
Prospective Epigenetic Actions of Organo-Sulfur Compounds against Cancer: Perspectives and Molecular Mechanisms

 , , , , , , , , , and
, , , , , , , , , and
Simple Summary
Abstract
1. Introduction
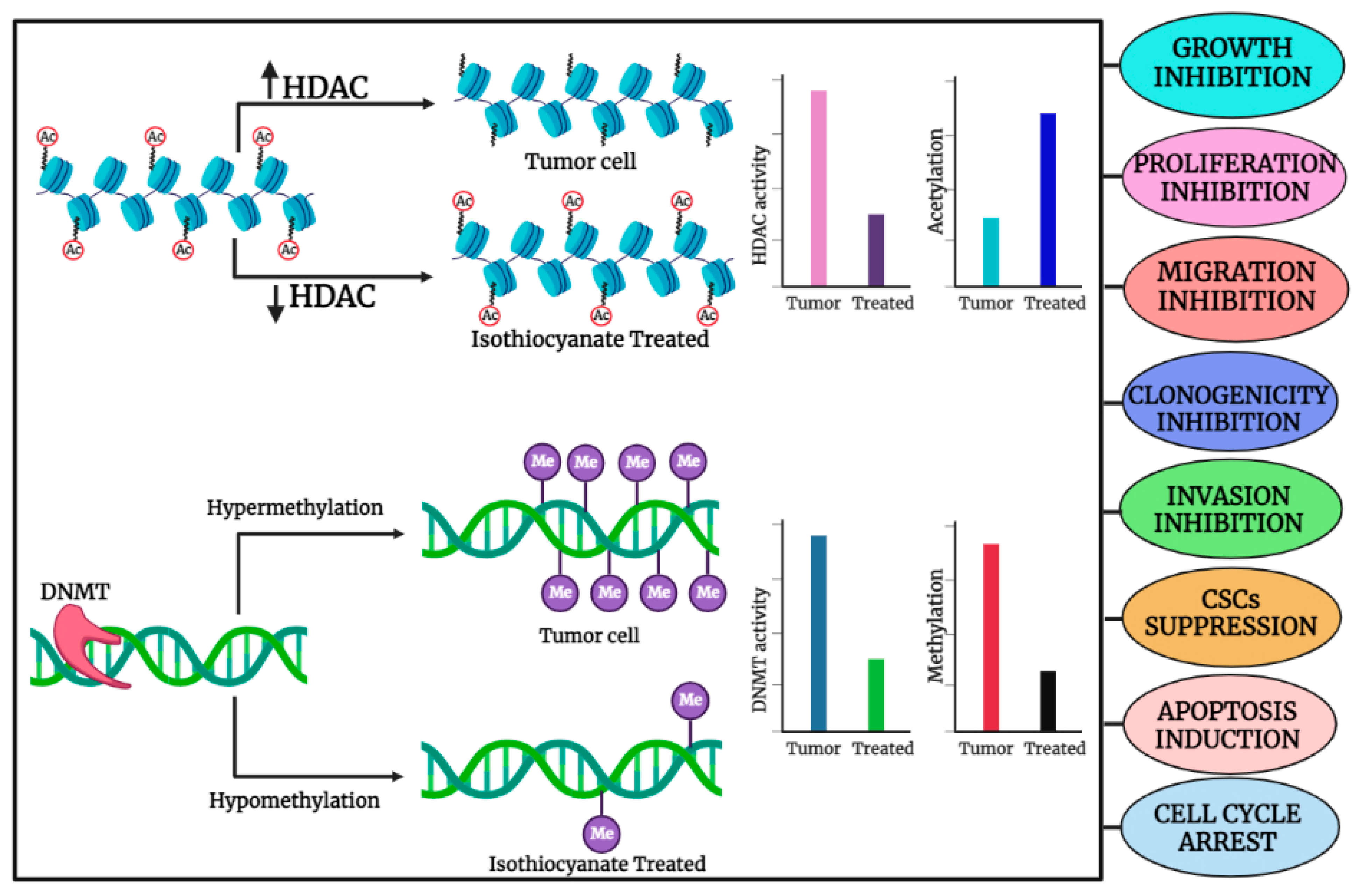
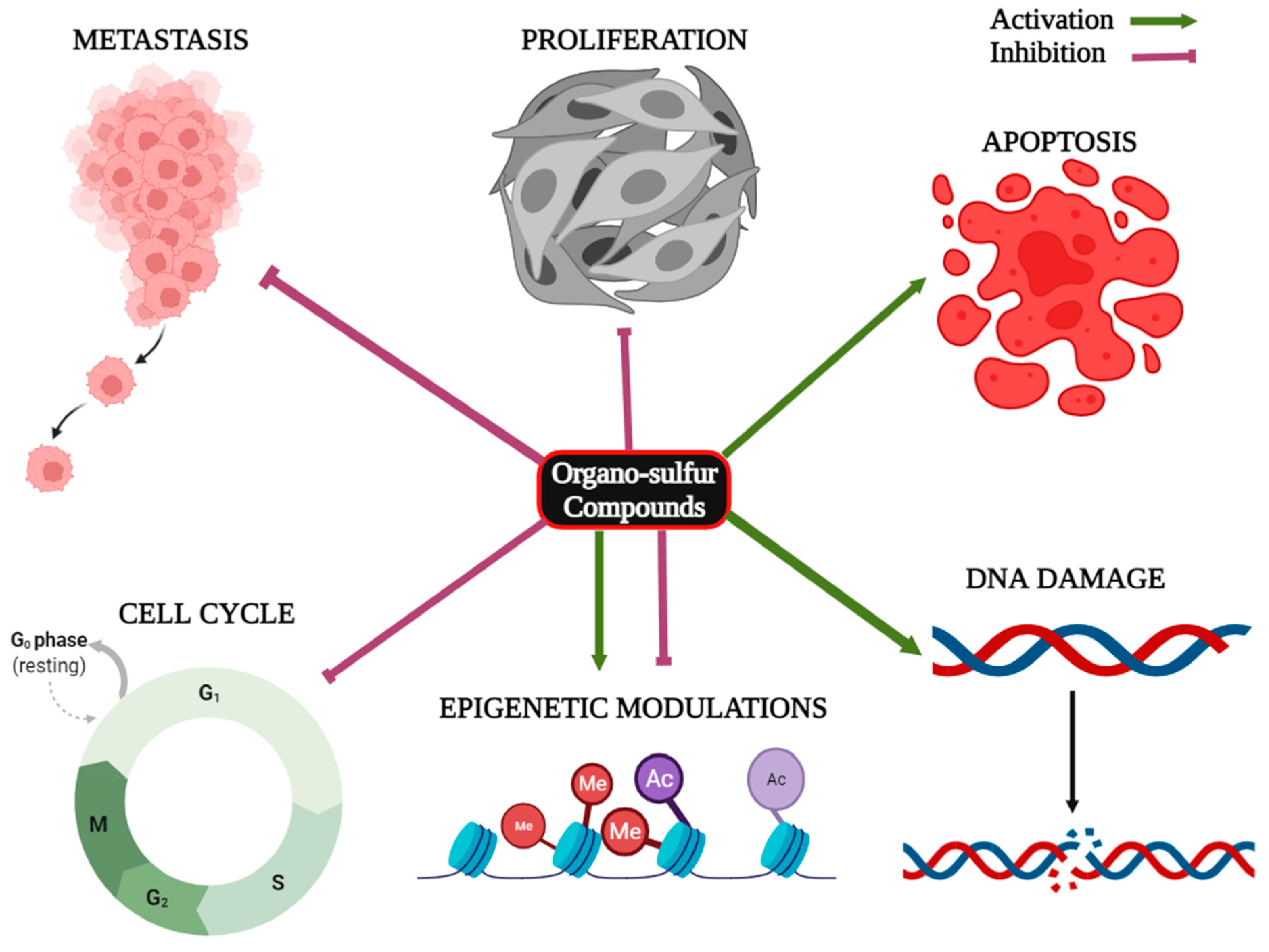
2. Organo-Sulfur Compounds and Their Epigenetic Modulatory Actions against Cancer
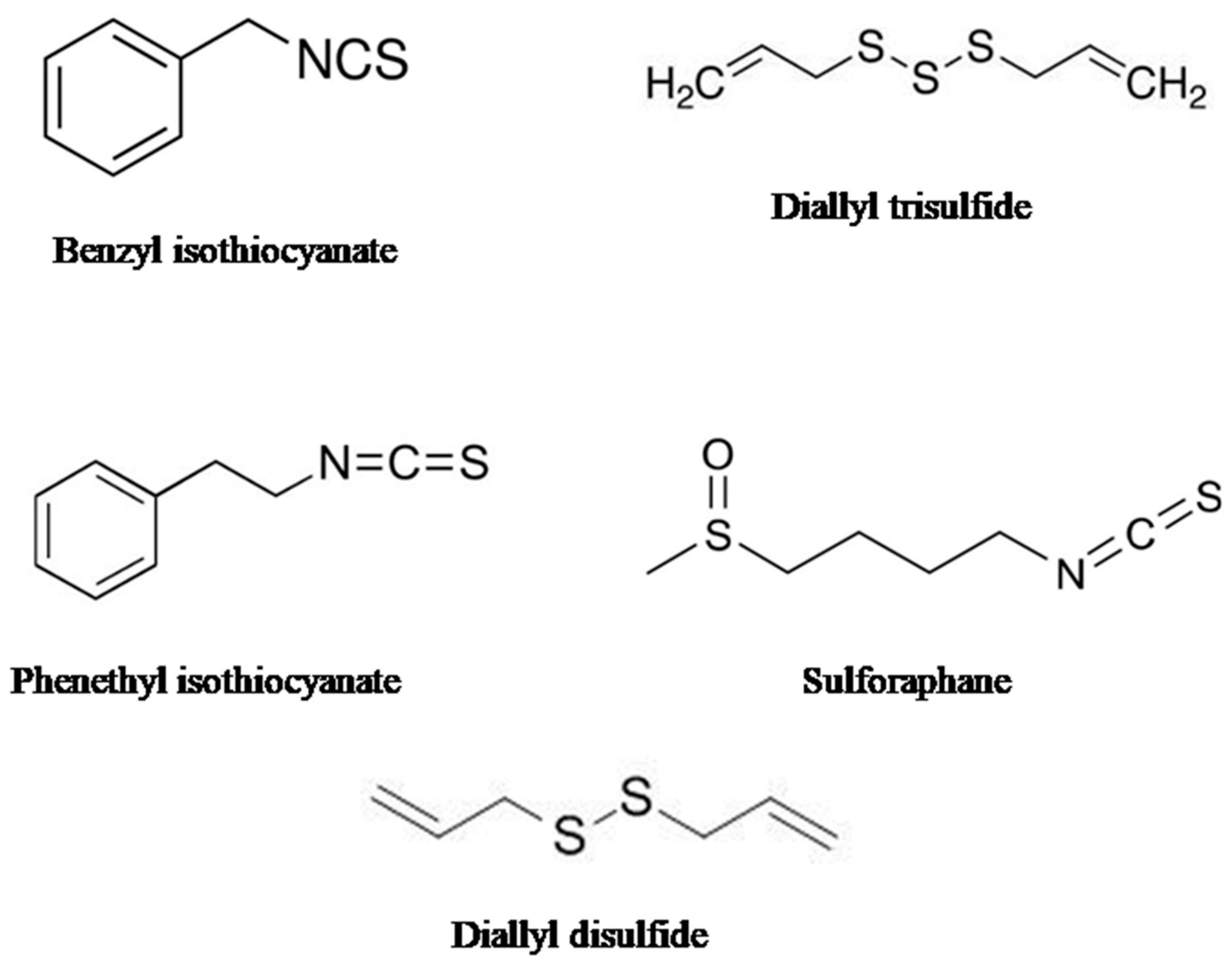
2.1. Sulforaphane (SFN)
2.2. Phenethyl Isothiocyanate (PEITC)
2.3. Diallyl Disulfide (DADS)
2.4. Benzyl Isothiocyanate (BITC)
2.5. Diallyl Thiosulfinate (Allicin)
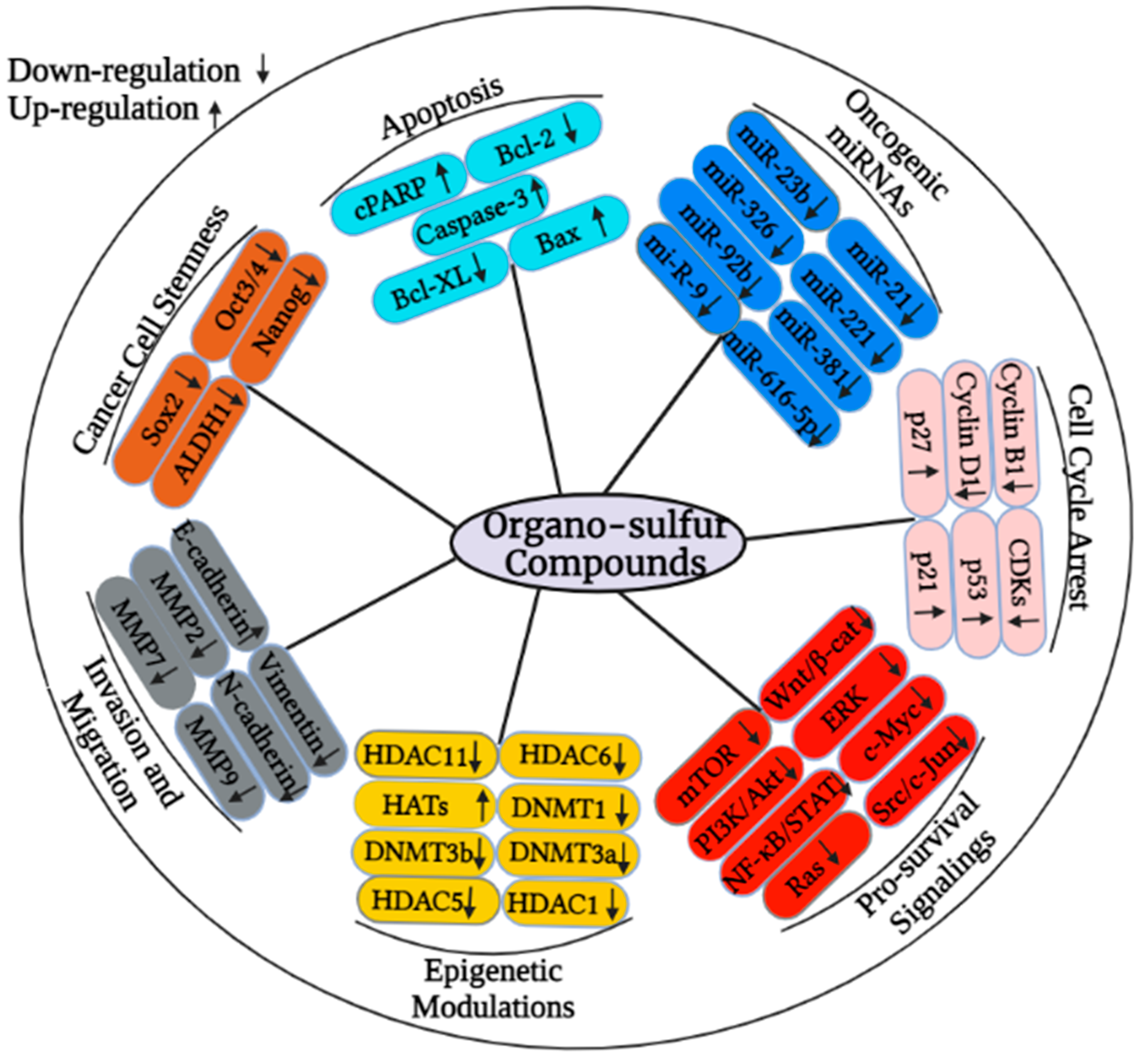
3. Organo-Sulfur Compounds and Their Synergistic Effects on Cancer Cells
4. Organo-Sulfur Compounds under Clinical Trials
5. Organo-Sulfur Compounds: Sources, Chemical Structures, and Pharmacological Importance
6. Research Methodology
7. Conclusions and Future Directions
Funding
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Globalcancerstatistics2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Baig, M.H.; Adil, M.; Khan, R.; Dhadi, S.; Ahmad, K.; Rabbani, G.; Bashir, T.; Imran, M.A.; Husain, F.M.; Lee, E.J.; et al. Enzyme targeting strategies for prevention and treatment of cancer: Implications for cancer therapy. In Seminars in Cancer Biology; Academic Press: Cambridge, MA, USA, 2019; Volume 56, pp. 1–11. [Google Scholar]
- Sokolenko, A.P.; Imyanitov, E.N. Molecular diagnostics in clinical oncology. Front. Mol. Biosci. 2018, 5, 76. [Google Scholar] [CrossRef] [PubMed]
- Ezzati, M.; Henley, S.J.; Lopez, A.D.; Thun, M.J. Role of smoking in global and regional cancer epidemiology: Current patterns and data needs. Int. J. Cancer 2005, 116, 963–971. [Google Scholar] [CrossRef] [PubMed]
- LoConte, N.K.; Brewster, A.M.; Kaur, J.S.; Merrill, J.K.; Alberg, A.J. Alcohol and cancer: A statement of the American Society of Clinical Oncology. J. Clin. Oncol. 2018, 36, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Avgerinos, K.I.; Spyrou, N.; Mantzoros, C.S.; Dalamaga, M. Obesity and cancer risk: Emerging biological mechanisms and perspectives. Metabolism 2019, 92, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Frank, S.A. Genetic predisposition to cancer—Insights from population genetics. Nat. Rev. Genet. 2004, 5, 764–772. [Google Scholar] [CrossRef]
- Carrillo-Infante, C.; Abbadessa, G.; Bagella, L.; Giordano, A. Viral infections as a cause of cancer. Int. J. Oncol. 2007, 30, 1521–1528. [Google Scholar] [CrossRef]
- Katz, M.H. No Smoke—Just Cancer-Causing Chemicals. JAMA Intern. Med. 2017, 177, 1052. [Google Scholar] [CrossRef][Green Version]
- Khandia, R.; Munjal, A. Interplay between inflammation and cancer. Adv. Protein Chem. Struct. Biol. 2020, 119, 199–245. [Google Scholar] [CrossRef]
- Schirrmacher, V. From chemotherapy to biological therapy: A review of novel concepts to reduce the side effects of systemic cancer treatment. Int. J. Oncol. 2019, 54, 407–419. [Google Scholar] [CrossRef]
- Fass, L. Imaging and cancer: A review. Mol. Oncol. 2008, 2, 115–152. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, B.B.; Ichikawa, H.; Garodia, P.; Weerasinghe, P.; Sethi, G.; Bhatt, I.D.; Pandey, M.K.; Shishodia, S.; Nair, M.G. From traditional Ayurvedic medicine to modern medicine: Identification of therapeutic targets for suppression of inflammation and cancer. Expert Opin. Ther. Targets 2006, 10, 87–118. [Google Scholar] [CrossRef] [PubMed]
- Multhoff, G.; Molls, M.; Radons, J. Chronic inflammation in cancer development. Front. Immunol. 2012, 2, 98. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.; Hebbar, V.; Shen, G.; Gopalakrishnan, A.; Khor, T.O.; Yu, S.; Xu, C.; Kong, A.-N. Synergistic effects of a combination of dietary factors sulforaphane and (−) epigallocatechin-3-gallate in HT-29 AP-1 human colon carcinoma cells. Pharm. Res. 2008, 25, 387–399. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Xu, J.; Brady, S.; Gao, H.; Yu, D.; Reuben, J.; Mehta, K. Tissue transglutaminase promotes drug resistance and invasion by inducing mesenchymal transition in mammary epithelial cells. PloS ONE 2010, 5, e13390. [Google Scholar] [CrossRef] [PubMed]
- Kawata, M.; Koinuma, D.; Ogami, T.; Umezawa, K.; Iwata, C.; Watabe, T.; Miyazono, K. TGF-β-induced epithelial-mesenchymal transition of A549 lung adenocarcinoma cells is enhanced by pro-inflammatory cytokines derived from RAW264.7 macrophage cells. J. Biochem. 2012, 151, 205–216. [Google Scholar] [CrossRef]
- Wiencke, J.K.; Zheng, S.; Morrison, Z.; Yeh, R.-F. Differentially expressed genes are marked by histone 3 lysine 9 trimethylation in human cancer cells. Oncogene 2008, 27, 2412–2421. [Google Scholar] [CrossRef]
- Singh, M.; Kumar, V.; Sehrawat, N.; Yadav, M.; Chaudhary, M.; Upadhyay, S.K.; Kumar, S.; Sharma, V.; Kumar, S.; Dilbaghi, N.; et al. Current paradigms in epigenetic anticancer therapeutics and future challenges. In Seminars in Cancer Biology; Academic Press: Cambridge, MA, USA, 2021. [Google Scholar] [CrossRef]
- Ohm, J.E.; McGarvey, K.M.; Yu, X.; Cheng, L.; Schuebel, K.E.; Cope, L.; Mohammad, H.P.; Chen, W.; Daniel, V.C.; Yu, W.; et al. A stem cell–like chromatin pattern may predispose tumor suppressor genes to DNA hypermethylation and heritable silencing. Nat. Genet. 2007, 39, 237–242. [Google Scholar] [CrossRef]
- Osanai, M.; Murata, M.; Nishikiori, N.; Chiba, H.; Kojima, T.; Sawada, N. Epigenetic silencing of occludin promotes tumorigenic and metastatic properties of cancer cells via modulations of unique sets of apoptosis-associated genes. Cancer Res. 2006, 66, 9125–9133. [Google Scholar] [CrossRef]
- Xu, X.; Chang, X.; Xu, Y.; Deng, P.; Wang, J.; Zhang, C.; Zhu, X.; Chen, S.; Dai, D. SAMD14 promoter methylation is strongly associated with gene expression and poor prognosis in gastric cancer. Int. J. Clin. Oncol. 2020, 25, 1105–1114. [Google Scholar] [CrossRef]
- Ogama, Y.; Ouchida, M.; Yoshino, T.; Ito, S.; Takimoto, H.; Shiote, Y.; Ishimaru, F.; Harada, M.; Tanimoto, M.; Shimizu, K. Prevalent hyper-methylation of the CDH13 gene promoter in malignant B cell lymphomas. Int. J. Oncol. 2004, 25, 685–691. [Google Scholar] [CrossRef]
- Tan, S.-H.; Ida, H.; Lau, Q.-C.; Goh, B.-C.; Chieng, W.-S.; Loh, M.; Ito, Y. Detection of promoter hypermethylation in serum samples of cancer patients by methylation-specific polymerase chain reaction for tumour suppressor genes including RUNX3. Oncol. Rep. 2007, 18, 1225–1230. [Google Scholar] [CrossRef]
- Rehman, I.; Cross, S.S.; Catto, J.W.; Leiblich, A.; Mukherjee, A.; Azzouzi, A.-R.; Leung, H.Y.; Hamdy, F.C. Promoter hyper-methylation of calcium binding proteins S100A6 and S100A2 in human prostate cancer. Prostate 2005, 65, 322–330. [Google Scholar] [CrossRef]
- Sabir, M.; Baig, R.M.; Ali, K.; Mahjabeen, I.; Saeed, M.; Kayani, M.A. Retinoblastoma (RB1) pocket domain mutations and promoter hyper-methylation in head and neck cancer. Cell. Oncol. 2014, 37, 203–213. [Google Scholar] [CrossRef]
- Xu, X.; Chang, X.; Li, Z.; Wang, J.; Deng, P.; Zhu, X.; Liu, J.; Zhang, C.-D.; Chen, S.; Dai, D. Aberrant SOX11 promoter methylation is associated with poor prognosis in gastric cancer. Cell. Oncol. 2015, 38, 183–194. [Google Scholar] [CrossRef]
- Piao, G.H.; Piao, W.H.; He, Y.; Zhang, H.H.; Wang, G.Q.; Piao, Z. Hyper-methylation of RIZ1 tumor suppressor gene is involved in the early tumorigenesis of hepatocellular carcinoma. Histology and Histopathology. Histol. Histopathol. 2008, 23, 1171–1175. [Google Scholar]
- Arnold, C.N.; Goel, A.; Niedzwiecki, D.; Dowell, J.M.; Wasserman, L.; Compton, C.; Mayer, R.J.; Bertagnolli, M.M.; Boland, C.R. APC promoter hypermethylation contributes to the loss of APC expression in colorectal cancers with allelic loss on 5q1. Cancer Biol. Ther. 2004, 3, 960–964. [Google Scholar] [CrossRef]
- Koinuma, K.; Yamashita, Y.; Liu, W.; Hatanaka, H.; Kurashina, K.; Wada, T.; Takada, S.; Kaneda, R.; Choi, Y.L.; Fujiwara, S.-I.; et al. Epigenetic silencing of AXIN2 in colorectal carcinoma with microsatellite instability. Oncogene 2006, 25, 139–146. [Google Scholar] [CrossRef]
- Qi, J.; Zhu, Y.-Q.; Luo, J.; Tao, W.-H. Hypermethylation and expression regulation of secreted frizzled-related protein genes in colorectal tumor. World J. Gastroenterol. WJG 2006, 12, 7113. [Google Scholar] [CrossRef]
- Samaei, N.M.; Yazdani, Y.; Alizadeh-Navaei, R.; Azadeh, H.; Farazmandfar, T. Promoter methylation analysis of Wnt/β-catenin pathway regulators and its association with expression of DNMT1 enzyme in colorectal cancer. J. Biomed. Sci. 2014, 21, 73. [Google Scholar] [CrossRef][Green Version]
- Debnath, T.; Nath, N.C.D.; Kim, E.-K.; Lee, K.-G. Role of phytochemicals in the modulation of miRNA expression in cancer. Food Funct. 2017, 8, 3432–3442. [Google Scholar] [CrossRef]
- Slaby, O.; Sachlova, M.; Brezkova, V.; Hezova, R.; Kovarikova, A.; Bischofová, S.; Sevcikova, S.; Bienertova-Vasku, J.; Vasku, A.; Svoboda, M.; et al. Identification of microRNAs regulated by isothiocyanates and association of polymorphisms inside their target sites with risk of sporadic colorectal cancer. Nutr. Cancer 2013, 65, 247–254. [Google Scholar] [CrossRef]
- Lubecka-Pietruszewska, K.; Kaufman-Szymczyk, A.; Stefanska, B.; Cebula-Obrzut, B.; Smolewski, P.; Fabianowska-Majewska, K. Sulforaphane alone and in combination with clofarabine epigenetically regulates the expression of DNA methylation-silenced tumour suppressor genes in human breast cancer cells. Lifestyle Genom. 2015, 8, 91–101. [Google Scholar] [CrossRef]
- Meeran, S.M.; Patel, S.N.; Tollefsbol, T.O. Sulforaphane causes epigenetic repression of hTERT expression in human breast cancer cell lines. PloS ONE 2010, 5, e11457. [Google Scholar] [CrossRef]
- Chen, L.; Chan, L.S.; Lung, H.L.; Yip, T.T.C.; Ngan, R.K.C.; Wong, J.W.C.; Lo, K.W.; Ng, W.T.; Lee, A.W.M.; Tsao, G.S.W.; et al. Crucifera sulforaphane (SFN) inhibits the growth of nasopharyngeal carcinoma through DNA methyltransferase1 (DNMT1)/Wnt inhibitory factor1(WIF1) axis. Phytomedicine 2019, 63, 153058. [Google Scholar] [CrossRef]
- Hsu, A.; Wong, C.P.; Yu, Z.; Williams, D.E.; Dashwood, R.H.; Ho, E. Promoter de-methylation of cyclin D2 by sulforaphane in prostate cancer cells. Clin. Epigenetics 2011, 3, 1–9. [Google Scholar] [CrossRef]
- dos Santos, P.W.d.S.; Machado, A.R.T.; De Grandis, R.A.; Ribeiro, D.L.; Tuttis, K.; Morselli, M.; Aissa, A.F.; Pellegrini, M.; Antunes, L.M.G. Transcriptome and DNA methylation changes modulated by sulforaphane induce cell cycle arrest, apoptosis, DNA damage, and suppression of proliferation in human liver cancer cells. Food Chem. Toxicol. 2020, 136, 111047. [Google Scholar] [CrossRef]
- Zhang, C.; Su, Z.-Y.; Khor, T.O.; Shu, L.; Kong, A.-N.T. Sulforaphane enhances Nrf2 expression in prostate cancer TRAMPC1 cells through epigenetic regulation. Biochem. Pharmacol. 2013, 85, 1398–1404. [Google Scholar] [CrossRef]
- Khan, M.A.; Sundaram, M.K.; Hamza, A.; Quraishi, U.; Gunasekera, D.; Ramesh, L.; Goala, P.; Al Alami, U.; Ansari, M.Z.; Rizvi, T.A.; et al. Sulforaphane reverses the expression of various tumor suppressor genes by targeting DNMT3B and HDAC1 in human cervical cancer cells. Evid. Based Complement. Altern. Med. 2015, 2015, 412149. [Google Scholar] [CrossRef]
- Su, Z.-Y.; Zhang, C.; Lee, J.H.; Shu, L.; Wu, T.-Y.; Khor, T.O.; Conney, A.H.; Lu, Y.-P.; Kong, A.-N.T. Requirement and epigenetics reprogramming of Nrf2 in suppression of tumor promoter TPA-induced mouse skin cell transformation by sulforaphane. Cancer Prev. Res. 2014, 7, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Mitsiogianni, M.; Trafalis, D.T.; Franco, R.; Zoumpourlis, V.; Pappa, A.; Panayiotidis, M.I. Sulforaphane and iberin are potent epigenetic modulators of histone acetylation and methylation in malignant melanoma. Eur. J. Nutr. 2021, 60, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Cao, C.; Wu, H.; Vasilatos, S.N.; Chandran, U.; Qin, Y.; Wan, Y.; Oesterreich, S.; Davidson, N.E.; Huang, Y. HDAC5–LSD1 axis regulates antineoplastic effect of natural HDAC inhibitor sulforaphane in human breast cancer cells. Int. J. Cancer 2018, 143, 1388–1401. [Google Scholar] [CrossRef]
- Jiang, L.-L.; Zhou, S.-J.; Zhang, X.-M.; Chen, H.-Q.; Liu, W. Sulforaphane suppresses in vitro and in vivo lung tumorigenesis through downregulation of HDAC activity. Biomed. Pharmacother. 2016, 78, 74–80. [Google Scholar] [CrossRef]
- Rajendran, P.; Dashwood, W.-M.; Li, L.; Kang, Y.; Kim, E.; Johnson, G.; Fischer, K.A.; Löhr, C.V.; Williams, D.E.; Ho, E.; et al. Nrf2 status affects tumor growth, HDAC3 gene promoter associations, and the response to sulforaphane in the colon. Clin. Epigenetics 2015, 7, 102. [Google Scholar] [CrossRef]
- Kawai, H.; Tomii, K.; Toyooka, S.; Yano, M.; Murakami, M.; Tsukuda, K.; Shimizu, N. Promoter methylation downregulates CDX2 expression in colorectal carcinomas. Oncol. Rep. 2005, 13, 547–551. [Google Scholar] [CrossRef]
- Kiani, S.; Akhavan-Niaki, H.; Fattahi, S.; Kavoosian, S.; Jelodar, N.B.; Bagheri, N.; Zarrini, H.N. Purified sulforaphane from broccoli (Brassica oleracea var. italica) leads to alterations of CDX1 and CDX2 expression and changes in miR-9 and miR-326 levels in human gastric cancer cells. Gene 2018, 678, 115–123. [Google Scholar] [CrossRef]
- Gao, L.; Cheng, D.; Yang, J.; Wu, R.; Li, W.; Kong, A.-N. Sulforaphane epigenetically demethylates the CpG sites of the miR-9-3 promoter and reactivates miR-9-3 expression in human lung cancer A549 cells. J. Nutr. Biochem. 2018, 56, 109–115. [Google Scholar] [CrossRef]
- Lewinska, A.; Adamczyk-Grochala, J.; Deręgowska, A.; Wnuk, M. Sulforaphane-induced cell cycle arrest and senescence are accompanied by DNA hypomethylation and changes in microRNA profile in breast cancer cells. Theranostics 2017, 7, 3461. [Google Scholar] [CrossRef]
- Martin, S.L.; Kala, R.; Tollefsbol, T.O. Mechanisms for the inhibition of colon cancer cells by sulforaphane through epigenetic modulation of microRNA-21 and human telomerase reverse transcriptase (hTERT) down-regulation. Curr. Cancer Drug Targets 2018, 18, 97–106. [Google Scholar] [CrossRef]
- Wang, D.-X.; Zou, Y.-J.; Zhuang, X.-B.; Chen, S.-X.; Lin, Y.; Li, W.-L.; Lin, J.-J.; Lin, Z.-Q. Sulforaphane suppresses EMT and metastasis in human lung cancer through miR-616-5p-mediated GSK3β/β-catenin signaling pathways. Acta Pharmacol. Sin. 2017, 38, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-M.; Peng, C.-Y.; Liao, Y.-W.; Lu, M.-Y.; Tsai, M.-L.; Yeh, J.-C.; Yu, C.-H.; Yu, C.-C. Sulforaphane targets cancer stemness and tumor initiating properties in oral squamous cell carcinomas via miR-200c induction. J. Formos. Med. Assoc. 2017, 116, 41–48. [Google Scholar] [CrossRef]
- Shan, Y.; Zhang, L.; Bao, Y.; Li, B.; He, C.; Gao, M.; Feng, X.; Xu, W.; Zhang, X.; Wang, S. Epithelial-mesenchymal transition, a novel target of sulforaphane viaCOX-2/MMP2, 9/Snail, ZEB1 and miR-200c/ZEB1 pathways in human bladder cancer cells. J. Nutr. Biochem. 2013, 24, 1062–1069. [Google Scholar] [CrossRef]
- Zhu, J.; Wang, S.; Chen, Y.; Li, X.; Jiang, Y.; Yang, X.; Li, Y.; Wang, X.; Meng, Y.; Zhu, M.; et al. miR-19 targeting of GSK3β mediates sulforaphane suppression of lung cancer stem cells. J. Nutr. Biochem. 2017, 44, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhao, Z.; Li, M.; Liu, M.; Bahena, A.; Zhang, Y.; Zhang, Y.; Nambiar, C.; Liu, G. Sulforaphane promotes apoptosis, and inhibits proliferation and self-renewal of nasopharyngeal cancer cells by targeting STAT signal through miRNA-124-3p. Biomed. Pharmacother. 2018, 103, 473–481. [Google Scholar] [CrossRef]
- Boyanapalli, S.S.; Li, W.; Fuentes, F.; Guo, Y.; Ramirez, C.N.; Gonzalez, X.-P.; Pung, D.; Kong, A.-N.T. Epigenetic reactivation of RASSF1A by phenethyl isothiocyanate(PEITC) and promotion of apoptosis in LNCaP cells. Pharmacol. Res. 2016, 114, 175–184. [Google Scholar] [CrossRef]
- Zhang, T.; Zhang, W.; Hao, M. Phenethylisothiocyanatereducesbreastcancerstemcell-likepropertiesbyepigeneticreactivationofCDH1. Oncol. Rep. 2021, 45, 337–348. [Google Scholar] [CrossRef]
- Park, J.E.; Sun, Y.; Lim, S.K.; Tam, J.P.; Dekker, M.; Chen, H.; Sze, S.K. Dietary phytochemical PEITC restricts tumor development via modulation of epigenetic writers and erasers. Sci. Rep. 2017, 7, 40569. [Google Scholar] [CrossRef]
- Wang, L.G.; Liu, X.M.; Fang, Y.; Dai, W.; Chiao, F.B.; Puccio, G.; Feng, J.; Liu, D.; Chiao, J.W. De-repression of the p21 promoter in prostate cancer cells by an isothiocyanate via inhibition of HDACs and c-Myc. Int. J. Oncol. 2008, 33, 375–380. [Google Scholar] [CrossRef]
- Mitsiogianni, M.; Anestopoulos, I.; Kyriakou, S.; Trafalis, D.T.; Franco, R.; Pappa, A.; Panayiotidis, M.I. Benzyl and phenethyl isothiocyanates as promising epigenetic drug compoundsbymodulatinghistoneacetylationandmethylationmarksinmalignantmelanoma. Investig. New Drugs 2021, 39, 1460–1468. [Google Scholar] [CrossRef]
- Wang, L.G.; Beklemisheva, A.; Liu, X.M.; Ferrari, A.C.; Feng, J.; Chiao, J.W. Dual action on promoter demethylation and chromatin by an isothiocyanate restored GSTP1 silenced in prostate cancer. Mol. Carcinog. 2007, 46, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Shu, L.; Zhang, C.; Li, W.; Wu, R.; Guo, Y.; Yang, Y.; Kong, A.-N. Histone methyl transferaseSetd7 regulates Nrf2 signaling pathway by phenethyl isothiocyanate and ursolic acid in human prostate cancer cells. Mol. Nutr. Food Res. 2018, 62, 1700840. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chakravarty, S.; Dey, M. Phenethylisothiocyanatealterssite-andpromoter-specifichistonetailmodificationsincancercells. PloS ONE 2013, 8, e64535. [Google Scholar]
- Zhang, C.; Shu, L.; Kim, H.; Khor, T.O.; Wu, R.; Li, W.; Kong, A.-N.T. Phenethyl isothiocyanate (PEITC) suppresses prostate cancer cell invasion epigenetically through regulating microRNA-194. Mol. Nutr. Food Res. 2016, 60, 1427–1436. [Google Scholar] [CrossRef]
- Yu, C.; Gong, A.Y.; Chen, D.; Solelo Leon, D.; Young, C.Y.; Chen, X.M. Phenethyl isothiocyanate inhibits androgen receptor-regulated transcriptional activity in prostate cancer cells through suppressing PCAF. Mol. Nutr. Food Res. 2013, 57, 1825–1833. [Google Scholar] [CrossRef]
- Zhang, T.; Shao, Y.; Chu, T.Y.; Huang, H.S.; Liou, Y.L.; Li, Q.; Zhou, H. miR-135 a and MRP1 play pivotal roles in the selective lethality of phenethyl isothiocyanate to malignant glioma cells. Am. J. Cancer Res. 2016, 6, 957. [Google Scholar]
- Su, B.; Xiang, S.L.; Su, J.; Tang, H.L.; Liao, Q.J.; Zhou, Y.J.; Su, Q. Diallyl disulfide increases histone acetylation and P21WAF1 expression in human gastric cancer cells in vivo and in vitro. Biochem. Pharmacol. 2012, 1, 7. [Google Scholar] [CrossRef]
- Altonsy, M.O.; Habib, T.N.; Andrews, S.C. Diallyl disulfide-induced apoptosis in a breast-cancer cell line (MCF-7) may be caused by inhibition of histone deacetylation. Nutr. Cancer 2012, 64, 1251–1260. [Google Scholar] [CrossRef]
- Druesne, N.; Pagniez, A.; Mayeur, C.; Thomas, M.; Cherbuy, C.; Duée, P.-H.; Latino-Martel, P.; Chaumontet, C. Diallyl disulfide (DADS) increases histone acetylation and p21waf1/cip1 expression in human colon tumor cell lines. Carcinogenesis 2004, 25, 1227–1236. [Google Scholar] [CrossRef]
- Nkrumah-Elie, Y.; Reuben, J.S.; Hudson, A.M.; Taka, E.; Badisa, R.; Ardley, T.; Israel, B.; Sadrud-Din, S.Y.; Oriaku, E.T.; Darling-Reed, S.F. The attenuation of early benzo(a)pyrene-induced carcinogenic insults by diallyl disulfide (DADS) in MCF-10A cells. Nutr. Cancer 2012, 64, 1112–1121. [Google Scholar] [CrossRef]
- Huang, N.J.; Liu, L.J.; Pan, Q.; Zhang, Z.W.; Tang, H.L. Diallyl Disulfide suppresses the proliferation and invasion of human gastric cancer cell line MGC-803 by down-regulating miR-222. Basic Clin. Med. 2015, 35, 1596. [Google Scholar]
- Wang, G.; Liu, G.; Ye, Y.; Fu, Y.; Zhang, X. Upregulation of miR-34a by diallyl disulfide suppresses invasion and induces apoptosis in SGC-7901 cells through inhibition of the PI3K/Akt signaling pathway. Oncol. Lett. 2016, 11, 2661–2667. [Google Scholar] [CrossRef]
- Tang, H.; Kong, Y.; Guo, J.; Tang, Y.; Xie, X.; Yang, L.; Su, Q.; Xie, X. Diallyl disulfide suppresses proliferation and induces apoptosis in human gastric cancer throughWnt-1 signaling pathway by up-regulation of miR-200b and miR-22. Cancer Lett. 2013, 340, 72–81. [Google Scholar] [CrossRef]
- Xiao, X.; Chen, B.; Liu, X.; Liu, P.; Zheng, G.; Ye, F.; Tang, H.; Xie, X. Diallyl disulfide suppresses SRC/Ras/ERK signaling-mediated proliferation and metastasis in human breastcancerby up-regulating miR-34a. PLoS ONE 2014, 9, e112720. [Google Scholar] [CrossRef]
- Li, Y.; Wang, Z.; Li, J.; Sang, X. Diallyl disulfide suppresses FOXM1-mediated proliferation and invasion in osteosarcoma by upregulating miR-134. J. Cell. Biochem. 2019, 120, 7286–7296. [Google Scholar] [CrossRef]
- Lin, J.-F.; Tsai, T.-F.; Lin, Y.-C.; Chen, H.-E.; Chou, K.-Y.; Hwang, T.I.-S. Benzyl isothiocyanate suppresses IGF1R, FGFR3 and mTOR expression by upregulation of miR-99a-5p in human bladder cancer cells. Int. J. Oncol. 2019, 54, 2106–2116. [Google Scholar] [CrossRef]
- Tsai, T.; Chen, P.; Lin, Y.; Chou, K.; Chen, H.; Ho, C.; Lin, J.; Hwang, T.I. Benzyl isothiocyanate promotes miR-99a expression through ERK/AP-1-dependent pathway in bladder cancer cells. Environ. Toxicol. 2020, 35, 47–54. [Google Scholar] [CrossRef]
- He, W.; Fu, Y.; Zheng, Y.; Wang, X.; Bin Liu, B.; Zeng, J. Diallyl thiosulfinate enhanced the anti-cancer activity of dexamethasone in the side population cells of multiple myeloma by promoting miR-127-3p and deactivating the PI3K/AKT signaling pathway. BMC Cancer 2021, 21, 125. [Google Scholar] [CrossRef]
- Wu, H.; Li, X.; Zhang, T.; Zhang, G.; Chen, J.; Chen, L.; He, M.; Hao, B.; Wang, C. Overexpression miR-486-3p promoted by allicin enhances temozolomide sensitivity in glioblastoma via targeting MGMT. Neuromolecular Med. 2020, 22, 359–369. [Google Scholar] [CrossRef]
- Abbas, A.; Hall, J.A.; Patterson, W.L.; Ho, E.; Hsu, A.; Al-Mulla, F.; Georgel, P.T. Sulforaphane modulates telomerase activity via epigenetic regulation in prostate cancer cell lines. Biochem. Cell Biol. 2016, 94, 71–81. [Google Scholar] [CrossRef]
- Rajendran, P.; Delage, B.; Dashwood, W.M.; Yu, T.-W.; Wuth, B.; E Williams, D.; Ho, E.; Dashwood, R.H. Histone deacetylase turn over and recovery in sulforaphane-treated colon cancer cells:Competingactionsof14-3-3andPin1inHDAC3/SMRTcorepressorcomplexdissociation/reassembly. Mol. Cancer 2011, 10, 68. [Google Scholar] [CrossRef]
- Druesne-Pecollo, N.; Pagniez, A.; Thomas, M.; Cherbuy, C.; Duée, P.-H.; Martel, P.; Chaumontet, C. Diallyl disulfide increases CDKN1A promoter-associated histone acetylation in human colon tumor cell lines. J. Agric. Food Chem. 2006, 54, 7503–7507. [Google Scholar] [CrossRef]
- Basu, A.; Alder, H.; Khiyami, A.; Leahy, P.; Croce, C.M.; Haldar, S. MicroRNA-375andmicroRNA-221:Potential noncoding RNAs associated with antiproliferative activity of benzyl isothiocyanate in pancreatic cancer. Genes Cancer 2011, 2, 108–119. [Google Scholar] [CrossRef]
- Weisbeck, A.; Jansen, R.J. Nutrients and the pancreas: An epigenetic perspective. Nutrients 2017, 9, 283. [Google Scholar] [CrossRef]
- Lv, Q.; Xia, Q.; Li, J.; Wang, Z. Allicin suppresses growth and metastasis of gastric carcinoma: The key role of micro RNA-383-5p-mediated inhibition of ERBB4 signaling. Biosci. Biotechnol. Biochem. 2020, 84, 1997–2004. [Google Scholar] [CrossRef]
- Royston, K.J.; Udayakumar, N.; Lewis, K.; Tollefsbol, T.O. A novel combination of withaferin A and sulforaphane inhibits epigenetic machinery, cellular viability and induces apoptosis of breast cancer cells. Int. J. Mol. Sci. 2017, 18, 1092. [Google Scholar] [CrossRef] [PubMed]
- Royston, K.J.; Paul, B.; Nozell, S.; Rajbhandari, R.; Tollefsbol, T.O. Withaferin A and sulforaphane regulate breast cancer cell cycle progression through epigenetic mechanisms. Exp. Cell Res. 2018, 368, 67–74. [Google Scholar] [CrossRef]
- Paul, B.; Li, Y.; Tollefsbol, T.O. The effects of combinatorial genistein and sulforaphane in breast tumor inhibition: Role in epigenetic regulation. Int. J. Mol. Sci. 2018, 19, 1754. [Google Scholar] [CrossRef]
- Wong, C.P.; Hsu, A.; Buchanan, A.; Palomera-Sanchez, Z.; Beaver, L.M.; Houseman, E.A.; Williams, D.E.; Dashwood, R.H.; Ho, E. Effects of sulforaphane and 3,3′-diindolylmethane on genome-wide promoter methylation in normal prostate epithelial cells and prostate cancer cells. PloS ONE 2014, 9, e86787. [Google Scholar] [CrossRef]
- Lubecka, K.; Kaufman-Szymczyk, A.; Fabianowska-Majewska, K. Inhibition of breast cancer cell growth by the combination of clofarabine and sulforaphane involves epigenetically mediated CDKN2A upregulation. Nucleosides Nucleotides Nucleic Acids 2018, 37, 280–289. [Google Scholar] [CrossRef]
- Kaboli, P.J.; Khoshkbejari, M.A.; Mohammadi, M.; Abiri, A.; Mokhtarian, R.; Vazifemand, R.; Amanollahi, S.; Sani, S.Y.; Li, M.; Zhao, Y.; et al. Targets and mechanisms of sulforaphane derivatives obtained from cruciferous plants with special focus on breast cancer–contradictory effects and future perspectives. Biomed. Pharmacother. 2020, 121, 109635. [Google Scholar] [CrossRef] [PubMed]
- Hutzen, B.; Willis, W.; Jones, S.; Cen, L.; Deangelis, S.; Fuh, B.; Lin, J. Dietary agent, benzyl isothiocyanate inhibits signal transducer and activator of transcription 3 phosphorylation and collaborates with sulforaphane in the growth suppression of PANC-1 cancer cells. Cancer Cell Int. 2009, 9, 24. [Google Scholar] [CrossRef]
- Gupta, R.; Bhatt, L.K.; Momin, M. Potent antitumor activity of Laccaic acid and Phenethyl isothiocyanate combination in colorectal cancer via dual inhibition of DNA methyltransferase-1 and Histone deacetylase-1. Toxicol. Appl. Pharmacol. 2019, 377, 114631. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Cang, S.; Ma, Y.; Chiao, J.W. Synergistic effect of paclitaxel and epigenetic agent phenethyl isothiocyanate on growth inhibition, cell cycle arrest and apoptosis in breast cancer cells. Cancer Cell Int. 2013, 13, 10. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Tollefsbol, T.O. Combinatorial epigenetic mechanisms of sulforaphane, genistein and sodium butyrate in breast cancer inhibition. Exp. Cell Res. 2022, 416, 113160. [Google Scholar] [CrossRef]
- Hu, Y.; Lu, W.; Chen, G.; Zhang, H.; Jia, Y.; Wei, Y.; Yang, H.; Zhang, W.; Fiskus, W.; Bhalla, K.; et al. Overcoming resistance to histone deacetylase inhibitors in human leukemia with the redox modulating compound β-phenylethyl isothiocyanate. Blood J. Am. Soc. Hematol. 2010, 116, 2732–2741. [Google Scholar] [CrossRef]
- Cornblatt, B.S.; Ye, L.; Dinkova-Kostova, A.; Erb, M.; Fahey, J.W.; Singh, N.K.; Chen, M.-S.A.; Stierer, T.; Garrett-Mayer, E.; Argani, P.; et al. Preclinical and clinical evaluation of sulforaphane for chemoprevention in the breast. Carcinogenesis 2007, 28, 1485–1490. [Google Scholar] [CrossRef]
- Cipolla, B.G.; Mandron, E.; Lefort, J.M.; Coadou, Y.; Della Negra, E.; Corbel, L.; Le Scodan, R.; Mottet, N. First double-blind placebo-controlled, multicenter, randomized trial of stabilized natural sulforaphane in men with rising PSA following radical prostatectomy. J. Clin. Oncol. 2014, 32, 5032. [Google Scholar] [CrossRef]
- Yanaka, A.; Fahey, J.W.; Fukumoto, A.; Nakayama, M.; Inoue, S.; Zhang, S.; Tauchi, M.; Suzuki, H.; Hyodo, I.; Yamamoto, M. Dietary sulforaphane-rich broccoli sprouts reduce colonization and attenuate gastritis in Helicobacter pylori–infected mice and humans. Cancer Prev. Res. 2009, 2, 353–360. [Google Scholar] [CrossRef]
- Yuan, J.-M.; Stepanov, I.; Murphy, S.E.; Wang, R.; Allen, S.; Jensen, J.; Strayer, L.; Adams-Haduch, J.; Upadhyaya, P.; Le, C.; et al. Clinical trial of 2-phenethyl isothiocyanate as an inhibitor of metabolic activation of a tobacco-specific lung carcinogen in cigarette smokers. Cancer Prev. Res. 2016, 9, 396–405. [Google Scholar] [CrossRef]
- Reiter, J.; Levina, N.; Van der Linden, M.; Gruhlke, M.; Martin, C.; Slusarenko, A.J. Diallyl thiosulfinate (Allicin), a volatile antimicrobial from garlic (Alliumsativum), kills human lung pathogenic bacteria, including MDR strains, as a vapor. Molecules 2017, 22, 1711. [Google Scholar] [CrossRef]
- Zhai, H.; Pan, J.; Pang, E.; Bai, B. Lavage with allicin in combination with vancomycin inhibits biofilm formation by Staphylococcus epidermidis in a rabbit model of prosthetic joint infection. PLoS ONE 2014, 9, e102760. [Google Scholar] [CrossRef]
- Lihua, L.; Jianhui, W.; Jialin, Y.; Yayin, L.; Guanxin, L. Effects of allicin on the formation of Pseudomonas aeruginosa biofilm and the production of quorum-sensing controlled virulence factors. Pol. J. Microbiol. 2013, 62, 243. [Google Scholar] [CrossRef]
- Ranjbar-Omid, M.; Arzanlou, M.; Amani, M.; Al-Hashem, S.K.S.; Mozafari, N.A.; Doghaheh, H.P. Allicin from garlic inhibits the biofilm formation and urease activity of Proteus mirabilis in vitro. FEMS Microbiol. Lett. 2015, 362, fnv049. [Google Scholar] [CrossRef] [PubMed]
- Dwivedi, V.P.; Bhattacharya, D.; Singh, M.; Bhaskar, A.; Kumar, S.; Fatima, S.; Sobia, P.; Van Kaer, L.; Das, G. Allicin enhances antimicrobial activity of macrophages during Mycobacterium tuberculosis infection. J. Ethnopharmacol. 2019, 243, 111634. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Daim, M.M.; Abushouk, A.I.; Donia, T.; Alarifi, S.; Alkahtani, S.; Aleya, L.; Bungau, S.G. The nephroprotective effects of allicin and ascorbic acid against cisplatin-induced toxicity in rats. Environ. Sci. Pollut. Res. 2019, 26, 13502–13509. [Google Scholar] [CrossRef] [PubMed]
- Rapa, S.F.; Di Iorio, B.R.; Campiglia, P.; Heidland, A.; Marzocco, S. Inflammation and oxidative stress in chronic kidney disease—Potential therapeutic role of minerals, vitamins and plant-derived metabolites. Int. J. Mol. Sci. 2019, 21, 263. [Google Scholar] [CrossRef]
- Coutinho-Wolino, K.S.; Almeida, P.P.; Mafra, D.; Stockler-Pinto, M.B. Bioactive compounds modulating Toll-like 4 receptor (TLR4)-mediated inflammation: Pathways involved and future perspectives. Nutr. Res. 2022, 107, 96–116. [Google Scholar] [CrossRef]
- Kumar, S. Dual inhibition of acetylcholinesterase and butyrylcholinesterase enzymes by allicin. Indian J. Pharmacol. 2015, 47, 444. [Google Scholar] [CrossRef]
- Edres, H.A.; Taha, N.M.; Lebda, M.A.; Elfeky, M.S. The potential neuroprotective effect of allicin and melatonin in acryl amide-induced brain damage in rats. Environ. Sci. Pollut. Res. 2021, 28, 58768–58780. [Google Scholar] [CrossRef]
- Yang, C.-X.; Wu, H.-T.; Li, X.-X.; Wu, H.-Y.; Niu, T.-X.; Wang, X.-N.; Lian, R.; Zhang, G.-L.; Hou, H.-M. Comparison of the inhibitory potential of benzyl isothiocyanate and phenethyl isothiocyanate on Shiga toxin-producing and enterotoxigenic Escherichia Coli. LWT 2020, 118, 108806. [Google Scholar] [CrossRef]
- Krause, K.; Pyrczak-Felczykowska, A.; Karczewska, M.; Narajczyk, M.; Herman-Antosiewicz, A.; Szalewska-Pałasz, A.; Nowicki, D. Dietary isothiocyanates, sulforaphane and 2-phenethyl isothiocyanate, effectively impair Vibrio cholerae virulence. Int. J. Mol. Sci. 2021, 22, 10187. [Google Scholar] [CrossRef] [PubMed]
- Gross-Steinmeyer, K.; Stapleton, P.L.; Tracy, J.H.; Bammler, T.K.; Strom, S.C.; Eaton, D.L. Sulforaphane- and phenethyl isothiocyanate–induced inhibition of aflatoxin B1–mediated genotoxicity in human hepatocytes: Role of GSTM1 genotype and CYP3A4 gene expression. Toxicol. Sci. 2010, 116, 422–432. [Google Scholar] [CrossRef] [PubMed]
- Latronico, T.; Larocca, M.; Milella, S.; Fasano, A.; Rossano, R.; Liuzzi, G.M. Neuroprotective potential of isothiocyanates in an in vitro model of neuroinflammation. Inflammopharmacology 2021, 29, 561–571. [Google Scholar] [CrossRef] [PubMed]
- Kamal, R.M.; Razis, A.F.A.; Sukri, N.S.M.; Perimal, E.K.; Ahmad, H.; Patrick, R.; Djedaini-Pilard, F.; Mazzon, E.; Rigaud, S. Beneficial health effects of glucosinolates-derived isothiocyanates on cardiovascular and neurodegenerative diseases. Molecules 2022, 27, 624. [Google Scholar] [CrossRef]
- Asif, M.; Kala, C.; Gilani, S.J.; Imam, S.S.; Mohamad, T.; Naaz, F.; Rahat, I.; Khan, N.A. Protective Effects of Isothiocyanates against Alzheimer’s Disease. Curr. Tradit. Med. 2022, 8, e091121197839. [Google Scholar] [CrossRef]
- Huang, C.S.; Lin, A.H.; Liu, C.T.; Tsai, C.W.; Chang, I.S.; Chen, H.W.; Lii, C.K. Isothiocyanates protect against oxidized LDL-induced endothelial dysfunction by upregulating Nrf2-dependent antioxidation and suppressing NFκB activation. Mol. Nutr. Food Res. 2013, 57, 1918–1930. [Google Scholar] [CrossRef]
- Wilson, A.E.; Bergaentzlé, M.; Bindler, F.; Marchioni, E.; Lintz, A.; Ennahar, S. In vitro efficacies of various isothiocyanates from cruciferous vegetables as antimicrobial agents against food borne pathogens and spoilage bacteria. Food Control 2013, 30, 318–324. [Google Scholar] [CrossRef]
- Yeh, Y.Y.; Liu, L. Cholesterol-lowering effect of garlic extracts and organosulfur compounds: Human and animal studies. J. Nutr. 2001, 131, 989S–993S. [Google Scholar] [CrossRef]
- Lee, H.H.; Han, M.H.; Hwang, H.J.; Kim, G.-Y.; Moon, S.-K.; Hyun, J.-W.; Kim, W.-J.; Choi, Y.H.H. Diallyl trisulfide exerts anti-inflammatory effects in lipopolysaccharide-stimulated RAW264.7 macrophages by suppressing the Toll-like receptor4/nuclear factor-κB pathway. Int. J. Mol. Med. 2015, 35, 487–495. [Google Scholar] [CrossRef]
- Predmore, B.L.; Kondo, K.; Bhushan, S.; Zlatopolsky, M.A.; King, A.L.; Aragon, J.P.; Grinsfelder, D.B.; Condit, M.E.; Lefer, D.J. The polysulfide diallyl trisulfide protects the ischemic myocardium by preservation of endogenous hydrogen sulfide and increasing nitric oxide bioavailability. Am. J. Physiol. -Heart Circ. Physiol. 2012, 302, H2410–H2418. [Google Scholar] [CrossRef]
- Hasan, H.F.; Abdel-Hamid, G.R.; Ebrahim, S.I. Antioxidant and anti-inflammatory effects of diallyl disulfide on hepatotoxicity induced by cyclophosphamide in rats. Nat. Prod. Commun. 2020, 15, 1934578X20969083. [Google Scholar] [CrossRef]
- Lee, H.J.; Lee, H.G.; Choi, K.S.; Surh, Y.J.; Na, H.K. Diallyl trisulfide suppresses dextran sodium sulfate-induced mouse colitis: NF-κB and STAT3 as potential targets. Biochem. Biophys. Res. Commun. 2013, 437, 267–273. [Google Scholar] [CrossRef]
- Kumar, M.M.; Tamizhselvi, R. Protective effect of diallyl disulfide against cerulein-induced acute pancreatitis and associated lung injury in mice. Int. Immunopharmacol. 2020, 80, 106136. [Google Scholar] [CrossRef]
- Zhang, H.; Shang, C.; Tian, Z.; Amin, H.K.; Kassab, R.B.; Abdel Moneim, A.E.; Zhang, Y. Diallyl disulfide suppresses inflammatory and oxidative machineries following carrageenan injection-induced pawedema in mice. Mediat. Inflamm. 2020, 2020, 8508906. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Organo-Sulfur Compound | Type of Cancer | Type of Study | Molecular Mechanism | References |
|---|---|---|---|---|
| SFN | Breast Cancer | In vitro | SFN caused significant improvement in the activities of p53, p21, and PTEN through epigenetic modulation of DNMT1, which resulted in the suppression of growth and proliferation of MCF-7 and MDA-MB-231 breast cancer cells. | [35] |
| SFN | Breast Cancer | In vitro | SFN inhibited hTERT activity through the down-regulation of DNMT1 and DNMT3, leading to the apoptosis induction in MCF-7 and MDA-MB-231 breast cancer cells. | [36] |
| SFN | Nasopharyngeal carcinoma | In vitroand in vivo | SFN inhibited tumor sphere formation and reduced the CSC-related proteins (SOX2 and ALDH). The mechanistic insights revealed restoration of WIF1 and down-regulation of DNMT1. | [37] |
| SFN | Prostate cancer | In vitro | SFN significantly decreased DNMT1 and DNMT3b expression in LNCaP prostate cancer cells, which were correlated with decreased methylation of cyclin D2 promoter regions containing c-Myc and Sp1 binding sites. | [38] |
| SFN | Liver cancer | In vitro | SFN reduced CPEB2 methylation and promoted the methylation of E2F3 and THAP1 along with the inhibition of HDAC5 and HDAC11, which led to decreased cell proliferation and apoptosis induction in HepG2 liver cancer cells. | [39] |
| SFN | Prostate cancer | In vitro | SFN treatment improved Nrf2 and NQO-1 activities in TRAMP C1 cells, which was linked with the reduced expression of HDACs DNMT1 and DNMT3a. | [40] |
| SFN | Cervical cancer | In vitro | SFN reverted the expression of CDH1, DAPK1, GSTP1, and RARβ, which was correlated with the decreased expression of DNMT3b and HDAC1. | [41] |
| SFN | Skin cancer | In vitro | SFN inhibited the expression of HDAC1, HDAC2, HDAC3, HDAC4, DNMT1, DNMT3a, and DNMT3b. SFN also augmented the expression of Nrf2, NQO-1, and HO-1 in skin cancer cells. | [42] |
| SFN | Prostate cancer | In vitro | SFN repressed the expression of hTERT in prostate cancer cell lines, which was linked with the acetylation of histone H3 lysine 18 and di-methylation of histone H3 lysine 4. | [43] |
| SFN | Breast cancer | In vitroand in vivo | SFN treatment led to the down-regulation of HDAC5 and repression of USF1 activity. Mechanistic insights also revealed elevated LSD1 ubiquitination in animals. | [44] |
| SFN | Lung cancer | In vitroand in vivo | SFN treatment resulted in the apoptosis induction and arrest at S-phase of the cell cycle in A549 and H1299 cells along with tumor growth inhibition in a mouse model, which was correlated with inhibition of HDAC activity and increased levels of acetylated histones H3 and H4. | [45] |
| SFN | Colon cancer | In vivo | SFN-rich diets reduced tumor burden in a mouse model, which was supposed to be due to the decreased expression of HDAC3 and augmented acetylation of histone H4. | [46] |
| SFN | Colon cancer | In vitro | SFN caused G/M cell cycle arrest in a time-dependent manner, which was further linked with the HDAC3 degradation. | [47] |
| SFN | Gastric cancer | In vitro | SFN treatment caused proliferation inhibition and apoptosis induction in AGS and MKN4 cells. Mechanistic insights revealed reduced expression of miR9 and miR-326, which further led to augmented levels of CDX1 and CDX2. | [48] |
| SFN | Lung cancer | In vitro | SFN treatment resulted in the increased expression of miR-9-3, which was due to the increased H3K4me1 enrichment and reduced CpG methylation in the miR-9-3 promoter. Further, they suggested attenuated expression of DNMT3a, HDAC1, HDAC3, and HDAC6. | [49] |
| SFN | Breast cancer | In vitro | SFN reduced the levels of DNMT1 and DNMT3b in MCF-7, MDA-MB-231, and SKBR3 cells. SFN also reduced the levels of miR23b, miR-92b, miR381, and miR-382. | [50] |
| SFN | Human colorectal cancer | In vitro | SFN down-regulated hTERT, HDAC1, and miR-21, which led to proliferation inhibition and apoptosis induction in HCT116 and RKO cells. | [51] |
| SFN | Non-small cell lung cancer | In vitro | SFN down-regulated miR-616-5p, vimentin, N-cadherin, and β-catenin and enhanced E-cadherin expression in 95D and H1299 cells. | [52] |
| SFN | Oral squamous cell carcinoma | In vitroand in vivo | SFN enhanced miR-200c expression and suppressed expression of ALDH1 and CD44. | [53] |
| SFN | Bladder cancer | In vitro | SFN increased expression of miR-200c, while it reduced ZEB1, COX-2, Snail, MMP-2, and MMP-9 in T24 cells. | [54] |
| SFN | Lung cancer | In vitro | SFN down-regulated miR19 and inhibited Wnt/β-catenin pathway activation in lung cancer cells. | [55] |
| SFN | Nasopharyngeal cancer | In vitro | SFN up-regulated miR-124-3p and decreased expression of STAT3, c-Myc, Oct-3/4 SOX2, Nanog, and β-catenin in HONE1 and SUN1 cells. | [56] |
| PEITC | Prostate cancer | In vitro | PEITC down-regulated DNMT3a, DNMT3b, HDAC1, HDAC2, HDAC4, and HDAC6 in LNCaP cells. | [57] |
| PEITC | Breast cancer | In vitro | PEITC decreased expression of DNMT1, DNMT3a, DNMT3b, HDAC1, and HDAC2 in breast cancer cells. PEITC also inhibited CSCs-related marker proteins including ALDH-1, Oct-4A, and SOX2. | [58] |
| PEITC | Colon cancer | In vitro | PEITC decreased methylation of PCDH10, VWC2, SPG20, HNF4A, and CDH6. | [59] |
| PEITC | Prostate cancer | In vitro | PEITC reduced c-Myc expression and up-regulated the expression of p21 and p27. | [60] |
| PEITC | Melanoma skin cancer | In vitro | PEITC diminished activities of HDAC1, HDAC2, HDAC4, and HDAC6 in A375 and Hs294T cells. PEITC treatment also reduced the expression of AcH4K12, AcH4K8, and AcH4K5 in melanoma skin cancer cells. | [61] |
| PEITC | Prostate cancer | In vitro | PEITC inhibited HDAC1 expression and promoted de-methylation at the promoter region of GSTP1 in LNCaP cells. | [62] |
| PEITC | Prostate cancer | In vitro | PEITC treatment improved Setd7 expression and enhanced H3K4me1 enrichment in PCa cells. | [63] |
| PEITC | Colon cancer | In vitro | PEITC at low doses increased the methylation of H3K27, leading to the attenuation of proinflammatory markers including IL8, CCL2, and CXCL10. | [64] |
| PEITC | Prostate cancer | In vitro | PEITC up-regulated miR-194, which led to the down-regulation of BMP1 in PC3 cells. | [64] |
| PEITC | Prostate cancer cells | In vitro | PEITC suppressed cell migration and invasion, which was due to the inhibition of MMP2 and MMP9. | [65] |
| PEITC | Prostate cancer | In vitro | PEITC suppressed cell growth and proliferation, which was linked with the increased expression of miR-17. | [66] |
| PEITC | Prostate cancer | In vitro | PEITC treatment increased miR194 expression, which resulted in the suppression of proliferation and invasion of LNCaP cells. | [67] |
| PEITC | Glioma | In vitro | PEITC up-regulated miR-135a and the down-regulation of anti-apoptotic proteins and caspase activation. | [66] |
| DADS | Gastric cancer | In vitroand in vivo | DADS augmented the acetylation of histones, H3, and H4 and up-regulated p21 in HGC803 cells and in mouse models as well. | [68] |
| DADS | Breast cancer | In vitro | DADS inhibited HDAC and caused the acetylation of histone H4 in MCF-7 cells. | [69] |
| DADS | Colon cancer | In vitro | DADS reduced HDAC activity and caused the acetylation of H4K12 and H4K27 in Caco2 and Ht-29 cells. | [70] |
| DADS | Colon cancer | In vitro | DADS caused the acetylation of histones, H3, and H4, which may lead to increased expression of CDKN1A at transcription level in Caco2 and HT-29 cells. | [71] |
| DADS | Gastric cancer | In vitro | DADS down-regulated miR-222, which further may result in the suppressed proliferation, invasion in MGC-803 cells. | [72] |
| DADS | Gastric cancer | In vitro | DADS up-regulated miR-34a, which may result in the reduction of cell viability and invasion and apoptosis induction in SGC-7901 cells. | [73] |
| DADS | Gastric cancer | In vitro | DADS caused miR-22 up-regulation, leading to reduced cell proliferation and apoptosis induction. | [74] |
| DADS | Breast cancer | In vitroand in vivo | DADS augmented miR-34a expression, resulting in the suppression of proliferation and metastasis in MDA-MB-231 cells and the inhibition of tumorigenicity in animal model through the attenuation of SRC/Ras/ERK signaling. | [75] |
| DADS | Osteosarcoma | In vitro and in vivo | DADS treatment significantly up-regulated miR-134, leading to the inhibition of proliferation and invasion in U2OS and MG-63 cells. DADS also exerted antitumor effects through the up-regulation of miR-134 in tumors. | [76] |
| BITC | Melanoma skin cancer | In vitro | BITC down-regulated AcH4K5, AcH4K8, and AcH4K12 and reduced acetylation of H3K56, H3K14, and H3K9 in A375 cells. | [61] |
| BITC | Bladder cancer | In vitro | BITC up-regulated miR-99a-5p, which was linked with the down-regulation of IGFR1, mTOR, FGFR3, and PARP cleavages. | [77] |
| BITC | Bladder cancer | In vitro | BITC treatment led to the over-expression of miR-99a, which may result in the activation of ERK/c-Jun signaling pathways. | [78] |
| BITC | Pancreatic cancer | In vitro | BITC suppressed the expression of miR-221 and miR-375, which may result in the proliferation of pancreatic cancer cells through the up-regulation of IGFBP5 and CAV-1. | [79] |
| DATS | Pancreatic cancer | In vitro | DATS treatment caused the up-regulation of miR-339-5p, which was possibly responsible for the inhibition of proliferation and metastasis and apoptosis induction in PANC-1 and GS799T cells. | [80] |
| DATS | Gastric cancer | In vitro | DATS over-expressed miR-383-5p and decreased cell viability, migration, and invasion of AGS and HGC27 cells. | [81] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shoaib, S.; Ansari, M.A.; Ghazwani, M.; Hani, U.; Jamous, Y.F.; Alali, Z.; Wahab, S.; Ahmad, W.; Weir, S.A.; Alomary, M.N.; et al. Prospective Epigenetic Actions of Organo-Sulfur Compounds against Cancer: Perspectives and Molecular Mechanisms. Cancers 2023, 15, 697. https://doi.org/10.3390/cancers15030697
Shoaib S, Ansari MA, Ghazwani M, Hani U, Jamous YF, Alali Z, Wahab S, Ahmad W, Weir SA, Alomary MN, et al. Prospective Epigenetic Actions of Organo-Sulfur Compounds against Cancer: Perspectives and Molecular Mechanisms. Cancers. 2023; 15(3):697. https://doi.org/10.3390/cancers15030697
Chicago/Turabian StyleShoaib, Shoaib, Mohammad Azam Ansari, Mohammed Ghazwani, Umme Hani, Yahya F. Jamous, Zahraa Alali, Shadma Wahab, Wasim Ahmad, Sydney A. Weir, Mohammad N. Alomary, and et al. 2023. "Prospective Epigenetic Actions of Organo-Sulfur Compounds against Cancer: Perspectives and Molecular Mechanisms" Cancers 15, no. 3: 697. https://doi.org/10.3390/cancers15030697
APA StyleShoaib, S., Ansari, M. A., Ghazwani, M., Hani, U., Jamous, Y. F., Alali, Z., Wahab, S., Ahmad, W., Weir, S. A., Alomary, M. N., Yusuf, N., & Islam, N. (2023). Prospective Epigenetic Actions of Organo-Sulfur Compounds against Cancer: Perspectives and Molecular Mechanisms. Cancers, 15(3), 697. https://doi.org/10.3390/cancers15030697