Simple Summary
Small cell lung cancer (SCLC) is a deadly cancer that is treated by chemo- and radiotherapy, which work by damaging DNA. However, most SCLC patients will develop resistance to these treatments, resulting in a poor outlook with few effective treatment options available. This review summarizes our understanding of the causes of treatment resistance and the treatments which have been studied to overcome resistance. While there is still much to be understood, new methods being developed may improve our ability to study this disease and find effective treatments.
Abstract
Background: Small cell lung cancer (SCLC) is a lethal form of lung cancer with few treatment options and a high rate of relapse. While SCLC is initially sensitive to first-line DNA-damaging chemo- and radiotherapy, relapse disease is almost universally therapy-resistant. As a result, there has been interest in understanding the mechanisms of therapeutic resistance in this disease. Conclusions: Progress has been made in elucidating these mechanisms, particularly as they relate to the DNA damage response and SCLC differentiation and transformation, leading to many clinical trials investigating new therapies and combinations. Yet there remain many gaps in our understanding, such as the effect of epigenetics or the tumor microenvironment on treatment response, and no single mechanism has been found to be ubiquitous, suggesting a significant heterogeneity in the mechanisms of acquired resistance. Nevertheless, the advancement of techniques in the laboratory and the clinic will improve our ability to study this disease, especially in patient populations, and identify methods to surmount therapeutic resistance.
1. Small Cell Lung Cancer
Small cell lung cancer (SCLC) is a lethal neuroendocrine carcinoma that accounts for about 15% of lung cancer cases [1]. Each year, there are approximately 250,000 new cases and 200,000 deaths globally, ranking it as the sixth most common cause of death from cancer [1]. Smoking is the main cause of SCLC development, with only about 2% of cases arising in never-smokers [2]. In addition, SCLC has a rapid doubling time, high growth fraction, and early metastasis, so the chance of early detection is low. This is especially the case because CT screening is not effective at detecting early-stage disease [3], and these patients are asymptomatic. Rather than the standard tumor, node, metastasis (TNM) staging system, SCLC is more commonly classified by the Veterans’ Administration Lung Study Group (VALSG) staging system into limited-stage and extensive-stage disease [4]. This staging system is based on whether the tumor can be safely confined to a single radiation port, and about two-thirds of patients will present with metastatic, extensive-stage disease.
1.1. Biology
1.1.1. Recurrent Alterations
SCLC exhibits a high mutational burden and enrichment for CG to AT transversions, a mutational signature commonly observed in smokers [5,6,7,8]. In particular, the biallelic loss of the tumor suppressors tumor protein p53 (TP53) and RB transcriptional corepressor 1 (RB1) is nearly universal, and results in the dysregulation of the DNA damage response and cell cycle. This is thought to be the main driver of SCLC, and genetically engineered mouse models (GEMMs) with this dual knockout (KO) develop tumors resembling SCLC [9]. Other recurrent mutations have also been identified at lower frequencies through genomic and transcriptomic profiling of SCLC patient samples [5,6,7,8,10,11]. These include the loss of other tumor suppressors: tumor protein p73 (TP73), a TP53 homolog; RB transcriptional corepressor like 1 (RBL1) and RB transcriptional corepressor like 2 (RBL2), which are paralogs of RB1; phosphatase and tensin homolog (PTEN); and Notch receptor (NOTCH) family genes. Loss of function mutations are also observed for genes involved in chromatin regulation such as lysine methyltransferase 2A (KMT2A), CREB binding protein (CREBBP), E1A binding protein p300 (EP300), and histone deacetylase 2 (HDAC2). In addition, activating mutations and amplifications of oncogenes have been observed in the MYC proto-oncogene (MYC) family genes (MYC, MYCN, MYCL), phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K) pathway genes, and RAS proto-oncogene (RAS) pathway genes. A small proportion of these mutations involved chimeric transcripts due to genomic rearrangements. SCLC is also clonally diverse, with multiple distinct clones contributing to intratumoral heterogeneity in each relapsed SCLC patient [11,12,13].
1.1.2. Subtypes
Consistent with its neuroendocrine phenotype, most SCLC tumors highly express markers of neuroendocrine differentiation, such as insulinoma-associated protein 1 (INSM1), neural cell adhesion molecule 1 (NCAM1), chromogranin-A (CHGA), and synaptophysin (SYP), and are considered neuroendocrine-high [14]. However, some tumors express lower levels of these markers and are considered neuroendocrine-low. SCLC can be classified into four subtypes based on elevated expression of key transcription factors [15]: achaete-scute family bHLH transcription factor 1 (ASCL1) in SCLC-A, neuronal differentiation 1 (NEUROD1) in SCLC-N, Yes1 associated transcriptional regulator (YAP1) in SCLC-Y, and POU class 2 homeobox 3 (POU2F3) in SCLC-P. SCLC-A and SCLC-N have a neuroendocrine-high phenotype, while SCLC-Y and SCLC-P are neuroendocrine-low. However, studies have suggested that SCLC can exhibit significant plasticity and subtype switching, and patient tumors may represent a heterogeneous population of various subtypes as the tumor adapts and evolves [12,13,16]. GEMMs of SCLC have shed light on possible drivers of such events, though they remain to be fully validated in human disease. MYC amplification is thought to drive SCLC-A switching to SCLC-N and SCLC-Y, as GEMMs with Trp53/Rb1 KO and hyperactive MYC develop invasive tumors of these other subtypes from primary SCLC-A tumors [17,18]. Furthermore, SCLC-A was found to correlate with MYCL amplification and negatively correlate with MYC amplification [19]. Similarly, activation of the NOTCH pathway can drive neuroendocrine-high to neuroendocrine-low transformation. GEMMs of SCLC overexpressing the intracellular domain of NOTCH1 develop neuroendocrine-low tumors from primary neuroendocrine-high tumors, and NOTCH pathway-mediated RE1 silencing transcription factor (Rest) and hes family bHLH transcription factor 1 (Hes1) expression represses neuroendocrine differentiation [20,21]. Indeed, SCLC-A negatively correlates with NOTCH family gene expression [8,20,22], and both SCLC-A and SCLC-N are enriched for delta like canonical Notch ligand 3 (DLL3), an inhibitory ligand of NOTCH proteins [14]. YAP1 expression has also been found to drive neuroendocrine-high to neuroendocrine-low transformation. ASCL1 inversely correlates with YAP1 signaling [22], and in GEMMs of SCLC, expression of constitutively active Yap1 resulted in tumor phenotype switching by upregulation of Notch2 and downstream Rest and Hes1 [21,23]. Moreover, Yap1 is upregulated by NOTCH signaling, suggesting that these pathways feed into each other to promote neuroendocrine-high to neuroendocrine-low transformation [21]. As opposed to these, POU2F3 expression is associated with chemosensory tuft cells [24], suggesting a different cell of origin, though whether SCLC-P can transform into other subtypes and vice versa is still unknown.
The existence of the SCLC-Y subtype is controversial. Though it was originally identified through transcriptomic profiling of SCLC cell lines and clinical samples, YAP1 protein expression in patient samples was found to be relatively low across tumors by immunohistochemistry [14]. A similar study classifying a biobank of circulating tumor cell-derived xenografts (CDXs) saw that SCLC-Y did not define a unique subtype of SCLC that was distinct from the others [25]. Given that SCLC-Y is more likely to retain RB1, studies have even suggested that cases of this subtype may belong to other cancers [26,27]. Another classification scheme replacing SCLC-Y with the inflammatory SCLC-I subtype has been proposed, characterized by low expression of the other transcription factors, an inflamed gene signature, infiltration by cluster of differentiation 8 (CD8)+/programmed cell death 1 ligand 1 (PD-L1)+ T-cells, and improved response to immunotherapy [28]. An alternative classification scheme based on transcriptomic analyses of clinical samples rather than transcription factors also identified immune-inflamed populations which respond best to immunotherapy, though these populations were neuroendocrine-high [29]. More work will be required to better classify SCLC and understand its phenotypic plasticity, as well as determine how this influences treatment outcomes or presents new therapeutic opportunities.
1.2. Treatment Options
In spite of advances in our understanding of its biology, SCLC is still treated clinically as a monolith, with differences based on disease stage [30,31,32]. Due to the lack of targetable driver mutations, the development of targeted therapies against SCLC has been slow. The high proliferation rate of SCLC sensitizes it to DNA damage, so the standard of care is a chemotherapy combination of a platinum-based DNA crosslinker, cisplatin or carboplatin, and the DNA topoisomerase II (TOP2) inhibitor, etoposide, across all stages. In rare cases of SCLC being detected early (T1-2N0M0), surgical resection followed by adjuvant chemotherapy is an option, though clinical evidence is limited [33]. For limited-stage disease, chemotherapy is given concurrently with thoracic radiotherapy. Patients who respond to this regimen are also given prophylactic cranial irradiation (PCI) to further improve clinical outcomes [34,35]. For extensive-stage disease, chemotherapy has remained the standard of care for over three decades. Addition of atezolizumab or durvalumab, which act as anti-PD-L1 agents, has received regulatory approval after demonstrating an overall survival (OS) benefit in large phase III trials [36,37]. Post hoc analyses further suggest that the addition of immunotherapy is effective against brain metastases [38,39]. While phase III trials of the anti-PD-1 antibodies, pembrolizumab and nivolumab, did not reach their primary endpoints of OS [40,41], a phase III trial of serplulimab showed similar efficacy to existing immunotherapies [42]. Interim results from the recent ADRIATIC phase III trial further demonstrated that consolidative durvalumab significantly improved OS and progression-free survival (PFS) in limited-stage patients, supporting its inclusion in the standard of care for these patients [43]. Additionally, a phase III trial by the Japanese Cooperative Oncology Group (JCOG) demonstrated a survival advantage when replacing etoposide with irinotecan [44], a TOP1 inhibitor. However, subsequent trials conducted by groups such as the Southwest Oncology Group (SWOG) were unable to replicate this improvement, though a different toxicity profile was observed [45,46]. The incorporation of radiotherapy for extensive-stage disease is also controversial, wherein a phase III trial investigating the addition of thoracic radiotherapy showed no statistically significant difference in OS at 1 year but a significant improvement at 2 years [47]. Similarly, while a European Organisation for Research and Treatment of Cancer (EORTC) study in the pre-MRI era found that addition of PCI for extensive-stage disease led to reductions in brain metastases with an increase in OS [48], no significant OS benefit was observed in a subsequent Japanese study mandating MRI screening [49].
While SCLC is quite sensitive to current first-line therapies, with objective response rates (ORR) of over 60% in patients with extensive-stage disease [50], more than 90% of patients will suffer from relapse within 2 years [51]. Generally, outcomes of relapsed disease differ based on the durability of response to first-line chemotherapy, with sensitive disease (commonly defined as duration ≥ 90 days) having better outcomes than resistant disease (duration < 90 days). Despite promising first-line outcomes, therapeutic options for the second-line and beyond remain limited and ineffective. Currently, the only FDA-approved second-line therapies for SCLC are topotecan, lurbinectedin, and tarlatamab. Topotecan, a TOP1 inhibitor, was approved following phase III trials demonstrating improved OS compared to best supportive care [52]. While topotecan was not superior to the combination of cyclophosphamide, doxorubicin, and vincristine (a chemotherapy regimen comparable to cisplatin and etoposide [53]), it did show improved symptom control [54]. Lurbinectedin, a DNA-alkylating agent, received accelerated approval after demonstrating efficacy against SCLC with a favorable toxicity profile in a phase II trial [55]. Tarlatamab, a bispecific T-cell engager targeting DLL3, similarly received accelerated approval due to favorable response rate and duration [56]. Confirmatory phase III trials are currently ongoing for both agents, pending full approval [57,58,59]. Patients with sensitive disease are typically retreated with platinum and etoposide, as a phase III trial demonstrated an improvement to the primary endpoint of PFS with carboplatin and etoposide compared to topotecan [60]. Similarly, a phase III trial of cisplatin, etoposide, and irinotecan demonstrated improved OS, PFS, and ORR compared to topotecan but had high rates of toxicity [61]. Several other agents like irinotecan, paclitaxel, temozolomide, and amrubicin have also shown activity against SCLC and are occasionally used in the second-line and beyond but are not superior to current therapies [62,63,64,65,66]. Immunotherapy with the anti-PD-1 antibodies nivolumab (alone or in combination with ipilimumab) and pembrolizumab have also demonstrated promising results in phase I/II trials and may be considered in the relapse setting [67,68], though a phase III trial of nivolumab alone as a second-line therapy did not reach its primary endpoint of OS [69].
2. Mechanisms of Resistance to DNA-Damaging Therapy in SCLC
One of the greatest challenges in SCLC treatment is the prevalence of treatment-resistant disease. The lack of targetable driver mutations limits the scope of effective targeted therapies, which is reflected in the current treatment paradigm consisting mostly of DNA-damaging chemotherapy. Although the rapid proliferation of SCLC causes increased sensitivity to DNA damage and replicative stress, recurrent disease no longer responds to this regimen, resulting in few effective therapeutic options available for treatment-resistant disease. This has spurred significant interest in understanding the mechanisms of therapeutic resistance in this disease, as well as identifying novel vulnerabilities and alternate approaches to treatment. Chemotherapy resistance has received more attention, given its broader application across SCLC treatment, though some work has also been conducted on radiotherapy resistance, and some mechanisms confer cross-resistance. The emphasis has been placed on enhancing DNA damage, such as by modulating repair and apoptosis pathways, as well as targeting SCLC differentiation and transformation pathways. Epigenetic regulation is another emerging area which has received increasing attention, and other mechanisms undergoing preclinical and clinical investigation include drug efflux, growth factor signaling pathways, metabolism, autophagy, and cell extrinsic factors such as the tumor microenvironment (TME). Some studies have also focused on sensitizers of chemo- and radiotherapy which are not linked to a particular resistance mechanism, such as cell cycle modulation. While new therapies have yet to reach the clinic, many new drugs are currently under investigation both preclinically and clinically (Table 1), including many combination therapies (Table 2), and this work has bolstered our understanding of SCLC biology.

Table 1.
Clinical investigations in overcoming SCLC therapeutic resistance. Outcomes of trials containing multiple cancer types only consider participants with SCLC.
Table 2.
Combination therapies under investigation to overcome SCLC therapeutic resistance.
2.1. Neuroendocrine-High to Neuroendocrine-Low Transformation
The pathways involved in the neuroendocrine-low differentiation of SCLC tumors (Figure 1), including YAP1, NOTCH, Wnt family (WNT), and MYC, have each been implicated in tumor progression and treatment resistance across various cancers. Indeed, neuroendocrine-low SCLC cells have been observed in GEMMs to be slower-growing but more chemoresistant [20,23]. As the master transcriptional regulator of the SCLC-Y subtype, YAP1 confers resistance to both chemotherapy and radiotherapy. For instance, YAP1 signaling has been correlated with drug efflux through breast cancer resistance protein (BCRP) and cancer cell stemness through CD133 [125,134]. It also feeds into the NOTCH pathway by upregulating NOTCH2 expression [21,23]. Overexpression of YAP1 in the YAP1-low cell line H69 resulted in resistance to cisplatin, etoposide, and radiation [114,134]. On the other hand, overexpression of a dominant-negative form of YAP1 (lacking its transactivation domain) in the YAP1-high cell line H446 had the opposite effect of sensitizing cells to treatment. In these cells, treatment with a YAP1 inhibitor, verteporfin, also sensitized cells to chemotherapy (Table 2). Similarly, ex vivo culture of mouse SCLC cells with increasing doses of etoposide generated resistant cells with upregulated YAP1 [23], and ex vivo cultures of two CDXs with high YAP1 from a biobank of 39 CDXs show resistance to cisplatin [25].
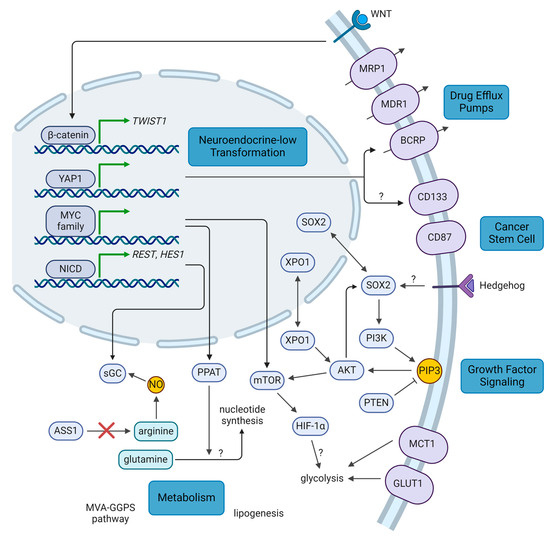
Figure 1.
Summary of key molecular pathways in therapeutic resistance. WNT, YAP1, NOTCH, and MYC signaling promote transformation to a resistant, neuroendocrine-low phenotype. Cancer stem cell factors such as SOX2, CD133, and CD87 promote a resistant cell state that survives treatment. Expression of drug efflux pumps may also be associated with this cell state. Growth factor signaling pathways promote cell proliferation and therapeutic resistance. Certain resistance pathways may have metabolic dependencies that can be targeted. Question marks: mechanisms which remain uncertain or unknown. AKT: AKT serine/threonine kinase, ASS1: argininosuccinate synthase 1, BCRP: breast cancer resistance protein, GLUT1: glucose transporter 1, HES1: hes family bHLH transcription factor 1, HIF-1α: hypoxia inducible factor 1α, MCT1: monocarboxylate transporter 1, MDR1: multidrug resistance protein 1, MRP1: multidrug resistance associated protein 1, mTOR: mechanistic target of rapamycin kinase, MVA-GGPS: mevalonate-geranylgeranyl pyrophosphate, MYC: MYC proto-oncogene, NICD: NOTCH intracellular domain, NO: nitrous oxide, NOTCH: Notch receptor, PI3K: phosphatidylinositol-4,5-bisphosphate 3-kinase, PIP3: phosphatidylinositol (3,4,5)-trisphosphate, PPAT: phosphoribosyl pyrophosphate amidotransferase, PTEN: phosphatase and tensin homolog, REST: RE1 silencing transcription factor, sGC: soluble guanylyl cyclase, SOX2: SRY-box transcription factor 2, TWIST1: twist family bHLH transcription factor 1, WNT: Wnt family, XPO1: exportin 1, YAP1: Yes1 associated transcriptional regulator (created with BioRender.com, accessed on 4 October 2024).
Another pathway found to drive neuroendocrine-low differentiation and chemoresistance is the NOTCH pathway. In GEMMs, NOTCH signaling upregulated Rest and Hes1 expression, two transcriptional repressors of neuroendocrine genes. Although tumor cells harboring Rest and Hes1 upregulation were slower-growing, they were more resistant to treatment with cisplatin and etoposide and acted as part of the TME in providing trophic support to other tumor cells [20,21]. Indeed, recent studies in patient samples and preclinical models have found that neuroendocrine-low cells have the capacity to remodel the extracellular matrix (ECM) [135] and secrete extracellular vesicles containing ECM factors which support the growth of neuroendocrine-high cells [136]. Furthermore, such remodeling mediates vasculogenic mimicry, which increased the ability of cisplatin to reach the tumor, but conversely promoted resistance to cisplatin [135,137,138]. NOTCH1 and HES1 levels were also observed to be higher in the multidrug-resistant H69AR cell line, where siRNA knockdown (KD) of HES1 sensitized cells to cisplatin and etoposide treatment [115]. Blocking NOTCH signaling with the NOTCH2/3 inhibitor tarextumab was able to delay onset of chemoresistance and synergize with chemotherapy in GEMMs [20], and inhibition of HES1 with FLI-06 was similarly able to synergize with chemotherapy in vivo [115]. In addition, NOTCH signaling has been shown to upregulate soluble guanylyl cyclase (sGC), a nitrous oxide (NO) sensor implicated in chemoresistance [139]. H196 cells with KO of a subunit of sGC are sensitized to etoposide treatment in vitro, and cell line-derived xenografts with this KO are more sensitive to cisplatin and etoposide. A randomized, multicenter phase Ib/II trial was undertaken comparing chemotherapy with or without tarextumab in 145 untreated patients with extensive-stage disease, but no significant improvement in PFS (5.5 vs. 5.5 months), OS (9.3 vs. 10.3 months), or ORR (68.6% vs. 70.8%) was observed and no biomarkers predicting efficacy were identified, resulting in the discontinuation of this drug’s development [70].
While the WNT pathway has not been directly implicated in neuroendocrine-low differentiation, ASCL1 has been shown to be antagonistic with WNT signaling in addition to YAP1 and NOTCH signaling [22]. Furthermore, whole-exome sequencing of treatment-naïve and relapse SCLC patient samples found that 80% of relapse samples had nonsynonymous mutations in canonical WNT pathway components that were not seen in naïve samples, resulting in higher levels of WNT activity [140]. Another study similarly identified mutations in APC regulator of WNT signaling pathway (APC), a negative regulator of canonical WNT signaling, across all relapse SCLC patient samples sequenced, as well as other WNT pathway mutations [11]. Correspondingly, cell lines with KD or KO of APC and elevated WNT activity were resistant to both cisplatin and etoposide and could be sensitized by re-expression of APC [140]. In addition, WNT signaling is known to upregulate the twist family bHLH transcription factor 1 (TWIST1) [141]. In patient-derived xenografts (PDXs) that were treated with cycles of chemotherapy to induce resistance, TWIST1 was one of the most highly upregulated genes in resistant models [142]. However, while TWIST1 has been observed to mediate chemoresistance across several cancer types [143,144,145], the authors did not see chemosensitization with TWIST1 KD. In a drug screen of SCLC cell lines and patient-derived cells, one of the top hits against cisplatin-resistant cells was the bromodomain and extraterminal domain (BET) family inhibitor, JQ1 [138]. JQ1 was predicted to target YAP1 signaling and the NOTCH, WNT, and transforming growth factor beta (TGFβ) pathways, and may be effective against tumors which leverage these resistance mechanisms. This agent may also be effective against cancer-associated fibroblasts (CAFs) which promote cisplatin resistance through TGFβ signaling [138], though this remains controversial as others have correlated CAFs with radiosensitivity and improved patient outcomes [146]. A combination between BET and mechanistic target of rapamycin kinase (mTOR) inhibition has also been proposed, as BET inhibitors can inadvertently promote tumor survival by upregulating the mTOR pathway [116].
MYC and MYCN, too, have been implicated in SCLC subtype transformation, and MYC is itself a target of both the NOTCH and WNT pathways [17,18]. Multiple studies have shown that high levels of MYC family proteins promote chemoresistance in SCLC both in vitro and in vivo [17,115,117,147,148,149], though MYC—and other classic oncogenes like RAS and Raf proto-oncogene (RAF)—were not found to confer radioresistance in SCLC [150]. In turn, MYC family proteins can promote NOTCH signaling to drive further tumor transformation [18]. Indeed, MYCN is capable of promoting chemoresistance through HES1 expression, while NOTCH inhibition abrogates this effect [115]. MYC-high SCLC tumors have been shown to be sensitive to aurora kinase (AURK), ubiquitin specific peptidase 7 (USP7), mTOR, and dual PI3K/HDAC inhibition using in vivo models [17,147,148,151]. The AURKA inhibitor alisertib has demonstrated some clinical efficacy against SCLC both alone and in combination with paclitaxel (Table 1), and MYC expression was found to be a biomarker of treatment efficacy [71,72]. A double-blind phase II trial of paclitaxel with or without alisertib in 178 relapsed/refractory patients saw a marginal increase in PFS (3.32 vs. 2.17 months), with a greater benefit seen among 33 patients positive for MYC expression (4.64 vs. 2.27 months) [72]. A phase II trial has also recently been launched to test alisertib in extensive-stage disease progressing after chemoimmunotherapy (NCT06095505). On the other hand, a phase II trial of the AURKB inhibitor barasertib-HQPA in 15 relapsed/refractory patients did not show any efficacy, with an ORR of 0%. [73]. One recent study [152] looked at targeted MYCN inhibition using a peptide nucleic acid based on the MYCN antigene. The authors showed that this agent downregulated MYCN targets, including the mTOR pathway, and was effective as a monotherapy against a mouse xenograft of the multidrug-resistant cell line H69AR.
2.2. Cancer Stem Cells
Cancer stem cells are a very small subpopulation of a tumor which possess self-renewal capabilities and are important in propagating the tumor [153]. They are more treatment-resistant and promote tumor repopulation following therapy. As noted previously, YAP1 promotes the expression of one such stem cell marker, CD133, which correlated with radioresistance in SCLC (Figure 1) [134]. CD133 has also been shown to mediate resistance to etoposide, and CD133+ cells have been observed to accumulate in a more chemoresistant, post-treatment patient sample [154,155]. CD133 correlated with levels of AKT serine/threonine kinase (AKT) and BCL2 apoptosis regulator (BCL2), and increased tumor propagation potential, corresponding with its role in cell stemness [154]. Sarvi et al. noted that CD133+ cells had increased expression of neuropeptide receptors, and neuropeptide antagonists could preferentially target this population [154]. Another stem cell marker, CD87, has also been shown to mediate chemoresistance in SCLC. CD87 conferred resistance to both cisplatin and etoposide and correlated with levels of the drug efflux pump, multidrug resistance protein 1 (MDR1), and another stem cell marker, CD44 [155,156].
One of the drivers of cell stemness is the transcription factor SRY-box transcription factor 2 (SOX2). Through genomic and transcriptomic profiling, SOX2 amplifications were identified in about 27% of patient samples and cell lines [6], as well as a proportion of relapsed patient samples [11]. Indeed, the key transcriptional regulator of the SCLC-A subtype, ASCL1, upregulates expression of SOX2, and this is important in both tumor establishment and MYC-dependent subtype regulation [19,157]. Upregulated SOX2 expression is correlated with both cisplatin and etoposide resistance in SCLC cell lines, and the signal transducer and activator of transcription 3 (STAT3) inhibitor napabucasin was shown to downregulate SOX2 and MYC, thereby resensitizing cisplatin-resistant cells both in vitro and in vivo (Table 2) [117,158,159]. Strikingly, an extensive-stage patient with a SOX2 amplification responded well to the Hedgehog inhibitor sonidegib in combination with chemotherapy in a phase I trial, remaining progression-free after 27 months on maintenance sonidegib despite the trial having a median PFS of only 5.5 months (Table 1) [74]. Hedgehog is known to regulate cell stemness through SOX2, suggesting that this signaling pathway may be involved in SCLC chemoresistance. However, a randomized phase II trial of chemotherapy with or without the Hedgehog inhibitor vismodegib in 155 untreated patients did not demonstrate a significant improvement in PFS (4.4 vs. 4.7 months) or OS (9.8 vs. 9.1 months), though selection for SOX2 expression may improve outcomes [75].
2.3. Growth Factor Signaling
The PI3K/AKT/mTOR pathway is a critical signaling pathway for cell growth and proliferation which has been found to be dysregulated in nearly all cancers, including SCLC (Figure 1) [5,6,7,8,10,160]. Indeed, it is upregulated downstream of several key pathways in SCLC, including WNT and MYC signaling [151,152], and has been found to form a positive feedback loop with SOX2, implicating this pathway in cell stemness [158,159]. Genomic and transcriptomic analyses found that PI3K/AKT was upregulated in chemoresistant SCLC cell lines and patient samples [161] and that mTOR signaling was upregulated in a panel of chemoresistant PDXs [149]. Interaction of PI3K/AKT with SOX2 also promoted resistance to both cisplatin and etoposide [158,159], and inhibitors of PI3K and mTOR reversed this chemoresistance and synergized with chemotherapy [161,162]. Furthermore, upregulation of the PI3K pathway and tyrosine kinase signaling have been observed to mediate resistance to chemotherapy and radiotherapy by promoting adhesion of SCLC cells to the ECM through integrins [162,163,164,165]. Correspondingly, inhibitors of PI3K, mTOR, and epidermal growth factor receptor (EGFR) could reverse therapeutic resistance. Inhibitors of the PI3K pathway also abrogated radioresistance by promoting glucose-6-phosphate dehydrogenase (G6PD) degradation and upregulating the production of reactive oxygen species, which synergized with radiation both in vitro and in vivo [118]. In addition, a 2022 study [119] found that an inhibitor of exportin 1 (XPO1), selinexor, synergized with chemotherapy to inhibit tumor growth in PDXs of chemoresistant relapse patients. Through transcriptomic analyses, the authors identified the PI3K/AKT/mTOR pathway as being upregulated by XPO1 and confirmed that selinexor inhibits the activity of this pathway. While several clinical trials of PI3K (NCT02194049) and mTOR [73,76,77,78] inhibitors have not demonstrated significant activity (Table 1), patient selection for PI3K pathway upregulation and combination with other chemotherapeutics may improve the efficacy of targeting this pathway. A phase II trial was also initiated for selinexor in relapsed SCLC, but the trial was terminated due to slow patient accrual (NCT02351505).
Downstream of PI3K signaling, hypoxia inducible factor 1 (HIF-1) has been found to be upregulated [161]. The HIF-1 pathway is a key survival mechanism activated by hypoxic conditions and is involved in resistance to both chemotherapy and radiotherapy across many cancers. While downregulation of HIF-1α via KD of the long non-coding RNA HIF1A-AS2 sensitizes H69AR cells to doxorubicin [166], the role of the HIF-1 pathway and hypoxia in resistance to radiotherapy and other chemotherapeutic agents in SCLC remains to be elucidated. On the other hand, PTEN is a negative regulator of the PI3K pathway which acts in direct opposition to PI3K. Downregulation of PTEN activates the PI3K pathway and promotes chemoresistance in SCLC [167], and mutations in PTEN have been shown to mediate differences in radiosensitivity [168].
2.4. DNA Damage Response
Given that current SCLC therapies focus on DNA damage, cells can acquire resistance by bolstering their DNA damage response to repair damage and avoid lethality (Figure 2). One of the key pathways implicated in SCLC is the ATR serine/threonine kinase (ATR)/checkpoint kinase 1 (CHK1) pathway, which is critical for activating both the S and G2/M checkpoints and promoting DNA repair. Transcriptomic and proteomic profiling have shown that levels of both ATR and CHK1 are higher in SCLC than in non-small cell lung cancer (NSCLC) [120,169,170]. Preclinical and clinical data indicate that inhibitors of ATR are effective against SCLC tumors both alone and in combination with second-line therapies [79,169,171,172]. A proof-of-concept phase II trial of the ATR inhibitor berzosertib in combination with topotecan in 25 relapsed SCLC patients reached its primary endpoint with an ORR of 36%, showing efficacy in patients with chemoresistant disease [79]. Further trials to test this combination are ongoing (NCT04768296, NCT03896503), as well as trials testing berzosertib in combination with lurbinectedin (NCT04802174). CHK1 inhibitors have also demonstrated efficacy against preclinical models of SCLC, especially in combination with cisplatin and etoposide [120,121,169]. In cells treated with increasing doses of cisplatin to generate chemoresistance, CHK1 inhibition was able to reverse resistance and synergize with cisplatin [120,121]. However, a phase II trial of the CHK1 inhibitor prexasertib in extensive-stage patients did not demonstrate efficacy in either platinum-sensitive (PFS = 1.41 months, OS = 5.42 months, ORR = 5.2%) or refractory patients (PFS = 1.36 months, OS = 3.15 months, ORR = 0%) [80]. Indeed, some SCLC cell lines are resistant to CHK1 inhibition, and in resistant cells generated through treatment with increasing doses of prexasertib, WEE1 G2 checkpoint kinase (WEE1) upregulation was identified as a mediator of resistance [173]. WEE1 is another regulator of cell cycle checkpoints, which supports a possible combination of CHK1 and WEE1 inhibitors. WEE1 itself is an interesting therapeutic target in SCLC. A recent study identified WEE1 as a mediator of DNA repair following radiation-induced damage, with high levels of WEE1 in SCLC conferring radioresistance [174]. However, SCLC cell lines have a range of sensitivities to the WEE1 inhibitor adavosertib [175], and clinical trials of adavosertib as a monotherapy show minimal efficacy in SCLC [73,81]. A phase II trial testing adavosertib in combination with carboplatin, among other therapies, has recently concluded, awaiting results [82]. In cell lines resistant to adavosertib, upregulation of CHK1 through AXL receptor tyrosine kinase (AXL) and mTOR was observed [175], further supporting the combination of CHK1 and WEE1 inhibition. Similar to ATR/CHK1, ATM serine/threonine kinase (ATM) is critical for promoting cell cycle arrest and DNA repair in response to DNA damage. ATM KO sensitized an SCLC cell line to radiation, and inhibition of ATM with AZD1390 synergized with radiation both in vitro and in vivo, including in PDX models (Table 2) [122].
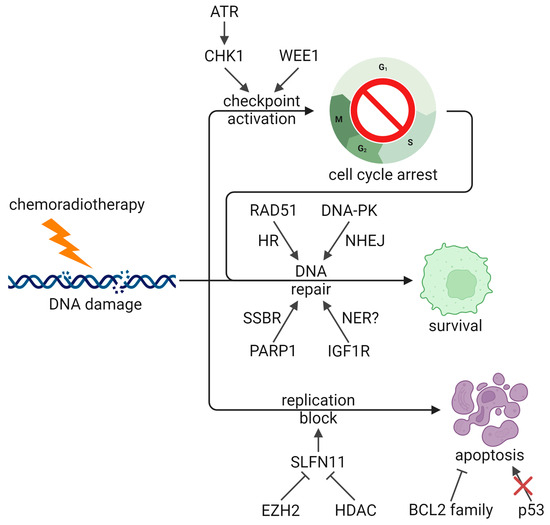
Figure 2.
DNA damage response and apoptosis pathways in therapeutic resistance. ATR/CHK1 and WEE1 promote cell cycle arrest to promote repair, and several DNA repair pathways are implicated in promoting treatment resistance and survival. EZH2 and HDACs inhibit SLFN11-mediated replication block and apoptosis, and both BCL2 activity and TP53 loss also block apoptosis. ATR: ATR serine/threonine kinase, BCL2: BCL2 apoptosis regulator, CHK1: checkpoint kinase 1, DNA-PK: DNA-dependent protein kinase, EZH2: enhancer of zeste 2 polycomb repressive complex 2 subunit, HDAC: histone deacetylase, HR: homologous recombination, IGF1R: insulin like growth factor 1 receptor, NER: nucleotide excision repair, NHEJ: non-homologous end joining, PARP1: poly (ADP-ribose) polymerase 1, RAD51: RAD51 recombinase, SLFN11: schlafen family member 11, SSBR: DNA single strand break repair, TP53: tumor protein p53, WEE1: WEE1 G2 checkpoint kinase (Created with BioRender.com, accessed 4 October 2024).
Another protein implicated in SCLC treatment resistance is schlafen family member 11 (SLFN11), an RNA-DNA helicase which promotes apoptosis in response to DNA damage at the S-phase checkpoint. In PDXs treated with cycles of chemotherapy, SLFN11 was greatly downregulated, while high SLFN11 expression correlated with chemosensitivity in SCLC patient samples [142]. Indeed, low SLFN11 was found to mediate resistance to cisplatin, topotecan, and lurbinectedin [171,176,177], and a combination of ATR inhibition and lurbinectedin overcame this resistance [171], potentially by enhancing cell cycle checkpoint dysregulation. SLFN11 expression is under epigenetic regulation and, in particular, the histone methyltransferase enhancer of zeste 2 polycomb repressive complex 2 subunit (EZH2) is responsible for silencing SLFN11 [142]. Correspondingly, EZH2 is overexpressed in SCLC compared to other cancers [170,178], and EZH2 inhibition is able to delay chemoresistance and synergize with chemotherapy [142]. Phase I trials of EZH2 inhibitors are currently underway (NCT05353439, NCT03460977). In contrast to these data, a different cohort of PDXs showed no correlation between SLFN11 mRNA or protein levels and chemosensitivity [149], indicating that other mechanisms of chemoresistance are at play.
Several other DNA repair proteins have also been implicated in SCLC therapeutic resistance. In a pair of cell lines derived from the same tumor, CPH-54A and CPH-54B, the more resistant cell line had higher levels of DNA-dependent protein kinase catalytic subunit (DNA-PKcs) and RAD51 recombinase (RAD51), as well as a higher rate of double-strand break repair, suggesting that both non-homologous end joining (NHEJ) and homologous recombination (HR) may be involved [179]. A phase Ib/II trial of the DNA-PK inhibitor M3814 with chemotherapy was terminated due to slow patient accrual (NCT03116971). On the other hand, two of six SCLC patients in a phase Ib trial showed an objective response to the multi-targeted tyrosine kinase and RAD51 inhibitor, amuvatinib, in combination with chemotherapy [83]. Although an open-label, multicenter phase II trial did not show significant efficacy (PFS = 68 days, OS = 119 days, ORR = 17.4%), two patients who responded had elevated KIT proto-oncogene (KIT), suggesting that this may be a biomarker for patient selection [84]. In the same vein, poly (ADP-ribose) polymerase (PARP) inhibitors sensitize SCLC cell lines and xenograft models to multiple DNA-damaging therapeutics, including platinum, etoposide, temozolomide, and radiation [180,181,182]. This effect was associated with DNA-PKcs downregulation by veliparib and PARP trapping by talazoparib. Clinical trials of PARP inhibitors both alone [85,86] and in combination with chemotherapy [87,88] have yielded mixed results (Table 1), which suggests that identification of biomarkers will improve outcomes. Several ongoing studies continue to investigate combinations of PARP inhibitor with chemoradiotherapy (NCT03532880, NCT04170946, NCT04624204, NCT04728230). The growth factor receptor, insulin like growth factor 1 receptor (IGF1R), is also upregulated in SCLC, and inhibition of IGF1R was associated with nucleotide excision repair (NER) downregulation which synergized with chemoradiotherapy [160]. However, in untreated patients, no significant improvement was observed in PFS (4.6 vs. 4.7 months), OS (10.1 vs. 9.1 months), or ORR (49% vs. 43%) in a randomized phase II trial of chemotherapy with or without the monoclonal antibody targeting IGF1R, cixutumumab [75]. Similarly, a randomized, multi-institution phase II trial in relapsed patients comparing topotecan to the IGF1R inhibitor linsitinib did not show a significant improvement to OS (5.3 vs. 3.4 months, p = 0.71), though a benefit was seen in PFS (3.0 vs. 1.2 months, p = 0.0001) [89]. Recently, the RNA helicase DEAD-box helicase 4 (DDX4) was found to mediate cisplatin resistance in SCLC, and proteomic profiling identified upregulation of DNA repair with overexpression of DDX4 [183]. Finally, deletions in the mismatch repair (MMR) genes mutS homolog 2 (MSH2) and MSH6 have been observed in SCLC [140], and an SCLC cell line deficient in MMR demonstrated resistance to an alkylating agent [184]. While MMR deficiencies have been observed to confer chemoresistance across several cancer types [185], evidence in SCLC is limited and more work will need to be conducted to determine the role of MMR in therapeutic resistance in this context.
2.5. Apoptosis Regulation
Apoptosis pathways are important in mediating response to DNA-damaging agents by promoting cell death. Therefore, tumor cells can promote therapeutic resistance and survival by upregulating anti-apoptotic pathways (Figure 2), such as through BCL2 family proteins. Indeed, SCLC has higher levels of BCL2 than NSCLC, and high BCL2 in SCLC correlates with cisplatin resistance and poor prognosis [186,187]. Targeting BCL2 family proteins like BCL2, BCL2 like 1 (BCL-xL), and MCL1 apoptosis regulator (MCL1) was found to promote apoptosis and synergize with chemotherapy and radiotherapy [187,188,189]. In addition, high BCL2 confers resistance to AURK inhibitors, and a combination of AURKB and BCL2 inhibition was effective in SCLC cell lines and PDXs [190]. However, challenges remain with BCL2-targeted therapies. Cells with low levels of BCL2 associated X (BAX) are resistant to BCL2 inhibition [188], and some studies have conversely found that low levels of BCL2 correlate with poorer outcomes [191,192]. In the latter case, since BCL2 expression is observed to be driven by ASCL1 and higher in SCLC-A [19,28,188], it is possible that cells with low BCL2 have alternate resistance mechanisms, especially those driven by subtype transformation. Correspondingly, SCLC-A cell lines have been observed to be most sensitive to BCL2 inhibition [28], suggesting that patient stratification is important. Several phase II trials have been conducted with various BCL2 family inhibitors both alone [90,91,92] and in combination with topotecan [93,94] or standard chemotherapy [95,96], showing little activity in unselected patients, though one phase I trial saw objective response in six of seven untreated extensive-stage patients [97]. Nevertheless, new BCL2 targeted therapies for SCLC are still being investigated, and a recent phase I trial of a dual BCL2/BCL-xL inhibitor in combination with paclitaxel in 28 relapsed/refractory SCLC patients had an ORR of 25%, indicating that this therapy shows some promise, especially with patient selection (Table 1) [98].
As noted previously, TP53 mutations are ubiquitous in SCLC. A proportion of these mutations abolish normal function, which promotes resistance to DNA-damaging therapies by attenuating apoptosis, and tumors with only missense TP53 mutations may acquire co-alterations in related genes like TP73 to achieve resistance [13]. Loss of p53 has been shown to confer resistance to both chemotherapy and radiotherapy, and accumulation of mutant p53 can have further gain-of-function effects on chemoresistance [193,194,195,196,197]. The p53 activator, APR-246/PRIMA-1Met, was shown to restore activity to mutant p53 and promote apoptosis, which demonstrated efficacy in SCLC cell lines and cell line-derived xenografts [193]. In addition, gain-of-function p53 mutations have been shown to mediate resistance to BCL2 inhibition, and heat shock protein 90 (HSP90) inhibition was able to destabilize mutant p53 and sensitize SCLC cell lines to BCL2 inhibition [195]. This combination was also found to downregulate MCL1 and nuclear factor kappa B (NF-κB) to further promote apoptosis [198]. Clinical trials of HSP90 inhibitors in combination with doxorubicin (terminated) [99] or carboplatin and paclitaxel (ORR = 0%) [100] have not demonstrated efficacy in SCLC, though other combinations, such as with BCL2 inhibition, may be more effective.
Another regulator of apoptosis is the JUN N-terminal kinase (JNK) pathway, which has been observed to have both pro- and anti-apoptotic effects in response to cell stress [199]. One group observed that JNK signaling in SCLC is generally anti-apoptotic and mediates resistance to cisplatin and UV radiation [200,201]. This suggests a potential synergy with JNK pathway inhibitors, though further work will be required to determine the efficacy of this combination.
2.6. Metabolism
Several studies have investigated the metabolic vulnerabilities of SCLC, as well as potential links to therapeutic resistance (Figure 1). Chalishazar et al. found that MYC-driven tumors, including chemoresistant ones, were dependent on arginine [151]. As noted previously, NOTCH and NO can promote chemoresistance through sGC [139], so considering that MYC cooperates with NOTCH in neuroendocrine-low transformation and arginine is a precursor in NO synthesis, arginine and MYC may cooperate to drive chemoresistance through sGC. SCLC cells have been found to harbor loss of argininosuccinate synthase 1 (ASS1), a critical enzyme in arginine synthesis, making them reliant on extrinsic sources of arginine. One study saw ASS1 loss in approximately half of SCLC patient samples and cell lines [202], and another observed that 83% of SCLC patient samples had low levels of ASS1 [203]. These arginine-dependent cells respond well to arginine depletion in preclinical models, by either arginine deiminase-based or arginase-based therapeutics [151,202,203,204]. A non-randomized, open-label phase II trial of arginine deiminase in ASS1-low SCLC patients was terminated due to slow patient accrual and lack of tumor response after 22 patients had been enrolled, though patient stratification based on factors such as MYC expression may be beneficial [101]. A phase I/II trial with arginase has also been completed, awaiting results (NCT03371979).
Another potential vulnerability of SCLC is glutamine, which functions at the nexus between the citric acid cycle and nucleotide synthesis. In SCLC, the purine synthesis pathway through phosphoribosyl pyrophosphate amidotransferase (PPAT) is favored over the citric acid cycle through glutaminase (GLS), potentially due to MYC since PPAT is one of its target genes [205]. Correspondingly, PPAT depletion reduced growth of SCLC cell lines, with similar effects from GLS overexpression. In NSCLC and ovarian cancer, some cisplatin-resistant cells have large nucleotide pools and are vulnerable to glutamine starvation and antimetabolites which interfere with nucleotide synthesis [206], though this has not been replicated in SCLC. Nevertheless, a recent study observed that SCLC cell lines treated with cycles of cisplatin to establish resistance had larger nucleotide pools and demonstrated that a combination of antimetabolites and an inhibitor of glutamine synthesis was effective in suppressing the growth of these cells both in vitro and in vivo (Table 2) [123].
Several other metabolic pathways have also been implicated in SCLC therapeutic resistance. Two studies have reported dysregulation of glucose metabolism in radioresistant SCLC. One group observed increased glucagon release and enolase 2 (ENO2) levels, suggesting increased glycolysis [207], and the other similarly observed higher glucose transporter 1 (GLUT1) levels and increased glycolytic activity [208]. Subsequent studies have shown that targeting glycolysis through inhibition of the lactate transporter, monocarboxylate transporter 1 (MCT1), is effective in preclinical models of SCLC and shows synergy with radiotherapy (Table 2) [124,209]. In addition, SCLC stem cells have higher levels of the glycolysis enzyme, 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 (PFKFB3) [125]. Inhibition of PFKFB3 by PFK158 reduced the levels of several stemness markers, such as CD133 and SOX2; impeded glycolysis; and attenuated tumor growth in SCLC cell line-derived xenografts. PFK158 was particularly effective in SCLC models overexpressing MYC due to the upregulation of aerobic glycolysis by MYC, consistent with the Warburg effect [210]. While the mechanism which links glycolytic activity to radioresistance in SCLC is unknown, studies in other cancer types have implicated improved redox stress control and DNA damage response [211]. The HIF-1 pathway is also known to promote both glycolysis and radioresistance, potentially linking these observations. More studies will be needed to establish the connection between the Warburg effect and radioresistance in the context of SCLC. A 2022 study investigating metabolic vulnerabilities in chemoresistant SCLC cell line-derived xenografts, established through cycles of cisplatin and etoposide treatment, identified reliance on the mevalonate–geranylgeranyl pyrophosphate (MVA-GGPS) pathway [212]. Combining chemotherapy with statins to target this pathway was effective against these xenograft models; however, clinical trials in untreated patients have not demonstrated efficacy of statins in combination with first-line chemotherapy or chemoradiotherapy (Table 1) [102,103,104]. Another study that year found increased lipogenesis in SCLC stem cells through the stabilization of fatty acid synthase (FASN) by the deubiquitinase USP13 [126]. FASN inhibition reduced lipogenesis and synergized with etoposide treatment in xenografts of the multidrug-resistant SCLC cell line H69AR (Table 2).
2.7. Drug Efflux Pumps
A common way through which tumor cells can develop therapeutic resistance is through the upregulation of ATP-binding cassette transporters which act as drug efflux pumps. In SCLC (Figure 1), several studies have correlated the expression of MDR1 and multidrug resistance associated protein 1 (MRP1) with chemoresistance and poor prognosis in SCLC patient samples and cell lines [213,214,215,216,217,218], and some relapse patients were found to have amplifications of ABCC1, which encodes MRP1 [140]. Indeed, one of the first studies to identify MRP1 used the multidrug-resistant H69AR cell line, which had a 100- to 200-fold increase in ABCC1 mRNA levels compared to H69 [213]. Upregulation of MRP1 was identified downstream of SOX2, suggesting a potential mechanism [158]. Interestingly, irradiation of the H69 cell line to establish radioresistance led to upregulation of both MRP1 and its homolog, MRP2 [192], suggesting shared transcriptional programs linking resistance to chemotherapy and radiotherapy. As noted previously, another drug efflux pump, BCRP, was also implicated in SCLC chemoresistance downstream of YAP1 [125]. Preclinical studies of MDR1 and BCRP inhibitors have demonstrated the ability to sensitize SCLC cell lines to topoisomerase inhibitors like etoposide in vitro [219]. However, a non-randomized, open-label, multicenter phase II trial in 36 relapsed SCLC patients adding the multi-target drug efflux pump inhibitor, biricodar, to doxorubicin and vincristine only showed an ORR of 19% [105]. Upregulation of drug efflux pumps appears to be uncommon in SCLC, and further work will be needed to ascertain their role in therapeutic resistance as well as the efficacy of drug efflux pump inhibitors with other regimens.
2.8. Epigenetics
Epigenetic programs have been implicated in therapeutic resistance across many cancers, including SCLC, and interact with other mechanisms as well. Similarly to EZH2, HDAC inhibitors have been observed to reverse chemoresistance by upregulating SLFN11, which synergizes with TOP1 inhibition [176,177]. Furthermore, MYC-high tumors are sensitive to dual PI3K/HDAC inhibitors [148]. In addition to these roles, HDAC inhibitors have been found to synergize with cisplatin through the induction of S-phase arrest [220], and dual PI3K/HDAC inhibitors have been identified as radiosensitizers, synergizing with irradiation by maintaining open chromatin and inhibiting the repair of DNA double-strand breaks [127]. However, HDAC inhibitors are generally inactive against SCLC as a monotherapy, with ORRs of 0% [106,107,108], while combinations with chemotherapy have high toxicity [109,110], except for belinostat with chemotherapy, which demonstrated acceptable toxicity and an ORR of 57% (Table 1) [111]. The latter study noted that patients with high copy numbers of specific UDP glucuronosyltransferase family 1 member A1 (UGT1A1) mutations (*28 and *60) had slower clearance of belinostat, suggesting that selection against patients with these mutations will reduce toxicity. DNA methyltransferase 1 (DNMT1) inhibitors are also important in the context of SCLC, as the combined inhibition of DNMT1 and pyrimidine synthesis was found to sidestep the need for p53-mediated apoptosis by promoting terminal neuroendocrine maturation, demonstrating efficacy against chemoresistant SCLC cell lines [194]. A phase I trial combining DNMT1 and HDAC inhibitors in pulmonary and pleural malignancies, including SCLC, has been completed, awaiting results (NCT00037817).
In a screen of cell lines representing various cancer types with a lysine demethylase 1A (LSD1) inhibitor, SCLC was identified as the most sensitive cancer type, with DNA hypomethylation signature acting as a biomarker for sensitivity [221]. Genomic analyses showed that cell lines with high MYCN were more sensitive to LSD1 inhibition, while cell lines with high MYC were resistant. Subsequent mechanistic studies showed that LSD1 inhibitors act by inhibiting ASCL1, through silencing of INSM1 or derepression of NOTCH1, as well as efficacy in chemoresistant PDXs of SCLC [222,223]. These findings suggest that LSD1 inhibition would be most effective against neuroendocrine-high subtypes of SCLC, especially SCLC-A, consistent with the observation that more mesenchymal-like SCLC cells are resistant to LSD1 inhibition [224]. While an open-label, multicenter phase I trial of the LSD1 inhibitor, GSK2879552, was terminated after recruiting 29 patients due to toxicity and lack of response [112], clinical trials investigating combination therapies with LSD1 inhibitors are ongoing (NCT03850067, NCT05191797, NCT05420636), and selection for neuroendocrine-high SCLC patients may improve response (Table 1). At the RNA level, adenosine methylation to form m6A has been correlated with chemoresistance, with a panel of seven m6A regulators being predictive of survival in a cohort of 200 patients [225]. In particular, methyltransferase 3 N6-adenosine-methyltransferase complex catalytic subunit (METTL3), which is responsible for m6A deposition on mRNA, was found to promote chemoresistance by activating mitophagy, and inhibition of METTL3 synergized with chemotherapy in H69AR xenografts (Table 2) [128].
Aside from these, recurrent mutations in epigenetic regulation genes such as KMT2A, CREBBP, and EP300 have been observed in SCLC [5,6,7,8,11]. A 2024 study looking at SCLC tumor evolution throughout treatment found that patients with mutations in the lysine acetyltransferase genes CREBBP and EP300 have an elevated risk of disease recurrence, implicating these genes in therapeutic resistance [13]. In particular, patients with CREBBP mutations have an increased sensitivity to HDAC inhibition [226], suggesting this alteration as a biomarker for HDAC inhibitors. A 2020 study of the SCLC epigenome further identified several epigenetic markers which are associated with sensitivity to agents such as ATR, AURK, BCL2, and mTOR inhibitors, though these associations require further validation [227]. Interestingly, the 5′ untranslated region of EZH2 was associated with sensitivity to AURK inhibitors and a fibroblast growth factor receptor (FGFR) inhibitor, suggesting that these therapies may be effective against SCLC cases where EZH2-mediated silencing of SLFN11 mediates chemoresistance. While mutations in other epigenetic regulation genes have been observed in SCLC, their impact on therapeutic resistance and the possibility of targeting them for treatment have not been investigated.
2.9. Other Resistance Mechanisms and Therapeutic Opportunities
Aside from these, several other factors have been implicated in SCLC therapeutic resistance. In both SCLC cell line-derived xenografts and PDXs, upregulation of cancer testis antigens has been observed following chemotherapy [142,228]. Cancer testis antigens are proteins normally found in male germ cells that can also have roles in cancers. In SCLC, the upregulation of two of these—PAGE family member 5 (PAGE5) and G antigen 2A (GAGE2A)—was found to promote chemoresistance, while PAGE5 and GAGE2A KD cell lines were sensitized to chemotherapy [228]. Autophagy has also been linked to chemoresistance in SCLC. One group observed that upregulating autophagy, through either BMX non-receptor tyrosine kinase (BMX) signaling or sequestosome 1 (SQSTM1), mediates chemoresistance, while inhibition of autophagy using chloroquine reverses resistance and synergizes with chemotherapy [229,230]. Furthermore, JNK signaling can promote autophagy to avoid apoptosis [199], which could explain its anti-apoptotic role in SCLC [200,201]. Unfortunately, clinical trials of chloroquine and its derivative, hydroxychloroquine, have been terminated due to slow patient accrual, with one showing toxicity and lack of response (NCT00969306, NCT01575782, NCT02722369). Another study identified a mutation in the translation factor, eukaryotic translation initiation factor 3 subunit A (eIF3a), as a mediator of chemoresistance by conducting genomic analyses on pre- and post-chemotherapy samples in a cohort of 52 patients [231]. This mutation conferred chemoresistance while attenuating the growth of SCLC cell lines, similar to the effects of neuroendocrine-low transformation.
Studies have also looked at chemo- and radiosensitizers which do not target or reverse a particular resistance mechanism. One approach has been to arrest the cell cycle to sensitize cells to DNA damage (Figure 2). Patel et al. found that the microtubule inhibitor albendazole arrested SCLC cell lines in the radiosensitive G2/M phase, synergizing with radiation [131]. A recent study identified the cell cycle regulator cell division cycle 7 (CDC7) as a target which synergizes with chemotherapy and found that CDC7 inhibition induced cell cycle arrest at the G1/S checkpoint and enhanced chemotherapy-induced DNA damage [132]. Two other microtubule inhibitors, paclitaxel and vinorelbine, have also been found to synergize with cisplatin, etoposide, and radiation in vitro, reversing resistance and sensitizing cells to treatment [129,130]. However, no difference has been seen in cell cycle distribution, suggesting a mechanism other than cell cycle arrest. Unfortunately, a randomized, prospective phase III trial in 587 untreated extensive-stage patients with chemotherapy with or without paclitaxel saw an increase in toxicity with no significant improvement in failure-free survival (6.4 vs. 5.9 months, p = 0.179) or OS (10.6 vs. 9.9 months, p = 0.169) [113], though many trials continue to investigate paclitaxel in combination with various therapies (NCT02769832, NCT05420636, NCT05856695, NCT06016270). A 2022 study tested a new approach to SCLC therapy with TAK-243, an inhibitor of ubiquitin like modifier activating enzyme 1 (UBA1), an E1 enzyme [133]. TAK-243 and other inhibitors of the ubiquitin–proteasome system attenuate protein degradation and therefore have diverse effects on cells, including disruption of DNA repair and the cell cycle [232]. While the mechanism of TAK-243 in SCLC remains unclear, it has been found to synergize with both chemotherapy and radiotherapy in PDXs (Table 2) [133].
3. Conclusions
SCLC is a challenging malignancy to treat owing to its rapid growth, propensity to metastasize, inter- and intratumoral heterogeneity, and lack of targetable drivers. Much work has been undertaken to tackle the considerable challenge of identifying effective treatments which can overcome the widespread resistance to current therapies. Progress has been made in elucidating the biology and resistance mechanisms of SCLC, especially as it relates to the DNA damage response and SCLC differentiation and transformation. These advancements have led to new treatments with diverse approaches, many of which are currently under clinical investigation and may lead to new avenues of treatment (Table 1).
Despite this, new therapies have yet to reach the clinic in a substantial way. Given the rapid proliferation of SCLC and initial sensitivity to DNA damage, agents which target DNA damage response, such as ATR and EZH2 inhibitors, are reasonable. As well, novel therapies which target neuroendocrine-low transformation hold potential in addressing chemoresistant populations. In particular, AURKA inhibition is promising as it targets MYC and may be especially effective against EZH2-high tumors [227]. However, no single mechanism has been found to be ubiquitous in mediating therapeutic resistance, demonstrating the inter- and intratumoral heterogeneity of SCLC. This suggests that precision medicine, through biomarker detection and patient selection, is critical to delivering more effective treatment by targeting the particular mechanisms that are relevant to a particular patient. Many preclinical studies have identified biomarkers, including epigenetic markers, which influence sensitivity to certain treatments, though these can be difficult to implement and study in a clinical setting and their relevance in patients requires further investigation. Nevertheless, several clinical trials have identified potential biomarkers for previously treated patients [72,74,77,84], and the lack of response in unselected patients across several trials may be improvable through selection for these biomarkers. This is particularly relevant for resistance mechanisms which only appear in a small subset of patients, as with drug efflux pumps. Furthermore, combination therapies targeting multiple pathways may be essential to prevent the development of resistance to particular agents (Table 2).
There also remain many gaps in our understanding of resistance mechanisms and how they may be overcome. For instance, while potential biomarkers have been identified clinically, the mechanisms through which they affect the treatment response are unknown, such as in elevated KIT sensitizing to amuvatinib [84]. Similarly, though several epigenetic markers have been associated with sensitivity to certain agents, the underlying mechanisms remain to be elucidated [227]. Additionally, most efforts have focused on chemoresistance, with less emphasis placed on radioresistance, since radiotherapy is given less frequently to SCLC patients. The identification of radiosensitizers in SCLC may enable broader application of radiotherapy as a treatment modality, particularly against extensive-stage disease where evidence is mixed. In addition, factors aside from the intrinsic genetic features of the tumor, such as epigenetics and the TME, are areas which require more attention in the context of SCLC. Given how rapidly SCLC tumors are capable of recurring and developing therapeutic resistance, it is likely that faster, epigenetic mechanisms of regulation play a critical role. Indeed, recurrent mutations in epigenetic regulation genes are observed in SCLC patients, and control other key resistance factors such as MYC and SLFN11 [142,148]. However, the clinical integration of HDAC inhibitors has proven challenging due to toxicity when combined with chemotherapy and warrants further investigation to minimize toxicity. Other agents targeting DNMT1 and LSD1 also require further investigation in combination with other therapies, as SCLC is not sensitive to monotherapies targeting epigenetic regulators [106,107,108,112]. TME modulation is another area which requires further study. The immune microenvironment of SCLC has received significant attention which has led to the approval of immunotherapies against PD-1 and DLL3, highlighting the potential that these avenues of investigation may hold. Yet limited focus has been placed on other microenvironment factors, such as hypoxia and CAFs, particularly in the context of modulating therapeutic sensitivity. Further exploration of these factors will inform the feasibility of implementing TME targeted therapies, such as HIF-1 antagonists, in SCLC.
An additional challenge is the paucity of patient samples for research. This limits our ability to investigate and understand both the heterogeneity of SCLC as well as epigenetic and TME features, which are difficult to capture using other preclinical models. The incorporation of new techniques in the clinic, such as liquid biopsy, may be a promising approach to tackle some of these challenges in the future. Detecting tumor cells and DNA in the blood is a much less invasive way to probe for biomarkers, which will simplify patient selection in the context of both treatment and clinical trials. It also facilitates SCLC patient sample collection, paving the way for larger-scale studies comparing untreated and relapse samples to identify resistance mechanisms, as well as studies investigating epigenetic features. Indeed, efforts are ongoing to leverage liquid biopsy to study clonal tumor evolution and the influence of genetic and epigenetic factors on therapeutic resistance. Ultimately, advancements in the laboratory and clinic will continue to deepen our understanding of SCLC biology to surmount treatment resistance and develop more effective therapeutic approaches.
Author Contributions
Conceptualization, T.Y. and B.H.L.; writing—original draft preparation, T.Y.; writing—review and editing, T.Y. and B.H.L.; supervision, B.H.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Acknowledgments
The authors thank the other members of the B.H.L. laboratory at the Princess Margaret Cancer Centre for their support in the preparation of this work. The B.H.L. laboratory acknowledges the funding support of the Terry Fox Research Institute, Canada Foundation for Innovation, Cancer Research Society, Canadian Institutes of Health Research, National Institute of Health/National Cancer Institute (U01CA253383), and the Clinical and Translational Science Center at Weill Cornell Medical Center, MSKCC (UL1TR00457). T.Y. is supported by funding from the Department of Medical Biophysics, Canadian Institutes of Health Research, and The Strategic Training in Transdisciplinary Radiation Science for the 21st Century (STARS21).
Conflicts of Interest
B.H.L. reports grants from Pfizer, personal fees from Daiichi Sankyo, and grants, personal fees, and non-financial support from AstraZeneca outside the submitted work. T.Y. declares no conflicts of interest.
References
- Rudin, C.M.; Brambilla, E.; Faivre-Finn, C.; Sage, J. Small-Cell Lung Cancer. Nat. Rev. Dis. Prim. 2021, 7, 3. [Google Scholar] [CrossRef]
- Pesch, B.; Kendzia, B.; Gustavsson, P.; Jöckel, K.-H.; Johnen, G.; Pohlabeln, H.; Olsson, A.; Ahrens, W.; Gross, I.M.; Brüske, I.; et al. Cigarette Smoking and Lung Cancer—Relative Risk Estimates for the Major Histological Types from a Pooled Analysis of Case–Control Studies. Int. J. Cancer 2012, 131, 1210–1219. [Google Scholar] [CrossRef]
- The National Lung Screening Trial Research Team. Reduced Lung-Cancer Mortality with Low-Dose Computed Tomographic Screening. N. Engl. J. Med. 2011, 365, 395–409. [Google Scholar] [CrossRef]
- Stahel, R.A.; Ginsberg, R.; Havemann, K.; Hirsch, F.R.; Ihde, D.C.; Jassem, J.; Karrer, K.; Herbert Maurer, L.; Osterlind, K.; Van Houtte, P. Staging and Prognostic Factors in Small Cell Lung Cancer: A Consensus Report. Lung Cancer 1989, 5, 119–126. [Google Scholar] [CrossRef]
- Peifer, M.; Fernández-Cuesta, L.; Sos, M.L.; George, J.; Seidel, D.; Kasper, L.H.; Plenker, D.; Leenders, F.; Sun, R.; Zander, T.; et al. Integrative Genome Analyses Identify Key Somatic Driver Mutations of Small-Cell Lung Cancer. Nat. Genet. 2012, 44, 1104–1110. [Google Scholar] [CrossRef]
- Rudin, C.M.; Durinck, S.; Stawiski, E.W.; Poirier, J.T.; Modrusan, Z.; Shames, D.S.; Bergbower, E.A.; Guan, Y.; Shin, J.; Guillory, J.; et al. Comprehensive Genomic Analysis Identifies SOX2 as a Frequently Amplified Gene in Small-Cell Lung Cancer. Nat. Genet. 2012, 44, 1111–1116. [Google Scholar] [CrossRef]
- Umemura, S.; Mimaki, S.; Makinoshima, H.; Tada, S.; Ishii, G.; Ohmatsu, H.; Niho, S.; Yoh, K.; Matsumoto, S.; Takahashi, A.; et al. Therapeutic Priority of the PI3K/AKT/MTOR Pathway in Small Cell Lung Cancers as Revealed by a Comprehensive Genomic Analysis. J. Thorac. Oncol. 2014, 9, 1324–1331. [Google Scholar] [CrossRef]
- George, J.; Lim, J.S.; Jang, S.J.; Cun, Y.; Ozretić, L.; Kong, G.; Leenders, F.; Lu, X.; Fernández-Cuesta, L.; Bosco, G.; et al. Comprehensive Genomic Profiles of Small Cell Lung Cancer. Nature 2015, 524, 47–53. [Google Scholar] [CrossRef]
- Meuwissen, R.; Linn, S.C.; Linnoila, R.I.; Zevenhoven, J.; Mooi, W.J.; Berns, A. Induction of Small Cell Lung Cancer by Somatic Inactivation of Both Trp53 and Rb1 in a Conditional Mouse Model. Cancer Cell 2003, 4, 181–189. [Google Scholar] [CrossRef]
- Udagawa, H.; Umemura, S.; Murakami, I.; Mimaki, S.; Makinoshima, H.; Ishii, G.; Miyoshi, T.; Kirita, K.; Matsumoto, S.; Yoh, K.; et al. Genetic Profiling-Based Prognostic Prediction of Patients with Advanced Small-Cell Lung Cancer in Large Scale Analysis. Lung Cancer 2018, 126, 182–188. [Google Scholar] [CrossRef]
- Chen, H.-Z.; Bonneville, R.; Paruchuri, A.; Reeser, J.W.; Wing, M.R.; Samorodnitsky, E.; Krook, M.A.; Smith, A.M.; Dao, T.; Miya, J.; et al. Genomic and Transcriptomic Characterization of Relapsed SCLC Through Rapid Research Autopsy. JTO Clin. Res. Rep. 2021, 2, 100164. [Google Scholar] [CrossRef] [PubMed]
- Stewart, C.A.; Gay, C.M.; Xi, Y.; Sivajothi, S.; Sivakamasundari, V.; Fujimoto, J.; Bolisetty, M.; Hartsfield, P.M.; Balasubramaniyan, V.; Chalishazar, M.D.; et al. Single-Cell Analyses Reveal Increased Intratumoral Heterogeneity after the Onset of Therapy Resistance in Small-Cell Lung Cancer. Nat. Cancer 2020, 1, 423–436. [Google Scholar] [CrossRef]
- George, J.; Maas, L.; Abedpour, N.; Cartolano, M.; Kaiser, L.; Fischer, R.N.; Scheel, A.H.; Weber, J.-P.; Hellmich, M.; Bosco, G.; et al. Evolutionary Trajectories of Small Cell Lung Cancer under Therapy. Nature 2024, 627, 880–889. [Google Scholar] [CrossRef]
- Baine, M.K.; Hsieh, M.-S.; Lai, W.V.; Egger, J.V.; Jungbluth, A.A.; Daneshbod, Y.; Beras, A.; Spencer, R.; Lopardo, J.; Bodd, F.; et al. SCLC Subtypes Defined by ASCL1, NEUROD1, POU2F3, and YAP1: A Comprehensive Immunohistochemical and Histopathologic Characterization. J. Thorac. Oncol. 2020, 15, 1823–1835. [Google Scholar] [CrossRef] [PubMed]
- Rudin, C.M.; Poirier, J.T.; Byers, L.A.; Dive, C.; Dowlati, A.; George, J.; Heymach, J.V.; Johnson, J.E.; Lehman, J.M.; MacPherson, D.; et al. Molecular Subtypes of Small Cell Lung Cancer: A Synthesis of Human and Mouse Model Data. Nat. Rev. Cancer 2019, 19, 289–297. [Google Scholar] [CrossRef]
- Heeke, S.; Gay, C.M.; Estecio, M.R.; Tran, H.; Morris, B.B.; Zhang, B.; Tang, X.; Raso, M.G.; Rocha, P.; Lai, S.; et al. Tumor- and Circulating-Free DNA Methylation Identifies Clinically Relevant Small Cell Lung Cancer Subtypes. Cancer Cell 2024, 42, 225–237. [Google Scholar] [CrossRef]
- Mollaoglu, G.; Guthrie, M.R.; Böhm, S.; Brägelmann, J.; Can, I.; Ballieu, P.M.; Marx, A.; George, J.; Heinen, C.; Chalishazar, M.D.; et al. MYC Drives Progression of Small Cell Lung Cancer to a Variant Neuroendocrine Subtype with Vulnerability to Aurora Kinase Inhibition. Cancer Cell 2017, 31, 270–285. [Google Scholar] [CrossRef] [PubMed]
- Ireland, A.S.; Micinski, A.M.; Kastner, D.W.; Guo, B.; Wait, S.J.; Spainhower, K.B.; Conley, C.C.; Chen, O.S.; Guthrie, M.R.; Soltero, D.; et al. MYC Drives Temporal Evolution of Small Cell Lung Cancer Subtypes by Reprogramming Neuroendocrine Fate. Cancer Cell 2020, 38, 60–78. [Google Scholar] [CrossRef]
- Borromeo, M.D.; Savage, T.K.; Kollipara, R.K.; He, M.; Augustyn, A.; Osborne, J.K.; Girard, L.; Minna, J.D.; Gazdar, A.F.; Cobb, M.H.; et al. ASCL1 and NEUROD1 Reveal Heterogeneity in Pulmonary Neuroendocrine Tumors and Regulate Distinct Genetic Programs. Cell Rep. 2016, 16, 1259–1272. [Google Scholar] [CrossRef]
- Lim, J.S.; Ibaseta, A.; Fischer, M.M.; Cancilla, B.; O’Young, G.; Cristea, S.; Luca, V.C.; Yang, D.; Jahchan, N.S.; Hamard, C.; et al. Intratumoural Heterogeneity Generated by Notch Signalling Promotes Small-Cell Lung Cancer. Nature 2017, 545, 360–364. [Google Scholar] [CrossRef]
- Shue, Y.T.; Drainas, A.P.; Li, N.Y.; Pearsall, S.M.; Morgan, D.; Sinnott-Armstrong, N.; Hipkins, S.Q.; Coles, G.L.; Lim, J.S.; Oro, A.E.; et al. A Conserved YAP/Notch/REST Network Controls the Neuroendocrine Cell Fate in the Lungs. Nat. Commun. 2022, 13, 2690. [Google Scholar] [CrossRef]
- Olsen, R.R.; Ireland, A.S.; Kastner, D.W.; Groves, S.M.; Spainhower, K.B.; Pozo, K.; Kelenis, D.P.; Whitney, C.P.; Guthrie, M.R.; Wait, S.J.; et al. ASCL1 Represses a SOX9(+) Neural Crest Stem-like State in Small Cell Lung Cancer. Genes. Dev. 2021, 35, 847–869. [Google Scholar] [CrossRef]
- Wu, Q.; Guo, J.; Liu, Y.; Zheng, Q.; Li, X.; Wu, C.; Fang, D.; Chen, X.; Ma, L.; Xu, P.; et al. YAP Drives Fate Conversion and Chemoresistance of Small Cell Lung Cancer. Sci. Adv. 2021, 7, eabg1850. [Google Scholar] [CrossRef]
- Huang, Y.-H.; Klingbeil, O.; He, X.-Y.; Wu, X.S.; Arun, G.; Lu, B.; Somerville, T.D.D.; Milazzo, J.P.; Wilkinson, J.E.; Demerdash, O.E.; et al. POU2F3 Is a Master Regulator of a Tuft Cell-like Variant of Small Cell Lung Cancer. Genes. Dev. 2018, 32, 915–928. [Google Scholar] [CrossRef]
- Pearsall, S.M.; Humphrey, S.; Revill, M.; Morgan, D.; Frese, K.K.; Galvin, M.; Kerr, A.; Carter, M.; Priest, L.; Blackhall, F.; et al. The Rare YAP1 Subtype of SCLC Revisited in a Biobank of 39 Circulating Tumor Cell Patient Derived Explant Models: A Brief Report. J. Thorac. Oncol. 2020, 15, 1836–1843. [Google Scholar] [CrossRef]
- Sonkin, D.; Thomas, A.; Teicher, B.A. Are Neuroendocrine Negative Small Cell Lung Cancer and Large Cell Neuroendocrine Carcinoma with WT RB1 Two Faces of the Same Entity? Lung Cancer Manag. 2019, 8, LMT13. [Google Scholar] [CrossRef]
- Ng, J.; Cai, L.; Girard, L.; Prall, O.W.J.; Rajan, N.; Khoo, C.; Batrouney, A.; Byrne, D.J.; Boyd, D.K.; Kersbergen, A.J.; et al. Molecular and Pathologic Characterization of YAP1-Expressing Small Cell Lung Cancer Cell Lines Leads to Reclassification as SMARCA4-Deficient Malignancies. Clin. Cancer Res. 2024, OF1–OF13. [Google Scholar] [CrossRef]
- Gay, C.M.; Stewart, C.A.; Park, E.M.; Diao, L.; Groves, S.M.; Heeke, S.; Nabet, B.Y.; Fujimoto, J.; Solis, L.M.; Lu, W.; et al. Patterns of Transcription Factor Programs and Immune Pathway Activation Define Four Major Subtypes of SCLC with Distinct Therapeutic Vulnerabilities. Cancer Cell 2021, 39, 346–360.e7. [Google Scholar] [CrossRef]
- Nabet, B.Y.; Hamidi, H.; Lee, M.C.; Banchereau, R.; Morris, S.; Adler, L.; Gayevskiy, V.; Elhossiny, A.M.; Srivastava, M.K.; Patil, N.S.; et al. Immune Heterogeneity in Small-Cell Lung Cancer and Vulnerability to Immune Checkpoint Blockade. Cancer Cell 2024, 42, 429–443.e4. [Google Scholar] [CrossRef]
- Johnson, B.E.; Grayson, J.; Makuch, R.W.; Linnoila, R.I.; Anderson, M.J.; Cohen, M.H.; Glatstein, E.; Minna, J.D.; Ihde, D.C. Ten-Year Survival of Patients with Small-Cell Lung Cancer Treated with Combination Chemotherapy with or without Irradiation. J. Clin. Oncol. 1990, 8, 396–401. [Google Scholar] [CrossRef]
- Warde, P.; Payne, D. Does Thoracic Irradiation Improve Survival and Local Control in Limited-Stage Small-Cell Carcinoma of the Lung? A Meta-Analysis. J. Clin. Oncol. 1992, 10, 890–895. [Google Scholar] [CrossRef] [PubMed]
- Pignon, J.-P.; Arriagada, R.; Ihde, D.C.; Johnson, D.H.; Perry, M.C.; Souhami, R.L.; Brodin, O.; Joss, R.A.; Kies, M.S.; Lebeau, B.; et al. A Meta-Analysis of Thoracic Radiotherapy for Small-Cell Lung Cancer. N. Engl. J. Med. 1992, 327, 1618–1624. [Google Scholar] [CrossRef]
- Martucci, N.; Morabito, A.; La Rocca, A.; De Luca, G.; De Cecio, R.; Botti, G.; Totaro, G.; Muto, P.; Picone, C.; Esposito, G.; et al. Surgery in Small-Cell Lung Cancer. Cancers 2021, 13, 390. [Google Scholar] [CrossRef] [PubMed]
- Nugent, J.L.; Bunn, P.A., Jr.; Matthews, M.J.; Ihde, D.C.; Cohen, M.H.; Gazdar, A.; Minna, J.D. CNS Metastases in Small Cell Bronchogenic Carcinoma. Increasing Frequency and Changing Pattern with Lengthening Survival. Cancer 1979, 44, 1885–1893. [Google Scholar] [CrossRef] [PubMed]
- Aupérin, A.; Arriagada, R.; Pignon, J.-P.; Le Péchoux, C.; Gregor, A.; Stephens, R.J.; Kristjansen, P.E.G.; Johnson, B.E.; Ueoka, H.; Wagner, H.; et al. Prophylactic Cranial Irradiation for Patients with Small-Cell Lung Cancer in Complete Remission. N. Engl. J. Med. 1999, 341, 476–484. [Google Scholar] [CrossRef]
- Horn, L.; Mansfield, A.S.; Szczęsna, A.; Havel, L.; Krzakowski, M.; Hochmair, M.J.; Huemer, F.; Losonczy, G.; Johnson, M.L.; Nishio, M.; et al. First-Line Atezolizumab plus Chemotherapy in Extensive-Stage Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 379, 2220–2229. [Google Scholar] [CrossRef]
- Paz-Ares, L.; Dvorkin, M.; Chen, Y.; Reinmuth, N.; Hotta, K.; Trukhin, D.; Statsenko, G.; Hochmair, M.J.; Özgüroğlu, M.; Ji, J.H.; et al. Durvalumab plus Platinum–Etoposide versus Platinum–Etoposide in First-Line Treatment of Extensive-Stage Small-Cell Lung Cancer (CASPIAN): A Randomised, Controlled, Open-Label, Phase 3 Trial. Lancet 2019, 394, 1929–1939. [Google Scholar] [CrossRef]
- Higgins, K.A.; Curran, W.J.; Liu, S.V.; Yu, W.; Brockman, M.; Johnson, A.; Bara, I.; Bradley, J.D. Patterns of Disease Progression after Carboplatin/Etoposide + Atezolizumab in Extensive-Stage Small-Cell Lung Cancer (ES-SCLC). Int. J. Radiat. Oncol. Biol. Phys. 2020, 108, 1398. [Google Scholar] [CrossRef]
- Chen, Y.; Paz-Ares, L.; Reinmuth, N.; Garassino, M.C.; Statsenko, G.; Hochmair, M.J.; Özgüroğlu, M.; Verderame, F.; Havel, L.; Losonczy, G.; et al. Impact of Brain Metastases on Treatment Patterns and Outcomes with First-Line Durvalumab Plus Platinum-Etoposide in Extensive-Stage SCLC (CASPIAN): A Brief Report. JTO Clin. Res. Rep. 2022, 3, 100330. [Google Scholar] [CrossRef]
- Rudin, C.M.; Awad, M.M.; Navarro, A.; Gottfried, M.; Peters, S.; Csőszi, T.; Cheema, P.K.; Rodriguez-Abreu, D.; Wollner, M.; Yang, J.C.-H.; et al. Pembrolizumab or Placebo Plus Etoposide and Platinum as First-Line Therapy for Extensive-Stage Small-Cell Lung Cancer: Randomized, Double-Blind, Phase III KEYNOTE-604 Study. J. Clin. Oncol. 2020, 38, 2369–2379. [Google Scholar] [CrossRef]
- Owonikoko, T.K.; Park, K.; Govindan, R.; Ready, N.; Reck, M.; Peters, S.; Dakhil, S.R.; Navarro, A.; Rodríguez-Cid, J.; Schenker, M.; et al. Nivolumab and Ipilimumab as Maintenance Therapy in Extensive-Disease Small-Cell Lung Cancer: CheckMate 451. J. Clin. Oncol. 2021, 39, 1349–1359. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Han, L.; Wu, L.; Chen, J.; Sun, H.; Wen, G.; Ji, Y.; Dvorkin, M.; Shi, J.; Pan, Z.; et al. Effect of First-Line Serplulimab vs Placebo Added to Chemotherapy on Survival in Patients with Extensive-Stage Small Cell Lung Cancer: The ASTRUM-005 Randomized Clinical Trial. JAMA 2022, 328, 1223–1232. [Google Scholar] [CrossRef]
- Spigel, D.R.; Cheng, Y.; Cho, B.C.; Laktionov, K.K.; Fang, J.; Chen, Y.; Zenke, Y.; Lee, K.H.; Wang, Q.; Navarro, A.; et al. ADRIATIC: Durvalumab (D) as Consolidation Treatment (Tx) for Patients (Pts) with Limited-Stage Small-Cell Lung Cancer (LS-SCLC). J. Clin. Oncol. 2024, 42 (Suppl. S17), LBA5. [Google Scholar] [CrossRef]
- Noda, K.; Nishiwaki, Y.; Kawahara, M.; Negoro, S.; Sugiura, T.; Yokoyama, A.; Fukuoka, M.; Mori, K.; Watanabe, K.; Tamura, T.; et al. Irinotecan plus Cisplatin Compared with Etoposide plus Cisplatin for Extensive Small-Cell Lung Cancer. N. Engl. J. Med. 2002, 346, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Hanna, N.; Bunn, P.A.; Langer, C.; Einhorn, L.; Guthrie, T.; Beck, T.; Ansari, R.; Ellis, P.; Byrne, M.; Morrison, M.; et al. Randomized Phase III Trial Comparing Irinotecan/Cisplatin with Etoposide/Cisplatin in Patients with Previously Untreated Extensive-Stage Disease Small-Cell Lung Cancer. J. Clin. Oncol. 2006, 24, 2038–2043. [Google Scholar] [CrossRef]
- Lara, P.N.; Natale, R.; Crowley, J.; Lenz, H.J.; Redman, M.W.; Carleton, J.E.; Jett, J.; Langer, C.J.; Kuebler, J.P.; Dakhil, S.R.; et al. Phase III Trial of Irinotecan/Cisplatin Compared with Etoposide/Cisplatin in Extensive-Stage Small-Cell Lung Cancer: Clinical and Pharmacogenomic Results from SWOG S0124. J. Clin. Oncol. 2009, 27, 2530–2535. [Google Scholar] [CrossRef]
- Slotman, B.J.; van Tinteren, H.; Praag, J.O.; Knegjens, J.L.; El Sharouni, S.Y.; Hatton, M.; Keijser, A.; Faivre-Finn, C.; Senan, S. Use of Thoracic Radiotherapy for Extensive Stage Small-Cell Lung Cancer: A Phase 3 Randomised Controlled Trial. Lancet 2015, 385, 36–42. [Google Scholar] [CrossRef]
- Slotman, B.; Faivre-Finn, C.; Kramer, G.; Rankin, E.; Snee, M.; Hatton, M.; Postmus, P.; Collette, L.; Musat, E.; Senan, S. Prophylactic Cranial Irradiation in Extensive Small-Cell Lung Cancer. N. Engl. J. Med. 2007, 357, 664–672. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Yamanaka, T.; Seto, T.; Harada, H.; Nokihara, H.; Saka, H.; Nishio, M.; Kaneda, H.; Takayama, K.; Ishimoto, O.; et al. Prophylactic Cranial Irradiation versus Observation in Patients with Extensive-Disease Small-Cell Lung Cancer: A Multicentre, Randomised, Open-Label, Phase 3 Trial. Lancet Oncol. 2017, 18, 663–671. [Google Scholar] [CrossRef]
- Lattuca-Truc, M.; Timsit, J.-F.; Levra, M.G.; Ruckly, S.; Villa, J.; Dumas, I.; Pinsolle, J.; Ferrer, L.; Guillem, P.; Moro-Sibilot, D.; et al. Trends in Response Rate and Survival in Small-Cell Lung Cancer Patients between 1997 and 2017. Lung Cancer 2019, 131, 122–127. [Google Scholar] [CrossRef]
- Karacz, C.M.; Yan, J.; Zhu, H.; Gerber, D.E. Timing, Sites, and Correlates of Lung Cancer Recurrence. Clin. Lung Cancer 2020, 21, 127–135.e3. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, M.E.R.; Ciuleanu, T.-E.; Tsekov, H.; Shparyk, Y.; Čučeviá, B.; Juhasz, G.; Thatcher, N.; Ross, G.A.; Dane, G.C.; Crofts, T. Phase III Trial Comparing Supportive Care Alone with Supportive Care With Oral Topotecan in Patients With Relapsed Small-Cell Lung Cancer. J. Clin. Oncol. 2006, 24, 5441–5447. [Google Scholar] [CrossRef] [PubMed]
- Roth, B.J.; Johnson, D.H.; Einhorn, L.H.; Schacter, L.P.; Cherng, N.C.; Cohen, H.J.; Crawford, J.; Randolph, J.A.; Goodlow, J.L.; Broun, G.O. Randomized Study of Cyclophosphamide, Doxorubicin, and Vincristine versus Etoposide and Cisplatin versus Alternation of These Two Regimens in Extensive Small-Cell Lung Cancer: A Phase III Trial of the Southeastern Cancer Study Group. J. Clin. Oncol. 1992, 10, 282–291. [Google Scholar] [CrossRef] [PubMed]
- von Pawel, J.; Schiller, J.H.; Shepherd, F.A.; Fields, S.Z.; Kleisbauer, J.P.; Chrysson, N.G.; Stewart, D.J.; Clark, P.I.; Palmer, M.C.; Depierre, A.; et al. Topotecan Versus Cyclophosphamide, Doxorubicin, and Vincristine for the Treatment of Recurrent Small-Cell Lung Cancer. J. Clin. Oncol. 1999, 17, 658. [Google Scholar] [CrossRef]
- Trigo, J.; Subbiah, V.; Besse, B.; Moreno, V.; López, R.; Sala, M.A.; Peters, S.; Ponce, S.; Fernández, C.; Alfaro, V.; et al. Lurbinectedin as Second-Line Treatment for Patients with Small-Cell Lung Cancer: A Single-Arm, Open-Label, Phase 2 Basket Trial. Lancet Oncol. 2020, 21, 645–654. [Google Scholar] [CrossRef]
- Myung-Ju, A.; Chul, C.B.; Enriqueta, F.; Ippokratis, K.; Kadoaki, O.; Margarita, M.; Oscar, J.-V.; Sabin, H.; Hiroki, I.; Jong-Seok, L.; et al. Tarlatamab for Patients with Previously Treated Small-Cell Lung Cancer. N. Engl. J. Med. 2023, 389, 2063–2075. [Google Scholar] [CrossRef]
- Besse, B.; Paz-Ares, L.G.; Peters, S.; Cappuzzo, F.; Reck, M.; Calles, A.; Califano, R.; Lopez-Vilariño, J.A.; Veramendi, S.; Kahatt, C.M.; et al. A Phase III Study of Lurbinectedin Alone or in Combination with Irinotecan vs Investigator’s Choice (Topotecan or Irinotecan) in Patients with Relapsed Small Cell Lung Cancer (SCLC; LAGOON Trial). J. Clin. Oncol. 2023, 41 (Suppl. S16), TPS8613. [Google Scholar] [CrossRef]
- Paz-Ares, L.; Reck, M.; Peters, S.; Borghaei, H.; Herbst, R.; Siddiqui, M.; Cuchelkar, V.; Bhatt, K.; Chakrabarti, D.; Wang, L.; et al. EP14.01-015 IMforte: A Phase III Study of Lurbinectedin and Atezolizumab Versus Atezolizumab as Maintenance Therapy in ES-SCLC. J. Thorac. Oncol. 2022, 17, S532–S533. [Google Scholar] [CrossRef]
- Paz-Ares, L.G.; Felip, E.; Ahn, M.-J.; Blackhall, F.H.; Borghaei, H.; Cho, B.C.; Johnson, M.L.; Ramalingam, S.S.; Reck, M.; Zhang, A.; et al. Randomized Phase 3 Study of Tarlatamab, a DLL3-Targeting Bispecific T-Cell Engager (BiTE), Compared to Standard of Care in Patients with Relapsed Small Cell Lung Cancer (DeLLphi-304). J. Clin. Oncol. 2023, 41 (Suppl. S16), TPS8611. [Google Scholar] [CrossRef]
- Baize, N.; Monnet, I.; Greillier, L.; Geier, M.; Lena, H.; Janicot, H.; Vergnenegre, A.; Crequit, J.; Lamy, R.; Auliac, J.-B.; et al. Carboplatin plus Etoposide versus Topotecan as Second-Line Treatment for Patients with Sensitive Relapsed Small-Cell Lung Cancer: An Open-Label, Multicentre, Randomised, Phase 3 Trial. Lancet Oncol. 2020, 21, 1224–1233. [Google Scholar] [CrossRef]
- Goto, K.; Ohe, Y.; Shibata, T.; Seto, T.; Takahashi, T.; Nakagawa, K.; Tanaka, H.; Takeda, K.; Nishio, M.; Mori, K.; et al. Combined Chemotherapy with Cisplatin, Etoposide, and Irinotecan versus Topotecan Alone as Second-Line Treatment for Patients with Sensitive Relapsed Small-Cell Lung Cancer (JCOG0605): A Multicentre, Open-Label, Randomised Phase 3 Trial. Lancet Oncol. 2016, 17, 1147–1157. [Google Scholar] [CrossRef]
- Masuda, N.; Fukuoka, M.; Kusunoki, Y.; Matsui, K.; Takifuji, N.; Kudoh, S.; Negoro, S.; Nishioka, M.; Nakagawa, K.; Takada, M. CPT-11: A New Derivative of Camptothecin for the Treatment of Refractory or Relapsed Small-Cell Lung Cancer. J. Clin. Oncol. 1992, 10, 1225–1229. [Google Scholar] [CrossRef] [PubMed]
- Smit, E.F.; Fokkema, E.; Biesma, B.; Groen, H.J.; Snoek, W.; Postmus, P.E. A Phase II Study of Paclitaxel in Heavily Pretreated Patients with Small-Cell Lung Cancer. Br. J. Cancer 1998, 77, 347–351. [Google Scholar] [CrossRef]
- Yamamoto, N.; Tsurutani, J.; Yoshimura, N.; Asai, G.Y.O.; Moriyama, A.; Nakagawa, K.; Kudoh, S.; Takada, M.; Minato, Y.; Fukuoka, M. Phase II Study of Weekly Paclitaxel for Relapsed and Refractory Small Cell Lung Cancer. Anticancer Res. 2006, 26, 777–781. [Google Scholar]
- Pietanza, M.C.; Kadota, K.; Huberman, K.; Sima, C.S.; Fiore, J.J.; Sumner, D.K.; Travis, W.D.; Heguy, A.; Ginsberg, M.S.; Holodny, A.I.; et al. Phase II Trial of Temozolomide in Patients with Relapsed Sensitive or Refractory Small Cell Lung Cancer, with Assessment of Methylguanine-DNA Methyltransferase as a Potential Biomarker. Clin. Cancer Res. 2012, 18, 1138–1145. [Google Scholar] [CrossRef] [PubMed]
- von Pawel, J.; Jotte, R.; Spigel, D.R.; O’Brien, M.E.R.; Socinski, M.A.; Mezger, J.; Steins, M.; Bosquée, L.; Bubis, J.; Nackaerts, K.; et al. Randomized Phase III Trial of Amrubicin Versus Topotecan as Second-Line Treatment for Patients with Small-Cell Lung Cancer. J. Clin. Oncol. 2014, 32, 4012–4019. [Google Scholar] [CrossRef]
- Antonia, S.J.; López-Martin, J.A.; Bendell, J.; Ott, P.A.; Taylor, M.; Eder, J.P.; Jäger, D.; Pietanza, M.C.; Le, D.T.; de Braud, F.; et al. Nivolumab Alone and Nivolumab plus Ipilimumab in Recurrent Small-Cell Lung Cancer (CheckMate 032): A Multicentre, Open-Label, Phase 1/2 Trial. Lancet Oncol. 2016, 17, 883–895. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.C.; Piha-Paul, S.A.; Lopez-Martin, J.; Schellens, J.H.M.; Kao, S.; Miller, W.H.; Delord, J.-P.; Gao, B.; Planchard, D.; Gottfried, M.; et al. Pembrolizumab After Two or More Lines of Previous Therapy in Patients with Recurrent or Metastatic SCLC: Results From the KEYNOTE-028 and KEYNOTE-158 Studies. J. Thorac. Oncol. 2020, 15, 618–627. [Google Scholar] [CrossRef]
- Spigel, D.R.; Vicente, D.; Ciuleanu, T.E.; Gettinger, S.; Peters, S.; Horn, L.; Audigier-Valette, C.; Pardo Aranda, N.; Juan-Vidal, O.; Cheng, Y.; et al. Second-Line Nivolumab in Relapsed Small-Cell Lung Cancer: CheckMate 331. Ann. Oncol. 2021, 32, 631–641. [Google Scholar] [CrossRef]
- Daniel, D.B.; Rudin, C.M.; Hart, L.; Spigel, D.R.; Edelman, M.J.; Goldschmidt, J.; Bordoni, R.; Glisson, B.; Burns, T.F.; Dowlati, A.; et al. Results of a Randomized, Placebo-Controlled, Phase 2 Study of Tarextumab (TRXT, Anti-Notch2/3) in Combination with Etoposide and Platinum (EP) in Patients (Pts) with Untreated Extensive-Stage Small-Cell Lung Cancer (ED-SCLC). Ann. Oncol. 2017, 28 (Suppl. S5), v540. [Google Scholar] [CrossRef]
- Melichar, B.; Adenis, A.; Lockhart, A.C.; Bennouna, J.; Dees, E.C.; Kayaleh, O.; Obermannova, R.; DeMichele, A.; Zatloukal, P.; Zhang, B.; et al. Safety and Activity of Alisertib, an Investigational Aurora Kinase A Inhibitor, in Patients with Breast Cancer, Small-Cell Lung Cancer, Non-Small-Cell Lung Cancer, Head and Neck Squamous-Cell Carcinoma, and Gastro-Oesophageal Adenocarcinoma: A Five-Arm Ph. Lancet Oncol. 2015, 16, 395–405. [Google Scholar] [CrossRef]
- Owonikoko, T.K.; Niu, H.; Nackaerts, K.; Csoszi, T.; Ostoros, G.; Mark, Z.; Baik, C.; Joy, A.A.; Chouaid, C.; Jaime, J.C.; et al. Randomized Phase II Study of Paclitaxel plus Alisertib versus Paclitaxel plus Placebo as Second-Line Therapy for SCLC: Primary and Correlative Biomarker Analyses. J. Thorac. Oncol. 2020, 15, 274–287. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Shim, J.; Mortimer, P.G.S.; Smith, S.A.; Godin, R.E.; Hollingsworth, S.J.; Kim, H.-J.; Jung, H.A.; Sun, J.-M.; Park, W.-Y.; et al. Biomarker-Driven Phase 2 Umbrella Trial Study for Patients with Recurrent Small Cell Lung Cancer Failing Platinum-Based Chemotherapy. Cancer 2020, 126, 4002–4012. [Google Scholar] [CrossRef] [PubMed]
- Pietanza, M.C.; Litvak, A.M.; Varghese, A.M.; Krug, L.M.; Fleisher, M.; Teitcher, J.B.; Holodny, A.I.; Sima, C.S.; Woo, K.M.; Ng, K.K.; et al. A Phase I Trial of the Hedgehog Inhibitor, Sonidegib (LDE225), in Combination with Etoposide and Cisplatin for the Initial Treatment of Extensive Stage Small Cell Lung Cancer. Lung Cancer 2016, 99, 23–30. [Google Scholar] [CrossRef]
- Belani, C.P.; Dahlberg, S.E.; Rudin, C.M.; Fleisher, M.; Chen, H.X.; Takebe, N.; Ramalingam, S.S.; Schiller, J.H. Three-Arm Randomized Phase II Study of Cisplatin and Etoposide (CE) versus CE with Either Vismodegib (V) or Cixutumumab (Cx) for Patients with Extensive Stage-Small Cell Lung Cancer (ES-SCLC) (ECOG 1508). J. Clin. Oncol. 2013, 31 (Suppl. S15), 7508. [Google Scholar] [CrossRef]
- Pandya, K.J.; Dahlberg, S.; Hidalgo, M.; Cohen, R.B.; Lee, M.W.; Schiller, J.H.; Johnson, D.H. A Randomized, Phase II Trial of Two Dose Levels of Temsirolimus (CCI-779) in Patients with Extensive-Stage Small-Cell Lung Cancer Who Have Responding or Stable Disease after Induction Chemotherapy: A Trial of the Eastern Cooperative Oncology Group (E1500). J. Thorac. Oncol. 2007, 2, 1036–1041. [Google Scholar] [CrossRef]
- Tarhini, A.; Kotsakis, A.; Gooding, W.; Shuai, Y.; Petro, D.; Friedland, D.; Belani, C.P.; Dacic, S.; Argiris, A. Phase II Study of Everolimus (RAD001) in Previously Treated Small Cell Lung Cancer. Clin. Cancer Res. 2010, 16, 5900–5907. [Google Scholar] [CrossRef]
- Sun, J.M.; Kim, J.R.; Do, I.G.; Lee, S.Y.; Lee, J.; Choi, Y.L.; Ahn, J.S.; Ahn, M.J.; Park, K. A Phase-1b Study of Everolimus plus Paclitaxel in Patients with Small-Cell Lung Cancer. Br. J. Cancer 2013, 109, 1482–1487. [Google Scholar] [CrossRef][Green Version]
- Thomas, A.; Takahashi, N.; Rajapakse, V.N.; Zhang, X.; Sun, Y.; Ceribelli, M.; Wilson, K.M.; Zhang, Y.; Beck, E.; Sciuto, L.; et al. Therapeutic Targeting of ATR Yields Durable Regressions in Small Cell Lung Cancers with High Replication Stress. Cancer Cell 2021, 39, 566–579.e7. [Google Scholar] [CrossRef]
- Byers, L.A.; Navarro, A.; Schaefer, E.; Johnson, M.; Özgüroğlu, M.; Han, J.-Y.; Bondarenko, I.; Cicin, I.; Dragnev, K.H.; Abel, A.; et al. A Phase II Trial of Prexasertib (LY2606368) in Patients with Extensive-Stage Small-Cell Lung Cancer. Clin. Lung Cancer 2021, 22, 531–540. [Google Scholar] [CrossRef] [PubMed]
- Bauer, T.M.; Moore, K.N.; Rader, J.S.; Simpkins, F.; Mita, A.C.; Beck, J.T.; Hart, L.; Chu, Q.; Oza, A.; Tinker, A.V.; et al. A Phase Ib Study Assessing the Safety, Tolerability, and Efficacy of the First-in-Class Wee1 Inhibitor Adavosertib (AZD1775) as Monotherapy in Patients with Advanced Solid Tumors. Target. Oncol. 2023, 18, 517–530. [Google Scholar] [CrossRef]
- von Pawel, J.; Vynnychenko, I.; Jiang, H.; Huang, Y.; Dennis, P.A. A Phase II, Open-Label, Multi-Arm Study of Novel Combinations of Immunotherapies or DDR Inhibitors in Platinum-Refractory, Extensive Disease Small-Cell Lung Cancer (ED-SCLC): BALTIC. J. Clin. Oncol. 2017, 35 (Suppl. S15), TPS8585. [Google Scholar] [CrossRef]
- Mita, M.; Gordon, M.; Rosen, L.; Kapoor, N.; Choy, G.; Redkar, S.; Taverna, P.; Oganesian, A.; Sahai, A.; Azab, M.; et al. Phase 1B Study of Amuvatinib in Combination with Five Standard Cancer Therapies in Adults with Advanced Solid Tumors. Cancer Chemother. Pharmacol. 2014, 74, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Byers, L.A.; Horn, L.; Ghandi, J.; Kloecker, G.; Owonikoko, T.; Naheed Waqar, S.; Krzakowski, M.; Cardnell, R.J.; Fujimoto, J.; Taverna, P.; et al. A Phase 2, Open-Label, Multi-Center Study of Amuvatinib in Combination with Platinum Etoposide Chemotherapy in Platinum-Refractory Small Cell Lung Cancer Patients. Oncotarget 2017, 8, 81441–81454. [Google Scholar] [CrossRef]
- de Bono, J.; Ramanathan, R.K.; Mina, L.; Chugh, R.; Glaspy, J.; Rafii, S.; Kaye, S.; Sachdev, J.; Heymach, J.; Smith, D.C.; et al. Phase I, Dose-Escalation, Two-Part Trial of the PARP Inhibitor Talazoparib in Patients with Advanced Germline BRCA1/2 Mutations and Selected Sporadic Cancers. Cancer Discov. 2017, 7, 620–629. [Google Scholar] [CrossRef]
- Park, S.; Kim, Y.J.; Min, Y.J.; Mortimer, P.G.S.; Kim, H.-J.; Smith, S.A.; Dean, E.; Jung, H.A.; Sun, J.-M.; Park, W.-Y.; et al. Biomarker-Driven Phase 2 Umbrella Trial: Clinical Efficacy of Olaparib Monotherapy and Combination with Ceralasertib (AZD6738) in Small Cell Lung Cancer. Cancer 2024, 130, 541–552. [Google Scholar] [CrossRef]
- Owonikoko, T.K.; Dahlberg, S.E.; Sica, G.L.; Wagner, L.I.; Wade, J.L.; Srkalovic, G.; Lash, B.W.; Leach, J.W.; Leal, T.B.; Aggarwal, C.; et al. Randomized Phase II Trial of Cisplatin and Etoposide in Combination with Veliparib or Placebo for Extensive-Stage Small-Cell Lung Cancer: ECOG-ACRIN 2511 Study. J. Clin. Oncol. 2018, 37, 222–229. [Google Scholar] [CrossRef]
- Byers, L.A.; Bentsion, D.; Gans, S.; Penkov, K.; Son, C.; Sibille, A.; Owonikoko, T.K.; Groen, H.J.M.; Gay, C.M.; Fujimoto, J.; et al. Veliparib in Combination with Carboplatin and Etoposide in Patients with Treatment-Naïve Extensive-Stage Small Cell Lung Cancer: A Phase 2 Randomized Study. Clin. Cancer Res. 2021, 27, 3884–3895. [Google Scholar] [CrossRef]
- Chiappori, A.A.; Otterson, G.A.; Dowlati, A.; Traynor, A.M.; Horn, L.; Owonikoko, T.K.; Ross, H.J.; Hann, C.L.; Abu Hejleh, T.; Nieva, J.; et al. A Randomized Phase II Study of Linsitinib (OSI-906) Versus Topotecan in Patients with Relapsed Small-Cell Lung Cancer. Oncologist 2016, 21, 1163–1164. [Google Scholar] [CrossRef]
- Lara, P.N.; Chansky, K.; Davies, A.M.; Franklin, W.A.; Gumerlock, P.H.; Guaglianone, P.P.; Atkins, J.N.; Farneth, N.; Mack, P.C.; Crowley, J.J.; et al. Bortezomib (PS-341) in Relapsed or Refractory Extensive Stage Small Cell Lung Cancer: A Southwest Oncology Group Phase II Trial (S0327). J. Thorac. Oncol. 2006, 1, 996–1001. [Google Scholar] [CrossRef]
- Baggstrom, M.Q.; Qi, Y.; Koczywas, M.; Argiris, A.; Johnson, E.A.; Millward, M.J.; Murphy, S.C.; Erlichman, C.; Rudin, C.M.; Govindan, R. A Phase II Study of AT-101 (Gossypol) in Chemotherapy-Sensitive Recurrent Extensive-Stage Small Cell Lung Cancer. J. Thorac. Oncol. 2011, 6, 1757–1760. [Google Scholar] [CrossRef] [PubMed]
- Rudin, C.M.; Hann, C.L.; Garon, E.B.; Ribeiro de Oliveira, M.; Bonomi, P.D.; Camidge, D.R.; Chu, Q.; Giaccone, G.; Khaira, D.; Ramalingam, S.S.; et al. Phase II Study of Single-Agent Navitoclax (ABT-263) and Biomarker Correlates in Patients with Relapsed Small Cell Lung Cancer. Clin. Cancer Res. 2012, 18, 3163–3169. [Google Scholar] [CrossRef] [PubMed]
- Suk Heist, R.; Fain, J.; Chinnasami, B.; Khan, W.; Molina, J.R.; Sequist, L.V.; Temel, J.S.; Fidias, P.; Brainerd, V.; Leopold, L.; et al. Phase I/II Study of AT-101 with Topotecan in Relapsed and Refractory Small Cell Lung Cancer. J. Thorac. Oncol. 2010, 5, 1637–1643. [Google Scholar] [CrossRef] [PubMed]
- Paik, P.K.; Rudin, C.M.; Pietanza, M.C.; Brown, A.; Rizvi, N.A.; Takebe, N.; Travis, W.; James, L.; Ginsberg, M.S.; Juergens, R.; et al. A Phase II Study of Obatoclax Mesylate, a Bcl-2 Antagonist, plus Topotecan in Relapsed Small Cell Lung Cancer. Lung Cancer 2011, 74, 481–485. [Google Scholar] [CrossRef]
- Rudin, C.M.; Salgia, R.; Wang, X.; Hodgson, L.D.; Masters, G.A.; Green, M.; Vokes, E.E. Randomized Phase II Study of Carboplatin and Etoposide with or Without the Bcl-2 Antisense Oligonucleotide Oblimersen for Extensive-Stage Small-Cell Lung Cancer: CALGB 30103. J. Clin. Oncol. 2008, 26, 870–876. [Google Scholar] [CrossRef]
- Langer, C.J.; Albert, I.; Ross, H.J.; Kovacs, P.; Blakely, L.J.; Pajkos, G.; Somfay, A.; Zatloukal, P.; Kazarnowicz, A.; Moezi, M.M.; et al. Randomized Phase II Study of Carboplatin and Etoposide with or without Obatoclax Mesylate in Extensive-Stage Small Cell Lung Cancer. Lung Cancer 2014, 85, 420–428. [Google Scholar] [CrossRef]
- Schelman, W.R.; Mohammed, T.A.; Traynor, A.M.; Kolesar, J.M.; Marnocha, R.M.; Eickhoff, J.; Keppen, M.; Alberti, D.B.; Wilding, G.; Takebe, N.; et al. A Phase I Study of AT-101 with Cisplatin and Etoposide in Patients with Advanced Solid Tumors with an Expanded Cohort in Extensive-Stage Small Cell Lung Cancer. Investig. New Drugs 2014, 32, 295–302. [Google Scholar] [CrossRef]
- Qin, A.; Kalemkerian, G.P.; Mohindra, N.A.; Patel, J.D.; Karapetis, C.S.; Carlisle, J.W.; Sands, J.; Spira, A.I.; Gao, B.; Amin, H.; et al. First-in-Human Study of Pelcitoclax (APG-1252) in Combination with Paclitaxel in Patients (Pts) with Relapsed/Refractory Small-Cell Lung Cancer (R/R SCLC). J. Clin. Oncol. 2022, 40 (Suppl. S16), e20612. [Google Scholar] [CrossRef]
- Lai, C.-H.; Park, K.-S.; Lee, D.-H.; Alberobello, A.T.; Raffeld, M.; Pierobon, M.; Pin, E.; Petricoin III, E.F.; Wang, Y.; Giaccone, G. HSP-90 Inhibitor Ganetespib Is Synergistic with Doxorubicin in Small Cell Lung Cancer. Oncogene 2014, 33, 4867–4876. [Google Scholar] [CrossRef]
- Gutierrez, M.; Guo, R.; Giaccone, G.; Liu, S.V.; Hao, Z.; Hilton, C.; Hinson, J.M.; Kris, M.G.; Orlemans, E.O.; Drilon, A. Phase 1 Multicenter Study of the HSP90 Inhibitor SNX-5422 plus Carboplatin and Paclitaxel in Patients with Lung Cancers. Lung Cancer 2021, 162, 23–28. [Google Scholar] [CrossRef]
- Hall, P.E.; Ready, N.; Johnston, A.; Bomalaski, J.S.; Venhaus, R.R.; Sheaff, M.; Krug, L.; Szlosarek, P.W. Phase II Study of Arginine Deprivation Therapy with Pegargiminase in Patients with Relapsed Sensitive or Refractory Small-Cell Lung Cancer. Clin. Lung Cancer 2020, 21, 527–533. [Google Scholar] [CrossRef]
- Han, J.-Y.; Lim, K.Y.; Yu, S.Y.; Yun, T.; Kim, H.T.; Lee, J.S. A Phase 2 Study of Irinotecan, Cisplatin, and Simvastatin for Untreated Extensive-Disease Small Cell Lung Cancer. Cancer 2011, 117, 2178–2185. [Google Scholar] [CrossRef] [PubMed]
- Seckl, M.J.; Ottensmeier, C.H.; Cullen, M.; Schmid, P.; Ngai, Y.; Muthukumar, D.; Thompson, J.; Harden, S.; Middleton, G.; Fife, K.M.; et al. Multicenter, Phase III, Randomized, Double-Blind, Placebo-Controlled Trial of Pravastatin Added to First-Line Standard Chemotherapy in Small-Cell Lung Cancer (LUNGSTAR). J. Clin. Oncol. 2017, 35, 1506–1514. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Lee, S.-H.; Lee, G.K.; Lim, E.J.; Han, J.-Y. A Randomized Phase II Study of Irinotecan Plus Cisplatin with or without Simvastatin in Ever-Smokers with Extended Disease Small Cell Lung Cancer. Cancer Res. Treat. 2023, 55, 885–893. [Google Scholar] [CrossRef]
- Gandhi, L.; Harding, M.W.; Neubauer, M.; Langer, C.J.; Moore, M.; Ross, H.J.; Johnson, B.E.; Lynch, T.J. A Phase II Study of the Safety and Efficacy of the Multidrug Resistance Inhibitor VX-710 Combined with Doxorubicin and Vincristine in Patients with Recurrent Small Cell Lung Cancer. Cancer 2007, 109, 924–932. [Google Scholar] [CrossRef]
- Schrump, D.S.; Fischette, M.R.; Nguyen, D.M.; Zhao, M.; Li, X.; Kunst, T.F.; Hancox, A.; Hong, J.A.; Chen, G.A.; Kruchin, E.; et al. Clinical and Molecular Responses in Lung Cancer Patients Receiving Romidepsin. Clin. Cancer Res. 2008, 14, 188–198. [Google Scholar] [CrossRef]
- Otterson, G.A.; Hodgson, L.; Pang, H.; Vokes, E.E. Phase II Study of the Histone Deacetylase Inhibitor Romidepsin in Relapsed Small Cell Lung Cancer (Cancer and Leukemia Group B 30304). J. Thorac. Oncol. 2010, 5, 1644–1648. [Google Scholar] [CrossRef]
- de Marinis, F.; Atmaca, A.; Tiseo, M.; Giuffreda, L.; Rossi, A.; Gebbia, V.; Antonio, C.D.; Zotto, L.D.; Al-Batran, S.-E.; Marsoni, S.; et al. A Phase II Study of the Histone Deacetylase Inhibitor Panobinostat (LBH589) in Pretreated Patients with Small-Cell Lung Cancer. J. Thorac. Oncol. 2013, 8, 1091–1094. [Google Scholar] [CrossRef]
- Tarhini, A.A.; Zahoor, H.; McLaughlin, B.; Gooding, W.E.; Schmitz, J.C.; Siegfried, J.M.; Socinski, M.A.; Argiris, A. Phase I Trial of Carboplatin and Etoposide in Combination with Panobinostat in Patients with Lung Cancer. Anticancer Res. 2013, 33, 4475–4481. [Google Scholar]
- Gentzler, R.D.; Villaruz, L.C.; Rhee, J.C.; Horton, B.; Mock, J.; Hanley, M.; Kim, K.; Rudek, M.A.; Phelps, M.A.; Carducci, M.A.; et al. Phase I Study of Entinostat, Atezolizumab, Carboplatin, and Etoposide in Previously Untreated Extensive-Stage Small Cell Lung Cancer, ETCTN 10399. Oncologist 2023, 28, 1007–e1107. [Google Scholar] [CrossRef]
- Balasubramaniam, S.; Redon, C.E.; Peer, C.J.; Bryla, C.; Lee, M.-J.; Trepel, J.B.; Tomita, Y.; Rajan, A.; Giaccone, G.; Bonner, W.M.; et al. Phase I Trial of Belinostat with Cisplatin and Etoposide in Advanced Solid Tumors, with a Focus on Neuroendocrine and Small Cell Cancers of the Lung. Anticancer Drugs 2018, 29, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Bauer, T.M.; Besse, B.; Martinez-Marti, A.; Trigo, J.M.; Moreno, V.; Garrido, P.; Ferron-Brady, G.; Wu, Y.; Park, J.; Collingwood, T.; et al. Phase I, Open-Label, Dose-Escalation Study of the Safety, Pharmacokinetics, Pharmacodynamics, and Efficacy of GSK2879552 in Relapsed/Refractory SCLC. J. Thorac. Oncol. 2019, 14, 1828–1838. [Google Scholar] [CrossRef] [PubMed]
- Niell, H.B.; Herndon, J.E.; Miller, A.A.; Watson, D.M.; Sandler, A.B.; Kelly, K.; Marks, R.S.; Perry, M.C.; Ansari, R.H.; Otterson, G.; et al. Randomized Phase III Intergroup Trial of Etoposide and Cisplatin with or Without Paclitaxel and Granulocyte Colony-Stimulating Factor in Patients With Extensive-Stage Small-Cell Lung Cancer: Cancer and Leukemia Group B Trial 9732. J. Clin. Oncol. 2005, 23, 3752–3759. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Sun, Y.; Lei, Y.; Yang, K.; Tang, R. YAP1 Promotes Multidrug Resistance of Small Cell Lung Cancer by CD74-Related Signaling Pathways. Cancer Med. 2020, 9, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Tong, Q.; Ouyang, S.; Chen, R.; Huang, J.; Guo, L. MYCN-Mediated Regulation of the HES1 Promoter Enhances the Chemoresistance of Small-Cell Lung Cancer by Modulating Apoptosis. Am. J. Cancer Res. 2019, 9, 1938–1956. [Google Scholar]
- Kumari, A.; Gesumaria, L.; Liu, Y.-J.; Hughitt, V.K.; Zhang, X.; Ceribelli, M.; Wilson, K.M.; Klumpp-Thomas, C.; Chen, L.; McKnight, C.; et al. MTOR Inhibition Overcomes RSK3-Mediated Resistance to BET Inhibitors in Small Cell Lung Cancer. JCI Insight 2023, 8, e156657. [Google Scholar] [CrossRef]
- Li, J.-M.; Hsu, P.-C.; Kuan, F.-C.; Shi, C.-S.; Yang, C.-T. The Cancer Stemness Inhibitor Napabucasin Suppresses Small Cell Lung Cancer Growth through SOX2 Expression. Am. J. Cancer Res. 2022, 12, 4637–4651. [Google Scholar]
- Deng, H.; Chen, Y.; Wang, L.; Zhang, Y.; Hang, Q.; Li, P.; Zhang, P.; Ji, J.; Song, H.; Chen, M.; et al. PI3K/MTOR Inhibitors Promote G6PD Autophagic Degradation and Exacerbate Oxidative Stress Damage to Radiosensitize Small Cell Lung Cancer. Cell Death Dis. 2023, 14, 652. [Google Scholar] [CrossRef]
- Quintanal-Villalonga, A.; Taniguchi, H.; Hao, Y.; Chow, A.; Zhan, Y.A.; Chavan, S.S.; Uddin, F.; Allaj, V.; Manoj, P.; Shah, N.S.; et al. Inhibition of XPO1 Sensitizes Small Cell Lung Cancer to First- and Second-Line Chemotherapy. Cancer Res. 2022, 82, 472–483. [Google Scholar] [CrossRef]
- Sen, T.; Tong, P.; Stewart, C.A.; Cristea, S.; Valliani, A.; Shames, D.S.; Redwood, A.B.; Fan, Y.H.; Li, L.; Glisson, B.S.; et al. CHK1 Inhibition in Small-Cell Lung Cancer Produces Single-Agent Activity in Biomarker-Defined Disease Subsets and Combination Activity with Cisplatin or Olaparib. Cancer Res. 2017, 77, 3870–3884. [Google Scholar] [CrossRef]
- Hsu, W.-H.; Zhao, X.; Zhu, J.; Kim, I.-K.; Rao, G.; McCutcheon, J.; Hsu, S.-T.; Teicher, B.; Kallakury, B.; Dowlati, A.; et al. Checkpoint Kinase 1 Inhibition Enhances Cisplatin Cytotoxicity and Overcomes Cisplatin Resistance in SCLC by Promoting Mitotic Cell Death. J. Thorac. Oncol. 2019, 14, 1032–1045. [Google Scholar] [CrossRef]
- Ran, X.; Wu, B.X.; Shi, M.; Song, L.; Nixon, K.; Philip, V.; He, H.H.; Tsao, M.-S.; Lok, B.H. CRISPR Screen of Druggable Targets in Small Cell Lung Cancer Identified ATM Inhibitor (AZD1390) as a Radiosensitizer. Int. J. Radiat. Oncol. Biol. Phys. 2024, 118, 1308–1314. [Google Scholar] [CrossRef]
- Kodama, M.; Toyokawa, G.; Sugahara, O.; Sugiyama, S.; Haratake, N.; Yamada, Y.; Wada, R.; Takamori, S.; Shimokawa, M.; Takenaka, T.; et al. Modulation of Host Glutamine Anabolism Enhances the Sensitivity of Small Cell Lung Cancer to Chemotherapy. Cell Rep. 2023, 42, 112899. [Google Scholar] [CrossRef]
- Bola, B.M.; Chadwick, A.L.; Michopoulos, F.; Blount, K.G.; Telfer, B.A.; Williams, K.J.; Smith, P.D.; Critchlow, S.E.; Stratford, I.J. Inhibition of Monocarboxylate Transporter-1 (MCT1) by AZD3965 Enhances Radiosensitivity by Reducing Lactate Transport. Mol. Cancer Ther. 2014, 13, 2805–2816. [Google Scholar] [CrossRef] [PubMed]
- Thirusangu, P.; Ray, U.; Sarkar Bhattacharya, S.; Oien, D.B.; Jin, L.; Staub, J.; Kannan, N.; Molina, J.R.; Shridhar, V. PFKFB3 Regulates Cancer Stemness through the Hippo Pathway in Small Cell Lung Carcinoma. Oncogene 2022, 41, 4003–4017. [Google Scholar] [CrossRef]
- Wang, J.; Lin, W.; Li, R.; Cheng, H.; Sun, S.; Shao, F.; Yang, Y.; Zhang, L.; Feng, X.; Gao, S.; et al. The Deubiquitinase USP13 Maintains Cancer Cell Stemness by Promoting FASN Stability in Small Cell Lung Cancer. Front. Oncol. 2022, 12, 899987. [Google Scholar] [CrossRef]
- Li, H.; Ma, L.; Bian, X.; Lv, Y.; Lin, W. FK228 Sensitizes Radioresistant Small Cell Lung Cancer Cells to Radiation. Clin. Epigenetics 2021, 13, 41. [Google Scholar] [CrossRef]
- Sun, Y.; Shen, W.; Hu, S.; Lyu, Q.; Wang, Q.; Wei, T.; Zhu, W.; Zhang, J. METTL3 Promotes Chemoresistance in Small Cell Lung Cancer by Inducing Mitophagy. J. Exp. Clin. Cancer Res. 2023, 42, 65. [Google Scholar] [CrossRef]
- Fukuoka, K.; Arioka, H.; Iwamoto, Y.; Fukumoto, H.; Kurokawa, H.; Ishida, T.; Tomonari, A.; Suzuki, T.; Usuda, J.; Kanzawa, F.; et al. Mechanism of Vinorelbine-Induced Radiosensitization of Human Small Cell Lung Cancer Cells. Cancer Chemother. Pharmacol. 2002, 49, 385–390. [Google Scholar] [CrossRef]
- Locke, V.L.; Davey, R.A.; Davey, M.W. Modulation of Drug and Radiation Resistance in Small Cell Lung Cancer Cells by Paclitaxel. Anticancer. Drugs 2003, 14, 523–531. [Google Scholar] [CrossRef]
- Patel, K.; Doudican, N.A.; Schiff, P.B.; Orlow, S.J. Albendazole Sensitizes Cancer Cells to Ionizing Radiation. Radiat. Oncol. 2011, 6, 160. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Yang, L.; Zhu, S.; Li, M.; Wang, Y.; Cao, X.; Wang, Q.; Guo, L. Identifying CDC7 as a Synergistic Target of Chemotherapy in Resistant Small-Cell Lung Cancer via CRISPR/Cas9 Screening. Cell Death Discov. 2023, 9, 40. [Google Scholar] [CrossRef]
- Majeed, S.; Aparnathi, M.K.; Nixon, K.C.J.; Venkatasubramanian, V.; Rahman, F.; Song, L.; Weiss, J.; Barayan, R.; Sugumar, V.; Barghout, S.H.; et al. Targeting the Ubiquitin–Proteasome System Using the UBA1 Inhibitor TAK-243 Is a Potential Therapeutic Strategy for Small-Cell Lung Cancer. Clin. Cancer Res. 2022, 28, 1966–1978. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Zhao, Y.; Du, Y.; Tang, R. Evaluation of Hippo Pathway and CD133 in Radiation Resistance in Small-Cell Lung Cancer. J. Oncol. 2021, 2021, 8842554. [Google Scholar] [CrossRef]
- Pearsall, S.M.; Williamson, S.C.; Humphrey, S.; Hughes, E.; Morgan, D.; García Marqués, F.J.; Awanis, G.; Carroll, R.; Burks, L.; Shue, Y.T.; et al. Lineage Plasticity in SCLC Generates Non-Neuroendocrine Cells Primed for Vasculogenic Mimicry. J. Thorac. Oncol. 2023, 18, 1362–1385. [Google Scholar] [CrossRef]
- Jimenez, L.; Stolzenbach, V.; Ozawa, P.M.M.; Ramirez-Solano, M.; Liu, Q.; Sage, J.; Weaver, A.M. Extracellular Vesicles from Non-Neuroendocrine SCLC Cells Promote Adhesion and Survival of Neuroendocrine SCLC Cells. Proteomics 2023, 2300030. [Google Scholar] [CrossRef]
- Williamson, S.C.; Metcalf, R.L.; Trapani, F.; Mohan, S.; Antonello, J.; Abbott, B.; Leong, H.S.; Chester, C.P.E.; Simms, N.; Polanski, R.; et al. Vasculogenic Mimicry in Small Cell Lung Cancer. Nat. Commun. 2016, 7, 13322. [Google Scholar] [CrossRef]
- Kim, J.; Kim, S.; Park, S.-Y.; Lee, G.K.; Lim, K.Y.; Kim, J.Y.; Hwang, J.-A.; Yu, N.; Kang, E.H.; Hwang, M.; et al. Molecular Subtypes and Tumor Microenvironment Characteristics of Small-Cell Lung Cancer Associated with Platinum-Resistance. Cancers 2023, 15, 3568. [Google Scholar] [CrossRef]
- Schenk, M.W.; Humphrey, S.; Hossain, A.S.M.M.; Revill, M.; Pearsall, S.; Lallo, A.; Brown, S.; Bratt, S.; Galvin, M.; Descamps, T.; et al. Soluble Guanylate Cyclase Signalling Mediates Etoposide Resistance in Progressing Small Cell Lung Cancer. Nat. Commun. 2021, 12, 6652. [Google Scholar] [CrossRef]
- Wagner, A.H.; Devarakonda, S.; Skidmore, Z.L.; Krysiak, K.; Ramu, A.; Trani, L.; Kunisaki, J.; Masood, A.; Waqar, S.N.; Spies, N.C.; et al. Recurrent WNT Pathway Alterations Are Frequent in Relapsed Small Cell Lung Cancer. Nat. Commun. 2018, 9, 3787. [Google Scholar] [CrossRef]
- Reinhold, M.I.; Kapadia, R.M.; Liao, Z.; Naski, M.C. The Wnt-Inducible Transcription Factor Twist1 Inhibits Chondrogenesis. J. Biol. Chem. 2006, 281, 1381–1388. [Google Scholar] [CrossRef] [PubMed]
- Gardner, E.E.; Lok, B.H.; Schneeberger, V.E.; Desmeules, P.; Miles, L.A.; Arnold, P.K.; Ni, A.; Khodos, I.; de Stanchina, E.; Nguyen, T.; et al. Chemosensitive Relapse in Small Cell Lung Cancer Proceeds through an EZH2-SLFN11 Axis. Cancer Cell 2017, 31, 286–299. [Google Scholar] [CrossRef] [PubMed]
- Fischer, K.R.; Durrans, A.; Lee, S.; Sheng, J.; Li, F.; Wong, S.T.C.; Choi, H.; El Rayes, T.; Ryu, S.; Troeger, J.; et al. Epithelial-to-Mesenchymal Transition Is Not Required for Lung Metastasis but Contributes to Chemoresistance. Nature 2015, 527, 472–476. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Carstens, J.L.; Kim, J.; Scheible, M.; Kaye, J.; Sugimoto, H.; Wu, C.-C.; LeBleu, V.S.; Kalluri, R. Epithelial-to-Mesenchymal Transition Is Dispensable for Metastasis but Induces Chemoresistance in Pancreatic Cancer. Nature 2015, 527, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Bahar, E.; Kim, J.-Y.; Kim, H.-S.; Yoon, H. Establishment of Acquired Cisplatin Resistance in Ovarian Cancer Cell Lines Characterized by Enriched Metastatic Properties with Increased Twist Expression. Int. J. Mol. Sci. 2020, 21, 7613. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Qi, J.; Wei, H.; Lei, Y.; Yu, H.; Liu, N.; Zhao, L.; Wang, P. TGFβ1 in Cancer-Associated Fibroblasts Is Associated with Progression and Radiosensitivity in Small-Cell Lung Cancer. Front. Cell Dev. Biol. 2021, 9, 667645. [Google Scholar] [CrossRef]
- Grunblatt, E.; Wu, N.; Zhang, H.; Liu, X.; Norton, J.P.; Ohol, Y.; Leger, P.; Hiatt, J.B.; Eastwood, E.C.; Thomas, R.; et al. MYCN Drives Chemoresistance in Small Cell Lung Cancer While USP7 Inhibition Can Restore Chemosensitivity. Genes Dev. 2020, 34, 1210–1226. [Google Scholar] [CrossRef]
- Chen, J.; Guanizo, A.C.; Jakasekara, W.S.N.; Inampudi, C.; Luong, Q.; Garama, D.J.; Alamgeer, M.; Thakur, N.; DeVeer, M.; Ganju, V.; et al. MYC Drives Platinum Resistant SCLC That Is Overcome by the Dual PI3K-HDAC Inhibitor Fimepinostat. J. Exp. Clin. Cancer Res. 2023, 42, 100. [Google Scholar] [CrossRef]
- Drapkin, B.J.; George, J.; Christensen, C.L.; Mino-Kenudson, M.; Dries, R.; Sundaresan, T.; Phat, S.; Myers, D.T.; Zhong, J.; Igo, P.; et al. Genomic and Functional Fidelity of Small Cell Lung Cancer Patient-Derived Xenografts. Cancer Discov. 2018, 8, 600–615. [Google Scholar] [CrossRef]
- Rygaard, K.; Slebos, R.J.C.; Spang-Thomsen, M. Radiosensitivity of Small-Cell Lung Cancer Xenografts Compared with Activity of c-Myc, N-Myc, L-Myc, c-Raf-1 and K-Ras Proto-Oncogenes. Int. J. Cancer 1991, 49, 279–284. [Google Scholar] [CrossRef]
- Chalishazar, M.D.; Wait, S.J.; Huang, F.; Ireland, A.S.; Mukhopadhyay, A.; Lee, Y.; Schuman, S.S.; Guthrie, M.R.; Berrett, K.C.; Vahrenkamp, J.M.; et al. MYC-Driven Small-Cell Lung Cancer Is Metabolically Distinct and Vulnerable to Arginine Depletion. Clin. Cancer Res. 2019, 25, 5107–5121. [Google Scholar] [CrossRef] [PubMed]
- Bortolotti, S.; Angelucci, S.; Montemurro, L.; Bartolucci, D.; Raieli, S.; Lampis, S.; Amadesi, C.; Scardovi, A.; Nieddu, G.; Cerisoli, L.; et al. Antigene MYCN Silencing by BGA002 Inhibits SCLC Progression Blocking MTOR Pathway and Overcomes Multidrug Resistance. Cancers 2023, 15, 990. [Google Scholar] [CrossRef] [PubMed]
- Nassar, D.; Blanpain, C. Cancer Stem Cells: Basic Concepts and Therapeutic Implications. Annu. Rev. Pathol. Mech. Dis. 2016, 11, 47–76. [Google Scholar] [CrossRef] [PubMed]
- Sarvi, S.; Mackinnon, A.C.; Avlonitis, N.; Bradley, M.; Rintoul, R.C.; Rassl, D.M.; Wang, W.; Forbes, S.J.; Gregory, C.D.; Sethi, T. CD133+ Cancer Stem-like Cells in Small Cell Lung Cancer Are Highly Tumorigenic and Chemoresistant but Sensitive to a Novel Neuropeptide Antagonist. Cancer Res. 2014, 74, 1554–1565. [Google Scholar] [CrossRef]
- Kubo, T.; Takigawa, N.; Osawa, M.; Harada, D.; Ninomiya, T.; Ochi, N.; Ichihara, E.; Yamane, H.; Tanimoto, M.; Kiura, K. Subpopulation of Small-Cell Lung Cancer Cells Expressing CD133 and CD87 Show Resistance to Chemotherapy. Cancer Sci. 2013, 104, 78–84. [Google Scholar] [CrossRef]
- Gutova, M.; Najbauer, J.; Gevorgyan, A.; Metz, M.Z.; Weng, Y.; Shih, C.-C.; Aboody, K.S. Identification of UPAR-Positive Chemoresistant Cells in Small Cell Lung Cancer. PLoS ONE 2007, 2, e243. [Google Scholar] [CrossRef]
- Voigt, E.; Wallenburg, M.; Wollenzien, H.; Thompson, E.; Kumar, K.; Feiner, J.; McNally, M.; Friesen, H.; Mukherjee, M.; Afeworki, Y.; et al. Sox2 Is an Oncogenic Driver of Small-Cell Lung Cancer and Promotes the Classic Neuroendocrine Subtype. Mol. Cancer Res. 2021, 19, 2015–2025. [Google Scholar] [CrossRef]
- Tripathi, S.C.; Fahrmann, J.F.; Celiktas, M.; Aguilar, M.; Marini, K.D.; Jolly, M.K.; Katayama, H.; Wang, H.; Murage, E.N.; Dennison, J.B.; et al. MCAM Mediates Chemoresistance in Small-Cell Lung Cancer via the PI3K/AKT/SOX2 Signaling Pathway. Cancer Res. 2017, 77, 4414–4425. [Google Scholar] [CrossRef]
- Liang, S.; Wang, Q.; Wen, Y.; Wang, Y.; Li, M.; Wang, Q.; Peng, J.; Guo, L. Ligand-Independent EphA2 Contributes to Chemoresistance in Small-Cell Lung Cancer by Enhancing PRMT1-Mediated SOX2 Methylation. Cancer Sci. 2023, 114, 921–936. [Google Scholar] [CrossRef]
- Ferté, C.; Loriot, Y.; Clémenson, C.; Commo, F.; Gombos, A.; Bibault, J.-E.; Fumagalli, I.; Hamama, S.; Auger, N.; Lahon, B.; et al. IGF-1R Targeting Increases the Antitumor Effects of DNA-Damaging Agents in SCLC Model: An Opportunity to Increase the Efficacy of Standard Therapy. Mol. Cancer Ther. 2013, 12, 1213–1222. [Google Scholar] [CrossRef]
- Jin, Y.; Chen, Y.; Tang, H.; Hu, X.; Hubert, S.M.; Li, Q.; Su, D.; Xu, H.; Fan, Y.; Yu, X.; et al. Activation of PI3K/AKT Pathway Is a Potential Mechanism of Treatment Resistance in Small Cell Lung Cancer. Clin. Cancer Res. 2022, 28, 526–539. [Google Scholar] [CrossRef] [PubMed]
- Tsurutani, J.; West, K.A.; Sayyah, J.; Gills, J.J.; Dennis, P.A. Inhibition of the Phosphatidylinositol 3-Kinase/Akt/Mammalian Target of Rapamycin Pathway but Not the MEK/ERK Pathway Attenuates Laminin-Mediated Small Cell Lung Cancer Cellular Survival and Resistance to Imatinib Mesylate or Chemotherapy. Cancer Res. 2005, 65, 8423–8432. [Google Scholar] [CrossRef] [PubMed]
- Fridman, R.; Giaccone, G.; Kanemoto, T.; Martin, G.R.; Gazdar, A.F.; Mulshine, J.L. Reconstituted Basement Membrane (Matrigel) and Laminin Can Enhance the Tumorigenicity and the Drug Resistance of Small Cell Lung Cancer Cell Lines. Proc. Natl. Acad. Sci. USA 1990, 87, 6698–6702. [Google Scholar] [CrossRef]
- Sethi, T.; Rintoul, R.C.; Moore, S.M.; MacKinnon, A.C.; Salter, D.; Choo, C.; Chilvers, E.R.; Dransfield, I.; Donnelly, S.C.; Strieter, R.; et al. Extracellular Matrix Proteins Protect Small Cell Lung Cancer Cells against Apoptosis: A Mechanism for Small Cell Lung Cancer Growth and Drug Resistance in Vivo. Nat. Med. 1999, 5, 662–668. [Google Scholar] [CrossRef] [PubMed]
- Hodkinson, P.S.; Elliott, T.; Wong, W.S.; Rintoul, R.C.; Mackinnon, A.C.; Haslett, C.; Sethi, T. ECM Overrides DNA Damage-Induced Cell Cycle Arrest and Apoptosis in Small-Cell Lung Cancer Cells through Β1 Integrin-Dependent Activation of PI3-Kinase. Cell Death Differ. 2006, 13, 1776–1788. [Google Scholar] [CrossRef]
- Güçlü, E.; Eroğlu Güneş, C.; Kurar, E.; Vural, H. Knockdown of LncRNA HIF1A-AS2 Increases Drug Sensitivity of SCLC Cells in Association with Autophagy. Med. Oncol. 2021, 38, 113. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Shan, W.; Hua, Y.; Chao, F.; Cui, Y.; Lv, L.; Dou, X.; Bian, X.; Zou, J.; Li, H.; et al. Exosomal MiR-92b-3p Promotes Chemoresistance of Small Cell Lung Cancer Through the PTEN/AKT Pathway. Front. Cell Dev. Biol. 2021, 9, 661602. [Google Scholar] [CrossRef]
- Wang, C.; Yang, D.; Zhang, X.; Zhang, X.; Yang, L.; Wang, P.; Zhou, W.; Li, H.; Li, Y.; Nie, H.; et al. Association of PTEN Gene SNPs Rs2299939 with PFS in Patients with Small Cell Lung Cancer Treated with Early Radiotherapy. Front. Genet. 2020, 11, 298. [Google Scholar] [CrossRef]
- Doerr, F.; George, J.; Schmitt, A.; Beleggia, F.; Rehkämper, T.; Hermann, S.; Walter, V.; Weber, J.-P.; Thomas, R.K.; Wittersheim, M.; et al. Targeting a Non-Oncogene Addiction to the ATR/CHK1 Axis for the Treatment of Small Cell Lung Cancer. Sci. Rep. 2017, 7, 15511. [Google Scholar] [CrossRef]
- Byers, L.A.; Wang, J.; Nilsson, M.B.; Fujimoto, J.; Saintigny, P.; Yordy, J.; Giri, U.; Peyton, M.; Fan, Y.H.; Diao, L.; et al. Proteomic Profiling Identifies Dysregulated Pathways in Small Cell Lung Cancer and Novel Therapeutic Targets Including PARP1. Cancer Discov. 2012, 2, 798–811. [Google Scholar] [CrossRef]
- Kundu, K.; Cardnell, R.J.; Zhang, B.; Shen, L.; Stewart, C.A.; Ramkumar, K.; Cargill, K.R.; Wang, J.; Gay, C.M.; Byers, L.A. SLFN11 Biomarker Status Predicts Response to Lurbinectedin as a Single Agent and in Combination with ATR Inhibition in Small Cell Lung Cancer. Transl. Lung Cancer Res. 2021, 10, 4095–4105. [Google Scholar] [CrossRef]
- Schultz, C.W.; Zhang, Y.; Elmeskini, R.; Zimmermann, A.; Fu, H.; Murai, Y.; Wangsa, D.; Kumar, S.; Takahashi, N.; Atkinson, D.; et al. ATR Inhibition Augments the Efficacy of Lurbinectedin in Small-cell Lung Cancer. EMBO Mol. Med. 2023, 15, e17313. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Kim, I.-K.; Kallakury, B.; Chahine, J.J.; Iwama, E.; Pierobon, M.; Petricoin, E.; McCutcheon, J.N.; Zhang, Y.-W.; Umemura, S.; et al. Acquired Small Cell Lung Cancer Resistance to Chk1 Inhibitors Involves Wee1 Up-Regulation. Mol. Oncol. 2021, 15, 1130–1145. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Wen, X.; Liu, B. Wee1 Epigenetically Modulates H2B Mono-Ubiquitination at K120 Lysine and DNA Double-Strand Break Repair through Phosphorylation of H2BY37-Dependent Manner in Small-Cell Lung Cancer. Thorac. Cancer 2023, 14, 1420–1429. [Google Scholar] [CrossRef]
- Sen, T.; Tong, P.; Diao, L.; Li, L.; Fan, Y.; Hoff, J.; Heymach, J.V.; Wang, J.; Byers, L.A. Targeting AXL and MTOR Pathway Overcomes Primary and Acquired Resistance to WEE1 Inhibition in Small-Cell Lung Cancer. Clin. Cancer Res. 2017, 23, 6239–6253. [Google Scholar] [CrossRef]
- Tang, S.-W.; Thomas, A.; Murai, J.; Trepel, J.B.; Bates, S.E.; Rajapakse, V.N.; Pommier, Y. Overcoming Resistance to DNA-Targeted Agents by Epigenetic Activation of Schlafen 11 (SLFN11) Expression with Class I Histone Deacetylase Inhibitors. Clin. Cancer Res. 2018, 24, 1944–1953. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Ma, L.; Cao, G.; Hua, J.; Lv, X.; Lin, W. FK228 Potentiates Topotecan Activity against Small Cell Lung Cancer Cells via Induction of SLFN11. Acta Pharmacol. Sin. 2022, 43, 2119–2127. [Google Scholar] [CrossRef]
- Poirier, J.T.; Gardner, E.E.; Connis, N.; Moreira, A.L.; de Stanchina, E.; Hann, C.L.; Rudin, C.M. DNA Methylation in Small Cell Lung Cancer Defines Distinct Disease Subtypes and Correlates with High Expression of EZH2. Oncogene 2015, 34, 5869–5878. [Google Scholar] [CrossRef]
- Hansen, L.T.; Lundin, C.; Helleday, T.; Poulsen, H.S.; Sørensen, C.S.; Petersen, L.N.; Spang-Thomsen, M. DNA Repair Rate and Etoposide (VP16) Resistance of Tumor Cell Subpopulations Derived from a Single Human Small Cell Lung Cancer. Lung Cancer 2003, 40, 157–164. [Google Scholar] [CrossRef]
- Owonikoko, T.K.; Zhang, G.; Deng, X.; Rossi, M.R.; Switchenko, J.M.; Doho, G.H.; Chen, Z.; Kim, S.; Strychor, S.; Christner, S.M.; et al. Poly (ADP) Ribose Polymerase Enzyme Inhibitor, Veliparib, Potentiates Chemotherapy and Radiation in Vitro and in Vivo in Small Cell Lung Cancer. Cancer Med. 2014, 3, 1579–1594. [Google Scholar] [CrossRef]
- Lok, B.H.; Gardner, E.E.; Schneeberger, V.E.; Ni, A.; Desmeules, P.; Rekhtman, N.; de Stanchina, E.; Teicher, B.A.; Riaz, N.; Powell, S.N.; et al. PARP Inhibitor Activity Correlates with SLFN11 Expression and Demonstrates Synergy with Temozolomide in Small Cell Lung Cancer. Clin. Cancer Res. 2017, 23, 523–535. [Google Scholar] [CrossRef] [PubMed]
- Laird, J.H.; Lok, B.H.; Ma, J.; Bell, A.; de Stanchina, E.; Poirier, J.T.; Rudin, C.M. Talazoparib Is a Potent Radiosensitizer in Small Cell Lung Cancer Cell Lines and Xenografts. Clin. Cancer Res. 2018, 24, 5143–5152. [Google Scholar] [CrossRef]
- Noyes, C.; Kitajima, S.; Li, F.; Suita, Y.; Miriyala, S.; Isaac, S.; Ahsan, N.; Knelson, E.; Vajdi, A.; Tani, T.; et al. The Germline Factor DDX4 Contributes to the Chemoresistance of Small Cell Lung Cancer Cells. Commun. Biol. 2023, 6, 65. [Google Scholar] [CrossRef]
- Hansen, L.T.; Thykjaer, T.; Ørntoft, T.F.; Rasmussen, L.J.; Keller, P.; Spang-Thomsen, M.; Bocker Edmonston, T.; Schmutte, C.; Fishel, R.; Nørgård Petersen, L. The Role of Mismatch Repair in Small-Cell Lung Cancer Cells. Eur. J. Cancer 2003, 39, 1456–1467. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Jiang, H.; Kang, Z.; Guan, M. Biomarkers for Chemotherapy and Drug Resistance in the Mismatch Repair Pathway. Clin. Chim. Acta 2023, 544, 117338. [Google Scholar] [CrossRef]
- Dingemans, A.M.; Witlox, M.A.; Stallaert, R.A.; van der Valk, P.; Postmus, P.E.; Giaccone, G. Expression of DNA Topoisomerase IIα and Topoisomerase IIβ Genes Predicts Survival and Response to Chemotherapy in Patients with Small Cell Lung Cancer. Clin. Cancer Res. 1999, 5, 2048–2058. [Google Scholar]
- Li, H.; Wang, H.; Deng, K.; Han, W.; Hong, B.; Lin, W. The Ratio of Bcl-2/Bim as a Predictor of Cisplatin Response Provides a Rational Combination of ABT-263 with Cisplatin or Radiation in Small Cell Lung Cancer. Cancer Biomark. 2019, 24, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Valko, Z.; Megyesfalvi, Z.; Schwendenwein, A.; Lang, C.; Paku, S.; Barany, N.; Ferencz, B.; Horvath-Rozsas, A.; Kovacs, I.; Schlegl, E.; et al. Dual Targeting of BCL-2 and MCL-1 in the Presence of BAX Breaks Venetoclax Resistance in Human Small Cell Lung Cancer. Br. J. Cancer 2023, 128, 1850–1861. [Google Scholar] [CrossRef]
- Loriot, Y.; Mordant, P.; Dugue, D.; Geneste, O.; Gombos, A.; Opolon, P.; Guegan, J.; Perfettini, J.-L.; Pierre, A.; Berthier, L.K.; et al. Radiosensitization by a Novel Bcl-2 and Bcl-XL Inhibitor S44563 in Small-Cell Lung Cancer. Cell Death Dis. 2014, 5, e1423. [Google Scholar] [CrossRef]
- Ramkumar, K.; Tanimoto, A.; Della Corte, C.M.; Stewart, C.A.; Wang, Q.; Shen, L.; Cardnell, R.J.; Wang, J.; Polanska, U.M.; Andersen, C.; et al. Targeting BCL2 Overcomes Resistance and Augments Response to Aurora Kinase B Inhibition by AZD2811 in Small Cell Lung Cancer. Clin. Cancer Res. 2023, 29, 3237–3249. [Google Scholar] [CrossRef]
- Kaiser, U.; Schilli, M.; Haag, U.; Neumann, K.; Kreipe, H.; Kogan, E.; Havemann, K. Expression of Bcl-2—Protein in Small Cell Lung Cancer. Lung Cancer 1996, 15, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Henness, S.; Davey, M.W.; Harvie, R.M.; Davey, R.A. Fractionated Irradiation of H69 Small-Cell Lung Cancer Cells Causes Stable Radiation and Drug Resistance with Increased MRP1, MRP2, and Topoisomerase IIα Expression. Int. J. Radiat. Oncol. Biol. Phys. 2002, 54, 895–902. [Google Scholar] [CrossRef]
- Zandi, R.; Selivanova, G.; Christensen, C.L.; Gerds, T.A.; Willumsen, B.M.; Poulsen, H.S. PRIMA-1Met/APR-246 Induces Apoptosis and Tumor Growth Delay in Small Cell Lung Cancer Expressing Mutant P53. Clin. Cancer Res. 2011, 17, 2830–2841. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.; Kang, K.; Ng, K.P.; Radivoyevitch, T.; Schalper, K.; Zhang, H.; Lindner, D.J.; Thomas, A.; MacPherson, D.; Gastman, B.; et al. Neuroendocrine Lineage Commitment of Small Cell Lung Cancers Can Be Leveraged into P53-Independent Non-Cytotoxic Therapy. Cell Rep. 2023, 42, 113016. [Google Scholar] [CrossRef]
- Neely, V.; Manchikalapudi, A.; Nguyen, K.; Dalton, K.; Hu, B.; Koblinski, J.E.; Faber, A.C.; Deb, S.; Harada, H. Targeting Oncogenic Mutant P53 and BCL-2 for Small Cell Lung Cancer Treatment. Int. J. Mol. Sci. 2023, 24, 13082. [Google Scholar] [CrossRef]
- Rodríguez-Salas, N.; Palacios, J.; Moreno, G.; de Castro, J.; González-Barón, M.; Gamallo, C. Correlation of P53 Oncoprotein Expression with Chemotherapy Response in Small Cell Lung Carcinomas. Lung Cancer 2001, 34, 67–74. [Google Scholar] [CrossRef]
- Akeno, N.; Reece, A.L.; Callahan, M.; Miller, A.L.; Kim, R.G.; He, D.; Lane, A.; Moulton, J.S.; Wikenheiser-Brokamp, K.A. TRP53 Mutants Drive Neuroendocrine Lung Cancer Through Loss-of-Function Mechanisms with Gain-of-Function Effects on Chemotherapy Response. Mol. Cancer Ther. 2017, 16, 2913–2926. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Lee, M.-H.; Park, I.; Jeon, H.; Choi, J.; Seo, S.; Kim, S.-W.; Koh, G.Y.; Park, K.-S.; Lee, D.H. HSP90 Inhibitor (NVP-AUY922) Enhances the Anti-Cancer Effect of BCL-2 Inhibitor (ABT-737) in Small Cell Lung Cancer Expressing BCL-2. Cancer Lett. 2017, 411, 19–26. [Google Scholar] [CrossRef]
- Wu, Q.; Wu, W.; Fu, B.; Shi, L.; Wang, X.; Kuca, K. JNK Signaling in Cancer Cell Survival. Med. Res. Rev. 2019, 39, 2082–2104. [Google Scholar] [CrossRef]
- Butterfield, L.; Storey, B.; Maas, L.; Heasley, L.E. C-Jun NH2-Terminal Kinase Regulation of the Apoptotic Response of Small Cell Lung Cancer Cells to Ultraviolet Radiation. J. Biol. Chem. 1997, 272, 10110–10116. [Google Scholar] [CrossRef]
- Levresse, V.; Marek, L.; Blumberg, D.; Heasley, L.E. Regulation of Platinum-Compound Cytotoxicity by the c-Jun N-Terminal Kinase and c-Jun Signaling Pathway in Small-Cell Lung Cancer Cells. Mol. Pharmacol. 2002, 62, 689–697. [Google Scholar] [CrossRef] [PubMed]
- Kelly, M.P.; Jungbluth, A.A.; Wu, B.-W.; Bomalaski, J.; Old, L.J.; Ritter, G. Arginine Deiminase PEG20 Inhibits Growth of Small Cell Lung Cancers Lacking Expression of Argininosuccinate Synthetase. Br. J. Cancer 2012, 106, 324–332. [Google Scholar] [CrossRef] [PubMed]
- Agnello, G.; Alters, S.E.; Rowlinson, S.W. Preclinical Safety and Antitumor Activity of the Arginine-Degrading Therapeutic Enzyme Pegzilarginase, a PEGylated, Cobalt-Substituted Recombinant Human Arginase 1. Transl. Res. 2020, 217, 11–22. [Google Scholar] [CrossRef]
- Xu, S.; Lam, S.-K.; Cheng, P.N.-M.; Ho, J.C.-M. Recombinant Human Arginase Induces Apoptosis through Oxidative Stress and Cell Cycle Arrest in Small Cell Lung Cancer. Cancer Sci. 2018, 109, 3471–3482. [Google Scholar] [CrossRef] [PubMed]
- Kodama, M.; Oshikawa, K.; Shimizu, H.; Yoshioka, S.; Takahashi, M.; Izumi, Y.; Bamba, T.; Tateishi, C.; Tomonaga, T.; Matsumoto, M.; et al. A Shift in Glutamine Nitrogen Metabolism Contributes to the Malignant Progression of Cancer. Nat. Commun. 2020, 11, 1320. [Google Scholar] [CrossRef]
- Obrist, F.; Michels, J.; Durand, S.; Chery, A.; Pol, J.; Levesque, S.; Joseph, A.; Astesana, V.; Pietrocola, F.; Wu, G.S.; et al. Metabolic Vulnerability of Cisplatin-Resistant Cancers. EMBO J. 2018, 37, e98597. [Google Scholar] [CrossRef]
- Brodin, O.; Arnberg, H.; Bergh, J.; Nilsson, S. Increased Radioresistance of an in Vitro Transformed Human Small Cell Lung Cancer Cell Line. Lung Cancer 1995, 12, 183–198. [Google Scholar] [CrossRef]
- Pedersen, M.W.B.; Holm, S.; Lund, E.L.; Hojgaard, L.; Kristjansen, P.E.G. Coregulation of Glucose Uptake and Vascular Endothelial Growth Factor (VEGF) in Two Small-Cell Lung Cancer (SCLC) Sublines In Vivo and In Vitro. Neoplasia 2001, 3, 80–87. [Google Scholar] [CrossRef]
- Polański, R.; Hodgkinson, C.L.; Fusi, A.; Nonaka, D.; Priest, L.; Kelly, P.; Trapani, F.; Bishop, P.W.; White, A.; Critchlow, S.E.; et al. Activity of the Monocarboxylate Transporter 1 Inhibitor AZD3965 in Small Cell Lung Cancer. Clin. Cancer Res. 2014, 20, 926–937. [Google Scholar] [CrossRef]
- Cargill, K.R.; Stewart, C.A.; Park, E.M.; Ramkumar, K.; Gay, C.M.; Cardnell, R.J.; Wang, Q.; Diao, L.; Shen, L.; Fan, Y.-H.; et al. Targeting MYC-Enhanced Glycolysis for the Treatment of Small Cell Lung Cancer. Cancer Metab. 2021, 9, 33. [Google Scholar] [CrossRef]
- Kang, H.; Kim, B.; Park, J.; Youn, H.; Youn, B. The Warburg Effect on Radioresistance: Survival beyond Growth. Biochim. Biophys. Acta - Rev. Cancer 2023, 1878, 188988. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Wan, R.; He, Y.; Lin, S.-H.; Cao, J.; Qiu, Y.; Zhang, T.; Zhao, Q.; Niu, Y.; Jin, Y.; et al. Therapeutic Targeting of the Mevalonate–Geranylgeranyl Diphosphate Pathway with Statins Overcomes Chemotherapy Resistance in Small Cell Lung Cancer. Nat. Cancer 2022, 3, 614–628. [Google Scholar] [CrossRef]
- Cole, S.P.C.; Bhardwaj, G.; Gerlach, J.H.; Mackie, J.E.; Grant, C.E.; Almquist, K.C.; Stewart, A.J.; Kurz, E.U.; Duncan, A.M.V.; Deeley, R.G. Overexpression of a Transporter Gene in a Multidrug-Resistant Human Lung Cancer Cell Line. Science 1992, 258, 1650–1654. [Google Scholar] [CrossRef] [PubMed]
- Holzmayer, T.A.; Hilsenbeck, S.; Von Hoff, D.D.; Roninson, I.B. Clinical Correlates of MDR1 (P-Glycoprotein) Gene Expression in Ovarian and Small-Cell Lung Carcinomas. JNCI J. Natl. Cancer Inst. 1992, 84, 1486–1491. [Google Scholar] [CrossRef]
- Poupon, M.F.; Arvelo, F.; Goguel, A.F.; Bourgeois, Y.; Jacrot, M.; Hanania, N.; Arriagada, R.; Chevalier, T.L. Response of Small-Cell Lung Cancer Xenografts to Chemotherapy: Multidrug Resistance and Direct Clinical Correlates. JNCI J. Natl. Cancer Inst. 1993, 85, 2023–2029. [Google Scholar] [CrossRef][Green Version]
- Campling, B.G.; Young, L.C.; Baer, K.A.; Lam, Y.M.; Deeley, R.G.; Cole, S.P.; Gerlach, J.H. Expression of the MRP and MDR1 Multidrug Resistance Genes in Small Cell Lung Cancer. Clin. Cancer Res. 1997, 3, 115–122. [Google Scholar]
- Savaraj, N.; Wu, C.J.; Xu, R.; Lampidis, T.; Lai, S.; Donnelly, E.; Solomon, J.; Feun, L.G. Multidrug-Resistant Gene Expression in Small-Cell Lung Cancer. Am. J. Clin. Oncol. 1997, 20, 398–403. [Google Scholar] [CrossRef]
- Yeh, J.-J.; Hsu, N.-Y.; Hsu, W.-H.; Tsai, C.-H.; Lin, C.-C.; Liang, J.-A. Comparison of Chemotherapy Response with P-Glycoprotein, Multidrug Resistance-Related Protein-1, and Lung Resistance-Related Protein Expression in Untreated Small Cell Lung Cancer. Lung 2005, 183, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Omori, M.; Noro, R.; Seike, M.; Matsuda, K.; Hirao, M.; Fukuizumi, A.; Takano, N.; Miyanaga, A.; Gemma, A. Inhibitors of ABCB1 and ABCG2 Overcame Resistance to Topoisomerase Inhibitors in Small Cell Lung Cancer. Thorac. Cancer 2022, 13, 2142–2151. [Google Scholar] [CrossRef]
- Solta, A.; Boettiger, K.; Kovács, I.; Lang, C.; Megyesfalvi, Z.; Ferk, F.; Mišík, M.; Hoetzenecker, K.; Aigner, C.; Kowol, C.R.; et al. Entinostat Enhances the Efficacy of Chemotherapy in Small Cell Lung Cancer Through S-Phase Arrest and Decreased Base Excision Repair. Clin. Cancer Res. 2023, 29, 4644–4659. [Google Scholar] [CrossRef]
- Mohammad, H.P.; Smitheman, K.N.; Kamat, C.D.; Soong, D.; Federowicz, K.E.; Van Aller, G.S.; Schneck, J.L.; Carson, J.D.; Liu, Y.; Butticello, M.; et al. A DNA Hypomethylation Signature Predicts Antitumor Activity of LSD1 Inhibitors in SCLC. Cancer Cell 2015, 28, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Takagi, S.; Ishikawa, Y.; Mizutani, A.; Iwasaki, S.; Matsumoto, S.; Kamada, Y.; Nomura, T.; Nakamura, K. LSD1 Inhibitor T-3775440 Inhibits SCLC Cell Proliferation by Disrupting LSD1 Interactions with SNAG Domain Proteins INSM1 and GFI1B. Cancer Res. 2017, 77, 4652–4662. [Google Scholar] [CrossRef] [PubMed]
- Augert, A.; Eastwood, E.; Ibrahim, A.H.; Wu, N.; Grunblatt, E.; Basom, R.; Liggitt, D.; Eaton, K.D.; Martins, R.; Poirier, J.T.; et al. Targeting NOTCH Activation in Small Cell Lung Cancer through LSD1 Inhibition. Sci. Signal. 2019, 12, eaau2922. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Chung, C.-Y.; Xie, T.; Ozeck, M.; Nichols, T.C.; Frey, J.; Udyavar, A.R.; Sharma, S.; Paul, T.A. Intrinsic and Acquired Drug Resistance to LSD1 Inhibitors in Small Cell Lung Cancer Occurs through a TEAD4-Driven Transcriptional State. Mol. Oncol. 2022, 16, 1309–1328. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, C.; Yang, Z.; Zhang, G.; Wu, P.; Luo, Y.; Zeng, Q.; Wang, L.; Xue, Q.; Zhang, Y.; et al. M6A Regulators as Predictive Biomarkers for Chemotherapy Benefit and Potential Therapeutic Targets for Overcoming Chemotherapy Resistance in Small-Cell Lung Cancer. J. Hematol. Oncol. 2021, 14, 190. [Google Scholar] [CrossRef]
- Jia, D.; Augert, A.; Kim, D.-W.; Eastwood, E.; Wu, N.; Ibrahim, A.H.; Kim, K.-B.; Dunn, C.T.; Pillai, S.P.S.; Gazdar, A.F.; et al. Crebbp Loss Drives Small Cell Lung Cancer and Increases Sensitivity to HDAC Inhibition. Cancer Discov. 2018, 8, 1422–1437. [Google Scholar] [CrossRef]
- Krushkal, J.; Silvers, T.; Reinhold, W.C.; Sonkin, D.; Vural, S.; Connelly, J.; Varma, S.; Meltzer, P.S.; Kunkel, M.; Rapisarda, A.; et al. Epigenome-Wide DNA Methylation Analysis of Small Cell Lung Cancer Cell Lines Suggests Potential Chemotherapy Targets. Clin. Epigenetics 2020, 12, 93. [Google Scholar] [CrossRef]
- Wollenzien, H.; Afeworki Tecleab, Y.; Szczepaniak-Sloane, R.; Restaino, A.; Kareta, M.S. Single-Cell Evolutionary Analysis Reveals Drivers of Plasticity and Mediators of Chemoresistance in Small Cell Lung Cancer. Mol. Cancer Res. 2023, 21, 892–907. [Google Scholar] [CrossRef]
- Wang, Q.; Zeng, F.; Sun, Y.; Qiu, Q.; Zhang, J.; Huang, W.; Huang, J.; Huang, X.; Guo, L. Etk Interaction with PFKFB4 Modulates Chemoresistance of Small-Cell Lung Cancer by Regulating Autophagy. Clin. Cancer Res. 2018, 24, 950–962. [Google Scholar] [CrossRef]
- Shen, W.; Luo, P.; Sun, Y.; Zhang, W.; Zhou, N.; Zhan, H.; Zhang, Q.; Shen, J.; Lin, A.; Cheng, Q.; et al. NRBF2 Regulates the Chemoresistance of Small Cell Lung Cancer by Interacting with the P62 Protein in the Autophagy Process. iScience 2022, 25, 104471. [Google Scholar] [CrossRef]
- Chen, Y.-X.; Wang, C.-J.; Xiao, D.-S.; He, B.-M.; Li, M.; Yi, X.-P.; Zhang, W.; Yin, J.-Y.; Liu, Z.-Q. EIF3a R803K Mutation Mediates Chemotherapy Resistance by Inducing Cellular Senescence in Small Cell Lung Cancer. Pharmacol. Res. 2021, 174, 105934. [Google Scholar] [CrossRef] [PubMed]
- Hyer, M.L.; Milhollen, M.A.; Ciavarri, J.; Fleming, P.; Traore, T.; Sappal, D.; Huck, J.; Shi, J.; Gavin, J.; Brownell, J.; et al. A Small-Molecule Inhibitor of the Ubiquitin Activating Enzyme for Cancer Treatment. Nat. Med. 2018, 24, 186–193. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).