Shin, S.S.; Lin, Y.; Jeong, B.S.; Mehnert, J.M.; Chen, S.; Cohen-Sola, K.A.; Goydos, J.S. Activation of the glutamate receptor GRM1 enhances angiogenic signaling to drive melanoma progression. Cancer Res. 2014, 74, 2499–2509. [Google Scholar] [CrossRef]
- Massoumi, R.; Kuphal, S.; Hellerbrand, C.; Haas, B.; Wild, P.; Spruss, T.; Pfeifer, A.; Fässler, R.; Bosserhoff, A.K. Down-regulation of CYLD expression by Snail promotes tumor progression in malignant melanoma. J. Exp. Med. 2009, 206, 221–232. [Google Scholar] [CrossRef]
- de Jel, M.M.; Schott, M.; Lamm, S.; Neuhuber, W.; Kuphal, S.; Bosserhoff, A.K. Loss of CYLD accelerates melanoma development and progression in the Tg(Grm1) melanoma mouse model. Oncogenesis 2019, 8, 56. [Google Scholar] [CrossRef]
- Xia, L.; Tan, S.; Zhou, Y.; Lin, J.; Wang, H.; Oyang, L.; Tian, Y.; Liu, L.; Su, M.; Wang, H.; et al. Role of the NFκB-signaling pathway in cancer. Onco Targets Ther. 2018, 11, 2063–2073. [Google Scholar] [CrossRef]
- Sun, S.C. CYLD: A tumor suppressor deubiquitinase regulating NF-kappaB activation and diverse biological processes. Cell Death Differ. 2010, 17, 25–34. [Google Scholar] [CrossRef]
- Marín, Y.E.; Wall, B.A.; Wang, S.; Namkoong, J.; Martino, J.J.; Suh, J.; Lee, H.J.; Rabson, A.B.; Yang, C.S.; Chen, S.; et al. Curcumin downregulates the constitutive activity of NF-kappaB and induces apoptosis in novel mouse melanoma cells. Melanoma Res. 2007, 17, 274–283. [Google Scholar] [CrossRef]
- Shah, R.; Singh, S.J.; Eddy, K.; Filipp, F.V.; Chen, S. Concurrent Targeting of Glutaminolysis and Metabotropic Glutamate Receptor 1 (GRM1) Reduces Glutamate Bioavailability in GRM1(+) Melanoma. Cancer Res. 2019, 79, 1799–1809. [Google Scholar] [CrossRef]
- Yip, D.; Le, M.N.; Chan, J.L.; Lee, J.H.; Mehnert, J.A.; Yudd, A.; Kempf, J.; Shih, W.J.; Chen, S.; Goydos, J.S. A phase 0 trial of riluzole in patients with resectable stage III and IV melanoma. Clin. Cancer Res. 2009, 15, 3896–3902. [Google Scholar] [CrossRef]
- Mehnert, J.M.; Silk, A.W.; Lee, J.H.; Dudek, L.; Jeong, B.S.; Li, J.; Schenkel, J.M.; Sadimin, E.; Kane, M.; Lin, H.; et al. A phase II trial of riluzole, an antagonist of metabotropic glutamate receptor 1 (GRM1) signaling, in patients with advanced melanoma. Pigment Cell Melanoma Res. 2018, 31, 534–540. [Google Scholar] [CrossRef] [PubMed]
- van Kan, H.J.; Groeneveld, G.J.; Kalmijn, S.; Spieksma, M.; van den Berg, L.H.; Guchelaar, H.J. Association between CYP1A2 activity and riluzole clearance in patients with amyotrophic lateral sclerosis. Br. J. Clin. Pharmacol. 2005, 59, 310–313. [Google Scholar] [CrossRef] [PubMed]
- Eddy, K.; Gupta, K.; Pelletier, J.C.; Isola, A.L.; Marinaro, C.; Rasheed, M.A.; Campagnolo, J.; Eddin, M.N.; Rossi, M.; Fateeva, A.; et al. A Spontaneous Melanoma Mouse Model Applicable for a longitudinal Chemotherapy and Immunotherapy Study. J. Investig. Dermatol. 2023, 143, 2007–2018.e6. [Google Scholar] [CrossRef]
- Wall, B.A.; Wangari-Talbot, J.; Shin, S.S.; Schiff, D.; Sierra, J.; Yu, L.J.; Khan, A.; Haffty, B.; Goydos, J.S.; Chen, S. Disruption of GRM1-mediated signalling using riluzole results in DNA damage in melanoma cells. Pigment Cell Melanoma Res. 2014, 27, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Cerchio, R., Jr.; Marinaro, C.; Foo, T.K.; Xia, B.; Chen, S. Nonhomologous end-joining repair is likely involved in the repair of double-stranded DNA breaks induced by riluzole in melanoma cells. Melanoma Res. 2020, 30, 303–308. [Google Scholar] [CrossRef]
- Liu, J.; Xia, X.; Huang, P. xCT: A Critical Molecule That Links Cancer Metabolism to Redox Signaling. Mol. Ther. 2020, 28, 2358–2366. [Google Scholar] [CrossRef]
- Shin, S.S.; Jeong, B.S.; Wall, B.A.; Li, J.; Shan, N.L.; Wen, Y.; Goydos, J.S.; Chen, S. Participation of xCT in melanoma cell proliferation in vitro and tumorigenesis in vivo. Oncogenesis 2018, 7, 86. [Google Scholar] [CrossRef]
- Eddy, K.; Gupta, K.; Eddin, M.N.; Marinaro, C.; Putta, S.; Sauer, J., Jr.; Chaly, A.; Freeman, K.B.; Pelletier, J.C.; Fateeva, A.; et al. Assessing Longitudinal Treatment Efficacies and Alterations in Molecular Markers Associated with Glutamatergic Signaling and Immune Checkpoint Inhibitors in a Spontaneous Melanoma Mouse Model. JID Innov. 2024, 4, 100262. [Google Scholar] [CrossRef]
- Silk, A.W.; Saraiya, B.; Groisberg, R.; Chan, N.; Spencer, K.; Girda, E.; Shih, W.; Palmeri, M.; Saunders, T.; Berman, R.M.; et al. A phase Ib dose-escalation study of troriluzole (BHV-4157), an oral glutamatergic signaling modulator, in combination with nivolumab in patients with advanced solid tumors. Eur. J. Med. Res. 2022, 27, 107. [Google Scholar] [CrossRef]
- Medikonda, R.; Choi, J.; Pant, A.; Saleh, L.; Routkevitch, D.; Tong, L.; Belcaid, Z.; Kim, Y.H.; Jackson, C.M.; Jackson, C.; et al. Synergy between glutamate modulation and anti-programmed cell death protein 1 immunotherapy for glioblastoma. J. Neurosu. 2022, 136, 379–388. [Google Scholar] [CrossRef]
- Blyufer, A.; Lhamo, S.; Tam, C.; Tariq, I.; Thavornwatanayong, T.; Mahajan, S.S. Riluzole: A neuroprotective drug with potential as a novel anticancer agent (Review). Int. J. Oncol. 2021, 59, 95. [Google Scholar] [CrossRef] [PubMed]
- Rizaner, N.; Uzun, S.; Fraser, S.P.; Djamgoz, M.B.A.; Altun, S. Riluzole: Anti-invasive effects on rat prostate cancer cells under normoxic and hypoxic conditions. Basic Clin. Pharmacol. Toxicol. 2020, 127, 254–264. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.K.; Ma, Y.; Lam, B.Q.; Shrivastava, A.; Srivastav, S.; Shankar, S.; Srivastava, R.K. Riluzole regulates pancreatic cancer cell metabolism by suppressing the Wnt-β-catenin pathway. Sci. Rep. 2022, 12, 11062. [Google Scholar] [CrossRef] [PubMed]
- Speyer, C.L.; Nassar, M.A.; Hachem, A.H.; Bukhsh, M.A.; Jafry, W.S.; Khansa, R.M.; Gorski, D.H. Riluzole mediates anti-tumor properties in breast cancer cells independent of metabotropic glutamate receptor-1. Breast Cancer Res. Treat. 2016, 157, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; He, X.; Jiang, X.; Tao, H. The new role of riluzole in the treatment of pancreatic cancer through the apoptosis and autophagy pathways. J. Cell Biochem. 2021, 122, 934–944. [Google Scholar] [CrossRef] [PubMed]
- Lemieszek, M.K.; Stepulak, A.; Sawa-Wejksza, K.; Czerwonka, A.; Ikonomidou, C.; Rzeski, W. Riluzole Inhibits Proliferation, Migration and Cell Cycle Progression and Induces Apoptosis in Tumor Cells of Various Origins. Anticancer. Agents Med. Chem. 2018, 18, 565–572. [Google Scholar] [CrossRef]
- Dolfi, S.C.; Medina, D.J.; Kareddula, A.; Paratala, B.; Rose, A.; Dhami, J.; Chen, S.; Ganesan, S.; Mackay, G.; Vazquez, A.; et al. Riluzole exerts distinct antitumor effects from a metabotropic glutamate receptor 1-specific inhibitor on breast cancer cells. Oncotarget 2017, 8, 44639–44653. [Google Scholar] [CrossRef] [PubMed]
- Akamatsu, K.; Shibata, M.A.; Ito, Y.; Sohma, Y.; Azuma, H.; Otsuki, Y. Riluzole induces apoptotic cell death in human prostate cancer cells via endoplasmic reticulum stress. Anticancer Res. 2009, 29, 2195–2204. [Google Scholar]
- Seol, H.S.; Lee, S.E.; Song, J.S.; Lee, H.Y.; Park, S.; Kim, I.; Singh, S.R.; Chang, S.; Jang, S.J. Glutamate release inhibitor, Riluzole, inhibited proliferation of human hepatocellular carcinoma cells by elevated ROS production. Cancer Lett. 2016, 382, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Wadosky, K.M.; Shourideh, M.; Goodrich, D.W.; Koochekpour, S. Riluzole induces AR degradation via endoplasmic reticulum stress pathway in androgen-dependent and castration-resistant prostate cancer cells. Prostate 2019, 79, 140–150. [Google Scholar] [CrossRef] [PubMed]
- Raghubir, M.; Rahman, C.N.; Fang, J.; Matsui, H.; Mahajan, S.S. Osteosarcoma growth suppression by riluzole delivery via iron oxide nanocage in nude mice. Oncol. Rep. 2020, 43, 169–176. [Google Scholar] [CrossRef]
- Raghubir, M.; Azeem, S.M.; Hasnat, R.; Rahman, C.N.; Wong, L.; Yan, S.; Huang, Y.Q.; Zhagui, R.; Blyufer, A.; Tariq, I.; et al. Riluzole-induced apoptosis in osteosarcoma is mediated through Yes-associated protein upon phosphorylation by c-Abl Kinase. Sci. Rep. 2021, 11, 20974. [Google Scholar] [CrossRef]
- Khan, A.J.; LaCava, S.; Mehta, M.; Schiff, D.; Thandoni, A.; Jhawar, S.; Danish, S.; Haffty, B.G.; Chen, S. The glutamate release inhibitor riluzole increases DNA damage and enhances cytotoxicity in human glioma cells, in vitro and in vivo. Oncotarget 2019, 10, 2824–2834. [Google Scholar] [CrossRef]
- Liao, S.; Ruiz, Y.; Gulzar, H.; Yelskaya, Z.; Ait Taouit, L.; Houssou, M.; Jaikaran, T.; Schvarts, Y.; Kozlitina, K.; Basu-Roy, U.; et al. Osteosarcoma cell proliferation and survival requires mGluR5 receptor activity and is blocked by Riluzole. PLoS ONE 2017, 12, e0171256. [Google Scholar] [CrossRef]
- Zhang, C.; Yuan, X.R.; Li, H.Y.; Zhao, Z.J.; Liao, Y.W.; Wang, X.Y.; Su, J.; Sang, S.S.; Liu, Q. Anti-cancer effect of metabotropic glutamate receptor 1 inhibition in human glioma U87 cells: Involvement of PI3K/Akt/mTOR pathway. Cell Physiol. Biochem. 2015, 35, 419–432. [Google Scholar] [CrossRef]
- Sperling, S.; Aung, T.; Martin, S.; Rohde, V.; Ninkovic, M. Riluzole: A potential therapeutic intervention in human brain tumor stem-like cells. Oncotarget 2017, 8, 96697–96709. [Google Scholar] [CrossRef]
- Yelskaya, Z.; Carrillo, V.; Dubisz, E.; Gulzar, H.; Morgan, D.; Mahajan, S.S. Synergistic inhibition of survival, proliferation, and migration of U87 cells with a combination of LY341495 and Iressa. PLoS ONE 2013, 8, e64588. [Google Scholar] [CrossRef]
- Yu, L.J.; Wall, B.A.; Chen, S. The current management of brain metastasis in melanoma: A focus on riluzole. Expert Rev. Neurother. 2015, 15, 779–792. [Google Scholar] [CrossRef]
- Wall, B.A.; Yu, L.J.; Khan, A.; Haffty, B.; Goydos, J.S.; Chen, S. Riluzole is a radio-sensitizing agent in an in vivo model of brain metastasis derived from GRM1 expressing human melanoma cells. Pigment Cell Melanoma Res. 2015, 28, 105–109. [Google Scholar] [CrossRef]
- Spencer, K.R.; Portal, D.E.; Aisner, J.; Stein, M.N.; Malhotra, J.; Shih, W.; Chan, N.; Silk, A.W.; Ganesan, S.; Goodin, S.; et al. A phase I trial of riluzole and sorafenib in patients with advanced solid tumors: CTEP #8850. Oncotarget 2023, 14, 302–315. [Google Scholar] [CrossRef]
- Speyer, C.L.; Bukhsh, M.A.; Jafry, W.S.; Sexton, R.E.; Bandyopadhyay, S.; Gorski, D.H. Riluzole synergizes with paclitaxel to inhibit cell growth and induce apoptosis in triple-negative breast cancer. Breast Cancer Res. Treat. 2017, 166, 407–419. [Google Scholar] [CrossRef]
- Fortunato, A. The role of hERG1 ion channels in epithelial-mesenchymal transition and the capacity of riluzole to reduce cisplatin resistance in colorectal cancer cells. Cell Oncol. 2017, 40, 367–378. [Google Scholar] [CrossRef]
- Benavides-Serrato, A.; Saunders, J.T.; Holmes, B.; Nishimura, R.N.; Lichtenstein, A.; Gera, J. Repurposing Potential of Riluzole as an ITAF Inhibitor in mTOR Therapy Resistant Glioblastoma. Int. J. Mol. Sci. 2020, 21, 344. [Google Scholar] [CrossRef]
- Yamada, T.; Tsuji, S.; Nakamura, S.; Egashira, Y.; Shimazawa, M.; Nakayama, N.; Yano, H.; Iwama, T.; Hara, H. Riluzole enhances the antitumor effects of temozolomide via suppression of MGMT expression in glioblastoma. J. Neurosurg. 2020, 134, 701–710. [Google Scholar] [CrossRef]
- Sun, L.; Wu, C.; Ming, J.; Nie, X.; Guo, E.; Zhang, W.; Hu, G. Riluzole Enhances the Response of Human Nasopharyngeal Carcinoma Cells to Ionizing Radiation via ATM/P53 Signalling Pathway. J. Cancer 2020, 11, 3089–3098. [Google Scholar] [CrossRef]
- Li, Z.; Qiao, X.; Liu, X.M.; Shi, S.H.; Qiao, X.; Xu, J.Y. Blocking xCT and PI3K/Akt pathway synergized with DNA damage of Riluzole-Pt(IV) prodrugs for cancer treatment. Eur. J. Med. Chem. 2023, 250, 115233. [Google Scholar] [CrossRef]
- Pasinelli, P.; Dena, J. Rethinking Drug Treatment Approaches in ALS by Targeting ABC Efflux Transporters; Annual Rept. Accession Number: ADA594930. Defenst Technical Information Center: Fort Belvoir, VA, USA, 2012.
- Mohamed, L.A.; Markandaiah, S.; Bonanno, S.; Pasinelli, P.; Trotti, D. Blood-Brain Barrier Driven Pharmacoresistance in Amyotrophic Lateral Sclerosis and Challenges for Effective Drug Therapies. AAPS J. 2017, 19, 1600–1614. [Google Scholar] [CrossRef]
- Jablonski, M.; Miller, D.S.; Pasinelli, P.; Trotti, D. ABC transporter-driven pharmacoresistance in Amyotrophic Lateral Sclerosis. Brain Res. 2015, 1607, 1–14. [Google Scholar] [CrossRef]
- Mohamed, L.A.; Markandaiah, S.S.; Bonanno, S.; Pasinelli, P.; Trotti, D. Excess glutamate secreted from astrocytes drives upregulation of P-glycoprotein in endothelial cells in amyotrophic lateral sclerosis. Exp. Neurol. 2019, 316, 27–38. [Google Scholar] [CrossRef]
- Chan, G.N.; Evans, R.A.; Banks, D.B.; Mesev, E.V.; Miller, D.S.; Cannon, R.E. Selective induction of P-glycoprotein at the CNS barriers during symptomatic stage of an ALS animal model. Neurosci. Lett. 2017, 639, 103–113. [Google Scholar] [CrossRef]
- Milane, A.; Fernandez, C.; Dupuis, L.; Buyse, M.; Loeffler, J.P.; Farinotti, R.; Meininger, V.; Bensimon, G. P-glycoprotein expression and function are increased in an animal model of amyotrophic lateral sclerosis. Neurosci. Lett. 2010, 472, 166–170. [Google Scholar] [CrossRef]
- Milane, A.; Fernandez, C.; Vautier, S.; Bensimon, G.; Meininger, V.; Farinotti, R. Minocycline and riluzole brain disposition: Interactions with p-glycoprotein at the blood-brain barrier. J. Neurochem. 2007, 103, 164–173. [Google Scholar] [CrossRef]
- Milane, A.; Vautier, S.; Chacun, H.; Meininger, V.; Bensimon, G.; Farinotti, R.; Fernandez, C. Interactions between riluzole and ABCG2/BCRP transporter. Neurosci. Lett. 2009, 452, 12–16. [Google Scholar] [CrossRef]
- Jablonski, M.R.; Markandaiah, S.S.; Jacob, D.; Meng, N.J.; Li, K.; Gennaro, V.; Lepore, A.C.; Trotti, D.; Pasinelli, P. Inhibiting drug efflux transporters improves efficacy of ALS therapeutics. Ann. Clin. Transl. Neurol. 2014, 1, 996–1005. [Google Scholar] [CrossRef]
- Mandalà, M.; Romano, E. Mechanisms of Drug Resistance in Cancer Therapy; Springer: Paris, France, 2018. [Google Scholar]
- Lito, P.; Rosen, N.; Solit, D.B. Tumor adaptation and resistance to RAF inhibitors. Nat. Med. 2013, 19, 1401–1409. [Google Scholar] [CrossRef]
- Nazarian, R.; Shi, H.; Wang, Q.; Kong, X.; Koya, R.C.; Lee, H.; Chen, Z.; Lee, M.K.; Attar, N.; Sazegar, H.; et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature 2010, 468, 973–977. [Google Scholar] [CrossRef]
- Maertens, O.; Johnson, B.; Hollstein, P.; Frederick, D.T.; Cooper, Z.A.; Messiaen, L.; Bronson, R.T.; McMahon, M.; Granter, S.; Flaherty, K.; et al. Elucidating distinct roles for NF1 in melanomagenesis. Cancer Discov. 2013, 3, 338–349. [Google Scholar] [CrossRef]
- Whittaker, S.R.; Theurillat, J.P.; Van Allen, E.; Wagle, N.; Hsiao, J.; Cowley, G.S.; Schadendorf, D.; Root, D.E.; Garraway, L.A. A genome-scale RNA interference screen implicates NF1 loss in resistance to RAF inhibition. Cancer Discov. 2013, 3, 350–362. [Google Scholar] [CrossRef]
- Solit, D.B.; Rosen, N. Resistance to BRAF inhibition in melanomas. N. Engl. J. Med. 2011, 364, 772–774. [Google Scholar] [CrossRef]
- Poulikakos, P.I.; Persaud, Y.; Janakiraman, M.; Kong, X.; Ng, C.; Moriceau, G.; Shi, H.; Atefi, M.; Titz, B.; Gabay, M.T.; et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Nature 2011, 480, 387–390. [Google Scholar] [CrossRef]
- Kemper, K.; Krijgsman, O.; Kong, X.; Cornelissen-Steijger, P.; Shahrabi, A.; Weeber, F.; van der Velden, D.L.; Bleijerveld, O.B.; Kuilman, T.; Kluin, R.J.C.; et al. BRAF(V600E) Kinase Domain Duplication Identified in Therapy-Refractory Melanoma Patient-Derived Xenografts. Cell Rep. 2016, 16, 263–277. [Google Scholar] [CrossRef] [PubMed]
- Wagle, N.; Emery, C.; Berger, M.F.; Davis, M.J.; Sawyer, A.; Pochanard, P.; Kehoe, S.M.; Johannessen, C.M.; Macconaill, L.E.; Hahn, W.C.; et al. Dissecting therapeutic resistance to RAF inhibition in melanoma by tumor genomic profiling. J. Clin. Oncol. 2011, 29, 3085–3096. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.H.; Kim, S.H.; Trousil, S.; Frederick, D.T.; Piris, A.; Yuan, P.; Cai, L.; Gu, L.; Li, M.; Lee, J.H.; et al. Loss of cohesin complex components STAG2 or STAG3 confers resistance to BRAF inhibition in melanoma. Nat. Med. 2016, 22, 1056–1061. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, J.; Vultur, A.; Lee, J.T.; Somasundaram, R.; Fukunaga-Kalabis, M.; Cipolla, A.K.; Wubbenhorst, B.; Xu, X.; Gimotty, P.A.; Kee, D.; et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell 2010, 18, 683–695. [Google Scholar] [CrossRef]
- Wilson, T.R.; Fridlyand, J.; Yan, Y.; Penuel, E.; Burton, L.; Chan, E.; Peng, J.; Lin, E.; Wang, Y.; Sosman, J.; et al. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature 2012, 487, 505–509. [Google Scholar] [CrossRef] [PubMed]
- Levy, C.; Khaled, M.; Fisher, D.E. MITF: Master regulator of melanocyte development and melanoma oncogene. Trends Mol. Med. 2006, 12, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Johannessen, C.M.; Johnson, L.A.; Piccioni, F.; Townes, A.; Frederick, D.T.; Donahue, M.K.; Narayan, R.; Flaherty, K.T.; Wargo, J.A.; Root, D.E.; et al. A melanocyte lineage program confers resistance to MAP kinase pathway inhibition. Nature 2013, 504, 138–142. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.P.; Brunton, H.; Rowling, E.J.; Ferguson, J.; Arozarena, I.; Miskolczi, Z.; Lee, J.L.; Girotti, M.R.; Marais, R.; Levesque, M.P.; et al. Inhibiting Drivers of Non-mutational Drug Tolerance Is a Salvage Strategy for Targeted Melanoma Therapy. Cancer Cell 2016, 29, 270–284. [Google Scholar] [CrossRef]
- Haq, R.; Yokoyama, S.; Hawryluk, E.B.; Jönsson, G.B.; Frederick, D.T.; McHenry, K.; Porter, D.; Tran, T.N.; Love, K.T.; Langer, R.; et al. BCL2A1 is a lineage-specific antiapoptotic melanoma oncogene that confers resistance to BRAF inhibition. Proc. Natl. Acad. Sci. USA 2013, 110, 4321–4326. [Google Scholar] [CrossRef] [PubMed]
- Van Allen, E.M.; Wagle, N.; Sucker, A.; Treacy, D.J.; Johannessen, C.M.; Goetz, E.M.; Place, C.S.; Taylor-Weiner, A.; Whittaker, S.; Kryukov, G.V.; et al. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov. 2014, 4, 94–109. [Google Scholar] [CrossRef]
- Smith, M.P.; Ferguson, J.; Arozarena, I.; Hayward, R.; Marais, R.; Chapman, A.; Hurlstone, A.; Wellbrock, C. Effect of SMURF2 targeting on susceptibility to MEK inhibitors in melanoma. J. Natl. Cancer Inst. 2013, 105, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Hugo, W.; Kong, X.; Hong, A.; Koya, R.C.; Moriceau, G.; Chodon, T.; Guo, R.; Johnson, D.B.; Dahlman, K.B.; et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov. 2014, 4, 80–93. [Google Scholar] [CrossRef]
- Müller, J.; Krijgsman, O.; Tsoi, J.; Robert, L.; Hugo, W.; Song, C.; Kong, X.; Possik, P.A.; Cornelissen-Steijger, P.D.; Geukes Foppen, M.H.; et al. Low MITF/AXL ratio predicts early resistance to multiple targeted drugs in melanoma. Nat. Commun. 2014, 5, 5712. [Google Scholar] [CrossRef]
- Konieczkowski, D.J.; Johannessen, C.M.; Abudayyeh, O.; Kim, J.W.; Cooper, Z.A.; Piris, A.; Frederick, D.T.; Barzily-Rokni, M.; Straussman, R.; Haq, R.; et al. A melanoma cell state distinction influences sensitivity to MAPK pathway inhibitors. Cancer Discov. 2014, 4, 816–827. [Google Scholar] [CrossRef]
- Kozar, I.; Margue, C.; Rothengatter, S.; Haan, C.; Kreis, S. Many ways to resistance: How melanoma cells evade targeted therapies. Biochim. Biophys. Acta Rev. Cancer 2019, 1871, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Pinner, S.; Jordan, P.; Sharrock, K.; Bazley, L.; Collinson, L.; Marais, R.; Bonvin, E.; Goding, C.; Sahai, E. Intravital imaging reveals transient changes in pigment production and Brn2 expression during metastatic melanoma dissemination. Cancer Res. 2009, 69, 7969–7977. [Google Scholar] [CrossRef] [PubMed]
- Ahn, A.; Chatterjee, A.; Eccles, M.R. The Slow Cycling Phenotype: A Growing Problem for Treatment Resistance in Melanoma. Mol. Cancer Ther. 2017, 16, 1002–1009. [Google Scholar] [CrossRef] [PubMed]
- Roesch, A.; Fukunaga-Kalabis, M.; Schmidt, E.C.; Zabierowski, S.E.; Brafford, P.A.; Vultur, A.; Basu, D.; Gimotty, P.; Vogt, T.; Herlyn, M. A temporarily distinct subpopulation of slow-cycling melanoma cells is required for continuous tumor growth. Cell 2010, 141, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Roesch, A.; Vultur, A.; Bogeski, I.; Wang, H.; Zimmermann, K.M.; Speicher, D.; Körbel, C.; Laschke, M.W.; Gimotty, P.A.; Philipp, S.E.; et al. Overcoming intrinsic multidrug resistance in melanoma by blocking the mitochondrial respiratory chain of slow-cycling JARID1B(high) cells. Cancer Cell 2013, 23, 811–825. [Google Scholar] [CrossRef] [PubMed]
- Tirosh, I.; Izar, B.; Prakadan, S.M.; Wadsworth, M.H., 2nd; Treacy, D.; Trombetta, J.J.; Rotem, A.; Rodman, C.; Lian, C.; Murphy, G.; et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 2016, 352, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Vlčková, K.; Vachtenheim, J.; Réda, J.; Horák, P.; Ondrušová, L. Inducibly decreased MITF levels do not affect proliferation and phenotype switching but reduce differentiation of melanoma cells. J. Cell Mol. Med. 2018, 22, 2240–2251. [Google Scholar] [CrossRef]
- Haq, R.; Shoag, J.; Andreu-Perez, P.; Yokoyama, S.; Edelman, H.; Rowe, G.C.; Frederick, D.T.; Hurley, A.D.; Nellore, A.; Kung, A.L.; et al. Oncogenic BRAF regulates oxidative metabolism via PGC1α and MITF. Cancer Cell 2013, 23, 302–315. [Google Scholar] [CrossRef]
- Bristot, I.J.; Kehl Dias, C.; Chapola, H.; Parsons, R.B.; Klamt, F. Metabolic rewiring in melanoma drug-resistant cells. Crit. Rev. Oncol. Hematol. 2020, 153, 102995. [Google Scholar] [CrossRef] [PubMed]
- Hugo, W.; Shi, H.; Sun, L.; Piva, M.; Song, C.; Kong, X.; Moriceau, G.; Hong, A.; Dahlman, K.B.; Johnson, D.B.; et al. Non-genomic and Immune Evolution of Melanoma Acquiring MAPKi Resistance. Cell 2015, 162, 1271–1285. [Google Scholar] [CrossRef] [PubMed]
- Xing, F.; Persaud, Y.; Pratilas, C.A.; Taylor, B.S.; Janakiraman, M.; She, Q.B.; Gallardo, H.; Liu, C.; Merghoub, T.; Hefter, B.; et al. Concurrent loss of the PTEN and RB1 tumor suppressors attenuates RAF dependence in melanomas harboring (V600E)BRAF. Oncogene 2012, 31, 446–457. [Google Scholar] [CrossRef] [PubMed]
- Greger, J.G.; Eastman, S.D.; Zhang, V.; Bleam, M.R.; Hughes, A.M.; Smitheman, K.N.; Dickerson, S.H.; Laquerre, S.G.; Liu, L.; Gilmer, T.M. Combinations of BRAF, MEK, and PI3K/mTOR inhibitors overcome acquired resistance to the BRAF inhibitor GSK2118436 dabrafenib, mediated by NRAS or MEK mutations. Mol. Cancer Ther. 2012, 11, 909–920. [Google Scholar] [CrossRef] [PubMed]
- Paraiso, K.H.; Das Thakur, M.; Fang, B.; Koomen, J.M.; Fedorenko, I.V.; John, J.K.; Tsao, H.; Flaherty, K.T.; Sondak, V.K.; Messina, J.L.; et al. Ligand-independent EPHA2 signaling drives the adoption of a targeted therapy-mediated metastatic melanoma phenotype. Cancer Discov. 2015, 5, 264–273. [Google Scholar] [CrossRef]
- Mulcahy Levy, J.M.; Thorburn, A. Autophagy in cancer: Moving from understanding mechanism to improving therapy responses in patients. Cell Death Differ. 2020, 27, 843–857. [Google Scholar] [CrossRef]
- Martin, S.; Dudek-Peric, A.M.; Garg, A.D.; Roose, H.; Demirsoy, S.; Van Eygen, S.; Mertens, F.; Vangheluwe, P.; Vankelecom, H.; Agostinis, P. An autophagy-driven pathway of ATP secretion supports the aggressive phenotype of BRAF(V600E) inhibitor-resistant metastatic melanoma cells. Autophagy 2017, 13, 1512–1527. [Google Scholar] [CrossRef]
- Xue, G.; Kohler, R.; Tang, F.; Hynx, D.; Wang, Y.; Orso, F.; Prêtre, V.; Ritschard, R.; Hirschmann, P.; Cron, P.; et al. mTORC1/autophagy-regulated MerTK in mutant BRAFV600 melanoma with acquired resistance to BRAF inhibition. Oncotarget 2017, 8, 69204–69218. [Google Scholar] [CrossRef][Green Version]
- Cerezo, M.; Lehraiki, A.; Millet, A.; Rouaud, F.; Plaisant, M.; Jaune, E.; Botton, T.; Ronco, C.; Abbe, P.; Amdouni, H.; et al. Compounds Triggering ER Stress Exert Anti-Melanoma Effects and Overcome BRAF Inhibitor Resistance. Cancer Cell 2016, 29, 805–819. [Google Scholar] [CrossRef]
- Mulcahy Levy, J.M.; Zahedi, S.; Griesinger, A.M.; Morin, A.; Davies, K.D.; Aisner, D.L.; Kleinschmidt-DeMasters, B.K.; Fitzwalter, B.E.; Goodall, M.L.; Thorburn, J.; et al. Autophagy inhibition overcomes multiple mechanisms of resistance to BRAF inhibition in brain tumors. eLife 2017, 6, e19671. [Google Scholar] [CrossRef]
- Fattore, L.; Ruggiero, C.F.; Pisanu, M.E.; Liguoro, D.; Cerri, A.; Costantini, S.; Capone, F.; Acunzo, M.; Romano, G.; Nigita, G.; et al. Reprogramming miRNAs global expression orchestrates development of drug resistance in BRAF mutated melanoma. Cell Death Differ. 2019, 26, 1267–1282. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Martínez, M.; Benito-Jardón, L.; Alonso, L.; Koetz-Ploch, L.; Hernando, E.; Teixidó, J. miR-204-5p and miR-211-5p Contribute to BRAF Inhibitor Resistance in Melanoma. Cancer Res. 2018, 78, 1017–1030. [Google Scholar] [CrossRef] [PubMed]
- Vitiello, M.; Tuccoli, A.; D’Aurizio, R.; Sarti, S.; Giannecchini, L.; Lubrano, S.; Marranci, A.; Evangelista, M.; Peppicelli, S.; Ippolito, C.; et al. Context-dependent miR-204 and miR-211 affect the biological properties of amelanotic and melanotic melanoma cells. Oncotarget 2017, 8, 25395–25417. [Google Scholar] [CrossRef] [PubMed]
- Olbryt, M.; Rajczykowski, M.; Widłak, W. Biological Factors behind Melanoma Response to Immune Checkpoint Inhibitors. Int. J. Mol. Sci. 2020, 21, 4071. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; Wolchok, J.D.; Schadendorf, D.; Larkin, J.; Long, G.V.; Qian, X.; Saci, A.; Young, T.C.; Srinivasan, S.; Chang, H.; et al. TMB and Inflammatory Gene Expression Associated with Clinical Outcomes following Immunotherapy in Advanced Melanoma. Cancer Immunol. Res. 2021, 9, 1202–1213. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.; Chen, X.; Zhang, H.; Liang, Y.; Li, L.; Wei, H.; Sun, W.; Wang, Y. Prognostic Role of Tumor Mutational Burden in Cancer Patients Treated with Immune Checkpoint Inhibitors: A Systematic Review and Meta-Analysis. Front. Oncol. 2021, 11, 706652. [Google Scholar] [CrossRef]
- Valero, C.; Lee, M.; Hoen, D.; Zehir, A.; Berger, M.F.; Seshan, V.E.; Chan, T.A.; Morris, L.G.T. Response Rates to Anti-PD-1 Immunotherapy in Microsatellite-Stable Solid Tumors With 10 or More Mutations per Megabase. JAMA Oncol. 2021, 7, 739–743. [Google Scholar] [CrossRef]
- Wang, L.; Chen, F.; Liu, R.; Shi, L.; Zhao, G.; Yan, Z. Gene expression and immune infiltration in melanoma patients with different mutation burden. BMC Cancer 2021, 21, 379. [Google Scholar] [CrossRef]
- Abbott, C.W.; Boyle, S.M.; Pyke, R.M.; McDaniel, L.D.; Levy, E.; Navarro, F.C.P.; Mellacheruvu, D.; Zhang, S.V.; Tan, M.; Santiago, R.; et al. Prediction of Immunotherapy Response in Melanoma through Combined Modeling of Neoantigen Burden and Immune-Related Resistance Mechanisms. Clin. Cancer Res. 2021, 27, 4265–4276. [Google Scholar] [CrossRef]
- Riaz, N.; Havel, J.J.; Makarov, V.; Desrichard, A.; Urba, W.J.; Sims, J.S.; Hodi, F.S.; Martín-Algarra, S.; Mandal, R.; Sharfman, W.H.; et al. Tumor and Microenvironment Evolution during Immunotherapy with Nivolumab. Cell 2017, 171, 934–949.e916. [Google Scholar] [CrossRef]
- Huo, J.L.; Wang, Y.T.; Fu, W.J.; Lu, N.; Liu, Z.S. The promising immune checkpoint LAG-3 in cancer immunotherapy: From basic research to clinical application. Front. Immunol. 2022, 13, 956090. [Google Scholar] [CrossRef] [PubMed]
- Sidaway, P. LAG3 inhibition improves outcomes. Nat. Rev. Clin. Oncol. 2022, 19, 149. [Google Scholar] [CrossRef] [PubMed]
- Shen, R.; Postow, M.A.; Adamow, M.; Arora, A.; Hannum, M.; Maher, C.; Wong, P.; Curran, M.A.; Hollmann, T.J.; Jia, L.; et al. LAG-3 expression on peripheral blood cells identifies patients with poorer outcomes after immune checkpoint blockade. Sci. Transl. Med. 2021, 13, eabf5107. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, V.; Khattak, A.; Haydon, A.; Eastgate, M.; Roy, A.; Prithviraj, P.; Mueller, C.; Brignone, C.; Triebel, F. Eftilagimod alpha, a soluble lymphocyte activation gene-3 (LAG-3) protein plus pembrolizumab in patients with metastatic melanoma. J. Immunother. Cancer 2020, 8, e001681. [Google Scholar] [CrossRef] [PubMed]
- Machiraju, D.; Schäfer, S.; Hassel, J.C. Potential Reasons for Unresponsiveness to Anti-PD1 Immunotherapy in Young Patients with Advanced Melanoma. Life 2021, 11, 1318. [Google Scholar] [CrossRef] [PubMed]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef]
- Bald, T.; Landsberg, J.; Lopez-Ramos, D.; Renn, M.; Glodde, N.; Jansen, P.; Gaffal, E.; Steitz, J.; Tolba, R.; Kalinke, U.; et al. Immune cell-poor melanomas benefit from PD-1 blockade after targeted type I IFN activation. Cancer Discov. 2014, 4, 674–687. [Google Scholar] [CrossRef]
- Monjazeb, A.M.; Kent, M.S.; Grossenbacher, S.K.; Mall, C.; Zamora, A.E.; Mirsoian, A.; Chen, M.; Kol, A.; Shiao, S.L.; Reddy, A.; et al. Blocking Indolamine-2,3-Dioxygenase Rebound Immune Suppression Boosts Antitumor Effects of Radio-Immunotherapy in Murine Models and Spontaneous Canine Malignancies. Clin. Cancer Res. 2016, 22, 4328–4340. [Google Scholar] [CrossRef] [PubMed]
- Spranger, S.; Spaapen, R.M.; Zha, Y.; Williams, J.; Meng, Y.; Ha, T.T.; Gajewski, T.F. Up-regulation of PD-L1, IDO, and T(regs) in the melanoma tumor microenvironment is driven by CD8(+) T cells. Sci. Transl. Med. 2013, 5, 200ra116. [Google Scholar] [CrossRef]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef]
- Parsa, A.T.; Waldron, J.S.; Panner, A.; Crane, C.A.; Parney, I.F.; Barry, J.J.; Cachola, K.E.; Murray, J.C.; Tihan, T.; Jensen, M.C.; et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat. Med. 2007, 13, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Mittendorf, E.A.; Philips, A.V.; Meric-Bernstam, F.; Qiao, N.; Wu, Y.; Harrington, S.; Su, X.; Wang, Y.; Gonzalez-Angulo, A.M.; Akcakanat, A.; et al. PD-L1 expression in triple-negative breast cancer. Cancer Immunol. Res. 2014, 2, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Marzec, M.; Zhang, Q.; Goradia, A.; Raghunath, P.N.; Liu, X.; Paessler, M.; Wang, H.Y.; Wysocka, M.; Cheng, M.; Ruggeri, B.A.; et al. Oncogenic kinase NPM/ALK induces through STAT3 expression of immunosuppressive protein CD274 (PD-L1, B7-H1). Proc. Natl. Acad. Sci. USA 2008, 105, 20852–20857. [Google Scholar] [CrossRef] [PubMed]
- Akbay, E.A.; Koyama, S.; Carretero, J.; Altabef, A.; Tchaicha, J.H.; Christensen, C.L.; Mikse, O.R.; Cherniack, A.D.; Beauchamp, E.M.; Pugh, T.J.; et al. Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Cancer Discov. 2013, 3, 1355–1363. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Tang, L.; Chen, S.; Yin, C.; Peng, S.; Li, X.; Liu, T.; Liu, W.; Han, C.; Stawski, L.; et al. Targeting the upstream transcriptional activator of PD-L1 as an alternative strategy in melanoma therapy. Oncogene 2018, 37, 4941–4954. [Google Scholar] [CrossRef]
- Huang, Y.H.; Zhu, C.; Kondo, Y.; Anderson, A.C.; Gandhi, A.; Russell, A.; Dougan, S.K.; Petersen, B.S.; Melum, E.; Pertel, T.; et al. CEACAM1 regulates TIM-3-mediated tolerance and exhaustion. Nature 2015, 517, 386–390. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.S.; Zaretsky, J.M.; Escuin-Ordinas, H.; Garcia-Diaz, A.; Hu-Lieskovan, S.; Kalbasi, A.; Grasso, C.S.; Hugo, W.; Sandoval, S.; Torrejon, D.Y.; et al. Primary Resistance to PD-1 Blockade Mediated by JAK1/2 Mutations. Cancer Discov. 2017, 7, 188–201. [Google Scholar] [CrossRef]
- Xia, T.; Konno, H.; Barber, G.N. Recurrent Loss of STING Signaling in Melanoma Correlates with Susceptibility to Viral Oncolysis. Cancer Res. 2016, 76, 6747–6759. [Google Scholar] [CrossRef] [PubMed]
- Falahat, R.; Perez-Villarroel, P.; Mailloux, A.W.; Zhu, G.; Pilon-Thomas, S.; Barber, G.N.; Mulé, J.J. STING Signaling in Melanoma Cells Shapes Antigenicity and Can Promote Antitumor T-cell Activity. Cancer Immunol. Res. 2019, 7, 1837–1848. [Google Scholar] [CrossRef]
- Demaria, O.; De Gassart, A.; Coso, S.; Gestermann, N.; Di Domizio, J.; Flatz, L.; Gaide, O.; Michielin, O.; Hwu, P.; Petrova, T.V.; et al. STING activation of tumor endothelial cells initiates spontaneous and therapeutic antitumor immunity. Proc. Natl. Acad. Sci. USA 2015, 112, 15408–15413. [Google Scholar] [CrossRef]
- Nakamura, T.; Sato, T.; Endo, R.; Sasaki, S.; Takahashi, N.; Sato, Y.; Hyodo, M.; Hayakawa, Y.; Harashima, H. STING agonist loaded lipid nanoparticles overcome anti-PD-1 resistance in melanoma lung metastasis via NK cell activation. J. Immunother. Cancer 2021, 9, e002852. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Dai, J.; Tang, L.; Yang, L.; Si, L.; Sheng, X.; Cui, C.; Chi, Z.; Kong, Y.; Guo, J. EZH2 Inhibitor Enhances the STING Agonist—Induced Antitumor Immunity in Melanoma. J. Investig. Dermatol. 2022, 142, 1158–1170.e1158. [Google Scholar] [CrossRef] [PubMed]
- Cartier, A.; Hla, T. Sphingosine 1-phosphate: Lipid signaling in pathology and therapy. Science 2019, 366, 323. [Google Scholar] [CrossRef] [PubMed]
- Imbert, C.; Montfort, A.; Fraisse, M.; Marcheteau, E.; Gilhodes, J.; Martin, E.; Bertrand, F.; Marcellin, M.; Burlet-Schiltz, O.; Peredo, A.G.; et al. Resistance of melanoma to immune checkpoint inhibitors is overcome by targeting the sphingosine kinase-1. Nat. Commun. 2020, 11, 437. [Google Scholar] [CrossRef]
- Theivanthiran, B.; Evans, K.S.; DeVito, N.C.; Plebanek, M.; Sturdivant, M.; Wachsmuth, L.P.; Salama, A.K.; Kang, Y.; Hsu, D.; Balko, J.M.; et al. A tumor-intrinsic PD-L1/NLRP3 inflammasome signaling pathway drives resistance to anti-PD-1 immunotherapy. J. Clin. Investig. 2020, 130, 2570–2586. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Tumeh, P.C. Cancer therapy: Tumours switch to resist. Nature 2012, 490, 347–348. [Google Scholar] [CrossRef] [PubMed]
- Thornton, J.; Chhabra, G.; Singh, C.K.; Guzmán-Pérez, G.; Shirley, C.A.; Ahmad, N. Mechanisms of Immunotherapy Resistance in Cutaneous Melanoma: Recognizing a Shapeshifter. Front. Oncol. 2022, 12, 880876. [Google Scholar] [CrossRef]
- Griffin, G.K.; Wu, J.; Iracheta-Vellve, A.; Patti, J.C.; Hsu, J.; Davis, T.; Dele-Oni, D.; Du, P.P.; Halawi, A.G.; Ishizuka, J.J.; et al. Epigenetic silencing by SETDB1 suppresses tumour intrinsic immunogenicity. Nature 2021, 595, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.M.; Cai, W.L.; Liu, X.; Thakral, D.; Luo, J.; Chan, L.H.; McGeary, M.K.; Song, E.; Blenman, K.R.M.; Micevic, G.; et al. KDM5B promotes immune evasion by recruiting SETDB1 to silence retroelements. Nature 2021, 598, 682–687. [Google Scholar] [CrossRef]
- Lazaro-Camp, V.J.; Salari, K.; Meng, X.; Yang, S. SETDB1 in cancer: Overexpression and its therapeutic implications. Am. J. Cancer Res. 2021, 11, 1803–1827. [Google Scholar]
- Strepkos, D.; Markouli, M.; Klonou, A.; Papavassiliou, A.G.; Piperi, C. Histone Methyltransferase SETDB1: A Common Denominator of Tumorigenesis with Therapeutic Potential. Cancer Res. 2021, 81, 525–534. [Google Scholar] [CrossRef] [PubMed]





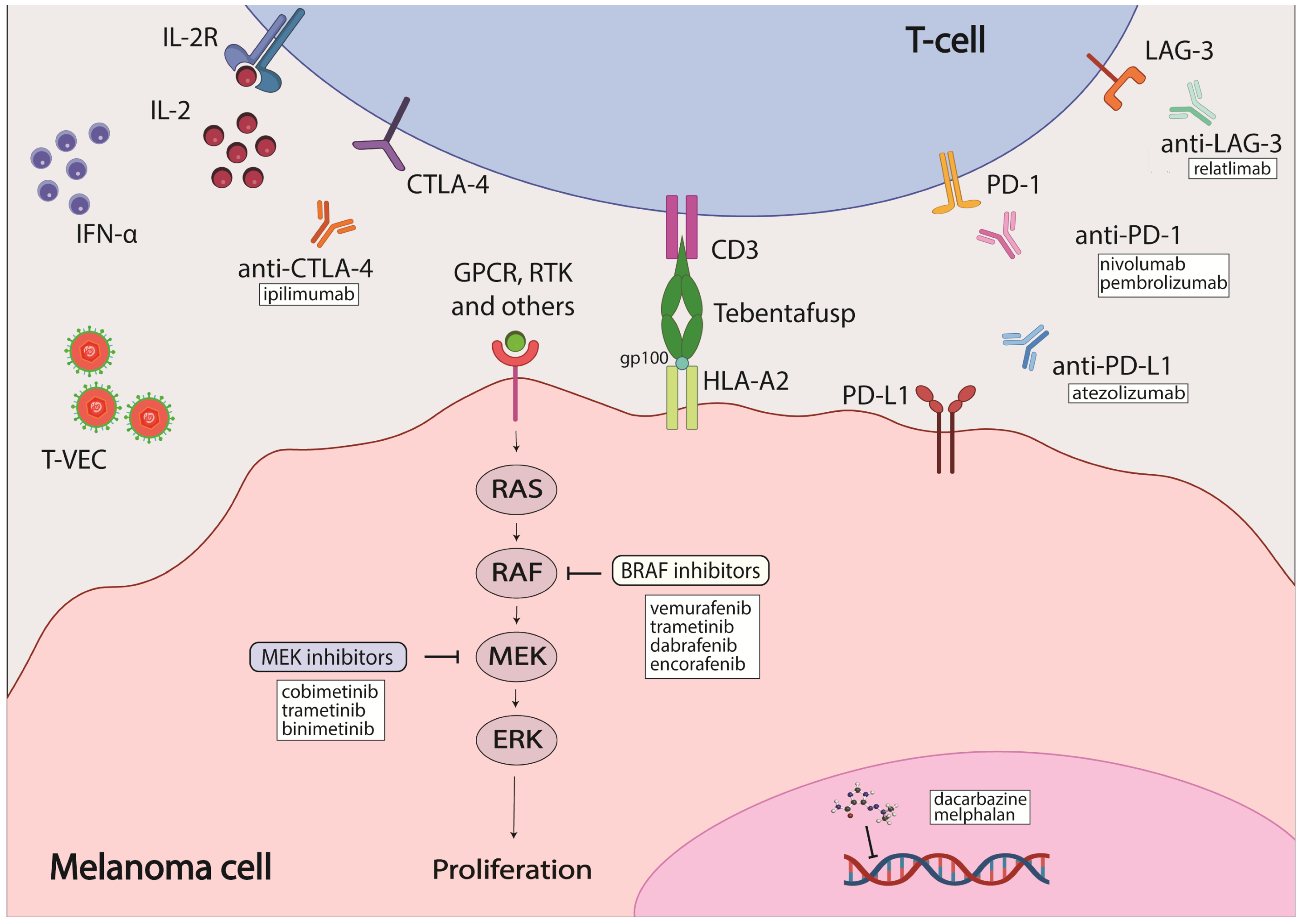{kind=link}
{kind=link}
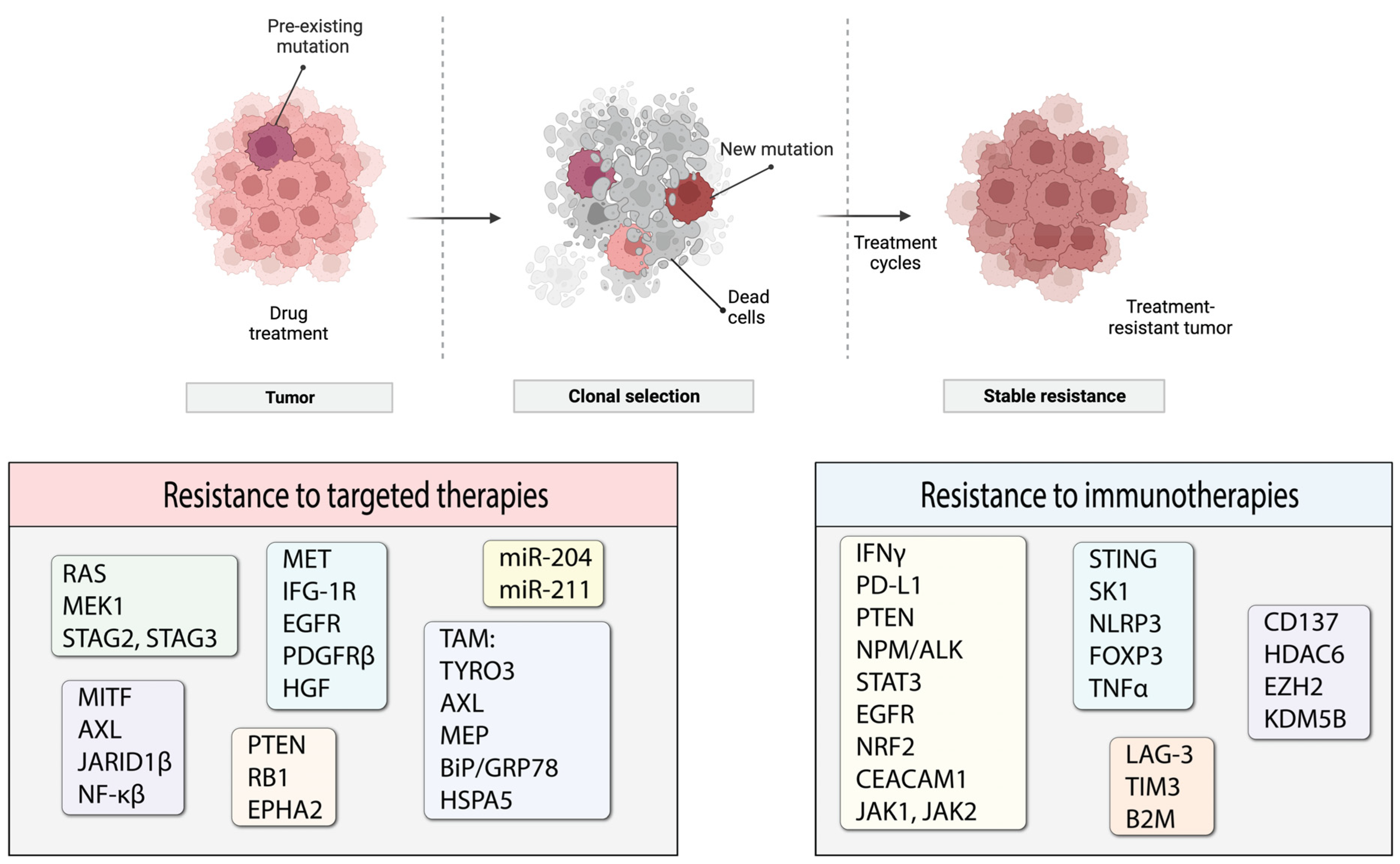{kind=link}
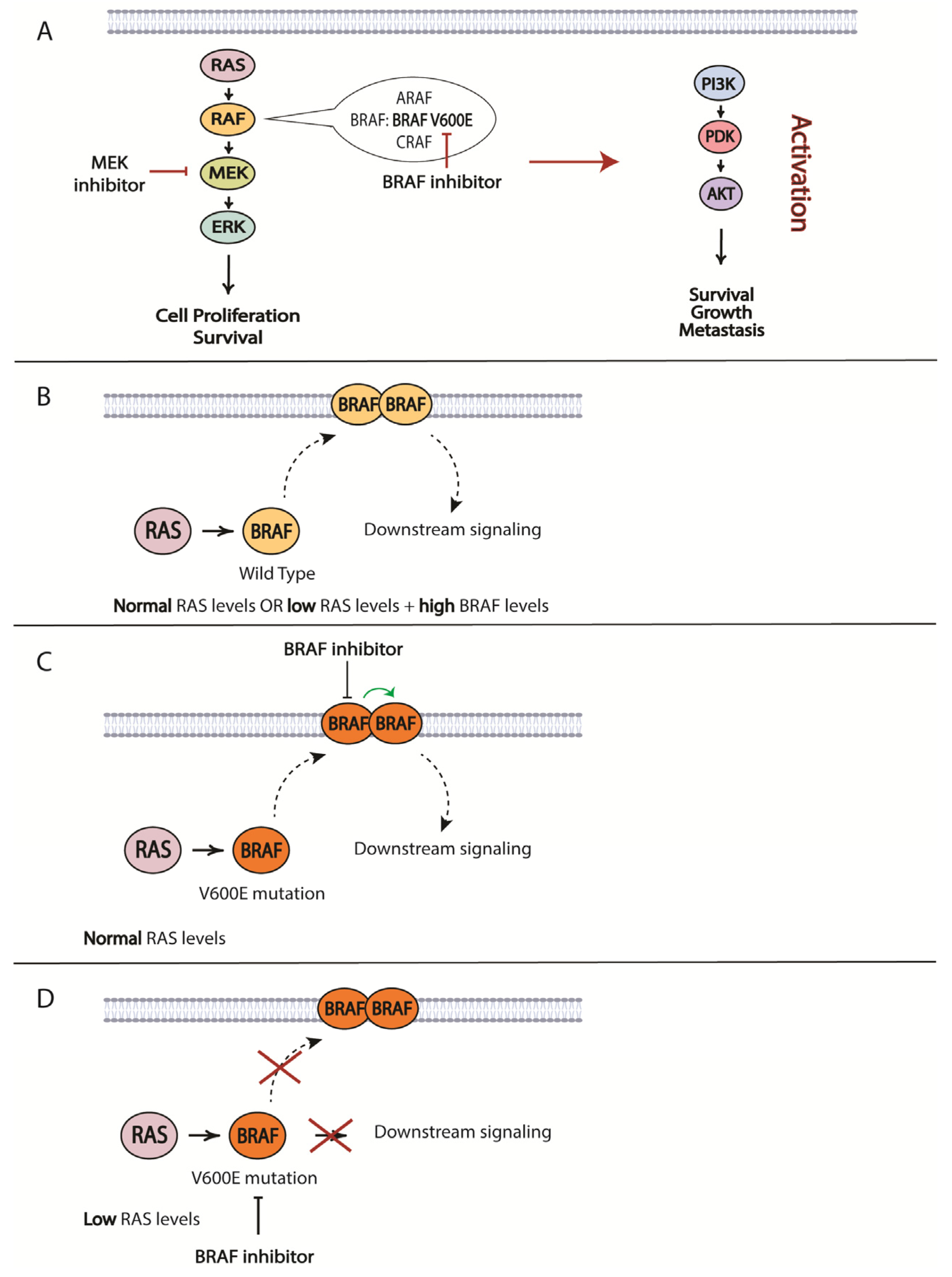{kind=link}
| Name | Therapy Type | Disease Conditions | Year of Approval | Details | Limitations | References |
|---|---|---|---|---|---|---|
| Dacarbazine | Chemotherapy |
| 1975 | DNA-alkylating agent |
| [16,19] |
| Recombinant Interferon Alfa-2b | Immunotherapy |
| 1995 | Recombinant form of the protein interferon alpha-2 |
| [20,21] |
| Aldesleukin | Immunotherapy |
| 1998 | Human recombinant interleukin-2 |
| [22,23] |
| Vemurafenib | Targeted therapy |
| 2011 | BRAF inhibitor |
| [24,25,26] |
| Ipilimumab | Immunotherapy |
| 2011 | Antibody against CTLA-4 |
| [27,28] |
| Nivolumab | Immunotherapy |
| 2014 | Antibody against PD-1 |
| [29,30] |
| Pembrolizumab | Immunotherapy |
| 2014 | Antibody against PD-1 |
| [31] |
| Cobimetinib + vemurafenib | Targeted therapy |
| 2015 | MEK inhibitor + BRAF inhibitor |
| [32] |
| Talimogene Laherparepvec (T-VEC) | Immunotherapy |
| 2015 | Oncolytic virus |
| [33] |
| Ipilimumab + nivolumab | Immunotherapy |
| 2015 | Antibody against CTLA-4 + antibody against PD-1 |
| [34] |
| Binimetinib + Encorafenib | Targeted therapy |
| 2018 | MEK inhibitor + BRAF inhibitor |
| [23] |
| Dabrafenib + trametinib | Targeted therapy |
| 2018 | BRAF inhibitor + MEK inhibitor |
| [35,36] |
| Atezolizumab + cobimetinib + vemurafenib | Immunotherapy + targeted therapy |
| 2020 | Antibody against PD-L1 + MEK inhibitor + BRAF inhibitor |
| [37] |
| Tebentafusp-tebn | Immunotherapy |
| 2022 | T-cell receptor-bispecific molecule that targets both glycoprotein 100 and CD3 |
| [38] |
| Nivolumab + relatlimab | Immunotherapy |
| 2022 | Antibody against PD-1 + antibody against LAG-3 |
| [39,40] |
| Melphalan | Chemotherapy |
| 2023 | DNA-alkylating agent |
| [41] |
| Lifileucel | Cellular therapy |
| 2024 | Tumor-derived autologous T-cell immunotherapy |
| [42] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fateeva, A.; Eddy, K.; Chen, S. Current State of Melanoma Therapy and Next Steps: Battling Therapeutic Resistance. Cancers 2024, 16, 1571. https://doi.org/10.3390/cancers16081571
Fateeva A, Eddy K, Chen S. Current State of Melanoma Therapy and Next Steps: Battling Therapeutic Resistance. Cancers. 2024; 16(8):1571. https://doi.org/10.3390/cancers16081571
Chicago/Turabian StyleFateeva, Anna, Kevinn Eddy, and Suzie Chen. 2024. "Current State of Melanoma Therapy and Next Steps: Battling Therapeutic Resistance" Cancers 16, no. 8: 1571. https://doi.org/10.3390/cancers16081571
APA StyleFateeva, A., Eddy, K., & Chen, S. (2024). Current State of Melanoma Therapy and Next Steps: Battling Therapeutic Resistance. Cancers, 16(8), 1571. https://doi.org/10.3390/cancers16081571