
Functional Homologous Recombination (HR) Screening Shows the Majority of BRCA1/2-Mutant Breast and Ovarian Cancer Cell Lines Are HR-Proficient

 , , , , and
, , , , and 
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. BC and OC Cell Lines
2.2. Whole Exome Sequencing
2.3. RAD51 Immunofluorescence
2.4. BRCA1/2 Western Blotting
2.5. DNA Methylation, Gene Expression and Mutational Signature Data
2.6. BRCA1 Δ11q Splice Variant Detection
2.7. Transient Transfection of GFP-SHLD2 cDNA and mScarlet-LMNA CRISPR/Cas9 Assay
2.8. Statistical Analyses
3. Results
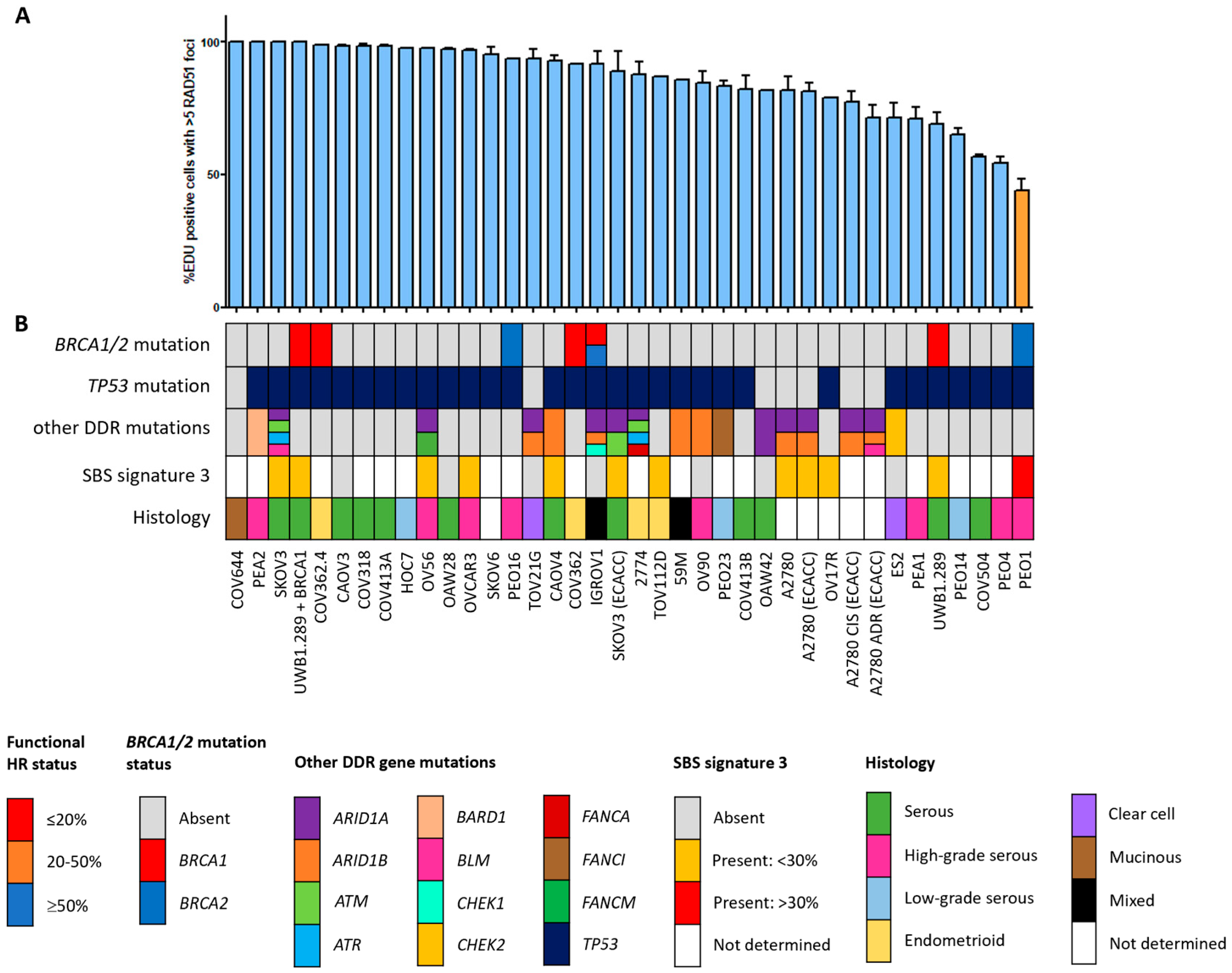
3.1. Correlation between Functional HR Phenotype and BRCA1/2 Mutations
3.2. The HRD Phenotype beyond BRCA1/2 Mutations
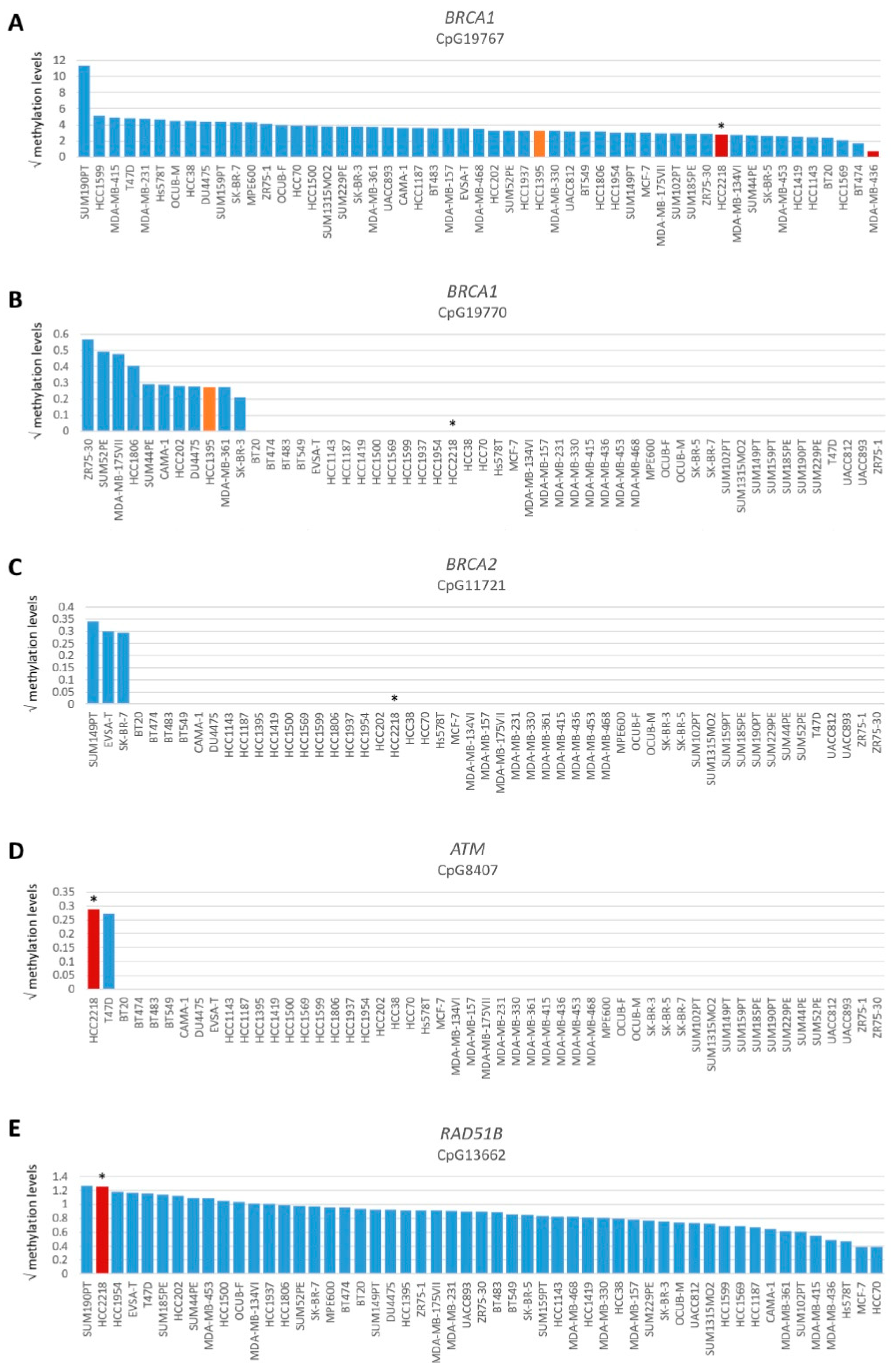
3.3. Restoration of HR in BRCA1/2-Mutant HRP Cell Lines
3.4. SBS Signature 3 and Functional HR Status
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hartman, A.; Kaldate, R.R.; Sailer, L.M.; Painter, L.; Grier, C.E.; Endsley, R.R.; Griffin, M.; Hamilton, S.A.; Frye, C.A.; Silberman, M.A.; et al. Prevalence of BRCA mutations in an unselected population of triple-negative breast Cancer. Cancer 2012, 118, 2787–2795. [Google Scholar] [CrossRef] [PubMed]
- Nelson, H.D.; Fu, R.; Goddard, K.; Mitchell, J.P.; Okinaka-Hu, L.; Pappas, M.; Zakher, B. Risk Assessment, Genetic Counseling, and Genetic Testing for BRCA-Related Cancer: Systematic Review to Update the U.S. Preventive Services Task Force Recommendation; Agency for Healthcare Research and Quality (US): Rockville, MD, USA, 2013.
- Ramus, S.J.; Gayther, S.A. The contribution of BRCA1 and BRCA2 to ovarian Cancer. Mol. Oncol. 2009, 3, 138–150. [Google Scholar] [CrossRef] [PubMed]
- Audeh, M.W.; Carmichael, J.; Penson, R.T.; Friedlander, M.; Powell, B.; Bell-McGuinn, K.M.; Scott, C.; Weitzel, J.N.; Oaknin, A.; Loman, N.; et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: A proof-of-concept trial. Lancet 2010, 376, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Ledermann, J.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.L.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: A preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol. 2014, 15, 852–861. [Google Scholar] [CrossRef]
- Robson, M.; Im, S.A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N. Engl. J. Med. 2017, 377, 523–533. [Google Scholar] [CrossRef] [PubMed]
- Tutt, A.; Robson, M.; Garber, J.E.; Domchek, S.M.; Audeh, M.W.; Weitzel, J.N.; Friedlander, M.; Arun, B.; Loman, N.; Schmutzler, R.K.; et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: A proof-of-concept trial. Lancet 2010, 376, 235–244. [Google Scholar] [CrossRef]
- da Cunha Colombo Bonadio, R.R.; Fogace, R.N.; Miranda, V.C.; Diz, M.P.E. Homologous recombination deficiency in ovarian cancer: A review of its epidemiology and management. Clinics 2018, 73, e450s. [Google Scholar] [CrossRef]
- Davies, H.; Glodzik, D.; Morganella, S.; Yates, L.R.; Staaf, J.; Zou, X.; Ramakrishna, M.; Martin, S.; Boyault, S.; Sieuwerts, A.M.; et al. HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat. Med. 2017, 23, 517–525. [Google Scholar] [CrossRef]
- Meijer, T.G.; Verkaik, N.S.; Sieuwerts, A.M.; van Riet, J.; Naipal, K.A.; van Deurzen, C.H.; Bakker, M.A.D.; Sleddens, H.F.; Dubbink, H.-J.; Toom, T.D.D.; et al. Functional Ex Vivo Assay Reveals Homologous Recombination Deficiency in Breast Cancer Beyond BRCA Gene Defects. Clin. Cancer Res. 2018, 24, 6277–6287. [Google Scholar] [CrossRef]
- Vollebergh, M.A.; Lips, E.H.; Nederlof, P.M.; Wessels, L.F.A.; Schmidt, M.K.; van Beers, E.H.; Cornelissen, S.; Holtkamp, M.; Froklage, F.E.; de Vries, E.G.E.; et al. An aCGH classifier derived from BRCA1-mutated breast cancer and benefit of high-dose platinum-based chemotherapy in HER2-negative breast cancer patients. Ann. Oncol. 2011, 22, 1561–1570. [Google Scholar] [CrossRef] [PubMed]
- Noordermeer, S.M.; van Attikum, H. PARP Inhibitor Resistance: A Tug-of-War in BRCA-Mutated Cells. Trends Cell Biol. 2019, 29, 820–834. [Google Scholar] [CrossRef]
- Gogola, E.; Rottenberg, S.; Jonkers, J. Resistance to PARP Inhibitors: Lessons from Preclinical Models of BRCA-Associated Cancer. Annu. Rev. Cancer Biol. 2019, 3, 235–254. [Google Scholar] [CrossRef]
- Fong, P.C.; Yap, T.A.; Boss, D.S.; Carden, C.P.; Mergui-Roelvink, M.; Gourley, C.; De Greve, J.; Lubinski, J.; Shanley, S.; Messiou, C.; et al. Poly(ADP)-ribose polymerase inhibition: Frequent durable responses in BRCA carrier ovarian cancer correlating with platinum-free interval. J. Clin. Oncol. 2010, 28, 2512–2519. [Google Scholar] [CrossRef]
- Bouwman, P.; Aly, A.; Escandell, J.M.; Pieterse, M.; Bartkova, J.; van der Gulden, H.; Hiddingh, S.; Thanasoula, M.; Kulkarni, A.; Yang, Q.; et al. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat. Struct. Mol. Biol. 2010, 17, 688–695. [Google Scholar] [CrossRef]
- Gogola, E.; Duarte, A.A.; de Ruiter, J.R.; Wiegant, W.W.; Schmid, J.A.; de Bruijn, R.; James, D.I.; Llobet, S.G.; Vis, D.J.; Annunziato, S.; et al. Selective Loss of PARG Restores PARylation and Counteracts PARP Inhibitor-Mediated Synthetic Lethality. Cancer Cell 2018, 33, 1078–1093. [Google Scholar] [CrossRef]
- Noordermeer, S.M.; Adam, S.; Setiaputra, D.; Barazas, M.; Pettitt, S.J.; Ling, A.K.; Olivieri, M.; Álvarez-Quilón, A.; Moatti, N.; Zimmermann, M.; et al. The shieldin complex mediates 53BP1-dependent DNA repair. Nature 2018, 560, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Banda, K.; Swisher, E.M.; Wu, D.; Pritchard, C.C.; Gadi, V.K. Somatic Reversion of Germline BRCA2 Mutation Confers Resistance to Poly(ADP-ribose) Polymerase Inhibitor Therapy. JCO Precis. Oncol. 2018, 2, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Norquist, B.; Wurz, K.A.; Pennil, C.C.; Garcia, R.; Gross, J.; Sakai, W.; Karlan, B.Y.; Taniguchi, T.; Swisher, E.M. Secondary somatic mutations restoring BRCA1/2 predict chemotherapy resistance in hereditary ovarian carcinomas. J. Clin. Oncol. 2011, 29, 3008–3015. [Google Scholar] [CrossRef]
- Nik-Zainal, S.; Alexandrov, L.B.; Wedge, D.C.; Van Loo, P.; Greenman, C.D.; Raine, K.; Jones, D.; Hinton, J.; Marshall, J.; Stebbings, L.A.; et al. Mutational processes molding the genomes of 21 breast cancers. Cell 2012, 149, 979–993. [Google Scholar] [CrossRef]
- Nik-Zainal, S.; Davies, H.; Staaf, J.; Ramakrishna, M.; Glodzik, D.; Zou, X.; Martincorena, I.; Alexandrov, L.B.; Martin, S.; Wedge, D.C.; et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 2016, 534, 47–54. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Ng, A.W.T.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The repertoire of mutational signatures in human Cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Gillet, J.-P.; Varma, S.; Gottesman, M.M. The clinical relevance of cancer cell lines. J. Natl. Cancer Inst. 2013, 105, 452–458. [Google Scholar] [CrossRef] [PubMed]
- Hollestelle, A.; Nagel, J.H.A.; Smid, M.; Lam, S.; Elstrodt, F.; Wasielewski, M.; Ng, S.S.; French, P.J.; Peeters, J.K.; Rozendaal, M.J.; et al. Distinct gene mutation profiles among luminal-type and basal-type breast cancer cell lines. Breast Cancer Res. Treat. 2010, 121, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Beaufort, C.M.; Helmijr, J.C.A.; Piskorz, A.M.; Hoogstraat, M.; Ruigrok-Ritstier, K.; Besselink, N.; Murtaza, M.; van Ijcken, W.; Heine, A.; Smid, M.; et al. Ovarian cancer cell line panel (OCCP): Clinical importance of in vitro morphological subtypes. PLoS ONE 2014, 9, e103988. [Google Scholar] [CrossRef] [PubMed]
- Naipal, K.A.; Verkaik, N.S.; Ameziane, N.; van Deurzen, C.H.; ter Brugge, P.; Meijers, M.; Sieuwerts, A.M.; Martens, J.W.; O’Connor, M.J.; Vrieling, H.; et al. Functional ex vivo assay to select homologous recombination-deficient breast tumors for PARP inhibitor treatment. Clin. Cancer Res. 2014, 20, 4816–4826. [Google Scholar] [CrossRef] [PubMed]
- Hollestelle, A.; Elstrodt, F.; Timmermans, M.; Sieuwerts, A.M.; Klijn, J.G.M.; Foekens, J.A.; Bakker, M.A.D.; Schutte, M. Four human breast cancer cell lines with biallelic inactivating alpha-catenin gene mutations. Breast Cancer Res. Treat. 2010, 122, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Riaz, M.; van Jaarsveld, M.T.; Hollestelle, A.; Prager-van der Smissen, W.J.; Heine, A.A.; Boersma, A.W.; Liu, J.; Helmijr, J.; Ozturk, B.; Smid, M.; et al. miRNA expression profiling of 51 human breast cancer cell lines reveals subtype and driver mutation-specific miRNAs. Breast Cancer Res. 2013, 15, R33. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Kim, S.; Scheffler, K.; Halpern, A.L.; Bekritsky, M.A.; Noh, E.; Källberg, M.; Chen, X.; Kim, Y.; Beyter, D.; Krusche, P.; et al. Strelka2: Fast and accurate calling of germline and somatic variants. Nat. Methods 2018, 15, 591–594. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Van Den Tempel, N.; Laffeber, C.; Odijk, H.; Van Cappellen, W.A.; Van Rhoon, G.C.; Franckena, M.; Kanaar, R. The effect of thermal dose on hyperthermia-mediated inhibition of DNA repair through homologous recombination. Oncotarget 2017, 8, 44593–44604. [Google Scholar] [CrossRef]
- Essers, J.; Hendriks, R.W.; Wesoly, J.; Beerens, C.E.; Smit, B.; Hoeijmakers, J.H.; Wyman, C.; Dronkert, M.L.; Kanaar, R. Analysis of mouse Rad54 expression and its implications for homologous recombination. DNA Repair 2002, 1, 779–793. [Google Scholar] [CrossRef] [PubMed]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Boers, R.; Boers, J.; de Hoon, B.; Kockx, C.; Ozgur, Z.; Molijn, A.; van Ijcken, W.; Laven, J.; Gribnau, J. Genome-wide DNA methylation profiling using the methylation-dependent restriction enzyme LpnPI. Genome Res. 2018, 28, 88–99. [Google Scholar] [CrossRef] [PubMed]
- Schutte, M.; Elstrodt, F.; Bralten, L.B.C.; Nagel, J.H.A.; Duijm, E.; Hollestelle, A.; Vuerhard, M.J.; Wasielewski, M.; Peeters, J.K.; van der Spek, P.; et al. Exon expression arrays as a tool to identify new cancer genes. PLoS ONE 2007, 3, e3007. [Google Scholar] [CrossRef]
- Johnson, W.E.; Li, C.; Rabinovic, A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 2007, 8, 118–127. [Google Scholar] [CrossRef]
- Jarvis, M.C.; Ebrahimi, D.; A Temiz, N.; Harris, R.S. Mutation Signatures Including APOBEC in Cancer Cell Lines. JNCI Cancer Spectr. 2018, 2, pky002. [Google Scholar] [CrossRef]
- Sieuwerts, A.M.; Gelder, M.E.M.-V.; Timmermans, M.; Trapman, A.M.; Garcia, R.R.; Arnold, M.; Goedheer, A.J.; Portengen, H.; Klijn, J.G.; Foekens, J.A. How ADAM-9 and ADAM-11 differentially from estrogen receptor predict response to tamoxifen treatment in patients with recurrent breast cancer: A retrospective study. Clin. Cancer Res. 2005, 11, 7311–7321. [Google Scholar] [CrossRef]
- Levesque, S.; Mayorga, D.; Fiset, J.-P.; Goupil, C.; Duringer, A.; Loiselle, A.; Bouchard, E.; Agudelo, D.; Doyon, Y. Marker-free co-selection for successive rounds of prime editing in human cells. Nat. Commun. 2022, 13, 5909. [Google Scholar] [CrossRef]
- Meijer, T.G.; Verkaik, N.S.; van Deurzen, C.H.; Dubbink, H.-J.; Toom, T.D.D.; Sleddens, H.F.; De Hoop, E.O.; Dinjens, W.N.; Kanaar, R.; van Gent, D.C.; et al. Direct Ex Vivo Observation of Homologous Recombination Defect Reversal After DNA-Damaging Chemotherapy in Patients with Metastatic Breast Cancer. JCO Precis Oncol. 2019, 3, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Bernhardy, A.J.; Cruz, C.; Krais, J.J.; Nacson, J.; Nicolas, E.; Peri, S.; van der Gulden, H.; van der Heijden, I.; O’Brien, S.W.; et al. The BRCA1-Delta11q Alternative Splice Isoform Bypasses Germline Mutations and Promotes Therapeutic Resistance to PARP Inhibition and Cisplatin. Cancer Res. 2016, 76, 2778–2790. [Google Scholar] [CrossRef] [PubMed]
- Drost, R.; Dhillon, K.K.; Van Der Gulden, H.; Van Der Heijden, I.; Brandsma, I.; Cruz, C.; Chondronasiou, D.; Castroviejo-Bermejo, M.; Boon, U.; Schut, E.; et al. BRCA1185delAG tumors may acquire therapy resistance through expression of RING-less BRCA1. J. Clin. Investig. 2016, 126, 2903–2918. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Krais, J.J.; Bernhardy, A.J.; Nicolas, E.; Cai, K.Q.; Harrell, M.I.; Kim, H.H.; George, E.; Swisher, E.M.; Simpkins, F.; et al. RING domain-deficient BRCA1 promotes PARP inhibitor and platinum resistance. J. Clin. Investig. 2016, 126, 3145–3157. [Google Scholar] [CrossRef] [PubMed]
- Polak, P.; Kim, J.; Braunstein, L.Z.; Karlic, R.; Haradhavala, N.J.; Tiao, G.; Rosebrock, D.; Livitz, D.; Kübler, K.; Mouw, K.W.; et al. A mutational signature reveals alterations underlying deficient homologous recombination repair in breast Cancer. Nat. Genet. 2017, 49, 1476–1486. [Google Scholar] [CrossRef] [PubMed]
- Weigelt, B.; Bi, R.; Kumar, R.; Blecua, P.; Mandelker, D.L.; Geyer, F.C.; Pareja, F.; A James, P.; Couch, F.J.; Eccles, D.M.; et al. The Landscape of Somatic Genetic Alterations in Breast Cancers From ATM Germline Mutation Carriers. J. Natl. Cancer Inst. 2018, 110, 1030–1034. [Google Scholar] [CrossRef]
- Pinder, J.; Salsman, J.; Dellaire, G. Nuclear domain ‘knock-in’ screen for the evaluation and identification of small molecule enhancers of CRISPR-based genome editing. Nucleic Acids Res. 2015, 43, 9379–9392. [Google Scholar] [CrossRef]
- Sato, K.; Brandsma, I.; E van Rossum-Fikkert, S.; Verkaik, N.; Oostra, A.B.; Dorsman, J.C.; van Gent, D.C.; Knipscheer, P.; Kanaar, R.; Zelensky, A.N. HSF2BP negatively regulates homologous recombination in DNA interstrand crosslink repair. Nucleic Acids Res. 2020, 48, 2442–2456. [Google Scholar] [CrossRef]
- Argunhan, B.; Iwasaki, H.; Tsubouchi, H. Post-translational modification of factors involved in homologous recombination. DNA Repair 2021, 104, 103114. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Pathogenic BRCA1/2 Mutation | HRD Status | BRCA1/2 Protein | Explanation for Discordance |
|---|---|---|---|---|
| MDA-MB-436 | BRCA1 c.5396+1G>A, p.E1731del28 and p.I1760ins*8 (VAF = 100%) | HRD | BRCA1 absent, BRCA2 present | HR and BRCA status are concordant |
| HCC2218 | Wild-type | HRD | Very low BRCA1/2 expression | Unexplained |
| HCC1395 | BRCA1 c.5251C>T, p.R1751* (VAF = 100%) and BRCA2 c.4777G>T, p.E1593* (VAF = 46.2%) | HRI | BRCA1 absent, BRCA2 present | In-frame TP53BP1 deletion? |
| PEO1 | BRCA2 c.4965C>G, p.Y1655* (VAF = 100%) and BRCA2 c.4964A>T, p.Y1655F (VAF = 29.3%) and BRCA2 c.682-2A>G (VAF = 10%) | HRI | BRCA1 and BRCA2 present | BRCA2 c.4964A>T reversion mutation |
| BT474 | BRCA2 c.9281C>A, p.S3094* (VAF = 46.6%) | HRP | BRCA1 and BRCA2 present | Heterozygous BRCA2 mutation |
| HCC202 | BRCA2 c.7033C>T, p.Q2345* (VAF = 4.3%) | HRP | BRCA2 present, low BRCA1 expression | Low VAF BRCA2 mutation |
| HCC1569 | BRCA2 c.5578delA, p.V1862* (VAF = 42%) | HRP | Low BRCA2 expression, BRCA1 absent | Heterozygous BRCA2 mutation |
| HCC1599 | BRCA2 c.4550_4559del, p.K1517Ifs*23 (VAF = 81%) | HRP | Very low BRCA2 expression, low BRCA1 expression | Incomplete BRCA2 protein loss |
| HCC1937 | BRCA1 c.5266dupC, p.Q1756Pfs*74 (VAF = 100%) | HRP | BRCA1 absent, BRCA2 present | Low SHLD2 mRNA expression |
| HCC1954 | BRCA1 c.5425_5426del, p.V1809Cfs*20 (VAF = 20.6%) | HRP | Low BRCA1 expression, BRCA2 present | Low VAF BRCA1 mutation |
| SUM149PT | BRCA1 c.2169delT, p.P724Lfs*12 (VAF = 96.7%) | HRP | BRCA1 absent, BRCA2 present | BRCA1-Δ11q alternative splice isoform [43] |
| SUM1315MO2 | BRCA1 c.66_67delAG, p.E23fs*17 (VAF = 100%) | HRP | Low BRCA1 expression, BRCA2 present | RING domain-deficient BRCA1 [44,45] |
| COV362 | BRCA1 c.2612_2613insT, p.F872Vfs*31 (VAF = 89.6%) and BRCA1 c.4096+1G>T (VAF = 100%) | HRP | BRCA1 absent, BRCA2 present | BRCA1-Δ11q alternative splice isoform |
| COV362.4 | BRCA1 c.2612_2613insT, p.F872Vfs*31 (VAF = 94.6%) and BRCA1 c.4096+1G>T (VAF = 100%) | HRP | BRCA1 absent, BRCA2 present | BRCA1-Δ11q alternative splice isoform |
| IGROV1 | BRCA1 c.1961delA, p.K654Sfs*47 (VAF = 19.8%) and BRCA2 c.3320delA, p.K1108Rfs*11 (VAF = 25%) | HRP | Low BRCA1 and BRCA2 expression | Low VAF BRCA1 and BRCA2 mutation |
| PEO16 | BRCA2 c.4141_4143del, p.K1381del (VAF = 94.7%) | HRP | BRCA1 and BRCA2 present | In-frame BRCA2 mutation |
| UWB1.289 | BRCA1 c.2475delC, p.D825Efs*21 (VAF = 96%) | HRP | BRCA1 absent, BRCA2 present | BRCA1-Δ11q alternative splice isoform [43] |
| UWB1.289+BRCA1 | BRCA1 c.2475delC, p.D825Efs*21 (VAF = 68.2%) | HRP | Low BRCA1 expression, BRCA2 present | Reconstitution of wild-type BRCA1 cDNA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meijer, T.G.; Martens, J.W.M.; Prager-van der Smissen, W.J.C.; Verkaik, N.S.; Beaufort, C.M.; van Herk, S.; Robert-Finestra, T.; Hoogenboezem, R.M.; Ruigrok-Ritstier, K.; Paul, M.W.; et al. Functional Homologous Recombination (HR) Screening Shows the Majority of BRCA1/2-Mutant Breast and Ovarian Cancer Cell Lines Are HR-Proficient. Cancers 2024, 16, 741. https://doi.org/10.3390/cancers16040741
Meijer TG, Martens JWM, Prager-van der Smissen WJC, Verkaik NS, Beaufort CM, van Herk S, Robert-Finestra T, Hoogenboezem RM, Ruigrok-Ritstier K, Paul MW, et al. Functional Homologous Recombination (HR) Screening Shows the Majority of BRCA1/2-Mutant Breast and Ovarian Cancer Cell Lines Are HR-Proficient. Cancers. 2024; 16(4):741. https://doi.org/10.3390/cancers16040741
Chicago/Turabian StyleMeijer, Titia G., John W. M. Martens, Wendy J. C. Prager-van der Smissen, Nicole S. Verkaik, Corine M. Beaufort, Stanley van Herk, Teresa Robert-Finestra, Remco M. Hoogenboezem, Kirsten Ruigrok-Ritstier, Maarten W. Paul, and et al. 2024. "Functional Homologous Recombination (HR) Screening Shows the Majority of BRCA1/2-Mutant Breast and Ovarian Cancer Cell Lines Are HR-Proficient" Cancers 16, no. 4: 741. https://doi.org/10.3390/cancers16040741
APA StyleMeijer, T. G., Martens, J. W. M., Prager-van der Smissen, W. J. C., Verkaik, N. S., Beaufort, C. M., van Herk, S., Robert-Finestra, T., Hoogenboezem, R. M., Ruigrok-Ritstier, K., Paul, M. W., Gribnau, J., Bindels, E. M. J., Kanaar, R., Jager, A., van Gent, D. C., & Hollestelle, A. (2024). Functional Homologous Recombination (HR) Screening Shows the Majority of BRCA1/2-Mutant Breast and Ovarian Cancer Cell Lines Are HR-Proficient. Cancers, 16(4), 741. https://doi.org/10.3390/cancers16040741