
Pre-Existing and Acquired Resistance to PARP Inhibitor-Induced Synthetic Lethality


Simple Summary
Abstract
1. Introduction
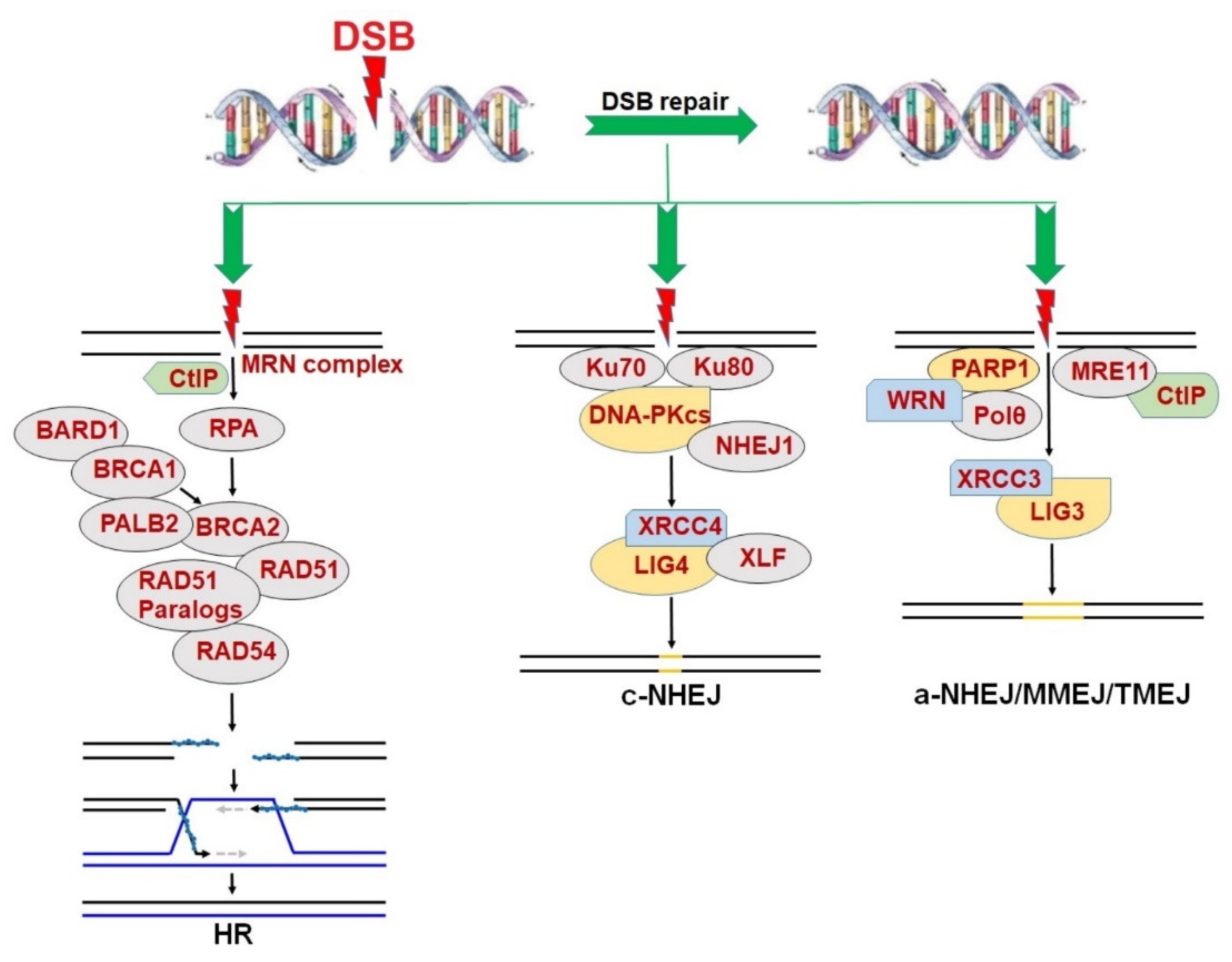
2. Synthetic Lethality in the Context of DNA Repair
3. PARPi-Induced Synthetic Lethality in BRCA1/2-Mutated Cancers
4. PARPi in Clinical Trials of BRCA1/2-Mutated Cancers
5. Therapeutic Potential of PARPi in Hematopoietic Malignancies
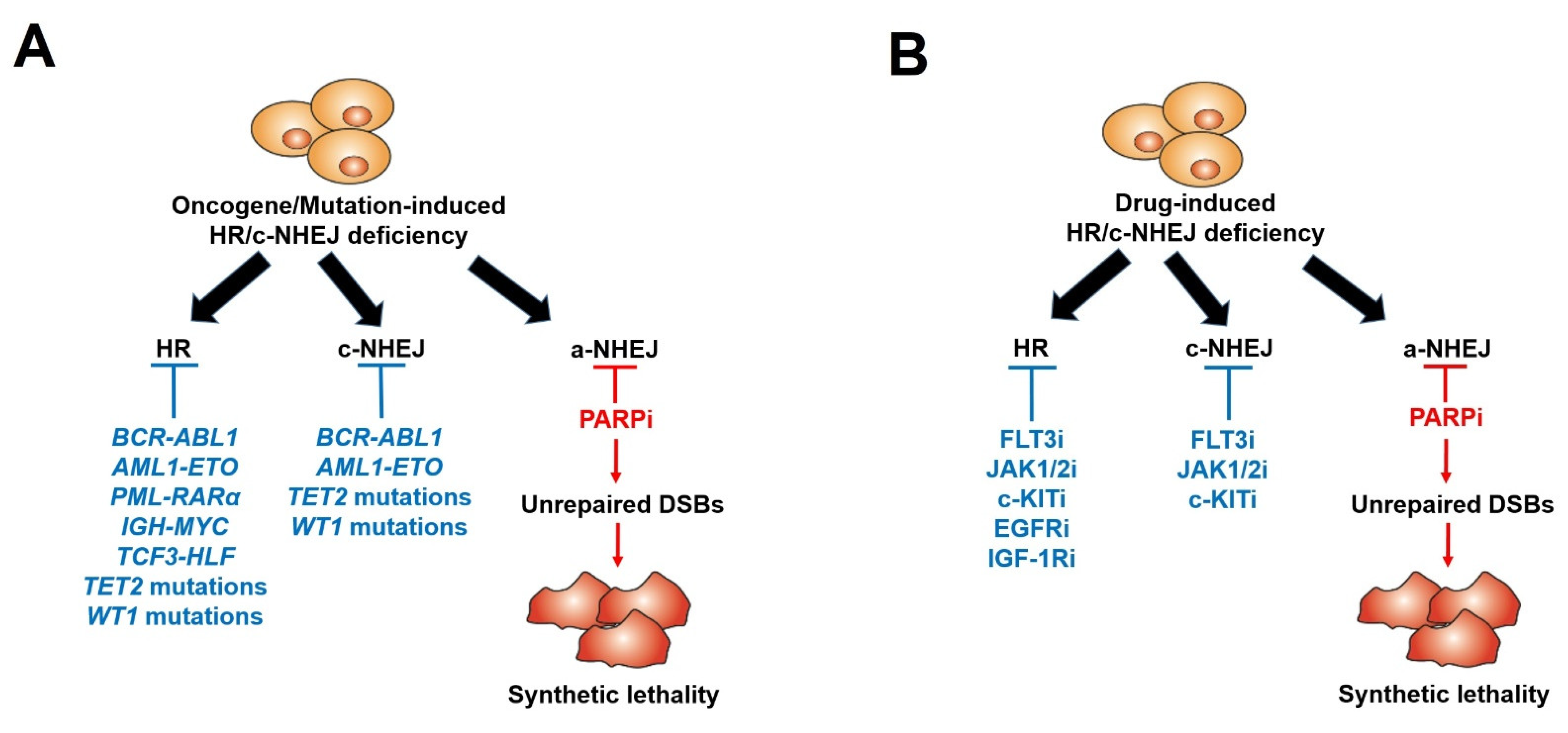
6. Acquired Resistance to PARPi-Mediated Synthetic Lethality
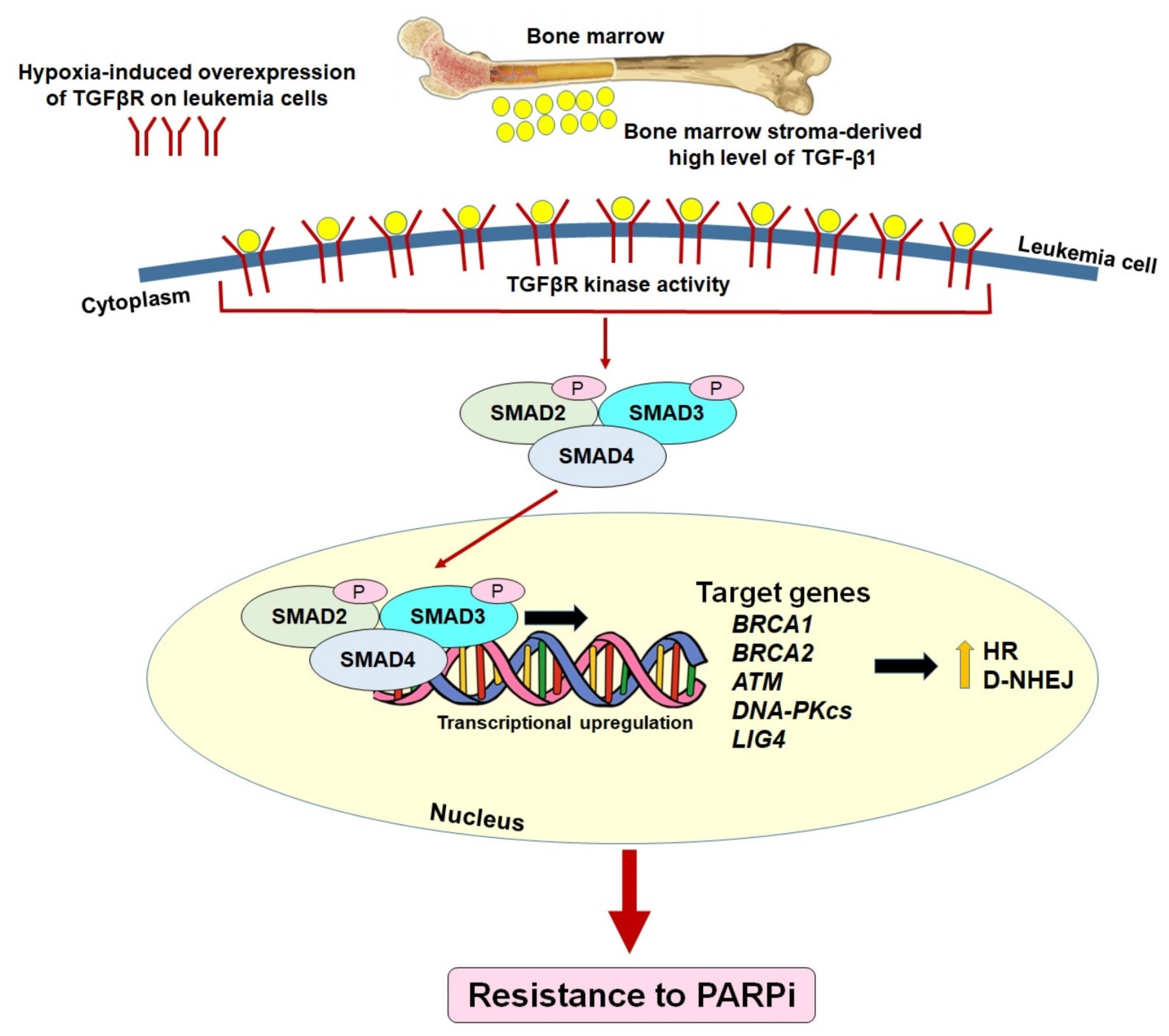
7. Pre-Existing Resistance to PARPi: The Bone Marrow Microenvironment
8. Pre-Existing Resistance to PARPi: Tumor-Inducing Mutations
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kaelin, W.G., Jr. The concept of synthetic lethality in the context of anticancer therapy. Nat. Rev. Cancer 2005, 5, 689–698. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Dudas, A.; Chovanec, M. DNA double-strand break repair by homologous recombination. Mutat. Res. 2004, 566, 131–167. [Google Scholar] [CrossRef]
- Yang, H.; Zhong, Y.; Peng, C.; Chen, J.Q.; Tian, D. Important role of indels in somatic mutations of human cancer genes. BMC Med. Genet. 2010, 11, 128. [Google Scholar] [CrossRef]
- Featherstone, C.; Jackson, S.P. DNA double-strand break repair. Curr. Biol. CB 1999, 9, R759–R761. [Google Scholar] [CrossRef]
- Mao, Z.; Bozzella, M.; Seluanov, A.; Gorbunova, V. Comparison of nonhomologous end joining and homologous recombination in human cells. DNA Repair 2008, 7, 1765–1771. [Google Scholar] [CrossRef]
- Humphryes, N.; Hochwagen, A. A non-sister act: Recombination template choice during meiosis. Exp. Cell Res. 2014, 329, 53–60. [Google Scholar] [CrossRef]
- Caldecott, K.W. Mammalian single-strand break repair: Mechanisms and links with chromatin. DNA Repair 2007, 6, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Dueva, R.; Iliakis, G. Alternative pathways of non-homologous end joining (NHEJ) in genomic instability and cancer. Transl. Cancer Res. 2013, 2, 163–177. [Google Scholar]
- Truong, L.N.; Li, Y.; Shi, L.Z.; Hwang, P.Y.-H.; He, J.; Wang, H.; Razavian, N.; Berns, M.W.; Wu, X. Microhomology-mediated End Joining and Homologous Recombination share the initial end resection step to repair DNA double-strand breaks in mammalian cells. Proc. Natl. Acad. Sci. USA 2013, 110, 7720–7725. [Google Scholar] [CrossRef] [PubMed]
- Kent, T.; Chandramouly, G.; McDevitt, S.M.; Ozdemir, A.Y.; Pomerantz, R.T. Mechanism of microhomology-mediated end-joining promoted by human DNA polymerase theta. Nat. Struct. Mol. Biol. 2015, 22, 230–237. [Google Scholar] [CrossRef]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef]
- Yap, T.A.; Sandhu, S.K.; Carden, C.P.; de Bono, J.S. Poly(ADP-ribose) polymerase (PARP) inhibitors: Exploiting a synthetic lethal strategy in the clinic. CA Cancer J. Clin. 2011, 61, 31–49. [Google Scholar] [CrossRef]
- Rottenberg, S.; Jaspers, J.E.; Kersbergen, A.; van der Burg, E.; Nygren, A.O.; Zander, S.A.; Derksen, P.W.; de Bruin, M.; Zevenhoven, J.; Lau, A.; et al. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc. Natl. Acad. Sci. USA 2008, 105, 17079–17084. [Google Scholar] [CrossRef] [PubMed]
- Fok, J.H.L.; Ramos-Montoya, A.; Vazquez-Chantada, M.; Wijnhoven, P.W.G.; Follia, V.; James, N.; Farrington, P.M.; Karmokar, A.; Willis, S.E.; Cairns, J.; et al. AZD7648 is a potent and selective DNA-PK inhibitor that enhances radiation, chemotherapy and olaparib activity. Nat. Commun. 2019, 10, 5065. [Google Scholar] [CrossRef]
- Chan, C.Y.; Tan, K.V.; Cornelissen, B. PARP Inhibitors in Cancer Diagnosis and Therapy. Clin. Cancer Res. 2021, 27, 1585–1594. [Google Scholar] [CrossRef] [PubMed]
- Nieborowska-Skorska, M.; Sullivan, K.; Dasgupta, Y.; Podszywalow-Bartnicka, P.; Hoser, G.; Maifrede, S.; Martinez, E.; Di Marcantonio, D.; Bolton-Gillespie, E.; Cramer-Morales, K.; et al. Gene expression and mutation-guided synthetic lethality eradicates proliferating and quiescent leukemia cells. J. Clin. Investig. 2017, 127, 2392–2406. [Google Scholar] [CrossRef] [PubMed]
- Fong, P.C.; Boss, D.S.; Yap, T.A.; Tutt, A.; Wu, P.; Mergui-Roelvink, M.; Mortimer, P.; Swaisland, H.; Lau, A.; O'Connor, M.J.; et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N. Engl. J. Med. 2009, 361, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Gunjur, A. Talazoparib for BRCA-mutated advanced breast cancer. Lancet Oncol. 2018, 19, e511. [Google Scholar] [CrossRef]
- Hurvitz, S.A.; Gonçalves, A.; Rugo, H.S.; Lee, K.H.; Fehrenbacher, L.; Mina, L.A.; Diab, S.; Blum, J.L.; Chakrabarti, J.; Elmeliegy, M.; et al. Talazoparib in Patients with a Germline BRCA-Mutated Advanced Breast Cancer: Detailed Safety Analyses from the Phase III EMBRACA Trial. Oncol. 2019, 25, e439–e450. [Google Scholar] [CrossRef] [PubMed]
- Litton, J.K.; Rugo, H.S.; Ettl, J.; Hurvitz, S.A.; Gonçalves, A.; Lee, K.H.; Fehrenbacher, L.; Yerushalmi, R.; Mina, L.A.; Martin, M.; et al. Talazoparib in Patients with Advanced Breast Cancer and a Germline BRCA Mutation. N. Engl. J. Med. 2018, 379, 753–763. [Google Scholar] [CrossRef] [PubMed]
- Podszywalow-Bartnicka, P.; Wolczyk, M.; Kusio-Kobialka, M.; Wolanin, K.; Skowronek, K.; Nieborowska-Skorska, M.; Dasgupta, Y.; Skorski, T.; Piwocka, K. Downregulation of BRCA1 protein in BCR-ABL1 leukemia cells depends on stress-triggered TIAR-mediated suppression of translation. Cell Cycle 2014, 13, 3727–3741. [Google Scholar] [CrossRef] [PubMed]
- Maifrede, S.; Martin, K.; Podszywalow-Bartnicka, P.; Sullivan-Reed, K.; Langer, S.K.; Nejati, R.; Dasgupta, Y.; Hulse, M.; Gritsyuk, D.; Nieborowska-Skorska, M.; et al. IGH/MYC Translocation Associates with BRCA2 Deficiency and Synthetic Lethality to PARP1 Inhibitors. Mol. Cancer Res. 2017, 15, 967–972. [Google Scholar] [CrossRef]
- Esposito, M.T.; Zhao, L.; Fung, T.K.; Rane, J.K.; Wilson, A.; Martin, N.; Gil, J.; Leung, A.Y.; Ashworth, A.; So, C.W. Synthetic lethal targeting of oncogenic transcription factors in acute leukemia by PARP inhibitors. Nat. Med. 2015, 21, 1481–1490. [Google Scholar] [CrossRef]
- Faraoni, I.; Giansanti, M.; Voso, M.T.; Lo-Coco, F.; Graziani, G. Targeting ADP-ribosylation by PARP inhibitors in acute myeloid leukaemia and related disorders. Biochem. Pharmacol. 2019, 167, 133–148. [Google Scholar] [CrossRef]
- Nieborowska-Skorska, M.; Maifrede, S.; Dasgupta, Y.; Sullivan, K.; Flis, S.; Le, B.V.; Solecka, M.; Belyaeva, E.A.; Kubovcakova, L.; Nawrocki, M.; et al. Ruxolitinib-induced defects in DNA repair cause sensitivity to PARP inhibitors in myeloproliferative neoplasms. Blood 2017, 130, 2848–2859. [Google Scholar] [CrossRef]
- Maifrede, S.; Nieborowska-Skorska, M.; Sullivan-Reed, K.; Dasgupta, Y.; Podszywalow-Bartnicka, P.; Le, B.V.; Solecka, M.; Lian, Z.; Belyaeva, E.A.; Nersesyan, A.; et al. Tyrosine kinase inhibitor-induced defects in DNA repair sensitize FLT3(ITD)-positive leukemia cells to PARP1 inhibitors. Blood 2018, 132, 67–77. [Google Scholar] [CrossRef]
- Nieborowska-Skorska, M.; Paietta, E.M.; Levine, R.L.; Fernandez, H.F.; Tallman, M.S.; Litzow, M.R.; Skorski, T. Inhibition of the mutated c-KIT kinase in AML1-ETO-positive leukemia cells restores sensitivity to PARP inhibitor. Blood Adv. 2019, 3, 4050–4054. [Google Scholar] [CrossRef]
- Nowsheen, S.; Cooper, T.; Stanley, J.A.; Yang, E.S. Synthetic Lethal Interactions between EGFR and PARP Inhibition in Human Triple Negative Breast Cancer Cells. PLoS ONE 2012, 7, e46614. [Google Scholar] [CrossRef] [PubMed]
- Amin, O.; Beauchamp, M.-C.; Nader, P.A.; Laskov, I.; Iqbal, S.; Philip, C.-A.; Yasmeen, A.; Gotlieb, W.H. Suppression of Homologous Recombination by insulin-like growth factor-1 inhibition sensitizes cancer cells to PARP inhibitors. BMC Cancer 2015, 15, 817. [Google Scholar] [CrossRef] [PubMed]
- D'Andrea, A.D. Mechanisms of PARP inhibitor sensitivity and resistance. DNA Repair 2018, 71, 172–176. [Google Scholar] [CrossRef] [PubMed]
- Sullivan-Reed, K.; Bolton-Gillespie, E.; Dasgupta, Y.; Langer, S.; Siciliano, M.; Nieborowska-Skorska, M.; Hanamshet, K.; Belyaeva, E.A.; Bernhardy, A.J.; Lee, J.; et al. Simultaneous Targeting of PARP1 and RAD52 Triggers Dual Synthetic Lethality in BRCA-Deficient Tumor Cells. Cell Rep. 2018, 23, 3127–3136. [Google Scholar] [CrossRef] [PubMed]
- Sakai, W.; Swisher, E.M.; Karlan, B.Y.; Agarwal, M.K.; Higgins, J.; Friedman, C.; Villegas, E.; Jacquemont, C.; Farrugia, D.J.; Couch, F.J.; et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature 2008, 451, 1116–1120. [Google Scholar] [CrossRef] [PubMed]
- Le, B.V.; Podszywalow-Bartnicka, P.; Maifrede, S.; Sullivan-Reed, K.; Nieborowska-Skorska, M.; Golovine, K.; Yao, J.C.; Nejati, R.; Cai, K.Q.; Caruso, L.B.; et al. TGFβR-SMAD3 Signaling Induces Resistance to PARP Inhibitors in the Bone Marrow Microenvironment. Cell Rep. 2020, 33, 108221. [Google Scholar] [CrossRef] [PubMed]
- Cooke, M.S.; Evans, M.D.; Dizdaroglu, M.; Lunec, J. Oxidative DNA damage: Mechanisms, mutation, and disease. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2003, 17, 1195–1214. [Google Scholar] [CrossRef] [PubMed]
- O'Neil, N.J.; Bailey, M.L.; Hieter, P. Synthetic lethality and cancer. Nat. Rev. Genet. 2017, 18, 613–623. [Google Scholar] [CrossRef] [PubMed]
- Nijman, S.M. Synthetic lethality: General principles, utility and detection using genetic screens in human cells. FEBS Lett. 2011, 585, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Dobzhansky, T. Genetics of natural populations; recombination and variability in populations of Drosophila pseudoobscura. Genetics 1946, 31, 269–290. [Google Scholar] [CrossRef] [PubMed]
- Grabarz, A.; Barascu, A.; Guirouilh-Barbat, J.; Lopez, B.S. Initiation of DNA double strand break repair: Signaling and single-stranded resection dictate the choice between homologous recombination, non-homologous end-joining and alternative end-joining. Am. J. Cancer Res. 2012, 2, 249–268. [Google Scholar] [PubMed]
- Mazin, A.V.; Bornarth, C.J.; Solinger, J.A.; Heyer, W.D.; Kowalczykowski, S.C. Rad54 protein is targeted to pairing loci by the Rad51 nucleoprotein filament. Mol. Cell 2000, 6, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Mazin, A.V.; Alexeev, A.A.; Kowalczykowski, S.C. A novel function of Rad54 protein. Stabilization of the Rad51 nucleoprotein filament. J. Biol. Chem. 2003, 278, 14029–14036. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495–506. [Google Scholar] [CrossRef] [PubMed]
- Howard, S.M.; Yanez, D.A.; Stark, J.M. DNA damage response factors from diverse pathways, including DNA crosslink repair, mediate alternative end joining. PLoS Genet. 2015, 11, e1004943. [Google Scholar] [CrossRef] [PubMed]
- Boboila, C.; Jankovic, M.; Yan, C.T.; Wang, J.H.; Wesemann, D.R.; Zhang, T.; Fazeli, A.; Feldman, L.; Nussenzweig, A.; Nussenzweig, M.; et al. Alternative end-joining catalyzes robust IgH locus deletions and translocations in the combined absence of ligase 4 and Ku70. Proc. Natl. Acad. Sci. USA 2010, 107, 3034–3039. [Google Scholar] [CrossRef] [PubMed]
- Keijzers, G.; Maynard, S.; Shamanna, R.A.; Rasmussen, L.J.; Croteau, D.L.; Bohr, V.A. The role of RecQ helicases in non-homologous end-joining. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 463–472. [Google Scholar] [CrossRef]
- Parsons, J.L.; Dianova, II; Allinson, S. L.; Dianov, G.L. Poly(ADP-ribose) polymerase-1 protects excessive DNA strand breaks from deterioration during repair in human cell extracts. FEBS J. 2005, 272, 2012–2021. [Google Scholar] [CrossRef] [PubMed]
- Bouchard, V.J.; Rouleau, M.; Poirier, G.G. PARP-1, a determinant of cell survival in response to DNA damage. Exp. Hematol. 2003, 31, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Kuzminov, A. Single-strand interruptions in replicating chromosomes cause double-strand breaks. Proc. Natl. Acad. Sci. USA 2001, 98, 8241–8246. [Google Scholar] [CrossRef] [PubMed]
- Iglehart, J.D.; Silver, D.P. Synthetic lethality--a new direction in cancer-drug development. N. Engl. J. Med. 2009, 361, 189–191. [Google Scholar] [CrossRef] [PubMed]
- McCann, K.E.; Hurvitz, S.A. Advances in the use of PARP inhibitor therapy for breast cancer. Drugs Context 2018, 7, 212540. [Google Scholar] [CrossRef] [PubMed]
- Mittica, G.; Ghisoni, E.; Giannone, G.; Genta, S.; Aglietta, M.; Sapino, A.; Valabrega, G. PARP Inhibitors in Ovarian Cancer. Recent Pat. Anticancer Drug Discov. 2018, 13, 392–410. [Google Scholar] [CrossRef] [PubMed]
- Cong, K.; Peng, M.; Kousholt, A.N.; Lee, W.T.C.; Lee, S.; Nayak, S.; Krais, J.; VanderVere-Carozza, P.S.; Pawelczak, K.S.; Calvo, J.; et al. Replication gaps are a key determinant of PARP inhibitor synthetic lethality with BRCA deficiency. Mol. Cell 2021, 81, 3128–3144.e3127. [Google Scholar] [CrossRef] [PubMed]
- Panzarino, N.J.; Krais, J.J.; Cong, K.; Peng, M.; Mosqueda, M.; Nayak, S.U.; Bond, S.M.; Calvo, J.A.; Doshi, M.B.; Bere, M.; et al. Replication Gaps Underlie BRCA Deficiency and Therapy Response. Cancer Res. 2021, 81, 1388–1397. [Google Scholar] [CrossRef]
- Czyz, M.; Toma, M.; Gajos-Michniewicz, A.; Majchrzak, K.; Hoser, G.; Szemraj, J.; Nieborowska-Skorska, M.; Cheng, P.; Gritsyuk, D.; Levesque, M.; et al. PARP1 inhibitor olaparib (Lynparza) exerts synthetic lethal effect against ligase 4-deficient melanomas. Oncotarget 2016, 7, 75551–75560. [Google Scholar] [CrossRef] [PubMed]
- Curtin, N.J.; Szabo, C. Therapeutic applications of PARP inhibitors: Anticancer therapy and beyond. Mol. Aspects Med. 2013, 34, 1217–1256. [Google Scholar] [CrossRef] [PubMed]
- Murai, J.; Huang, S.Y.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, T.A.; Ainsworth, W.B.; Ellis, P.A.; Donawho, C.K.; DiGiammarino, E.L.; Panchal, S.C.; Abraham, V.C.; Algire, M.A.; Shi, Y.; Olson, A.M.; et al. PARP1 Trapping by PARP Inhibitors Drives Cytotoxicity in Both Cancer Cells and Healthy Bone Marrow. Mol. Cancer Res. 2019, 17, 409–419. [Google Scholar] [CrossRef]
- Murai, J.; Huang, S.Y.; Renaud, A.; Zhang, Y.; Ji, J.; Takeda, S.; Morris, J.; Teicher, B.; Doroshow, J.H.; Pommier, Y. Stereospecific PARP trapping by BMN 673 and comparison with olaparib and rucaparib. Mol. Cancer Ther. 2014, 13, 433–443. [Google Scholar] [CrossRef]
- Meehan, R.S.; Chen, A.P. New treatment option for ovarian cancer: PARP inhibitors. Gynecol. Oncol. Res. Pract. 2016, 3, 3. [Google Scholar] [CrossRef]
- Kim, G.; Ison, G.; McKee, A.E.; Zhang, H.; Tang, S.; Gwise, T.; Sridhara, R.; Lee, E.; Tzou, A.; Philip, R.; et al. FDA Approval Summary: Olaparib Monotherapy in Patients with Deleterious Germline BRCA-Mutated Advanced Ovarian Cancer Treated with Three or More Lines of Chemotherapy. Clin. Cancer Res. 2015, 21, 4257–4261. [Google Scholar] [CrossRef] [PubMed]
- Pujade-Lauraine, E.; Ledermann, J.A.; Selle, F.; Gebski, V.; Penson, R.T.; Oza, A.M.; Korach, J.; Huzarski, T.; Poveda, A.; Pignata, S.; et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): A double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2017, 18, 1274–1284. [Google Scholar] [CrossRef]
- Friedlander, M.; Matulonis, U.; Gourley, C.; du Bois, A.; Vergote, I.; Rustin, G.; Scott, C.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Long-term efficacy, tolerability and overall survival in patients with platinum-sensitive, recurrent high-grade serous ovarian cancer treated with maintenance olaparib capsules following response to chemotherapy. Br. J. Cancer 2018, 119, 1075–1085. [Google Scholar] [CrossRef] [PubMed]
- Robson, M.; Im, S.-A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N. Engl. J. Med. 2017, 377, 523–533. [Google Scholar] [CrossRef] [PubMed]
- Golan, T.; Hammel, P.; Reni, M.; van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.O.; Hochhauser, D.; Arnold, D.; Oh, D.Y.; et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Ray-Coquard, I.; Pautier, P.; Pignata, S.; Pérol, D.; González-Martín, A.; Berger, R.; Fujiwara, K.; Vergote, I.; Colombo, N.; Mäenpää, J.; et al. Olaparib plus Bevacizumab as First-Line Maintenance in Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2416–2428. [Google Scholar] [CrossRef]
- De Bono, J.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Olaparib for Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2020, 382, 2091–2102. [Google Scholar] [CrossRef]
- Oza, A.M.; Tinker, A.V.; Oaknin, A.; Shapira-Frommer, R.; McNeish, I.A.; Swisher, E.M.; Ray-Coquard, I.; Bell-McGuinn, K.; Coleman, R.L.; O'Malley, D.M.; et al. Antitumor activity and safety of the PARP inhibitor rucaparib in patients with high-grade ovarian carcinoma and a germline or somatic BRCA1 or BRCA2 mutation: Integrated analysis of data from Study 10 and ARIEL2. Gynecol. Oncol. 2017, 147, 267–275. [Google Scholar] [CrossRef]
- Coleman, R.L.; Oza, A.M.; Lorusso, D.; Aghajanian, C.; Oaknin, A.; Dean, A.; Colombo, N.; Weberpals, J.I.; Clamp, A.; Scambia, G.; et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 390, 1949–1961. [Google Scholar] [CrossRef]
- Abida, W.; Campbell, D.; Patnaik, A.; Sautois, B.; Shapiro, J.; Vogelzang, N.J.; Bryce, A.H.; McDermott, R.; Ricci, F.; Rowe, J.; et al. 846PD - Preliminary results from the TRITON2 study of rucaparib in patients (pts) with DNA damage repair (DDR)-deficient metastatic castration-resistant prostate cancer (mCRPC): Updated analyses. Ann. Oncol. 2019, 30, v327–v328. [Google Scholar] [CrossRef]
- Mirza, M.R.; Monk, B.J.; Herrstedt, J.; Oza, A.M.; Mahner, S.; Redondo, A.; Fabbro, M.; Ledermann, J.A.; Lorusso, D.; Vergote, I.; et al. Niraparib Maintenance Therapy in Platinum-Sensitive, Recurrent Ovarian Cancer. N. Engl. J. Med. 2016, 375, 2154–2164. [Google Scholar] [CrossRef]
- Moore, K.N.; Secord, A.A.; Geller, M.A.; Miller, D.S.; Cloven, N.; Fleming, G.F.; Wahner Hendrickson, A.E.; Azodi, M.; DiSilvestro, P.; Oza, A.M.; et al. Niraparib monotherapy for late-line treatment of ovarian cancer (QUADRA): A multicentre, open-label, single-arm, phase 2 trial. Lancet Oncol. 2019, 20, 636–648. [Google Scholar] [CrossRef]
- González-Martín, A.; Pothuri, B.; Vergote, I.; DePont Christensen, R.; Graybill, W.; Mirza, M.R.; McCormick, C.; Lorusso, D.; Hoskins, P.; Freyer, G.; et al. Niraparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2391–2402. [Google Scholar] [CrossRef]
- De Bono, J.; Ramanathan, R.K.; Mina, L.; Chugh, R.; Glaspy, J.; Rafii, S.; Kaye, S.; Sachdev, J.; Heymach, J.; Smith, D.C.; et al. Phase I, Dose-Escalation, Two-Part Trial of the PARP Inhibitor Talazoparib in Patients with Advanced Germline BRCA1/2 Mutations and Selected Sporadic Cancers. Cancer Discov. 2017, 7, 620–629. [Google Scholar] [CrossRef]
- Exman, P.; Barroso-Sousa, R.; Tolaney, S.M. Evidence to date: Talazoparib in the treatment of breast cancer. Onco Targets Ther. 2019, 12, 5177–5187. [Google Scholar] [CrossRef] [PubMed]
- Ettl, J.; Quek, R.G.W.; Lee, K.H.; Rugo, H.S.; Hurvitz, S.; Gonçalves, A.; Fehrenbacher, L.; Yerushalmi, R.; Mina, L.A.; Martin, M.; et al. Quality of life with talazoparib versus physician's choice of chemotherapy in patients with advanced breast cancer and germline BRCA1/2 mutation: Patient-reported outcomes from the EMBRACA phase III trial. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2018, 29, 1939–1947. [Google Scholar] [CrossRef]
- Coleman, R.L.; Fleming, G.F.; Brady, M.F.; Swisher, E.M.; Steffensen, K.D.; Friedlander, M.; Okamoto, A.; Moore, K.N.; Efrat Ben-Baruch, N.; Werner, T.L.; et al. Veliparib with First-Line Chemotherapy and as Maintenance Therapy in Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2403–2415. [Google Scholar] [CrossRef] [PubMed]
- Murai, J.; Pommier, Y. Classification of PARP Inhibitors Based on PARP Trapping and Catalytic Inhibition, and Rationale for Combinations with Topoisomerase I Inhibitors and Alkylating Agents. In PARP Inhibitors for Cancer Therapy; Curtin, N.J., Sharma, R.A., Eds.; Springer International Publishing: Cham, Switzerland, 2015; pp. 261–274. [Google Scholar]
- Maifrede, S.; Martinez, E.; Nieborowska-Skorska, M.; Di Marcantonio, D.; Hulse, M.; Le, B.V.; Zhao, H.; Piwocka, K.; Tempera, I.; Sykes, S.M.; et al. MLL-AF9 leukemias are sensitive to PARP1 inhibitors combined with cytotoxic drugs. Blood Adv. 2017, 1, 1467–1472. [Google Scholar] [CrossRef] [PubMed]
- Piao, J.; Takai, S.; Kamiya, T.; Inukai, T.; Sugita, K.; Ohyashiki, K.; Delia, D.; Masutani, M.; Mizutani, S.; Takagi, M. Poly (ADP-ribose) polymerase inhibitors selectively induce cytotoxicity in TCF3-HLF-positive leukemic cells. Cancer Lett. 2017, 386, 131–140. [Google Scholar] [CrossRef]
- Molenaar, R.J.; Radivoyevitch, T.; Nagata, Y.; Khurshed, M.; Przychodzen, B.; Makishima, H.; Xu, M.; Bleeker, F.E.; Wilmink, J.W.; Carraway, H.E.; et al. IDH1/2 Mutations Sensitize Acute Myeloid Leukemia to PARP Inhibition and This Is Reversed by IDH1/2-Mutant Inhibitors. Clin. Cancer Res. 2018, 24, 1705–1715. [Google Scholar] [CrossRef]
- Deutsch, E.; Jarrousse, S.; Buet, D.; Dugray, A.; Bonnet, M.L.; Vozenin-Brotons, M.C.; Guilhot, F.; Turhan, A.G.; Feunteun, J.; Bourhis, J. Down-regulation of BRCA1 in BCR-ABL-expressing hematopoietic cells. Blood 2003, 101, 4583–4588. [Google Scholar] [CrossRef] [PubMed]
- Cimmino, L.; Dolgalev, I.; Wang, Y.; Yoshimi, A.; Martin, G.H.; Wang, J.; Ng, V.; Xia, B.; Witkowski, M.T.; Mitchell-Flack, M.; et al. Restoration of TET2 Function Blocks Aberrant Self-Renewal and Leukemia Progression. Cell 2017, 170, 1079–1095.e1020. [Google Scholar] [CrossRef]
- Inoue, S.; Li, W.Y.; Tseng, A.; Beerman, I.; Elia, A.J.; Bendall, S.C.; Lemonnier, F.; Kron, K.J.; Cescon, D.W.; Hao, Z.; et al. Mutant IDH1 Downregulates ATM and Alters DNA Repair and Sensitivity to DNA Damage Independent of TET2. Cancer Cell 2016, 30, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Maifrede, S.; Le, B.V.; Nieborowska-Skorska, M.; Golovine, K.; Sullivan-Reed, K.; Dunuwille, W.M.B.; Nacson, J.; Hulse, M.; Keith, K.; Madzo, J.; et al. TET2 and DNMT3A Mutations Exert Divergent Effects on DNA Repair and Sensitivity of Leukemia Cells to PARP Inhibitors. Cancer Res. 2021, 81, 5089–5101. [Google Scholar] [CrossRef] [PubMed]
- Dkhissi, F.; Aggoune, D.; Pontis, J.; Sorel, N.; Piccirilli, N.; LeCorf, A.; Guilhot, F.; Chomel, J.C.; Ait-Si-Ali, S.; Turhan, A.G. The downregulation of BAP1 expression by BCR-ABL reduces the stability of BRCA1 in chronic myeloid leukemia. Exp. Hematol. 2015, 43, 775–780. [Google Scholar] [CrossRef]
- Alcalay, M.; Meani, N.; Gelmetti, V.; Fantozzi, A.; Fagioli, M.; Orleth, A.; Riganelli, D.; Sebastiani, C.; Cappelli, E.; Casciari, C.; et al. Acute myeloid leukemia fusion proteins deregulate genes involved in stem cell maintenance and DNA repair. J. Clin. Investig. 2003, 112, 1751–1761. [Google Scholar] [CrossRef] [PubMed]
- Sulkowski Parker, L.; Corso Christopher, D.; Robinson Nathaniel, D.; Scanlon Susan, E.; Purshouse Karin, R.; Bai, H.; Liu, Y.; Sundaram Ranjini, K.; Hegan Denise, C.; Fons Nathan, R.; et al. 2-Hydroxyglutarate produced by neomorphic IDH mutations suppresses homologous recombination and induces PARP inhibitor sensitivity. Sci. Transl. Med. 2017, 9, eaal2463. [Google Scholar] [CrossRef]
- Sulkowski, P.L.; Oeck, S.; Dow, J.; Economos, N.G.; Mirfakhraie, L.; Liu, Y.; Noronha, K.; Bao, X.; Li, J.; Shuch, B.M.; et al. Oncometabolites suppress DNA repair by disrupting local chromatin signalling. Nature 2020, 582, 586–591. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.L.; Lin, H.P.; Zhou, W.J.; He, C.X.; Zhang, Z.Y.; Cheng, Z.L.; Song, J.B.; Liu, P.; Chen, X.Y.; Xia, Y.K.; et al. SNIP1 Recruits TET2 to Regulate c-MYC Target Genes and Cellular DNA Damage Response. Cell Rep. 2018, 25, 1485–1500.e1484. [Google Scholar] [CrossRef]
- Deutsch, E.; Dugray, A.; AbdulKarim, B.; Marangoni, E.; Maggiorella, L.; Vaganay, S.; M'Kacher, R.; Rasy, S.D.; Eschwege, F.; Vainchenker, W.; et al. BCR-ABL down-regulates the DNA repair protein DNA-PKcs. Blood 2001, 97, 2084–2090. [Google Scholar] [CrossRef]
- Tothova, Z.; Valton, A.-L.; Gorelov, R.A.; Vallurupalli, M.; Krill-Burger, J.M.; Holmes, A.; Landers, C.C.; Haydu, J.E.; Malolepsza, E.; Hartigan, C.; et al. Cohesin mutations alter DNA damage repair and chromatin structure and create therapeutic vulnerabilities in MDS/AML. JCI Insight 2021, 6, e142149. [Google Scholar] [CrossRef] [PubMed]
- Nowicki, M.O.; Falinski, R.; Koptyra, M.; Slupianek, A.; Stoklosa, T.; Gloc, E.; Nieborowska-Skorska, M.; Blasiak, J.; Skorski, T. BCR/ABL oncogenic kinase promotes unfaithful repair of the reactive oxygen species-dependent DNA double-strand breaks. Blood 2004, 104, 3746–3753. [Google Scholar] [CrossRef]
- Nieborowska-Skorska, M.; Kopinski, P.K.; Ray, R.; Hoser, G.; Ngaba, D.; Flis, S.; Cramer, K.; Reddy, M.M.; Koptyra, M.; Penserga, T.; et al. Rac2-MRC-cIII-generated ROS cause genomic instability in chronic myeloid leukemia stem cells and primitive progenitors. Blood 2012, 119, 4253–4263. [Google Scholar] [CrossRef] [PubMed]
- Marty, C.; Lacout, C.; Droin, N.; Le Couédic, J.P.; Ribrag, V.; Solary, E.; Vainchenker, W.; Villeval, J.L.; Plo, I. A role for reactive oxygen species in JAK2 V617F myeloproliferative neoplasm progression. Leukemia 2013, 27, 2187–2195. [Google Scholar] [CrossRef] [PubMed]
- Mufti, G.; Estey, E.; Popat, R.; Mattison, R.; Menne, T.; Azar, J.; Bloor, A.; Gaymes, T.; Khwaja, A.; Juckett, M. Results of a phase 1 study of BMN 673, a potent and specific PARP-1/2 inhibitor, in patients with advanced hematological malignancies. In Haematologica, 2014; Ferrata Storti Foundation: Pavia, Italy, 2014; pp. 33–34. [Google Scholar]
- Gojo, I.; Beumer, J.H.; Pratz, K.W.; McDevitt, M.A.; Baer, M.R.; Blackford, A.L.; Smith, B.D.; Gore, S.D.; Carraway, H.E.; Showel, M.M.; et al. A Phase 1 Study of the PARP Inhibitor Veliparib in Combination with Temozolomide in Acute Myeloid Leukemia. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 697–706. [Google Scholar] [CrossRef] [PubMed]
- Pratz, K.W.; Rudek, M.A.; Gojo, I.; Litzow, M.R.; McDevitt, M.A.; Ji, J.; Karnitz, L.M.; Herman, J.G.; Kinders, R.J.; Smith, B.D.; et al. A Phase I Study of Topotecan, Carboplatin and the PARP Inhibitor Veliparib in Acute Leukemias, Aggressive Myeloproliferative Neoplasms, and Chronic Myelomonocytic Leukemia. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 899–907. [Google Scholar] [CrossRef] [PubMed]
- Pratt, G.; Yap, C.; Oldreive, C.; Slade, D.; Bishop, R.; Griffiths, M.; Dyer, M.J.S.; Fegan, C.; Oscier, D.; Pettitt, A.; et al. A multi-centre phase I trial of the PARP inhibitor olaparib in patients with relapsed chronic lymphocytic leukaemia, T-prolymphocytic leukaemia or mantle cell lymphoma. Br. J. Haematol. 2018, 182, 429–433. [Google Scholar] [CrossRef] [PubMed]
- Morice, P.M.; Leary, A.; Dolladille, C.; Chrétien, B.; Poulain, L.; González-Martín, A.; Moore, K.; O'Reilly, E.M.; Ray-Coquard, I.; Alexandre, J. Myelodysplastic syndrome and acute myeloid leukaemia in patients treated with PARP inhibitors: A safety meta-analysis of randomised controlled trials and a retrospective study of the WHO pharmacovigilance database. Lancet Haematol. 2021, 8, e122–e134. [Google Scholar] [CrossRef]
- Baer, M.R.; Kogan, A.A.; Bentzen, S.M.; Mi, T.; Lapidus, R.G.; Duong, V.H.; Emadi, A.; Niyongere, S.; O'Connell, C.L.; Youngblood, B.A.; et al. Phase I Clinical Trial of DNA Methyltransferase Inhibitor Decitabine and PARP Inhibitor Talazoparib Combination Therapy in Relapsed/Refractory Acute Myeloid Leukemia. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2022, 28, 1313–1322. [Google Scholar] [CrossRef]
- Quigley, D.; Alumkal, J.J.; Wyatt, A.W.; Kothari, V.; Foye, A.; Lloyd, P.; Aggarwal, R.; Kim, W.; Lu, E.; Schwartzman, J.; et al. Analysis of Circulating Cell-Free DNA Identifies Multiclonal Heterogeneity of BRCA2 Reversion Mutations Associated with Resistance to PARP Inhibitors. Cancer Discov. 2017, 7, 999–1005. [Google Scholar] [CrossRef]
- Edwards, S.L.; Brough, R.; Lord, C.J.; Natrajan, R.; Vatcheva, R.; Levine, D.A.; Boyd, J.; Reis-Filho, J.S.; Ashworth, A. Resistance to therapy caused by intragenic deletion in BRCA2. Nature 2008, 451, 1111–1115. [Google Scholar] [CrossRef] [PubMed]
- Pettitt, S.J.; Frankum, J.R.; Punta, M.; Lise, S.; Alexander, J.; Chen, Y.; Yap, T.A.; Haider, S.; Tutt, A.N.J.; Lord, C.J. Clinical BRCA1/2 Reversion Analysis Identifies Hotspot Mutations and Predicted Neoantigens Associated with Therapy Resistance. Cancer Discov. 2020, 10, 1475–1488. [Google Scholar] [CrossRef] [PubMed]
- Jacot, W.; Thezenas, S.; Senal, R.; Viglianti, C.; Laberenne, A.C.; Lopez-Crapez, E.; Bibeau, F.; Bleuse, J.P.; Romieu, G.; Lamy, P.J. BRCA1 promoter hypermethylation, 53BP1 protein expression and PARP-1 activity as biomarkers of DNA repair deficit in breast cancer. BMC Cancer 2013, 13, 523. [Google Scholar] [CrossRef] [PubMed]
- Marzio, A.; Puccini, J.; Kwon, Y.; Maverakis, N.K.; Arbini, A.; Sung, P.; Bar-Sagi, D.; Pagano, M. The F-Box Domain-Dependent Activity of EMI1 Regulates PARPi Sensitivity in Triple-Negative Breast Cancers. Mol. Cell 2019, 73, 224–237.e226. [Google Scholar] [CrossRef] [PubMed]
- Marzio, A.; Kurz, E.; Sahni, J.M.; Di Feo, G.; Puccini, J.; Jiang, S.; Hirsch, C.A.; Arbini, A.A.; Wu, W.L.; Pass, H.I.; et al. EMSY inhibits homologous recombination repair and the interferon response, promoting lung cancer immune evasion. Cell 2022, 185, 169–183.e119. [Google Scholar] [CrossRef] [PubMed]
- Venugopal, K.; Feng, Y.; Nowialis, P.; Xu, H.; Shabashvili, D.E.; Berntsen, C.M.; Kaur, P.; Krajcik, K.I.; Taragjini, C.; Zaroogian, Z.; et al. DNMT3A Harboring Leukemia-Associated Mutations Directs Sensitivity to DNA Damage at Replication Forks. Clin. Cancer Res. 2022, 28, 756–769. [Google Scholar] [CrossRef]
- Liu, X.; Han, E.K.; Anderson, M.; Shi, Y.; Semizarov, D.; Wang, G.; McGonigal, T.; Roberts, L.; Lasko, L.; Palma, J.; et al. Acquired resistance to combination treatment with temozolomide and ABT-888 is mediated by both base excision repair and homologous recombination DNA repair pathways. Mol. Cancer Res. 2009, 7, 1686–1692. [Google Scholar] [CrossRef] [PubMed]
- Pettitt, S.J.; Krastev, D.B.; Brandsma, I.; Drean, A.; Song, F.; Aleksandrov, R.; Harrell, M.I.; Menon, M.; Brough, R.; Campbell, J.; et al. Genome-wide and high-density CRISPR-Cas9 screens identify point mutations in PARP1 causing PARP inhibitor resistance. Nat. Commun. 2018, 9, 1849. [Google Scholar] [CrossRef]
- Mirman, Z.; Lottersberger, F.; Takai, H.; Kibe, T.; Gong, Y.; Takai, K.; Bianchi, A.; Zimmermann, M.; Durocher, D.; de Lange, T. 53BP1–RIF1–shieldin counteracts DSB resection through CST- and Polα-dependent fill-in. Nature 2018, 560, 112–116. [Google Scholar] [CrossRef]
- Ghezraoui, H.; Oliveira, C.; Becker, J.R.; Bilham, K.; Moralli, D.; Anzilotti, C.; Fischer, R.; Deobagkar-Lele, M.; Sanchiz-Calvo, M.; Fueyo-Marcos, E.; et al. 53BP1 cooperation with the REV7-shieldin complex underpins DNA structure-specific NHEJ. Nature 2018, 560, 122–127. [Google Scholar] [CrossRef]
- Jaspers, J.E.; Kersbergen, A.; Boon, U.; Sol, W.; van Deemter, L.; Zander, S.A.; Drost, R.; Wientjens, E.; Ji, J.; Aly, A.; et al. Loss of 53BP1 causes PARP inhibitor resistance in Brca1-mutated mouse mammary tumors. Cancer Discov. 2013, 3, 68–81. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.-m.; Liao, X.-m.; Chen, Y.; Shen, Y.-y.; Yang, X.-y.; Su, Y.; Sun, Y.-m.; Gao, Y.-l.; Ding, J.; Zhang, A.; et al. Combining 53BP1 with BRCA1 as a biomarker to predict the sensitivity of poly(ADP-ribose) polymerase (PARP) inhibitors. Acta Pharmacol. Sin. 2017, 38, 1038–1047. [Google Scholar] [CrossRef] [PubMed]
- Nacson, J.; Krais, J.J.; Bernhardy, A.J.; Clausen, E.; Feng, W.; Wang, Y.; Nicolas, E.; Cai, K.Q.; Tricarico, R.; Hua, X.; et al. BRCA1 Mutation-Specific Responses to 53BP1 Loss-Induced Homologous Recombination and PARP Inhibitor Resistance. Cell Rep. 2018, 24, 3513–3527.e3517. [Google Scholar] [CrossRef] [PubMed]
- Drané, P.; Brault, M.-E.; Cui, G.; Meghani, K.; Chaubey, S.; Detappe, A.; Parnandi, N.; He, Y.; Zheng, X.-F.; Botuyan, M.V.; et al. TIRR regulates 53BP1 by masking its histone methyl-lysine binding function. Nature 2017, 543, 211–216. [Google Scholar] [CrossRef] [PubMed]
- Dev, H.; Chiang, T.-W.W.; Lescale, C.; de Krijger, I.; Martin, A.G.; Pilger, D.; Coates, J.; Sczaniecka-Clift, M.; Wei, W.; Ostermaier, M.; et al. Shieldin complex promotes DNA end-joining and counters homologous recombination in BRCA1-null cells. Nat. Cell Biol. 2018, 20, 954–965. [Google Scholar] [CrossRef]
- Xu, G.; Chapman, J.R.; Brandsma, I.; Yuan, J.; Mistrik, M.; Bouwman, P.; Bartkova, J.; Gogola, E.; Warmerdam, D.; Barazas, M.; et al. REV7 counteracts DNA double-strand break resection and affects PARP inhibition. Nature 2015, 521, 541–544. [Google Scholar] [CrossRef]
- Tomida, J.; Takata, K.-i.; Bhetawal, S.; Person, M.D.; Chao, H.-P.; Tang, D.G.; Wood, R.D. FAM35A associates with REV7 and modulates DNA damage responses of normal and BRCA1-defective cells. EMBO J. 2018, 37, e99543. [Google Scholar] [CrossRef]
- Sarangi, P.; Clairmont, C.S.; D'Andrea, A.D. Disassembly of the Shieldin Complex by TRIP13. Cell Cycle 2020, 19, 1565–1575. [Google Scholar] [CrossRef]
- Xie, W.; Wang, S.; Wang, J.; de la Cruz, M.J.; Xu, G.; Scaltriti, M.; Patel, D.J. Molecular mechanisms of assembly and TRIP13-mediated remodeling of the human Shieldin complex. Proc. Natl. Acad. Sci. USA 2021, 118, e2024512118. [Google Scholar] [CrossRef]
- Clairmont, C.S.; Sarangi, P.; Ponnienselvan, K.; Galli, L.D.; Csete, I.; Moreau, L.; Adelmant, G.; Chowdhury, D.; Marto, J.A.; D’Andrea, A.D. TRIP13 regulates DNA repair pathway choice through REV7 conformational change. Nat. Cell Biol. 2020, 22, 87–96. [Google Scholar] [CrossRef]
- He, Y.J.; Meghani, K.; Caron, M.-C.; Yang, C.; Ronato, D.A.; Bian, J.; Sharma, A.; Moore, J.; Niraj, J.; Detappe, A.; et al. DYNLL1 binds to MRE11 to limit DNA end resection in BRCA1-deficient cells. Nature 2018, 563, 522–526. [Google Scholar] [CrossRef]
- Fujita, H.; Ikeda, M.; Ui, A.; Ouchi, Y.; Mikami, Y.; Kanno, S.-i.; Yasui, A.; Tanaka, K. CHAMP1-POGZ counteracts the inhibitory effect of 53BP1 on homologous recombination and affects PARP inhibitor resistance. Oncogene 2022, 41, 2706–2718. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Sarangi, P.; Iyer, D.R.; Feng, H.; Moreau, L.; Nguyen, H.; Clairmont, C.; D’Andrea, A.D. CHAMP1 binds to REV7/FANCV and promotes homologous recombination repair. Cell Rep. 2022, 40, 111297. [Google Scholar] [CrossRef] [PubMed]
- Lomonosov, M.; Anand, S.; Sangrithi, M.; Davies, R.; Venkitaraman, A.R. Stabilization of stalled DNA replication forks by the BRCA2 breast cancer susceptibility protein. Genes Dev. 2003, 17, 3017–3022. [Google Scholar] [CrossRef] [PubMed]
- Rondinelli, B.; Gogola, E.; Yucel, H.; Duarte, A.A.; van de Ven, M.; van der Sluijs, R.; Konstantinopoulos, P.A.; Jonkers, J.; Ceccaldi, R.; Rottenberg, S.; et al. EZH2 promotes degradation of stalled replication forks by recruiting MUS81 through histone H3 trimethylation. Nat. Cell Biol. 2017, 19, 1371–1378. [Google Scholar] [CrossRef] [PubMed]
- Ray Chaudhuri, A.; Callen, E.; Ding, X.; Gogola, E.; Duarte, A.A.; Lee, J.-E.; Wong, N.; Lafarga, V.; Calvo, J.A.; Panzarino, N.J.; et al. Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature 2016, 535, 382–387. [Google Scholar] [CrossRef]
- Lok, B.H.; Gardner, E.E.; Schneeberger, V.E.; Ni, A.; Desmeules, P.; Rekhtman, N.; de Stanchina, E.; Teicher, B.A.; Riaz, N.; Powell, S.N.; et al. PARP Inhibitor Activity Correlates with SLFN11 Expression and Demonstrates Synergy with Temozolomide in Small Cell Lung Cancer. Clin. Cancer Res. 2017, 23, 523–535. [Google Scholar] [CrossRef]
- Zatreanu, D.; Robinson, H.M.R.; Alkhatib, O.; Boursier, M.; Finch, H.; Geo, L.; Grande, D.; Grinkevich, V.; Heald, R.A.; Langdon, S.; et al. Polθ inhibitors elicit BRCA-gene synthetic lethality and target PARP inhibitor resistance. Nat. Commun. 2021, 12, 3636. [Google Scholar] [CrossRef]
- Zatreanu, D.; Robinson, H.; Alkhatib, O.; Boursier, M.; Finch, H.; Geo, L.; Grande, D.; Grinkevich, V.; Heald, R.; Langdon, S.; et al. Abstract 5697: Targeting PARP inhibitor resistance with Polθ inhibitors. Cancer Res. 2022, 82, 5697. [Google Scholar] [CrossRef]
- Chapman, J.R.; Barral, P.; Vannier, J.B.; Borel, V.; Steger, M.; Tomas-Loba, A.; Sartori, A.A.; Adams, I.R.; Batista, F.D.; Boulton, S.J. RIF1 is essential for 53BP1-dependent nonhomologous end joining and suppression of DNA double-strand break resection. Mol. Cell 2013, 49, 858–871. [Google Scholar] [CrossRef]
- Schlacher, K.; Wu, H.; Jasin, M. A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell 2012, 22, 106–116. [Google Scholar] [CrossRef]
- Sung, P.; Klein, H. Mechanism of homologous recombination: Mediators and helicases take on regulatory functions. Nat. Rev. Mol. Cell Biol. 2006, 7, 739–750. [Google Scholar] [CrossRef]
- Rossi, M.J.; DiDomenico, S.F.; Patel, M.; Mazin, A.V. RAD52: Paradigm of Synthetic Lethality and New Developments. Front. Genet. 2021, 12, 780293. [Google Scholar] [CrossRef] [PubMed]
- Motycka, T.A.; Bessho, T.; Post, S.M.; Sung, P.; Tomkinson, A.E. Physical and functional interaction between the XPF/ERCC1 endonuclease and hRad52. J. Biol. Chem. 2004, 279, 13634–13639. [Google Scholar] [CrossRef] [PubMed]
- Bhargava, R.; Onyango, D.O.; Stark, J.M. Regulation of Single-Strand Annealing and its Role in Genome Maintenance. Trends Genet. TIG 2016, 32, 566–575. [Google Scholar] [CrossRef] [PubMed]
- Ceccaldi, R.; Liu, J.C.; Amunugama, R.; Hajdu, I.; Primack, B.; Petalcorin, M.I.; O'Connor, K.W.; Konstantinopoulos, P.A.; Elledge, S.J.; Boulton, S.J.; et al. Homologous-recombination-deficient tumours are dependent on Poltheta-mediated repair. Nature 2015, 518, 258–262. [Google Scholar] [CrossRef] [PubMed]
- Mateos-Gomez, P.A.; Gong, F.; Nair, N.; Miller, K.M.; Lazzerini-Denchi, E.; Sfeir, A. Mammalian polymerase theta promotes alternative NHEJ and suppresses recombination. Nature 2015, 518, 254–257. [Google Scholar] [CrossRef]
- Higgins, G.S.; Boulton, S.J. Beyond PARP-POLtheta as an anticancer target. Science 2018, 359, 1217–1218. [Google Scholar] [CrossRef]
- Yousefzadeh, M.J.; Wyatt, D.W.; Takata, K.; Mu, Y.; Hensley, S.C.; Tomida, J.; Bylund, G.O.; Doublié, S.; Johansson, E.; Ramsden, D.A.; et al. Mechanism of suppression of chromosomal instability by DNA polymerase POLQ. PLoS Genet. 2014, 10, e1004654. [Google Scholar] [CrossRef] [PubMed]
- Higgins, G.S.; Prevo, R.; Lee, Y.F.; Helleday, T.; Muschel, R.J.; Taylor, S.; Yoshimura, M.; Hickson, I.D.; Bernhard, E.J.; McKenna, W.G. A small interfering RNA screen of genes involved in DNA repair identifies tumor-specific radiosensitization by POLQ knockdown. Cancer Res. 2010, 70, 2984–2993. [Google Scholar] [CrossRef]
- Schofield, R. The relationship between the spleen colony-forming cell and the haemopoietic stem cell. Blood Cells 1978, 4, 7–25. [Google Scholar] [PubMed]
- Morrison, S.J.; Scadden, D.T. The bone marrow niche for haematopoietic stem cells. Nature 2014, 505, 327–334. [Google Scholar] [CrossRef]
- Konopleva, M.Y.; Jordan, C.T. Leukemia stem cells and microenvironment: Biology and therapeutic targeting. J. Clin. Oncol. 2011, 29, 591–599. [Google Scholar] [CrossRef] [PubMed]
- Hanoun, M.; Frenette, P.S. This niche is a maze; an amazing niche. Cell Stem Cell 2013, 12, 391–392. [Google Scholar] [CrossRef] [PubMed]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Konopleva, M.; Konoplev, S.; Hu, W.; Zaritskey, A.Y.; Afanasiev, B.V.; Andreeff, M. Stromal cells prevent apoptosis of AML cells by up-regulation of anti-apoptotic proteins. Leukemia 2002, 16, 1713–1724. [Google Scholar] [CrossRef]
- Tabe, Y.; Konopleva, M. Advances in understanding the leukaemia microenvironment. Br. J. Haematol. 2014, 164, 767–778. [Google Scholar] [CrossRef]
- Colmone, A.; Amorim, M.; Pontier, A.L.; Wang, S.; Jablonski, E.; Sipkins, D.A. Leukemic cells create bone marrow niches that disrupt the behavior of normal hematopoietic progenitor cells. Science 2008, 322, 1861–1865. [Google Scholar] [CrossRef]
- Kode, A.; Manavalan, J.S.; Mosialou, I.; Bhagat, G.; Rathinam, C.V.; Luo, N.; Khiabanian, H.; Lee, A.; Murty, V.V.; Friedman, R.; et al. Leukaemogenesis induced by an activating beta-catenin mutation in osteoblasts. Nature 2014, 506, 240–244. [Google Scholar] [CrossRef]
- Kode, A.; Mosialou, I.; Manavalan, S.J.; Rathinam, C.V.; Friedman, R.A.; Teruya-Feldstein, J.; Bhagat, G.; Berman, E.; Kousteni, S. FoxO1-dependent induction of acute myeloid leukemia by osteoblasts in mice. Leukemia 2016, 30, 1–13. [Google Scholar] [CrossRef]
- Herbertz, S.; Sawyer, J.S.; Stauber, A.J.; Gueorguieva, I.; Driscoll, K.E.; Estrem, S.T.; Cleverly, A.L.; Desaiah, D.; Guba, S.C.; Benhadji, K.A.; et al. Clinical development of galunisertib (LY2157299 monohydrate), a small molecule inhibitor of transforming growth factor-beta signaling pathway. Drug Des. Devel. Ther. 2015, 9, 4479–4499. [Google Scholar] [CrossRef]
- Azaro, A.; Rodon, J.; Carducci, M.; Sepulveda-Sanchez, J.M.; Gueorguieva, I.; Cleverly, A.L.; Desaiah, D.; Namaseevayam, S.P.; Holdhoff, M.; Lahn, M.M. Case Series of Cancer Patients Treated with Galunisertib, a Transforming Growth Factor-Beta Receptor I Kinase Inhibitor in a First-in-Human Dose Study. J. Med. Cases 2014, 5, 603–609. [Google Scholar] [CrossRef]
- Wick, A.; Desjardins, A.; Suarez, C.; Forsyth, P.; Gueorguieva, I.; Burkholder, T.; Cleverly, A.L.; Estrem, S.T.; Wang, S.; Lahn, M.M.; et al. Phase 1b/2a study of galunisertib, a small molecule inhibitor of transforming growth factor-beta receptor I, in combination with standard temozolomide-based radiochemotherapy in patients with newly diagnosed malignant glioma. Investig. New Drugs 2020, 38, 1570–1579. [Google Scholar] [CrossRef] [PubMed]
- Melisi, D.; Garcia-Carbonero, R.; Macarulla, T.; Pezet, D.; Deplanque, G.; Fuchs, M.; Trojan, J.; Oettle, H.; Kozloff, M.; Cleverly, A.; et al. Galunisertib plus gemcitabine vs. gemcitabine for first-line treatment of patients with unresectable pancreatic cancer. Br. J. Cancer 2018, 119, 1208–1214. [Google Scholar] [CrossRef]
- Santini, V.; Valcarcel, D.; Platzbecker, U.; Komrokji, R.S.; Cleverly, A.L.; Lahn, M.M.; Janssen, J.; Zhao, Y.; Chiang, A.; Giagounidis, A.; et al. Phase II Study of the ALK5 Inhibitor Galunisertib in Very Low-, Low-, and Intermediate-Risk Myelodysplastic Syndromes. Clin. Cancer Res. 2019, 25, 6976–6985. [Google Scholar] [CrossRef]
- Kelley, R.K.; Gane, E.; Assenat, E.; Siebler, J.; Galle, P.R.; Merle, P.; Hourmand, I.O.; Cleverly, A.; Zhao, Y.; Gueorguieva, I.; et al. A Phase 2 Study of Galunisertib (TGF-beta1 Receptor Type I Inhibitor) and Sorafenib in Patients With Advanced Hepatocellular Carcinoma. Clin. Transl. Gastroenterol. 2019, 10, e00056. [Google Scholar] [CrossRef]
- Zhou, J.; Gelot, C.; Pantelidou, C.; Li, A.; Yücel, H.; Davis, R.E.; Färkkilä, A.; Kochupurakkal, B.; Syed, A.; Shapiro, G.I.; et al. A first-in-class polymerase theta inhibitor selectively targets homologous-recombination-deficient tumors. Nat. Cancer 2021, 2, 598–610. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Disease | Oncogene/Mutation-Induced HR/c-NHEJ Deficiency | Deregulated Protein | References |
|---|---|---|---|
| CML | BCR-ABL1 | BRCA1, DNA-PKcs | [18,23,82,86,91] |
| AML | AML1-ETO | BRCA1, BRCA2, Ku70 | [25] |
| AML | PML-RARα | BRCA2, RAD51C | [25,87] |
| Burkitt lymphoma | IGH-MYC | BRCA2 | [24] |
| AML | IDH1/IDH2 mutants | ATM | [84,88,89] |
| AML/ALL | TCF3-HLF | BRCA1, BRCA2 | [80] |
| AML | FLT3ITD+TET2 mutant | BRCA1, LIG4 | [85] |
| AML | FLT3ITD+WT1 mutant | BRCA1, LIG4 | [85] |
| AML/MDS | TET2 mutant | BRCA1 | [90] |
| Disease | Drug-Induced HR/c-NHEJ Deficiency | Deregulated Protein | References |
|---|---|---|---|
| Myeloproliferative neoplams [JAK2(V617F)] | JAK1/2 kinase inhibitor (Ruxolitinib) | BRCA1, RAD51C, LIG4 | [27] |
| AML [FLT3(ITD)] | FLT3 kinase inhibitor (Quizartinib) | BRCA1, BRCA2, PALB2, RAD51, LIG4 | [28] |
| AML [c-KIT(N822K)] | c-KIT kinase inhibitor (Avapritinib) | BRCA1, BRCA2, DNA-PKcs | [29] |
| Breast cancer | EGFR kinase inhibitor (Lapatinib) | BRCA1 | [30] |
| Breast and ovarian cancers | IGF-1R kinase inhibitor | RAD51 | [31] |
| Pre-Existing PARPi Resistance | Mechanism | References |
|---|---|---|
| TGFβ1—TGFβR—SMAD2/3 signaling in a hypoxic bone marrow microenvironment | Restoration of HR/c-NHEJ | [35] |
| Loss-of-function mutations in DNMT3A | Enhanced HR/c-NHEJ Deregulation of PARP1 | [85,108] |
| Activation of FLT3 kinase [FLT3(ITD)] | Enhanced HR/c-NHEJ | [28] |
| Activation of JAK2 kinase [JAK2(V617F)] | Enhanced HR/c-NHEJ | [27] |
| Activation of c-KIT kinase [c-KIT(N822K)] | Enhanced HR/c-NHEJ | [29] |
| Activation of EGFR kinase | Increased BRCA1 | [30] |
| Activation of IGF-1R kinase | Increased RAD51 | [31] |
| Acquired PARPi Resistance | Mechanism | References |
| Secondary mutations in BRCA1/2 | Restoration of HR | [34,102,103,104] |
| Reduced BRCA1 promoter methylation | Restoration of HR | [105] |
| Deregulation of EMI1 | Restoration of HR | [106] |
| Overexpression of KEAP1, decrease in EMSY | Restoration of HR | [107] |
| Upregulation of P-glycoprotein efflux pumps | Enhanced drug efflux | [15] |
| Decrease in PARP1 | Loss of PARP1 expression | [109] |
| Mutations in PARP1 | Loss of PARP1 expression | [110] |
| Elevated activity of 53BP1-RIF1 complex | Enhanced c-NHEJ | [111,112] |
| Loss of 53BP1 | Enhanced DSB end resection Restoration of HR | [113,114,115] |
| Increase in TIRR | Blockage of 53BP1 localization to DSBs | [116] |
| Decrease in RIF1, REV7 and REV7-SHLD1/2/3 complex | Enhanced DSB end resection Restoration of HR | [117,118,119] |
| Overexpression of TRIP13 | Dissociation of REV7-SHLD1/2/3 complex | [120,121,122] |
| Decrease in DNLL1 | Enhanced DSB end resection Restoration of HR | [123] |
| Overexpression of CHAMP1-POGZ complex | Decreased 53BP1 and REV7-SHLD1/2/3 complex | [124,125] |
| Decrease in MRE1, MUS81 and EZH2 | Replication fork protection | [126,127] |
| Decrease in PTIP and SLFN11 | Prevention of fork degradation | [128,129] |
| Increase in XRCC1-LIG3 | Restoration of OFP Decrease/loss of replication gaps | [53] |
| Overexpression of RAD52 | Alternative factor: HR backup | [33] |
| Overexpression of Polθ | Alternative factor: MMEJ/TMEJ | [130,131] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Le, B.V.; Podszywałow-Bartnicka, P.; Piwocka, K.; Skorski, T. Pre-Existing and Acquired Resistance to PARP Inhibitor-Induced Synthetic Lethality. Cancers 2022, 14, 5795. https://doi.org/10.3390/cancers14235795
Le BV, Podszywałow-Bartnicka P, Piwocka K, Skorski T. Pre-Existing and Acquired Resistance to PARP Inhibitor-Induced Synthetic Lethality. Cancers. 2022; 14(23):5795. https://doi.org/10.3390/cancers14235795
Chicago/Turabian StyleLe, Bac Viet, Paulina Podszywałow-Bartnicka, Katarzyna Piwocka, and Tomasz Skorski. 2022. "Pre-Existing and Acquired Resistance to PARP Inhibitor-Induced Synthetic Lethality" Cancers 14, no. 23: 5795. https://doi.org/10.3390/cancers14235795
APA StyleLe, B. V., Podszywałow-Bartnicka, P., Piwocka, K., & Skorski, T. (2022). Pre-Existing and Acquired Resistance to PARP Inhibitor-Induced Synthetic Lethality. Cancers, 14(23), 5795. https://doi.org/10.3390/cancers14235795