

Molecular Landscape and Therapeutic Strategies against Colorectal Cancer


Abstract
Simple Summary
Abstract
1. Introduction
2. Molecular Diagnostic Techniques
2.1. Genomic Sequencing
2.2. Transcriptomics
2.3. Proteomics
2.4. Multi-Omics
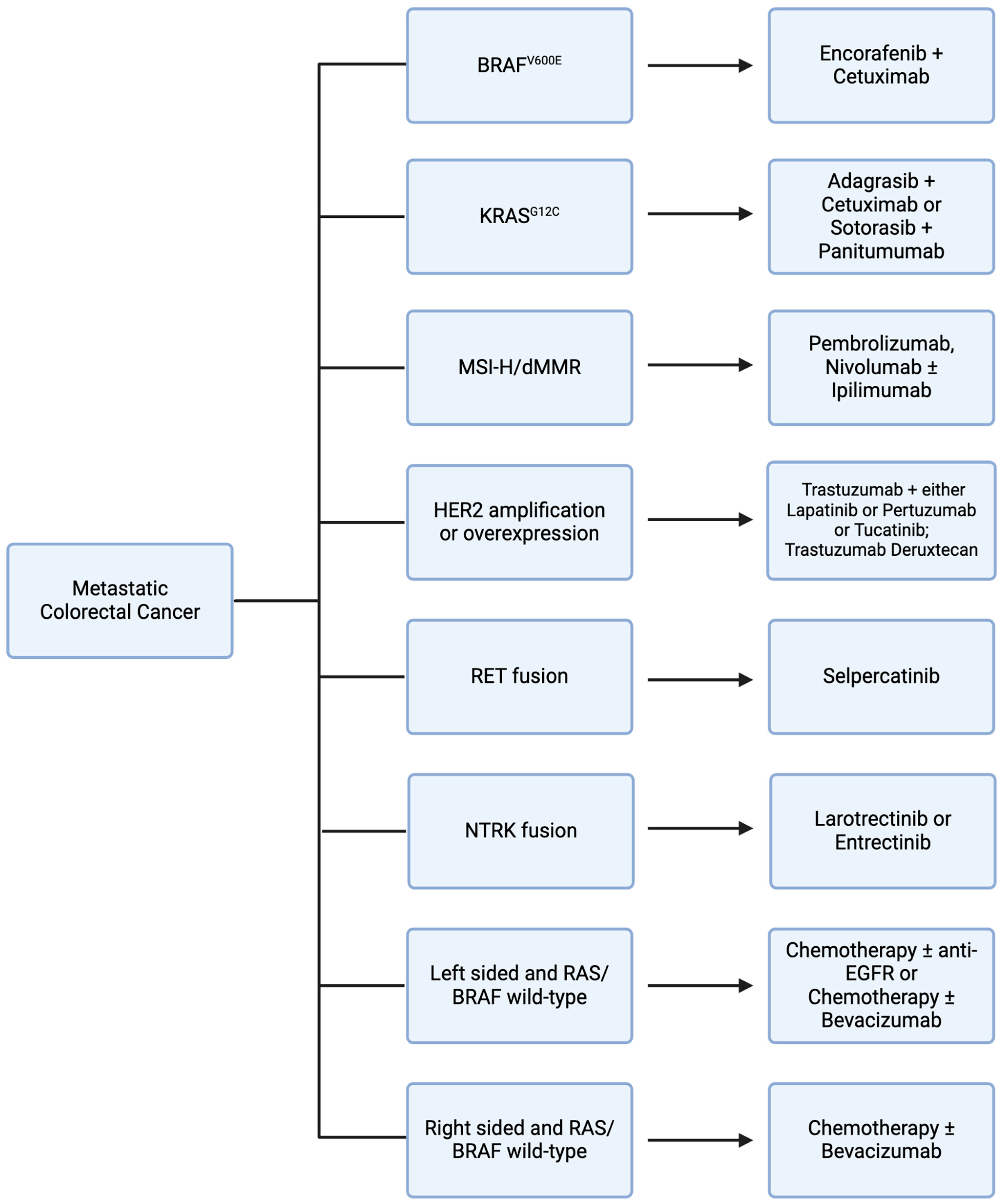
3. Potentially Targetable Genomic Alterations
3.1. VEGF
3.2. EGFR
3.3. BRAF
3.4. KRAS
3.5. MSI/MMR
3.6. HER2
3.7. RET
3.8. NTRK

{kind=link}
{kind=link}
| Therapeutic Class | Therapeutic Agent | Molecular Target | Reference |
|---|---|---|---|
| Targeted Therapy | Bevacizumab | VEGF | [24,25] |
| Regorafenib | Multiple kinases: VEGFR1-3, PDGFR, RAF, FGFR1-2, among others | [29] | |
| Fruquintinib | VEGFR1-3 kinases | [31] | |
| Cetuximab, Panitumumab | EGFR | [36,38] | |
| Encorafenib | BRAFV600E | [47] | |
| Adagrasib, Sotorasib | KRASG12C | [68,70] | |
| Trastuzumab | HER2 extracellular domain (ECD) IV; inhibits ligand-independent HER2 signaling | [110] | |
| Pertuzumab | HER2 ECD II; inhibits ligand-dependent HER2 signaling | [111] | |
| Trastuzumab Deruxtecan | HER2-directed antibody-drug (chemotherapy) conjugate (ADC) | [115] | |
| Lapatinib | EGFR and HER2 kinases | [110] | |
| Tucatinib | HER2 kinase | [113] | |
| Selpercatinib | RET | [127] | |
| Larotrectinib, Entrectinib | NTRK | [133,135] | |
| Immunotherapy (MSI-H/dMMR) | Pembrolizumab, Nivolumab | PD-1 | [87,92] |
| Ipilimumab | CTLA-4 | [89] | |
| Chemotherapy | CAPOX, FOLFOX, FOLFIRI, FOLFOXIRI | [24,26,36,38] | |
| Molecular Alteration Targeted | ClinicalTrials.gov Identifier and Trial Name (If Applicable) | Trial Phase | Experimental Agent(s) | Disease Stage(s) | Line of Treatment | Estimated Study Completion Date |
|---|---|---|---|---|---|---|
| BRAF | NCT05510895 (NeoBRAF) | II | Encorafenib plus Binimetinib plus Cetuximab | Resectable T3-4,N−/+,M0 | Neoadjuvant and Adjuvant | January 2025 |
| NCT05743036 | I/II | ZN-c3 plus Encorafenib plus Cetuximab | Stage IV | 2L+ | September 2026 | |
| NCT05308446 | II | Cetuximab plus Encorafenib plus Nivolumab | Stage IV | 2L+ | August 2024 | |
| NCT05127759 | II | HLX208 | Stage IV | 2L+ | February 2025 | |
| KRAS | NCT05593328 | II | Onvansertib plus FOLFIRI plus Bevacizumab | Stage IV | 2L+ | April 2026 |
| NCT05631574 | I | BMF-219 | Advanced | 2L+ | October 2026 | |
| NCT04117087 | I | KRAS peptide vaccine plus Nivolumab plus Ipilimumab | Stage IV | 3L+ | December 2024 | |
| NCT05379985 | I | RMC-6236 | Advanced | 2L+ | December 2025 | |
| NCT05737706 | I/II | MRTX1133 | Advanced | 2L+ | August 2026 | |
| MSI-H/dMMR | NCT04895722 | II | Pembrolizumab plus Quavonlimab or Favezelimab or Vibostolimab or MK-4830 | Stage IV | 1L+ | October 2025 |
| NCT05652894 | III | HX008 | Stage IV | 1L | October 2028 | |
| NCT04988191 | I/II | Toripalimab plus Bevacizumab plus Irinotecan | Resectable T3-4 rectal or T1-2 rectal within 12cm of anal verge or T4a-b colon | Neoadjuvant and Adjuvant | December 2023 | |
| NCT05371197 | II | Envafolimab | Resectable T3-4,N1-2, M0 | Neoadjuvant | December 2024 | |
| HER2 | NCT05578287 (DETECT) | II | Disitamab Vedotin, Tislelizumab, Capecitabine, and Celecoxib | Stage IV | 2L+ | December 2025 |
| NCT05350917 | II | Disitamab Vedotin, Tislelizumab, and Pyrotinib Maleate | Advanced | 2L+ | June 2026 | |
| NCT05785325 | II | RC48-ADC plus Bevacizumab | Stage IV | 2L+ | December 2024 | |
| NCT03929666 | II | Zanidatamab plus chemotherapy | Advanced | 1L | April 2024 | |
| NCT05673512 | II/III | IAH0968 plus CAPEOX | Advanced | 1L | March 2026 | |
| NCT05253651 (MOUNTAINEER-03) | III | Tucatinib plus trastuzumab plus mFOLFOX6 | Stage IV | 1L | April 2028 | |
| NCT05356897 (3T Study) | II | Tucatinib plus Trastuzumab plus TAS-102 | Stage IV | 2L+ | May 2029 |
3.9. Other Targeted Therapies
4. Young-Onset CRC
5. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer Statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Keum, N.; Giovannucci, E. Global Burden of Colorectal Cancer: Emerging Trends, Risk Factors and Prevention Strategies. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 713–732. [Google Scholar] [CrossRef]
- Sinicrope, F.A. Increasing Incidence of Early-Onset Colorectal Cancer. N. Engl. J. Med. 2022, 386, 1547–1558. [Google Scholar] [CrossRef] [PubMed]
- Holch, J.W.; Ricard, I.; Stintzing, S.; Modest, D.P.; Heinemann, V. The Relevance of Primary Tumour Location in Patients with Metastatic Colorectal Cancer: A Meta-Analysis of First-Line Clinical Trials. Eur. J. Cancer 2017, 70, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Kapiteijn, E.; Liefers, G.J.; Los, L.C.; Kranenbarg, E.K.; Hermans, J.; Tollenaar, R.A.; Moriya, Y.; van de Velde, C.J.; van Krieken, J.H. Mechanisms of Oncogenesis in Colon versus Rectal Cancer. J. Pathol. 2001, 195, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Chakravarty, D.; Johnson, A.; Sklar, J.; Lindeman, N.I.; Moore, K.; Ganesan, S.; Lovly, C.M.; Perlmutter, J.; Gray, S.W.; Hwang, J.; et al. Somatic Genomic Testing in Patients with Metastatic or Advanced Cancer: ASCO Provisional Clinical Opinion. J. Clin. Oncol. 2022, 40, 1231–1258. [Google Scholar] [CrossRef] [PubMed]
- NCCN Clinical Practice Guidelines in Oncology Colon Cancer Version 1.2023; NCCN: Philadelphia, PA, USA, 2023.
- Malla, M.; Loree, J.M.; Kasi, P.M.; Parikh, A.R. Using Circulating Tumor DNA in Colorectal Cancer: Current and Evolving Practices. J. Clin. Oncol. 2022, 40, 2846–2857. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, B.G.; Lo, A.; Yu, J.; Gonda, A.; Dehkordi-Vakil, F.; Dayyani, F.; Senthil, M. Circulating Tumor DNA Is Unreliable to Detect Somatic Gene Alterations in Gastrointestinal Peritoneal Carcinomatosis. Ann. Surg. Oncol. 2023, 30, 278–284. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Taniguchi, H.; Ikeda, M.; Bando, H.; Kato, K.; Morizane, C.; Esaki, T.; Komatsu, Y.; Kawamoto, Y.; Takahashi, N.; et al. Clinical Utility of Circulating Tumor DNA Sequencing in Advanced Gastrointestinal Cancer: SCRUM-Japan GI-SCREEN and GOZILA Studies. Nat. Med. 2020, 26, 1859–1864. [Google Scholar] [CrossRef] [PubMed]
- Parikh, A.R.; Van Seventer, E.E.; Siravegna, G.; Hartwig, A.V.; Jaimovich, A.; He, Y.; Kanter, K.; Fish, M.G.; Fosbenner, K.D.; Miao, B.; et al. Minimal Residual Disease Detection Using a Plasma-Only Circulating Tumor DNA Assay in Patients with Colorectal Cancer. Clin. Cancer Res. 2021, 27, 5586–5594. [Google Scholar] [CrossRef] [PubMed]
- Ten Hoorn, S.; de Back, T.R.; Sommeijer, D.W.; Vermeulen, L. Clinical Value of Consensus Molecular Subtypes in Colorectal Cancer: A Systematic Review and Meta-Analysis. J. Natl. Cancer Inst. 2022, 114, 503–516. [Google Scholar] [CrossRef] [PubMed]
- Guinney, J.; Dienstmann, R.; Wang, X.; de Reyniès, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The Consensus Molecular Subtypes of Colorectal Cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Wang, J.; Wang, X.; Zhu, J.; Liu, Q.; Shi, Z.; Chambers, M.C.; Zimmerman, L.J.; Shaddox, K.F.; Kim, S.; et al. Proteogenomic Characterization of Human Colon and Rectal Cancer. Nature 2014, 513, 382–387. [Google Scholar] [CrossRef]
- Vasaikar, S.; Huang, C.; Wang, X.; Petyuk, V.A.; Savage, S.R.; Wen, B.; Dou, Y.; Zhang, Y.; Shi, Z.; Arshad, O.A.; et al. Proteogenomic Analysis of Human Colon Cancer Reveals New Therapeutic Opportunities. Cell 2019, 177, 1035–1049.e19. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Zeng, J.; Roychoudhury, S.; Biswas, P.; Mohapatra, B.; Ray, S.; Dowlatshahi, K.; Wang, J.; Band, V.; Talmon, G.; et al. Targeting Histone Chaperone FACT Complex Overcomes 5-Fluorouracil Resistance in Colon Cancer. Mol. Cancer Ther. 2020, 19, 258–269. [Google Scholar] [CrossRef] [PubMed]
- Bhakat, K.K.; Ray, S. The FAcilitates Chromatin Transcription (FACT) Complex: Its Roles in DNA Repair and Implications for Cancer Therapy. DNA Repair 2022, 109, 103246. [Google Scholar] [CrossRef] [PubMed]
- Garcia, H.; Miecznikowski, J.C.; Safina, A.; Commane, M.; Ruusulehto, A.; Kilpinen, S.; Leach, R.W.; Attwood, K.; Li, Y.; Degan, S.; et al. Facilitates Chromatin Transcription Complex Is an “Accelerator” of Tumor Transformation and Potential Marker and Target of Aggressive Cancers. Cell Rep. 2013, 4, 159–173. [Google Scholar] [CrossRef] [PubMed]
- Gasparian, A.V.; Burkhart, C.A.; Purmal, A.A.; Brodsky, L.; Pal, M.; Saranadasa, M.; Bosykh, D.A.; Commane, M.; Guryanova, O.A.; Pal, S.; et al. Curaxins: Anticancer Compounds that Simultaneously Suppress NF-κB and Activate p53 by Targeting FACT. Sci. Transl. Med. 2011, 3, 95ra74. [Google Scholar] [CrossRef] [PubMed]
- Roelands, J.; Kuppen, P.J.K.; Ahmed, E.I.; Mall, R.; Masoodi, T.; Singh, P.; Monaco, G.; Raynaud, C.; de Miranda, N.F.C.C.; Ferraro, L.; et al. An Integrated Tumor, Immune and Microbiome Atlas of Colon Cancer. Nat. Med. 2023, 29, 1273–1286. [Google Scholar] [CrossRef] [PubMed]
- Pelka, K.; Hofree, M.; Chen, J.H.; Sarkizova, S.; Pirl, J.D.; Jorgji, V.; Bejnood, A.; Dionne, D.; Ge, W.H.; Xu, K.H.; et al. Spatially Organized Multicellular Immune Hubs in Human Colorectal Cancer. Cell 2021, 184, 4734–4752.e20. [Google Scholar] [CrossRef]
- Carmeliet, P. VEGF as a Key Mediator of Angiogenesis in Cancer. Oncology 2005, 69 (Suppl. S3), 4–10. [Google Scholar] [CrossRef]
- Hurwitz, H.; Fehrenbacher, L.; Novotny, W.; Cartwright, T.; Hainsworth, J.; Heim, W.; Berlin, J.; Baron, A.; Griffing, S.; Holmgren, E.; et al. Bevacizumab plus Irinotecan, Fluorouracil, and Leucovorin for Metastatic Colorectal Cancer. N. Engl. J. Med. 2004, 350, 2335–2342. [Google Scholar] [CrossRef]
- Kabbinavar, F.F.; Hambleton, J.; Mass, R.D.; Hurwitz, H.I.; Bergsland, E.; Sarkar, S. Combined Analysis of Efficacy: The Addition of Bevacizumab to Fluorouracil/leucovorin Improves Survival for Patients with Metastatic Colorectal Cancer. J. Clin. Oncol. 2005, 23, 3706–3712. [Google Scholar] [CrossRef] [PubMed]
- Goey, K.K.H.; Elias, S.G.; van Tinteren, H.; Laclé, M.M.; Willems, S.M.; Offerhaus, G.J.A.; de Leng, W.W.J.; Strengman, E.; Ten Tije, A.J.; Creemers, G.-J.M.; et al. Maintenance Treatment with Capecitabine and Bevacizumab versus Observation in Metastatic Colorectal Cancer: Updated Results and Molecular Subgroup Analyses of the Phase 3 CAIRO3 Study. Ann. Oncol. 2017, 28, 2128–2134. [Google Scholar] [CrossRef]
- Garcia, J.; Hurwitz, H.I.; Sandler, A.B.; Miles, D.; Coleman, R.L.; Deurloo, R.; Chinot, O.L. Bevacizumab (Avastin®) in Cancer Treatment: A Review of 15 Years of Clinical Experience and Future Outlook. Cancer Treat. Rev. 2020, 86, 102017. [Google Scholar] [CrossRef] [PubMed]
- Bennouna, J.; Sastre, J.; Arnold, D.; Österlund, P.; Greil, R.; Van Cutsem, E.; von Moos, R.; Viéitez, J.M.; Bouché, O.; Borg, C.; et al. Continuation of Bevacizumab after First Progression in Metastatic Colorectal Cancer (ML18147): A Randomised Phase 3 Trial. Lancet Oncol. 2013, 14, 29–37. [Google Scholar] [CrossRef]
- Grothey, A.; Van Cutsem, E.; Sobrero, A.; Siena, S.; Falcone, A.; Ychou, M.; Humblet, Y.; Bouché, O.; Mineur, L.; Barone, C.; et al. Regorafenib Monotherapy for Previously Treated Metastatic Colorectal Cancer (CORRECT): An International, Multicentre, Randomised, Placebo-Controlled, Phase 3 Trial. Lancet 2013, 381, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Goel, G. Evolution of Regorafenib from Bench to Bedside in Colorectal Cancer: Is It an Attractive Option or Merely a “Me Too” Drug? Cancer Manag. Res. 2018, 10, 425–437. [Google Scholar] [CrossRef]
- Dasari, A.; Lonardi, S.; Garcia-Carbonero, R.; Elez, E.; Yoshino, T.; Sobrero, A.; Yao, J.; García-Alfonso, P.; Kocsis, J.; Cubillo Gracian, A.; et al. Fruquintinib versus Placebo in Patients with Refractory Metastatic Colorectal Cancer (FRESCO-2): An International, Multicentre, Randomised, Double-Blind, Phase 3 Study. Lancet 2023, 402, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Sigismund, S.; Avanzato, D.; Lanzetti, L. Emerging Functions of the EGFR in Cancer. Mol. Oncol. 2018, 12, 3–20. [Google Scholar] [CrossRef]
- Cann, C.G.; LaPelusa, M.B.; Cimino, S.K.; Eng, C. Molecular and Genetic Targets within Metastatic Colorectal Cancer and Associated Novel Treatment Advancements. Front. Oncol. 2023, 13, 1176950. [Google Scholar] [CrossRef] [PubMed]
- Karapetis, C.S.; Khambata-Ford, S.; Jonker, D.J.; O’Callaghan, C.J.; Tu, D.; Tebbutt, N.C.; Simes, R.J.; Chalchal, H.; Shapiro, J.D.; Robitaille, S.; et al. K-Ras Mutations and Benefit from Cetuximab in Advanced Colorectal Cancer. N. Engl. J. Med. 2008, 359, 1757–1765. [Google Scholar] [CrossRef] [PubMed]
- Grothey, A.; Fakih, M.; Tabernero, J. Management of BRAF-Mutant Metastatic Colorectal Cancer: A Review of Treatment Options and Evidence-Based Guidelines. Ann. Oncol. 2021, 32, 959–967. [Google Scholar] [CrossRef] [PubMed]
- Douillard, J.-Y.; Siena, S.; Cassidy, J.; Tabernero, J.; Burkes, R.; Barugel, M.; Humblet, Y.; Bodoky, G.; Cunningham, D.; Jassem, J.; et al. Randomized, Phase III Trial of Panitumumab with Infusional Fluorouracil, Leucovorin, and Oxaliplatin (FOLFOX4) versus FOLFOX4 Alone as First-Line Treatment in Patients with Previously Untreated Metastatic Colorectal Cancer: The PRIME Study. J. Clin. Oncol. 2010, 28, 4697–4705. [Google Scholar] [CrossRef] [PubMed]
- Yaeger, R.; Chatila, W.K.; Lipsyc, M.D.; Hechtman, J.F.; Cercek, A.; Sanchez-Vega, F.; Jayakumaran, G.; Middha, S.; Zehir, A.; Donoghue, M.T.A.; et al. Clinical Sequencing Defines the Genomic Landscape of Metastatic Colorectal Cancer. Cancer Cell 2018, 33, 125–136.e3. [Google Scholar] [CrossRef] [PubMed]
- Tejpar, S.; Stintzing, S.; Ciardiello, F.; Tabernero, J.; Van Cutsem, E.; Beier, F.; Esser, R.; Lenz, H.-J.; Heinemann, V. Prognostic and Predictive Relevance of Primary Tumor Location in Patients with RAS Wild-Type Metastatic Colorectal Cancer: Retrospective Analyses of the CRYSTAL and FIRE-3 Trials. JAMA Oncol. 2017, 3, 194–201. [Google Scholar] [CrossRef] [PubMed]
- Siravegna, G.; Mussolin, B.; Buscarino, M.; Corti, G.; Cassingena, A.; Crisafulli, G.; Ponzetti, A.; Cremolini, C.; Amatu, A.; Lauricella, C.; et al. Clonal Evolution and Resistance to EGFR Blockade in the Blood of Colorectal Cancer Patients. Nat. Med. 2015, 21, 795–801. [Google Scholar] [CrossRef] [PubMed]
- Cremolini, C.; Rossini, D.; Dell’Aquila, E.; Lonardi, S.; Conca, E.; Del Re, M.; Busico, A.; Pietrantonio, F.; Danesi, R.; Aprile, G.; et al. Rechallenge for Patients with RAS and BRAF Wild-Type Metastatic Colorectal Cancer with Acquired Resistance to First-Line Cetuximab and Irinotecan: A Phase 2 Single-Arm Clinical Trial. JAMA Oncol. 2019, 5, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Sartore-Bianchi, A.; Pietrantonio, F.; Lonardi, S.; Mussolin, B.; Rua, F.; Crisafulli, G.; Bartolini, A.; Fenocchio, E.; Amatu, A.; Manca, P.; et al. Circulating Tumor DNA to Guide Rechallenge with Panitumumab in Metastatic Colorectal Cancer: The Phase 2 CHRONOS Trial. Nat. Med. 2022, 28, 1612–1618. [Google Scholar] [CrossRef] [PubMed]
- Parseghian, C.M.; Sun, R.; Woods, M.; Napolitano, S.; Lee, H.M.; Alshenaifi, J.; Willis, J.; Nunez, S.; Raghav, K.P.; Morris, V.K.; et al. Resistance Mechanisms to Anti-Epidermal Growth Factor Receptor Therapy in RAS/RAF Wild-Type Colorectal Cancer Vary by Regimen and Line of Therapy. J. Clin. Oncol. 2023, 41, 460–471. [Google Scholar] [CrossRef] [PubMed]
- Grassi, E.; Corbelli, J.; Papiani, G.; Barbera, M.A.; Gazzaneo, F.; Tamberi, S. Current Therapeutic Strategies in BRAF-Mutant Metastatic Colorectal Cancer. Front. Oncol. 2021, 11, 601722. [Google Scholar] [CrossRef] [PubMed]
- Tabernero, J.; Ros, J.; Élez, E. The Evolving Treatment Landscape in BRAF-V600E–Mutated Metastatic Colorectal Cancer. Am. Soc. Clin. Oncol. Educ. Book 2022, 42, 254–263. [Google Scholar] [CrossRef] [PubMed]
- Chu, J.E.; Johnson, B.; Kugathasan, L.; Morris, V.K.; Raghav, K.; Swanson, L.; Lim, H.J.; Renouf, D.J.; Gill, S.; Wolber, R.; et al. Population-Based Screening for BRAF V600E in Metastatic Colorectal Cancer Reveals Increased Prevalence and Poor Prognosis. Clin. Cancer Res. 2020, 26, 4599–4605. [Google Scholar] [CrossRef] [PubMed]
- Venderbosch, S.; Nagtegaal, I.D.; Maughan, T.S.; Smith, C.G.; Cheadle, J.P.; Fisher, D.; Kaplan, R.; Quirke, P.; Seymour, M.T.; Richman, S.D.; et al. Mismatch Repair Status and BRAF Mutation Status in Metastatic Colorectal Cancer Patients: A Pooled Analysis of the CAIRO, CAIRO2, COIN, and FOCUS Studies. Clin. Cancer Res. 2014, 20, 5322–5330. [Google Scholar] [CrossRef] [PubMed]
- Kopetz, S.; Grothey, A.; Yaeger, R.; Van Cutsem, E.; Desai, J.; Yoshino, T.; Wasan, H.; Ciardiello, F.; Loupakis, F.; Hong, Y.S.; et al. Encorafenib, Binimetinib, and Cetuximab in BRAF V600E-Mutated Colorectal Cancer. N. Engl. J. Med. 2019, 381, 1632–1643. [Google Scholar] [CrossRef] [PubMed]
- Tabernero, J.; Grothey, A.; Van Cutsem, E.; Yaeger, R.; Wasan, H.; Yoshino, T.; Desai, J.; Ciardiello, F.; Loupakis, F.; Hong, Y.S.; et al. Encorafenib Plus Cetuximab as a New Standard of Care for Previously Treated BRAF V600E-Mutant Metastatic Colorectal Cancer: Updated Survival Results and Subgroup Analyses from the BEACON Study. J. Clin. Oncol. 2021, 39, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Study of Encorafenib + Cetuximab Plus or Minus Binimetinib vs. Irinotecan/Cetuximab or Infusional 5-Fluorouracil (5-FU)/Folinic Acid (FA)/Irinotecan (FOLFIRI)/Cetuximab with a Safety Lead-in of Encorafenib + Binimetinib + Cetuximab in Patients with BRAF V600E-Mutant Metastatic Colorectal Cancer—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02928224 (accessed on 26 April 2023).
- Kopetz, S.; Grothey, A.; Yaeger, R.; Ciardiello, F.; Desai, J.; Kim, T.W.; Maughan, T.; Van Cutsem, E.; Wasan, H.S.; Yoshino, T.; et al. BREAKWATER: Randomized Phase 3 Study of Encorafenib (enco) + Cetuximab (cetux) ± Chemotherapy for First-Line (1L) Treatment (tx) of BRAF V600E-Mutant (BRAFV600E) Metastatic Colorectal Cancer (mCRC). J. Clin. Oncol. 2021, 39, TPS3619. [Google Scholar] [CrossRef]
- Kopetz, S. BREAKWATER Safety Lead-in (SLI): Encorafenib (E) + Cetuximab (C) + Chemotherapy for BRAFV600E Metastatic Colorectal Cancer (mCRC). J. Clin. Oncol. 2023, 41, 119. [Google Scholar] [CrossRef]
- Testing the Addition of Nivolumab to Standard Treatment for Patients with Metastatic or Unresectable Colorectal Cancer that Have a BRAF Mutation. Available online: https://clinicaltrials.gov/ct2/show/NCT05308446?term=BRAF&recrs=ab&cond=Colorectal+Cancer&draw=2&rank=31 (accessed on 26 April 2023).
- Encorafenib, Cetuximab, and Nivolumab in Treating Patients with Microsatellite Stable, BRAFV600E Mutated Unresectable or Metastatic Colorectal Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT04017650?term=NCT04017650&draw=2&rank=1 (accessed on 26 April 2023).
- Tolerability and Safety of Vemurafenib, Cetuximab Combined with Camrelizumab for BRAF V600E-Mutated/MSS Metastatic Colorectal Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT05019534?term=NCT05019534&draw=2&rank=1 (accessed on 26 April 2023).
- A Study of Encorafenib Plus Cetuximab Taken Together with Pembrolizumab Compared to Pembrolizumab Alone in People with Previously Untreated Metastatic Colorectal Cancer—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT05217446?term=NCT05217446&draw=2&rank=1 (accessed on 26 April 2023).
- Sullivan, R.J.; Infante, J.R.; Janku, F.; Wong, D.J.L.; Sosman, J.A.; Keedy, V.; Patel, M.R.; Shapiro, G.I.; Mier, J.W.; Tolcher, A.W.; et al. First-in-Class ERK1/2 Inhibitor Ulixertinib (BVD-523) in Patients with MAPK Mutant Advanced Solid Tumors: Results of a Phase I Dose-Escalation and Expansion Study. Cancer Discov. 2018, 8, 184–195. [Google Scholar] [CrossRef] [PubMed]
- Ciombor, K.K.; Strickler, J.H.; Bekaii-Saab, T.S.; Yaeger, R. BRAF-Mutated Advanced Colorectal Cancer: A Rapidly Changing Therapeutic Landscape. J. Clin. Oncol. 2022, 40, 2706–2715. [Google Scholar] [CrossRef] [PubMed]
- Neoadjuvant Encorafenib, Binimetinib and Cetuximab for Patients with BRAF V600E Mutated/pMMR Localized Colorectal Cancer—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT05510895?term=BRAF&recrs=ab&cond=Colorectal+Cancer&draw=2&rank=17 (accessed on 26 April 2023).
- Kopetz, S.; Murphy, D.A.; Pu, J.; Yaeger, R.; Ciardiello, F.; Desai, J.; Van Cutsem, E.; Wasan, H.S.; Yoshino, T.; Alkuzweny, B.; et al. Genomic Mechanisms of Acquired Resistance of Patients (pts) with BRAF V600E-Mutant (mt) Metastatic Colorectal Cancer (mCRC) Treated in the BEACON Study. Ann. Oncol. 2022, 33, S681–S682. [Google Scholar] [CrossRef]
- Liu, C.; Lu, H.; Wang, H.; Loo, A.; Zhang, X.; Yang, G.; Kowal, C.; Delach, S.; Wang, Y.; Goldoni, S.; et al. Combinations with Allosteric SHP2 Inhibitor TNO155 to Block Receptor Tyrosine Kinase Signaling. Clin. Cancer Res. 2021, 27, 342–354. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, Y.; Yamashita, N.; Hirose, H.; Fushimi, A.; Haratake, N.; Daimon, T.; Bhattacharya, A.; Ahmad, R.; Suzuki, Y.; Takahashi, H.; et al. MUC1-C Is Necessary for SHP2 Activation and BRAF Inhibitor Resistance in BRAF(V600E) Mutant Colorectal Cancer. Cancer Lett. 2023, 559, 216116. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Pei, L.; Xia, H.; Tang, Q.; Bi, F. Role of Oncogenic KRAS in the Prognosis, Diagnosis and Treatment of Colorectal Cancer. Mol. Cancer 2021, 20, 143. [Google Scholar] [CrossRef] [PubMed]
- Taieb, J.; Le Malicot, K.; Shi, Q.; Penault-Llorca, F.; Bouché, O.; Tabernero, J.; Mini, E.; Goldberg, R.M.; Folprecht, G.; Luc Van Laethem, J.; et al. Prognostic Value of BRAF and KRAS Mutations in MSI and MSS Stage III Colon Cancer. J. Natl. Cancer Inst. 2017, 109, djw272. [Google Scholar] [CrossRef]
- Ji, J.; Wang, C.; Fakih, M. Targeting KRASG12C-Mutated Advanced Colorectal Cancer: Research and Clinical Developments. OncoTargets Ther. 2022, 15, 747–756. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.S.; Fakih, M.G.; Strickler, J.H.; Desai, J.; Durm, G.A.; Shapiro, G.I.; Falchook, G.S.; Price, T.J.; Sacher, A.; Denlinger, C.S.; et al. KRASG12C Inhibition with Sotorasib in Advanced Solid Tumors. N. Engl. J. Med. 2020, 383, 1207–1217. [Google Scholar] [CrossRef] [PubMed]
- Fakih, M.G.; Kopetz, S.; Kuboki, Y.; Kim, T.W.; Munster, P.N.; Krauss, J.C.; Falchook, G.S.; Han, S.-W.; Heinemann, V.; Muro, K.; et al. Sotorasib for Previously Treated Colorectal Cancers with KRASG12C Mutation (CodeBreaK100): A Prespecified Analysis of a Single-Arm, Phase 2 Trial. Lancet Oncol. 2022, 23, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Amodio, V.; Yaeger, R.; Arcella, P.; Cancelliere, C.; Lamba, S.; Lorenzato, A.; Arena, S.; Montone, M.; Mussolin, B.; Bian, Y.; et al. EGFR Blockade Reverts Resistance to KRASG12C Inhibition in Colorectal Cancer. Cancer Discov. 2020, 10, 1129–1139. [Google Scholar] [CrossRef] [PubMed]
- Yaeger, R.; Weiss, J.; Pelster, M.S.; Spira, A.I.; Barve, M.; Ou, S.-H.I.; Leal, T.A.; Bekaii-Saab, T.S.; Paweletz, C.P.; Heavey, G.A.; et al. Adagrasib with or without Cetuximab in Colorectal Cancer with Mutated KRAS G12C. N. Engl. J. Med. 2023, 388, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Phase 3 Study of MRTX849 with Cetuximab vs Chemotherapy in Patients with Advanced Colorectal Cancer with KRAS G12C Mutation (KRYSTAL-10). Available online: https://clinicaltrials.gov/ct2/show/NCT04793958?term=NCT04793958&draw=2&rank=1 (accessed on 26 April 2023).
- Sotorasib and Panitumumab Versus Investigator’s Choice for Participants with Kirsten Rat Sarcoma (KRAS) p.G12C Mutation—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT05198934?term=NCT05198934&draw=2&rank=1 (accessed on 26 April 2023).
- Study of Onvansertib in Combination with FOLFIRI and Bevacizumab Versus FOLFIRI and Bevacizumab for Second Line Treatment of Metastatic Colorectal Cancer in Participants with a Kirsten Rat Sarcoma Virus Gene (KRAS) or Neuroblastoma-RAS (NRAS) Mutation. Available online: https://clinicaltrials.gov/ct2/show/NCT05593328?term=KRAS&recrs=ab&cond=Colorectal+Cancer&draw=2&rank=3 (accessed on 26 April 2023).
- Study of Covalent Menin Inhibitor BMF-219 in Adult Patients with KRAS Driven Non-Small Cell Lung Cancer, Pancreatic Cancer, and Colorectal Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT05631574?term=KRAS&recrs=ab&cond=Colorectal+Cancer&draw=2&rank=8 (accessed on 26 April 2023).
- Evaluation of RMC-6236 in Subjects with Advanced Solid Tumors Harboring Specific Mutations in KRAS. Available online: https://clinicaltrials.gov/ct2/show/NCT05379985?term=KRAS&recrs=ab&cond=Colorectal+Cancer&draw=2&rank=18 (accessed on 26 April 2023).
- Study of MRTX1133 in Patients with Advanced Solid Tumors Harboring a KRAS G12D Mutation. Available online: https://clinicaltrials.gov/ct2/show/NCT05737706?term=KRAS&recrs=ab&cond=Colorectal+Cancer&draw=2&rank=31 (accessed on 26 April 2023).
- Lietman, C.D.; Johnson, M.L.; McCormick, F.; Lindsay, C.R. More to the RAS Story: KRASG12C Inhibition, Resistance Mechanisms, and Moving Beyond KRASG12C. Am. Soc. Clin. Oncol. Educ. Book 2022, 42, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Sabari, J.K.; Park, H.; Tolcher, A.W.; Ou, S.-H.I.; Garon, E.B.; George, B.; Janne, P.A.; Moody, S.E.; Tan, E.Y.; Sen, S.K.; et al. KRYSTAL-2: A Phase I/II Trial of Adagrasib (MRTX849) in Combination with TNO155 in Patients with Advanced Solid Tumors with KRAS G12C Mutation. J. Clin. Oncol. 2021, 39, TPS146. [Google Scholar] [CrossRef]
- Molina-Arcas, M.; Moore, C.; Rana, S.; van Maldegem, F.; Mugarza, E.; Romero-Clavijo, P.; Herbert, E.; Horswell, S.; Li, L.-S.; Janes, M.R.; et al. Development of Combination Therapies to Maximize the Impact of KRAS-G12C Inhibitors in Lung Cancer. Sci. Transl. Med. 2019, 11, eaaw7999. [Google Scholar] [CrossRef]
- Lou, K.; Steri, V.; Ge, A.Y.; Hwang, Y.C.; Yogodzinski, C.H.; Shkedi, A.R.; Choi, A.L.M.; Mitchell, D.C.; Swaney, D.L.; Hann, B.; et al. KRASG12C Inhibition Produces a Driver-Limited State Revealing Collateral Dependencies. Sci. Signal. 2019, 12, eaaw9450. [Google Scholar] [CrossRef] [PubMed]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The Clinical KRAS(G12C) Inhibitor AMG 510 Drives Anti-Tumour Immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Fishel, R. Mismatch Repair. J. Biol. Chem. 2015, 290, 26395–26403. [Google Scholar] [CrossRef] [PubMed]
- De’Angelis, G.L.; Bottarelli, L.; Azzoni, C.; De’Angelis, N.; Leandro, G.; Di Mario, F.; Gaiani, F.; Negri, F. Microsatellite Instability in Colorectal Cancer. Acta Biomed. 2018, 89, 97–101. [Google Scholar]
- Jin, Z.; Sinicrope, F.A. Mismatch Repair-Deficient Colorectal Cancer: Building on Checkpoint Blockade. J. Clin. Oncol. 2022, 40, 2735–2750. [Google Scholar] [CrossRef] [PubMed]
- Jardim, D.L.; Goodman, A.; de Melo Gagliato, D.; Kurzrock, R. The Challenges of Tumor Mutational Burden as an Immunotherapy Biomarker. Cancer Cell 2021, 39, 154–173. [Google Scholar] [CrossRef]
- Koopman, M.; Kortman, G.A.M.; Mekenkamp, L.; Ligtenberg, M.J.L.; Hoogerbrugge, N.; Antonini, N.F.; Punt, C.J.A.; van Krieken, J.H.J.M. Deficient Mismatch Repair System in Patients with Sporadic Advanced Colorectal Cancer. Br. J. Cancer 2009, 100, 266–273. [Google Scholar] [CrossRef] [PubMed]
- Heinemann, V.; Kraemer, N.; Buchner, H.; Fischer von Weikersthal, L.; Decker, T.; Kiani, A.; Vehling-Kaiser, U.; Al-Batran, S.-E.; Heintges, T.; Lerchenmuller, C.A.; et al. Somatic DNA Mutations, Tumor Mutational Burden (TMB), and MSI Status: Association with Efficacy in Patients (pts) with Metastatic Colorectal Cancer (mCRC) of FIRE-3 (AIO KRK-0306). J. Clin. Oncol. 2018, 36, 3591. [Google Scholar] [CrossRef]
- Boland, C.R.; Goel, A. Microsatellite Instability in Colorectal Cancer. Gastroenterology 2010, 138, 2073–2087.e3. [Google Scholar] [CrossRef] [PubMed]
- Overman, M.J.; McDermott, R.; Leach, J.L.; Lonardi, S.; Lenz, H.-J.; Morse, M.A.; Desai, J.; Hill, A.; Axelson, M.; Moss, R.A.; et al. Nivolumab in Patients with Metastatic DNA Mismatch Repair-Deficient or Microsatellite Instability-High Colorectal Cancer (CheckMate 142): An Open-Label, Multicentre, Phase 2 Study. Lancet Oncol. 2017, 18, 1182–1191. [Google Scholar] [CrossRef] [PubMed]
- Center for Drug Evaluation. Research FDA Grants Nivolumab Accelerated Approval for MSI-H or dMMR Colorectal Cancer. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-nivolumab-accelerated-approval-msi-h-or-dmmr-colorectal-cancer (accessed on 20 April 2023).
- Overman, M.J.; Lonardi, S.; Wong, K.Y.M.; Lenz, H.-J.; Gelsomino, F.; Aglietta, M.; Morse, M.A.; Van Cutsem, E.; McDermott, R.; Hill, A.; et al. Durable Clinical Benefit with Nivolumab Plus Ipilimumab in DNA Mismatch Repair-Deficient/Microsatellite Instability-High Metastatic Colorectal Cancer. J. Clin. Oncol. 2018, 36, 773–779. [Google Scholar] [CrossRef] [PubMed]
- Center for Drug Evaluation. Research FDA Grants Accelerated Approval to Ipilimumab for MSI-H or dMMR Metastatic Colorectal Cancer. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-ipilimumab-msi-h-or-dmmr-metastatic-colorectal-cancer (accessed on 20 April 2023).
- Wang, D.Y.; Salem, J.-E.; Cohen, J.V.; Chandra, S.; Menzer, C.; Ye, F.; Zhao, S.; Das, S.; Beckermann, K.E.; Ha, L.; et al. Fatal Toxic Effects Associated with Immune Checkpoint Inhibitors: A Systematic Review and Meta-Analysis. JAMA Oncol. 2018, 4, 1721–1728. [Google Scholar] [CrossRef]
- Office of the Commissioner FDA Approves First Cancer Treatment for Any Solid Tumor with a Specific Genetic Feature. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-first-cancer-treatment-any-solid-tumor-specific-genetic-feature (accessed on 20 April 2023).
- Study of MK-3475 in Patients with Microsatellite Unstable (MSI) Tumors (Cohorts A, B and C). Available online: https://clinicaltrials.gov/ct2/show/NCT01876511 (accessed on 26 April 2023).
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [PubMed]
- André, T.; Shiu, K.-K.; Kim, T.W.; Jensen, B.V.; Jensen, L.H.; Punt, C.; Smith, D.; Garcia-Carbonero, R.; Benavides, M.; Gibbs, P.; et al. Pembrolizumab in Microsatellite-Instability-High Advanced Colorectal Cancer. N. Engl. J. Med. 2020, 383, 2207–2218. [Google Scholar] [CrossRef] [PubMed]
- Verschoor, Y.L.; van den Berg, J.; Beets, G.; Sikorska, K.; Aalbers, A.; van Lent, A.; Grootscholten, C.; Huibregtse, I.; Marsman, H.; Oosterling, S.; et al. Neoadjuvant Nivolumab, Ipilimumab, and Celecoxib in MMR-Proficient and MMR-Deficient Colon Cancers: Final Clinical Analysis of the NICHE Study. J. Clin. Oncol. 2022, 40, 3511. [Google Scholar] [CrossRef]
- Chalabi, M.; Verschoor, Y.L.; Van Den Berg, J.; Sikorska, K.; Beets, G.; Lent, A.V.; Grootscholten, M.C.; Aalbers, A.; Buller, N.; Marsman, H.; et al. Neoadjuvant Immune Checkpoint Inhibition in Locally Advanced MMR-Deficient Colon Cancer: The NICHE-2 Study. Ann. Oncol. 2022, 33, S1389. [Google Scholar] [CrossRef]
- Cercek, A.; Lumish, M.; Sinopoli, J.; Weiss, J.; Shia, J.; Lamendola-Essel, M.; El Dika, I.H.; Segal, N.; Shcherba, M.; Sugarman, R.; et al. PD-1 Blockade in Mismatch Repair-Deficient, Locally Advanced Rectal Cancer. N. Engl. J. Med. 2022, 386, 2363–2376. [Google Scholar] [CrossRef] [PubMed]
- Sinicrope, F.A.; Ou, F.-S.; Zemla, T.; Nixon, A.B.; Mody, K.; Levasseur, A.; Dueck, A.C.; Dhanarajan, A.R.; Lieu, C.H.; Cohen, D.J.; et al. Randomized Trial of Standard Chemotherapy Alone or Combined with Atezolizumab as Adjuvant Therapy for Patients with Stage III Colon Cancer and Deficient Mismatch Repair (ATOMIC, Alliance A021502). J. Clin. Oncol. 2019, 37, e15169. [Google Scholar] [CrossRef]
- Evaluation of Co-Formulated Pembrolizumab/Quavonlimab (MK-1308A) Versus Other Treatments in Participants with Microsatellite Instability-High (MSI-H) or Mismatch Repair Deficient (dMMR) Stage IV Colorectal Cancer (CRC) (MK-1308A-008). Available online: https://clinicaltrials.gov/ct2/show/NCT04895722?term=dmmr&recrs=ab&cond=Colorectal+Cancer&draw=2&rank=7 (accessed on 26 April 2023).
- A Study of HX008 Compared to Chemotherapy in the First-Line Treatment of Subjects with MSI-H/dMMR Metastatic Colorectal Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT05652894?term=dmmr&recrs=ab&cond=Colorectal+Cancer&draw=2&rank=8 (accessed on 26 April 2023).
- Toripalimab Plus Bevacizumab and Chemotherapy as Neoadjuvant Therapy in Advanced MSI-H or dMMR Colorectal Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT04988191?term=dmmr&recrs=ab&cond=Colorectal+Cancer&draw=2&rank=12 (accessed on 26 April 2023).
- Envafolimab as Neoadjuvant Immunotherapy in Resectable Locally Advanced dMMR/MSI-H Colorectal Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT05371197?term=dmmr&recrs=ab&cond=Colorectal+Cancer&draw=2&rank=14 (accessed on 26 April 2023).
- Sahin, I.H.; Ciombor, K.K.; Diaz, L.A.; Yu, J.; Kim, R. Immunotherapy for Microsatellite Stable Colorectal Cancers: Challenges and Novel Therapeutic Avenues. Am. Soc. Clin. Oncol. Educ. Book 2022, 42, 242–253. [Google Scholar] [CrossRef] [PubMed]
- Luke, J.J.; Bao, R.; Sweis, R.F.; Spranger, S.; Gajewski, T.F. WNT/β-Catenin Pathway Activation Correlates with Immune Exclusion across Human Cancers. Clin. Cancer Res. 2019, 25, 3074–3083. [Google Scholar] [CrossRef]
- Seo, A.N.; Kwak, Y.; Kim, D.-W.; Kang, S.-B.; Choe, G.; Kim, W.H.; Lee, H.S. HER2 Status in Colorectal Cancer: Its Clinical Significance and the Relationship between HER2 Gene Amplification and Expression. PLoS ONE 2014, 9, e98528. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, A.A.; Lau, D.K.; Chau, I. HER2 Targeted Therapy in Colorectal Cancer: New Horizons. Cancer Treat. Rev. 2022, 105, 102363. [Google Scholar]
- Ahcene Djaballah, S.; Daniel, F.; Milani, A.; Ricagno, G.; Lonardi, S. HER2 in Colorectal Cancer: The Long and Winding Road From Negative Predictive Factor to Positive Actionable Target. Am. Soc. Clin. Oncol. Educ. Book 2022, 42, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, A.; Nakamura, Y. Molecular Basis of HER2-Targeted Therapy for HER2-Positive Colorectal Cancer. Cancers 2022, 15, 183. [Google Scholar] [CrossRef] [PubMed]
- Siena, S.; Sartore-Bianchi, A.; Trusolino, L.; Martino, C.; Bencardino, K.; Lonardi, S.; Zagonel, V.; Leone, F.; Martinelli, E.; Ciardiello, F.; et al. Final Results of the HERACLES Trial in HER2 Amplified Colorectal Cancer. Ann. Oncol. 2016, 27, iv39. [Google Scholar] [CrossRef]
- Meric-Bernstam, F.; Hurwitz, H.; Raghav, K.P.S.; McWilliams, R.R.; Fakih, M.; VanderWalde, A.; Swanton, C.; Kurzrock, R.; Burris, H.; Sweeney, C.; et al. Pertuzumab plus Trastuzumab for HER2-Amplified Metastatic Colorectal Cancer (MyPathway): An Updated Report from a Multicentre, Open-Label, Phase 2a, Multiple Basket Study. Lancet Oncol. 2019, 20, 518–530. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Garrett-Mayer, E.; Halabi, S.; Mangat, P.K.; D’Andre, S.D.; Meiri, E.; Shrestha, S.; Warren, S.L.; Ranasinghe, S.; Schilsky, R.L. Pertuzumab plus Trastuzumab (P+T) in Patients (Pts) with Colorectal Cancer (CRC) with ERBB2 Amplification or Overexpression: Results from the TAPUR Study. J. Clin. Oncol. 2020, 38, 132. [Google Scholar] [CrossRef]
- Strickler, J.H.; Cercek, A.; Siena, S.; André, T.; Ng, K.; Van Cutsem, E.; Wu, C.; Paulson, A.S.; Hubbard, J.M.; Coveler, A.L.; et al. Tucatinib plus Trastuzumab for Chemotherapy-Refractory, HER2-Positive, RAS Wild-Type Unresectable or Metastatic Colorectal Cancer (MOUNTAINEER): A Multicentre, Open-Label, Phase 2 Study. Lancet Oncol. 2023, 24, 496–508. [Google Scholar] [CrossRef] [PubMed]
- Vernieri, C.; Milano, M.; Brambilla, M.; Mennitto, A.; Maggi, C.; Cona, M.S.; Prisciandaro, M.; Fabbroni, C.; Celio, L.; Mariani, G.; et al. Resistance Mechanisms to Anti-HER2 Therapies in HER2-Positive Breast Cancer: Current Knowledge, New Research Directions and Therapeutic Perspectives. Crit. Rev. Oncol. Hematol. 2019, 139, 53–66. [Google Scholar] [CrossRef] [PubMed]
- Siena, S.; Di Bartolomeo, M.; Raghav, K.; Masuishi, T.; Loupakis, F.; Kawakami, H.; Yamaguchi, K.; Nishina, T.; Fakih, M.; Elez, E.; et al. Trastuzumab Deruxtecan (DS-8201) in Patients with HER2-Expressing Metastatic Colorectal Cancer (DESTINY-CRC01): A Multicentre, Open-Label, Phase 2 Trial. Lancet Oncol. 2021, 22, 779–789. [Google Scholar] [CrossRef] [PubMed]
- RC48 Plus Tislelizumab, Low-Dose Capecitabine and Celecoxib for HER2-Positive Metastatic Colorectal Cancer—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT05578287?term=HER2&recrs=ab&cond=Colorectal+Cancer&draw=2&rank=1 (accessed on 25 April 2023).
- Study of Tislelizumab Combined with DisitamabVedotin and Pyrotinib Maleate in HER2-Positive or Mutated Advanced Colorectal Cancer Who Failed Standard Therapy. Available online: https://clinicaltrials.gov/ct2/show/NCT05350917?term=HER2&recrs=ab&cond=Colorectal+Cancer&draw=2&rank=2 (accessed on 25 April 2023).
- RC48-ADC Combined with Bevacizumab in HER2-Positive Advanced Colorectal Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT05785325?term=HER2&recrs=ab&cond=Colorectal+Cancer&draw=2&rank=3 (accessed on 25 April 2023).
- A Safety and Efficacy Study of ZW25 (Zanidatamab) Plus Combination Chemotherapy in HER2-Expressing Gastrointestinal Cancers, Including Gastroesophageal Adenocarcinoma, Biliary Tract Cancer, and Colorectal Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT03929666?term=HER2&recrs=ab&cond=Colorectal+Cancer&draw=2&rank=4 (accessed on 25 April 2023).
- To Evaluate IAH0968 in Combination with CAPEOX in HER2-Positive Metastatic Colorectal Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT05673512?term=HER2&recrs=ab&cond=Colorectal+Cancer&draw=2&rank=7 (accessed on 26 April 2023).
- A Study of Tucatinib with Trastuzumab and mFOLFOX6 Versus Standard of Care Treatment in First-Line HER2+ Metastatic Colorectal Cancer—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT05253651?term=HER2&recrs=ab&cond=Colorectal+Cancer&draw=2&rank=8 (accessed on 26 April 2023).
- Tucatinib Combined with Trastuzumab and TAS-102 for the Treatment of HER2 Positive Metastatic Colorectal Cancer in Molecularly Selected Patients, 3T Study. Available online: https://clinicaltrials.gov/ct2/show/NCT05356897?term=HER2&recrs=ab&cond=Colorectal+Cancer&draw=2&rank=9 (accessed on 26 April 2023).
- Thein, K.Z.; Velcheti, V.; Mooers, B.H.M.; Wu, J.; Subbiah, V. Precision Therapy for RET-Altered Cancers with RET Inhibitors. Trends Cancer Res. 2021, 7, 1074–1088. [Google Scholar] [CrossRef] [PubMed]
- Nagasaka, M.; Brazel, D.; Baca, Y.; Xiu, J.; Al-Hallak, M.N.; Kim, C.; Nieva, J.; Swensen, J.J.; Spetzler, D.; Korn, W.M.; et al. Pan-Tumor Survey of RET Fusions as Detected by next-Generation RNA Sequencing Identified RET Fusion Positive Colorectal Carcinoma as a Unique Molecular Subset. Transl. Oncol. 2023, 36, 101744. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Wang, S.; Xie, W.; Chang, J.; Gan, Y. The RET Fusion Gene and Its Correlation with Demographic and Clinicopathological Features of Non-Small Cell Lung Cancer: A Meta-Analysis. Cancer Biol. Ther. 2015, 16, 1019–1028. [Google Scholar] [CrossRef] [PubMed]
- Pietrantonio, F.; Di Nicolantonio, F.; Schrock, A.B.; Lee, J.; Morano, F.; Fucà, G.; Nikolinakos, P.; Drilon, A.; Hechtman, J.F.; Christiansen, J.; et al. RET Fusions in a Small Subset of Advanced Colorectal Cancers at Risk of Being Neglected. Ann. Oncol. 2018, 29, 1394–1401. [Google Scholar] [CrossRef]
- FDA Approves Selpercatinib for Locally Advanced or Metastatic RET Fusion-Positive Solid Tumors. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-selpercatinib-locally-advanced-or-metastatic-ret-fusion-positive-solid-tumors (accessed on 26 April 2023).
- Subbiah, V.; Wolf, J.; Konda, B.; Kang, H.; Spira, A.; Weiss, J.; Takeda, M.; Ohe, Y.; Khan, S.; Ohashi, K.; et al. Tumour-Agnostic Efficacy and Safety of Selpercatinib in Patients with RET Fusion-Positive Solid Tumours Other than Lung or Thyroid Tumours (LIBRETTO-001): A Phase 1/2, Open-Label, Basket Trial. Lancet Oncol. 2022, 23, 1261–1273. [Google Scholar] [CrossRef] [PubMed]
- Lange, A.M.; Lo, H.-W. Inhibiting TRK Proteins in Clinical Cancer Therapy. Cancers 2018, 10, 105. [Google Scholar] [CrossRef]
- Wang, H.; Li, Z.-W.; Ou, Q.; Wu, X.; Nagasaka, M.; Shao, Y.; Ou, S.-H.I.; Yang, Y. NTRK Fusion Positive Colorectal Cancer Is a Unique Subset of CRC with High TMB and Microsatellite Instability. Cancer Med. 2022, 11, 2541–2549. [Google Scholar] [CrossRef] [PubMed]
- Ratti, M.; Grizzi, G.; Passalacqua, R.; Lampis, A.; Cereatti, F.; Grassia, R.; Hahne, J.C. NTRK Fusions in Colorectal Cancer: Clinical Meaning and Future Perspective. Expert Opin. Ther. Targets 2021, 25, 677–683. [Google Scholar] [CrossRef] [PubMed]
- Hua, H.; He, W.; Chen, N.; He, Y.; Wu, G.; Ye, F.; Zhou, X.; Li, Y.; Ding, Y.; Zhong, W.; et al. Genomic and Transcriptomic Analysis of MSI-H Colorectal Cancer Patients with Targetable Alterations Identifies Clinical Implications for Immunotherapy. Front. Immunol. 2022, 13, 974793. [Google Scholar] [CrossRef] [PubMed]
- Drilon, A.; Laetsch, T.W.; Kummar, S.; DuBois, S.G.; Lassen, U.N.; Demetri, G.D.; Nathenson, M.; Doebele, R.C.; Farago, A.F.; Pappo, A.S.; et al. Efficacy of Larotrectinib in TRK Fusion-Positive Cancers in Adults and Children. N. Engl. J. Med. 2018, 378, 731–739. [Google Scholar] [CrossRef] [PubMed]
- Center for Drug Evaluation. Research FDA Approves Larotrectinib for Solid Tumors with NTRK Gene Fusions. Available online: https://www.fda.gov/drugs/fda-approves-larotrectinib-solid-tumors-ntrk-gene-fusions-0 (accessed on 15 April 2023).
- Doebele, R.C.; Drilon, A.; Paz-Ares, L.; Siena, S.; Shaw, A.T.; Farago, A.F.; Blakely, C.M.; Seto, T.; Cho, B.C.; Tosi, D.; et al. Entrectinib in Patients with Advanced or Metastatic NTRK Fusion-Positive Solid Tumours: Integrated Analysis of Three Phase 1-2 Trials. Lancet Oncol. 2020, 21, 271–282. [Google Scholar] [CrossRef] [PubMed]
- FDA Approves Entrectinib for NTRK Solid Tumors and ROS-1 NSCLC. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-entrectinib-ntrk-solid-tumors-and-ros-1-nsclc (accessed on 15 April 2023).
- Zheng, G.; Tseng, L.-H.; Haley, L.; Ibrahim, J.; Bynum, J.; Xian, R.; Gocke, C.D.; Eshleman, J.R.; Lin, M.-T. Clinical Validation of Coexisting Driver Mutations in Colorectal Cancers. Hum. Pathol. 2019, 86, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Cecchini, M.; Sokol, E.; Vasan, N.; Pavlick, D.C.; Huang, R.S.P.; Pelletier, M.; Levy, M.A.; Pusztai, L.; Lacy, J.; Eder, J.P.; et al. Molecular Characteristics of Advanced Colorectal Cancer and Multi-Hit PIK3CA Mutations. J. Clin. Oncol. 2022, 40, 3535. [Google Scholar] [CrossRef]
- André, F.; Ciruelos, E.; Rubovszky, G.; Campone, M.; Loibl, S.; Rugo, H.S.; Iwata, H.; Conte, P.; Mayer, I.A.; Kaufman, B.; et al. Alpelisib for PIK3CA-Mutated, Hormone Receptor-Positive Advanced Breast Cancer. N. Engl. J. Med. 2019, 380, 1929–1940. [Google Scholar] [CrossRef] [PubMed]
- Tabernero, J.; Van Geel, R.; Guren, T.K.; Yaeger, R.D.; Spreafico, A.; Faris, J.E.; Yoshino, T.; Yamada, Y.; Kim, T.W.; Bendell, J.C.; et al. Phase 2 Results: Encorafenib (ENCO) and Cetuximab (CETUX) with or without Alpelisib (ALP) in Patients with Advanced BRAF-Mutant Colorectal Cancer (BRAFm CRC). J. Clin. Oncol. 2016, 34, 3544. [Google Scholar] [CrossRef]
- Juric, D.; Rodon, J.; Tabernero, J.; Janku, F.; Burris, H.A.; Schellens, J.H.M.; Middleton, M.R.; Berlin, J.; Schuler, M.; Gil-Martin, M.; et al. Phosphatidylinositol 3-Kinase α-Selective Inhibition with Alpelisib (BYL719) in PIK3CA-Altered Solid Tumors: Results From the First-in-Human Study. J. Clin. Oncol. 2018, 36, 1291–1299. [Google Scholar] [CrossRef] [PubMed]
- Ciombor, K.K.; Zemla, T.J.; Hubbard, J.M.; Jia, J.; Gbolahan, O.B.; Sousa, A.; Wilson, L.; Waechter, B.; Ou, F.-S.; Nixon, A.B.; et al. A Phase II Single-Arm Study of the FGFR Inhibitor Pemigatinib in Patients with Metastatic Colorectal Cancer (mCRC) Harboring FGF/FGFR Alterations. J. Clin. Oncol. 2023, 41, 139. [Google Scholar] [CrossRef]
- Hussung, S.; Akhoundova, D.; Sivakumar, S.; Kahraman, A.; Zoche, M.; Rechsteiner, M.; Angst, F.; Britschgi, C.; Töpfer, A.; Moch, H.; et al. Frequency, Molecular Characteristics, and Therapeutic Targeting of ROS1 Oncogenic Fusions in Colorectal Cancer. J. Clin. Oncol. 2022, 40, 160. [Google Scholar] [CrossRef]
- Hsiao, S.-Y.; He, H.-L.; Weng, T.-S.; Lin, C.-Y.; Chao, C.-M.; Huang, W.-T.; Tsao, C.-J. Colorectal Cancer with EML4-ALK Fusion Gene Response to Alectinib: A Case Report and Review of the Literature. Case Rep. Oncol. 2021, 14, 232–238. [Google Scholar] [CrossRef] [PubMed]
- Eng, C.; Jácome, A.A.; Agarwal, R.; Hayat, M.H.; Byndloss, M.X.; Holowatyj, A.N.; Bailey, C.; Lieu, C.H. A Comprehensive Framework for Early-Onset Colorectal Cancer Research. Lancet Oncol. 2022, 23, e116–e128. [Google Scholar] [CrossRef] [PubMed]
- Akimoto, N.; Ugai, T.; Zhong, R.; Hamada, T.; Fujiyoshi, K.; Giannakis, M.; Wu, K.; Cao, Y.; Ng, K.; Ogino, S. Rising Incidence of Early-Onset Colorectal Cancer—A Call to Action. Nat. Rev. Clin. Oncol. 2021, 18, 230–243. [Google Scholar] [CrossRef] [PubMed]
- Pearlman, R.; Frankel, W.L.; Swanson, B.; Zhao, W.; Yilmaz, A.; Miller, K.; Bacher, J.; Bigley, C.; Nelsen, L.; Goodfellow, P.J.; et al. Prevalence and Spectrum of Germline Cancer Susceptibility Gene Mutations Among Patients with Early-Onset Colorectal Cancer. JAMA Oncol. 2017, 3, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Sinicrope, F.A. Lynch Syndrome-Associated Colorectal Cancer. N. Engl. J. Med. 2018, 379, 764–773. [Google Scholar] [CrossRef] [PubMed]
- Mauri, G.; Sartore-Bianchi, A.; Russo, A.-G.; Marsoni, S.; Bardelli, A.; Siena, S. Early-Onset Colorectal Cancer in Young Individuals. Mol. Oncol. 2019, 13, 109–131. [Google Scholar] [CrossRef] [PubMed]
- Lundqvist, E.; Kuchinskaya, E.; Landerholm, K.; Assarsson, J.; Benckert, A.; Myrelid, P.; Haapaniemi, S. Hereditary Evaluation and Genetic Counselling in Young Individuals with Colorectal Cancer in a Population-Based Cohort. Surg. Oncol. 2022, 41, 101741. [Google Scholar] [CrossRef] [PubMed]
- Khalessi Hosseini, S.A.; Morrissey, C.; Nakamura, T.; Becker, D.J. Exploring the Cancer Genome Atlas (TCGA) for the Molecular Profile of Young Onset Colorectal Cancers. J. Clin. Oncol. 2020, 38, 3548. [Google Scholar] [CrossRef]
- Holowatyj, A.N.; Gigic, B.; Herpel, E.; Scalbert, A.; Schneider, M.; Ulrich, C.M.; MetaboCCC Consortium; ColoCare Study. Distinct Molecular Phenotype of Sporadic Colorectal Cancers Among Young Patients Based on Multiomics Analysis. Gastroenterology 2020, 158, 1155–1158.e2. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patel, A.; Gulhati, P. Molecular Landscape and Therapeutic Strategies against Colorectal Cancer. Cancers 2024, 16, 1551. https://doi.org/10.3390/cancers16081551
Patel A, Gulhati P. Molecular Landscape and Therapeutic Strategies against Colorectal Cancer. Cancers. 2024; 16(8):1551. https://doi.org/10.3390/cancers16081551
Chicago/Turabian StylePatel, Aakash, and Pat Gulhati. 2024. "Molecular Landscape and Therapeutic Strategies against Colorectal Cancer" Cancers 16, no. 8: 1551. https://doi.org/10.3390/cancers16081551
APA StylePatel, A., & Gulhati, P. (2024). Molecular Landscape and Therapeutic Strategies against Colorectal Cancer. Cancers, 16(8), 1551. https://doi.org/10.3390/cancers16081551