
Identification of Src Family Kinases as Potential Therapeutic Targets for Chemotherapy-Resistant Triple Negative Breast Cancer

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
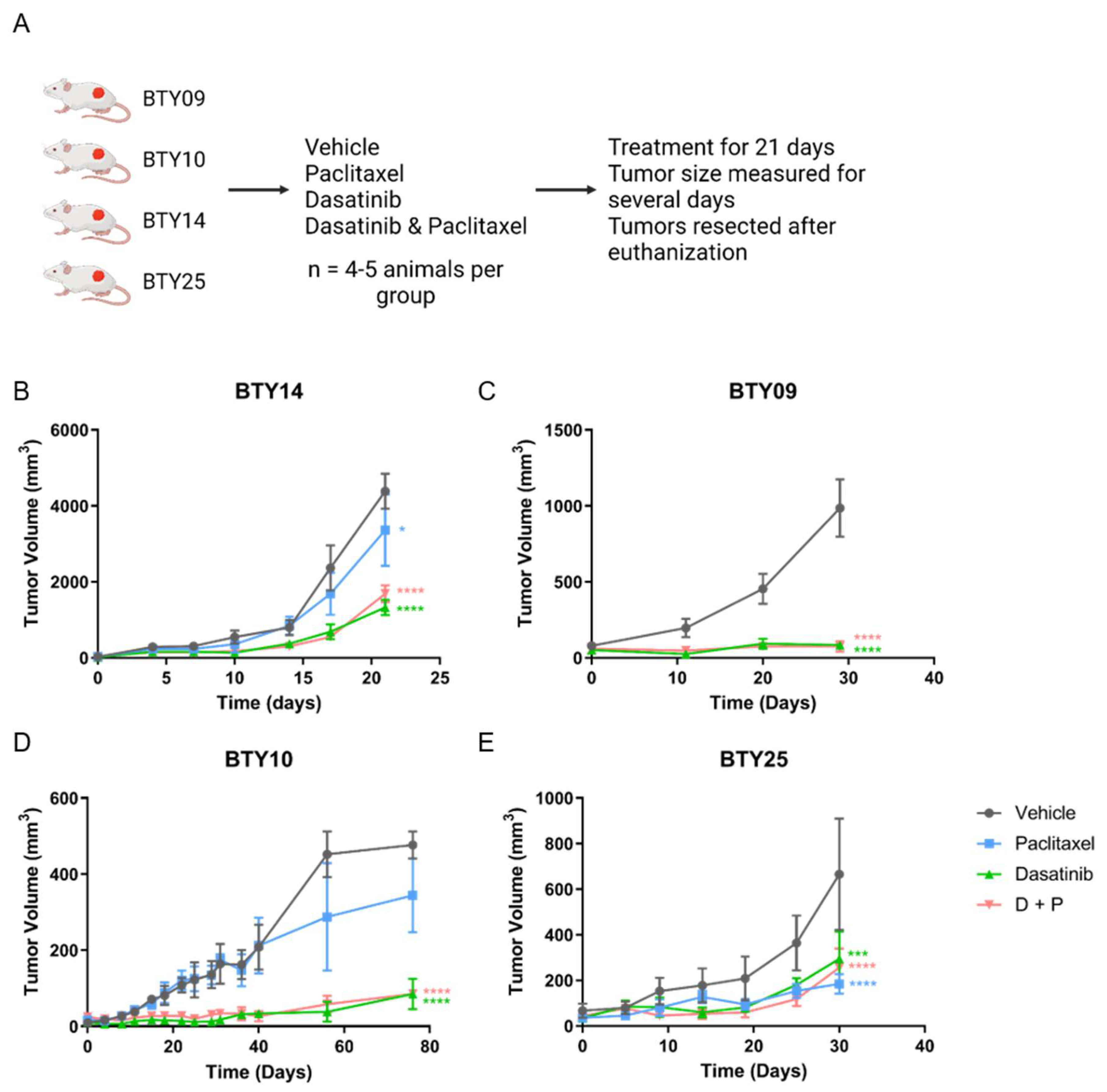
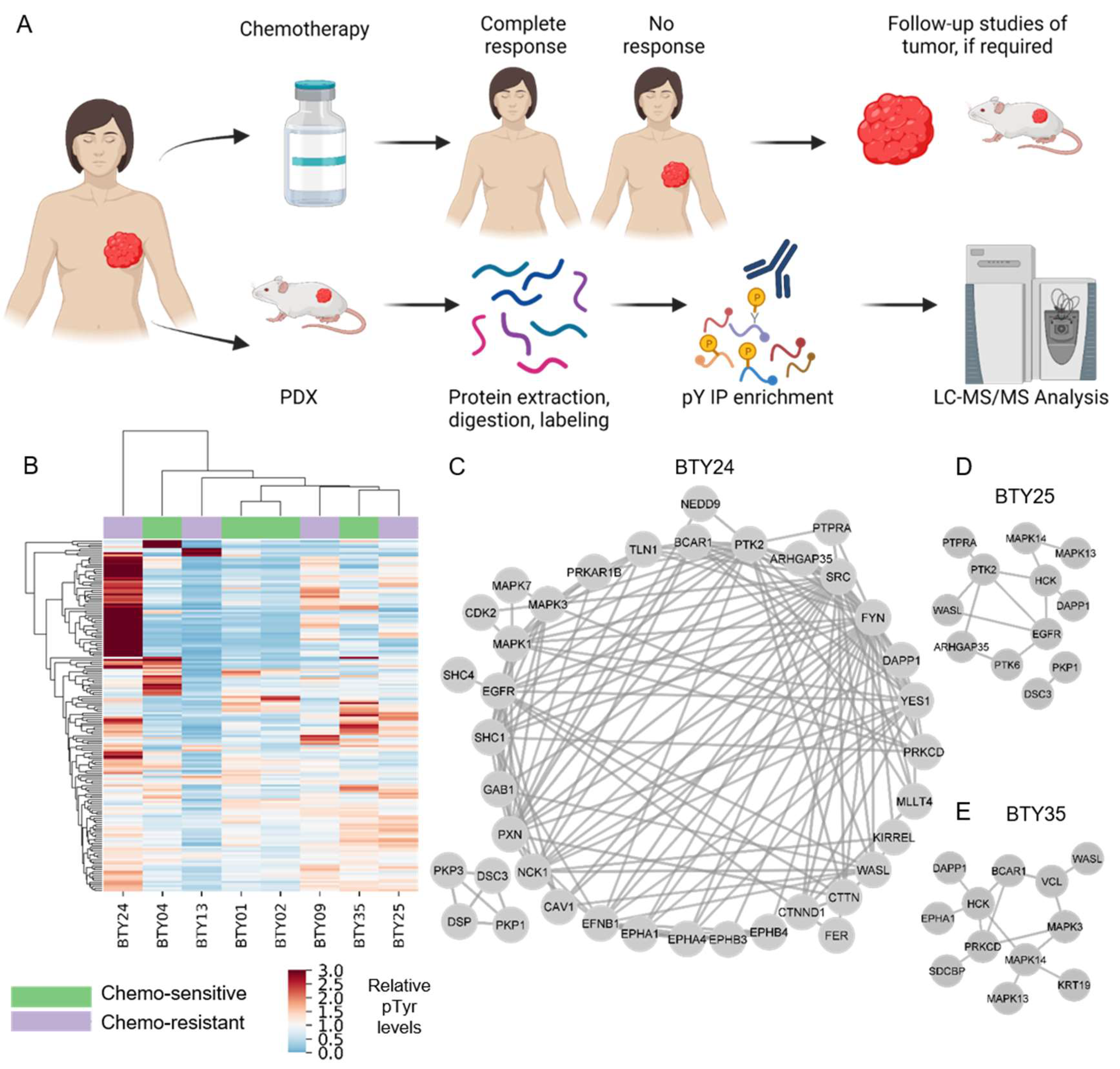
2.1. Animal Studies
2.2. Clinical Samples
2.3. Tumor Tissue Processing for Phosphoproteomics
2.4. Mass Spectrometry Data Analysis
2.5. RNAseq
2.6. Data Availability
3. Results
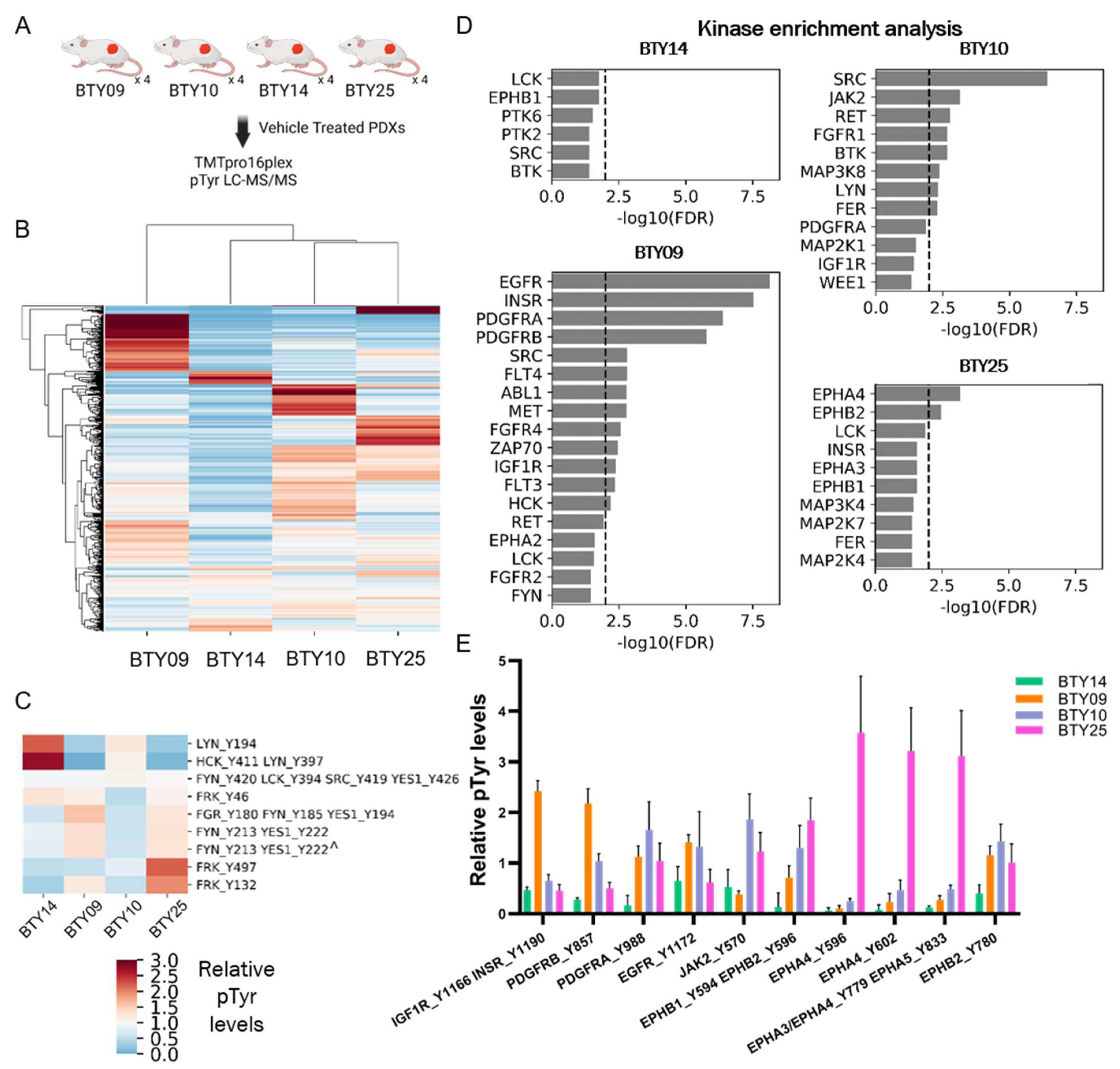
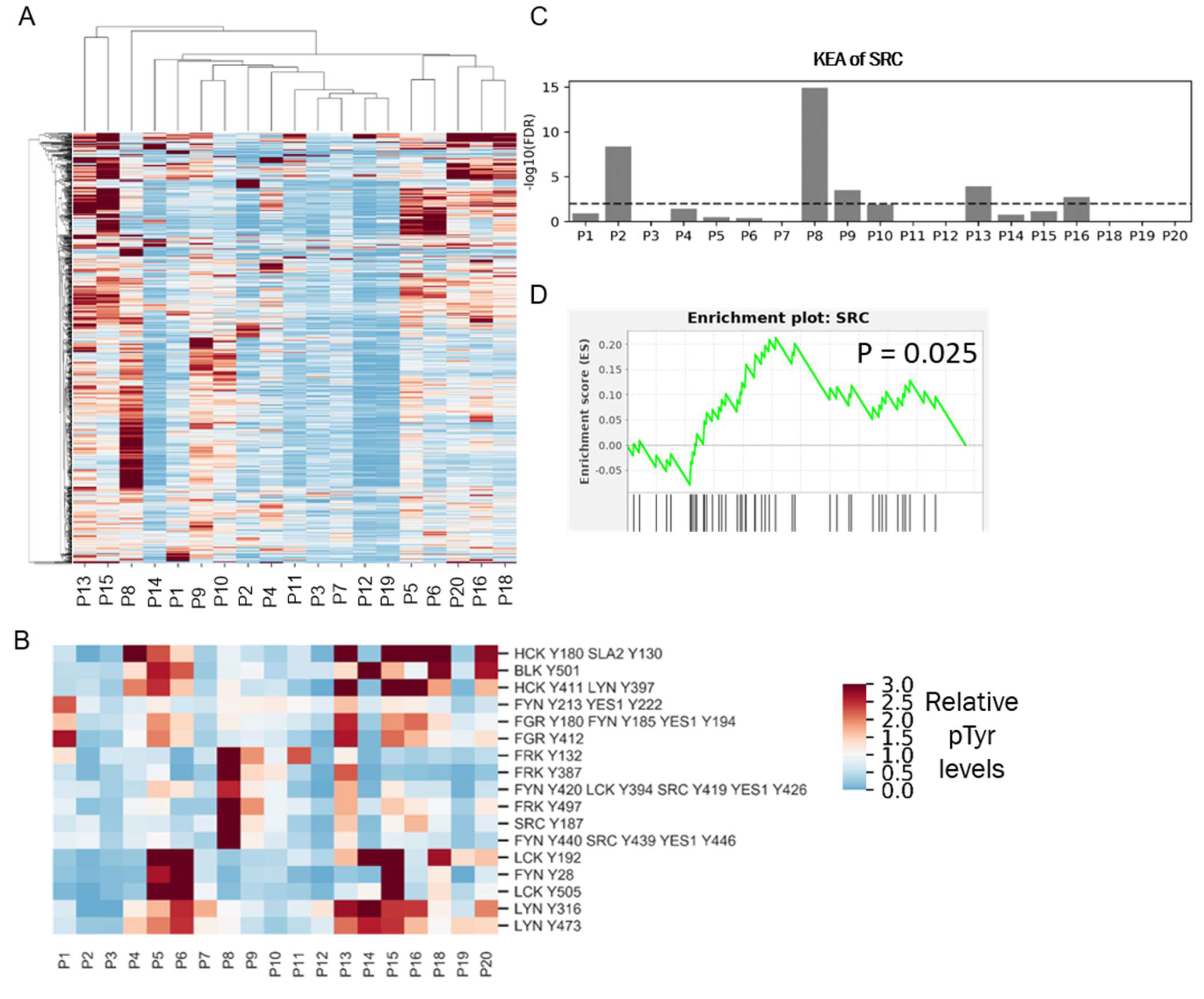
3.1. Analysis of PDXs Reveal Patient-Specific Phosphotyrosine Signatures
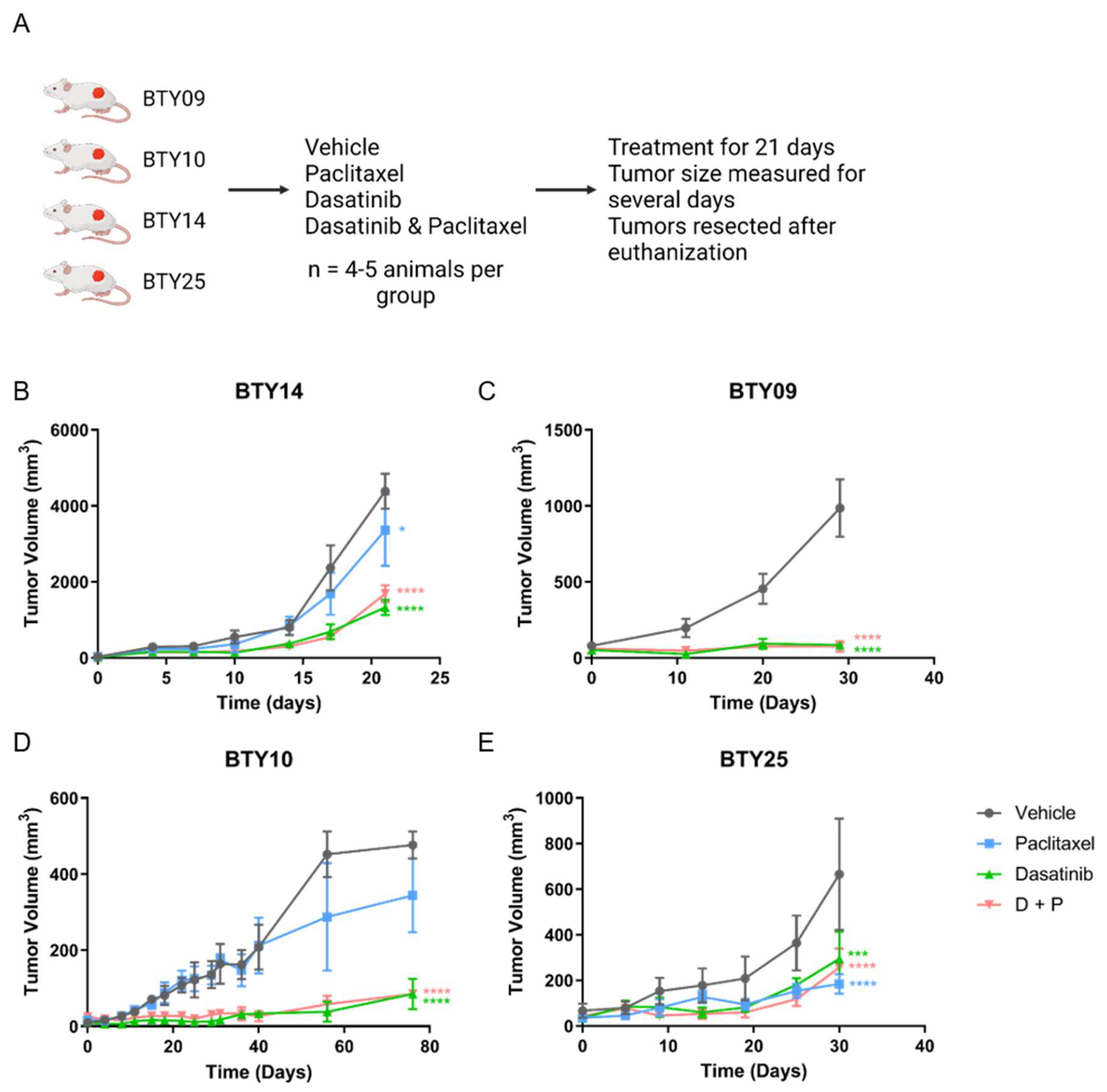
3.2. Treatment with SFK Inhibitor Leads to Tumor Growth Arrest In Vivo
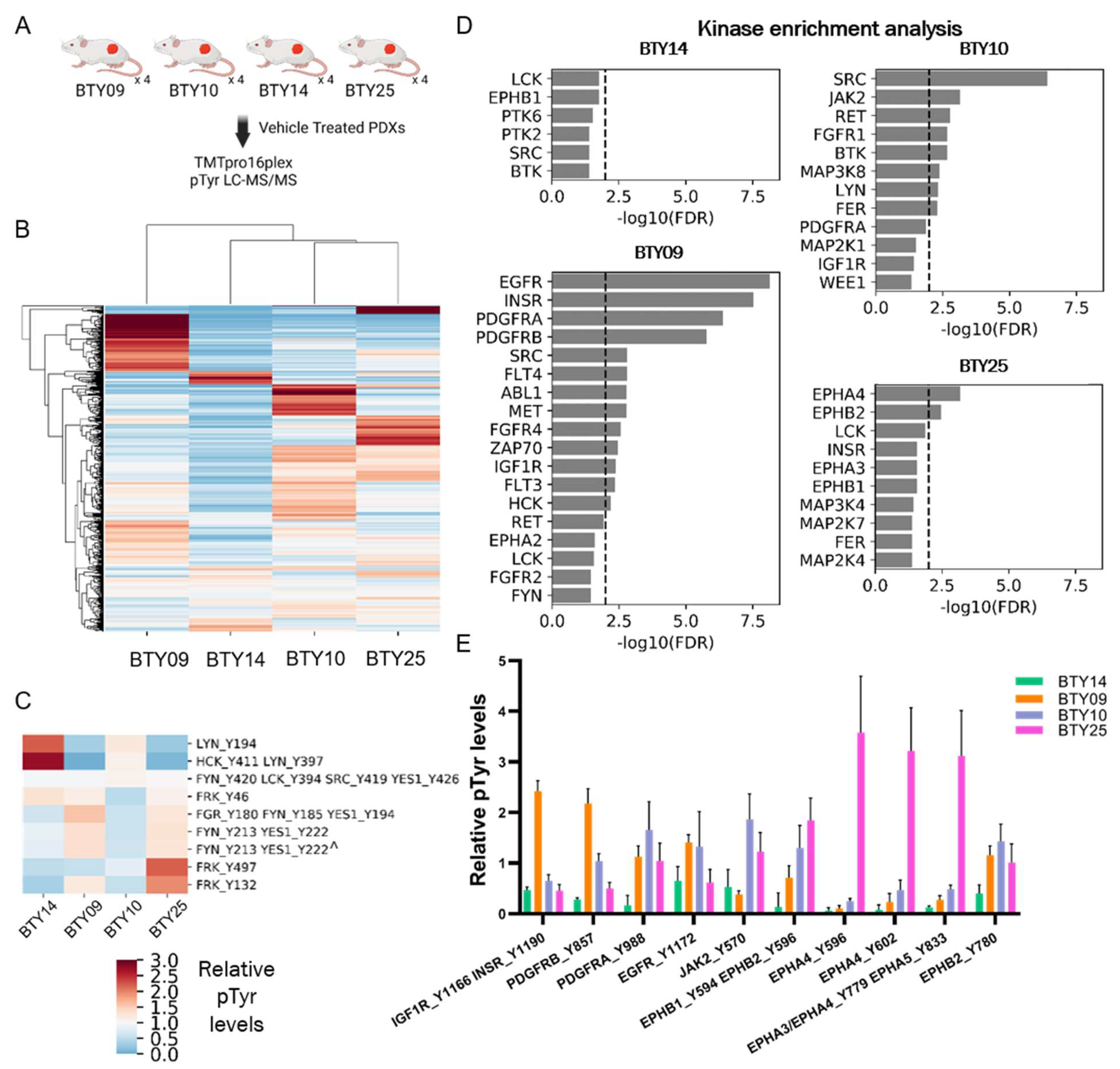
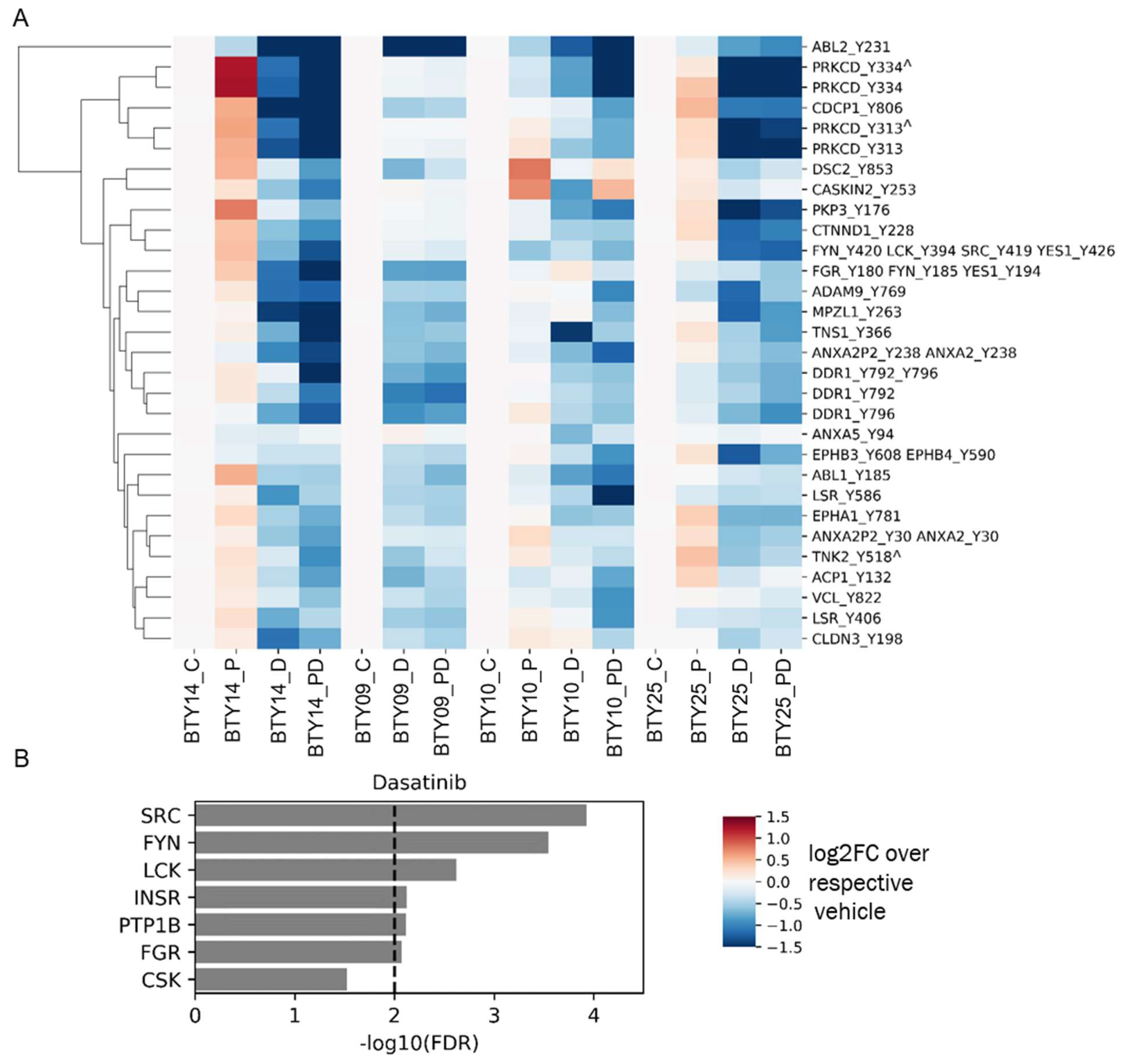
3.3. Phosphotyrosine Profiles Are Correlated with Sensitivity to SFK Inhibitor
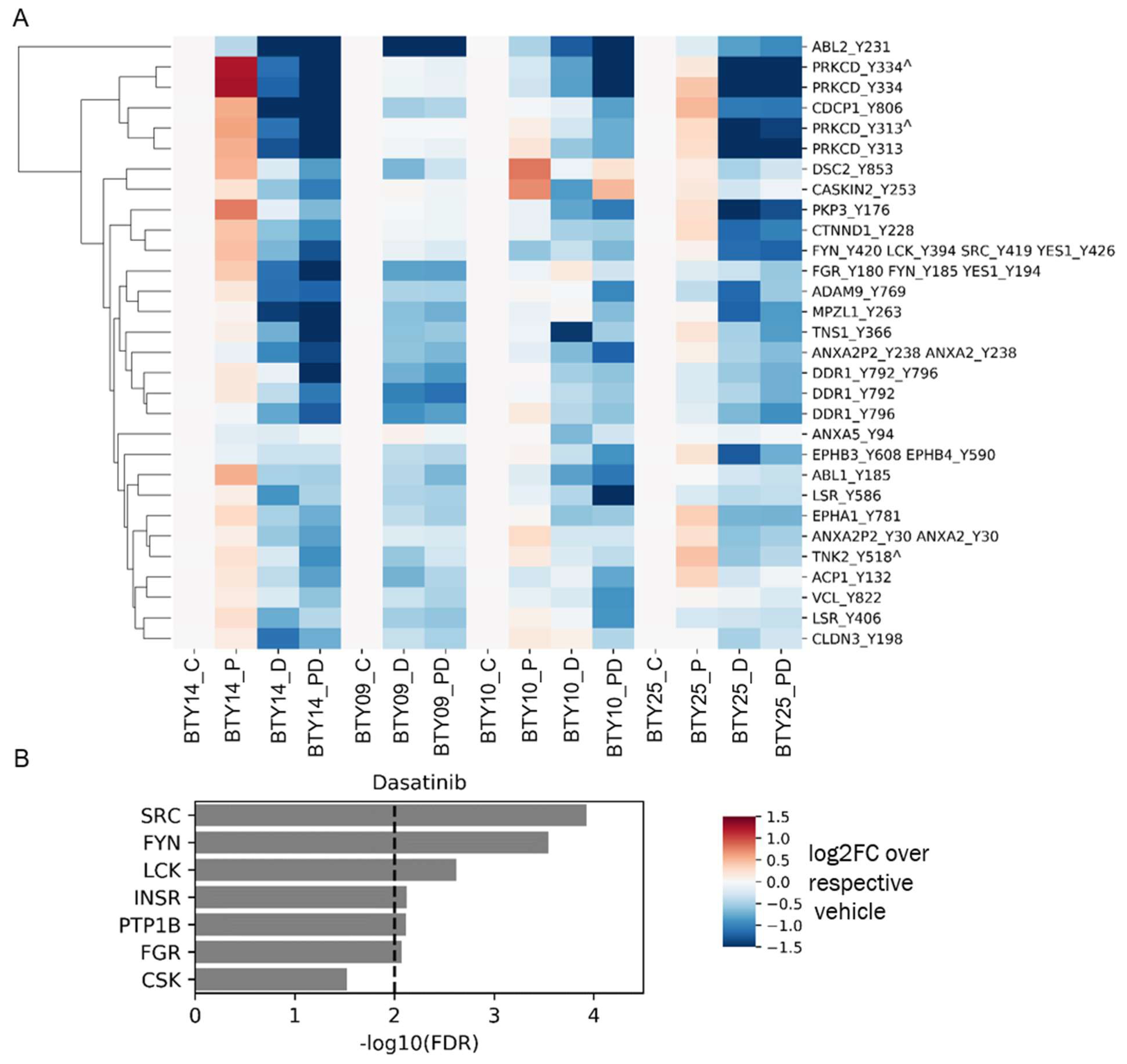
3.4. Tumors Undergo Adaptive Response to Therapy
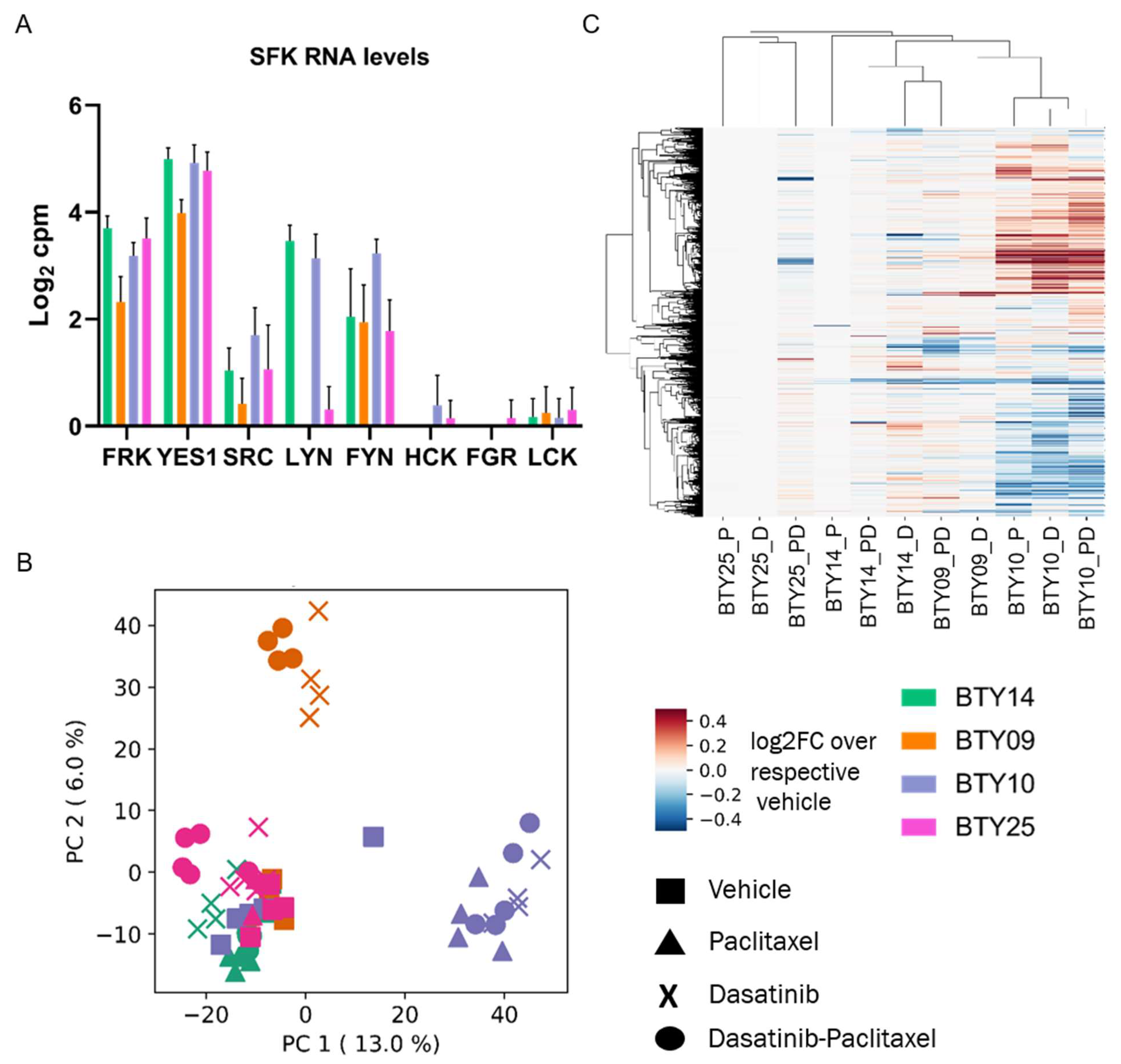
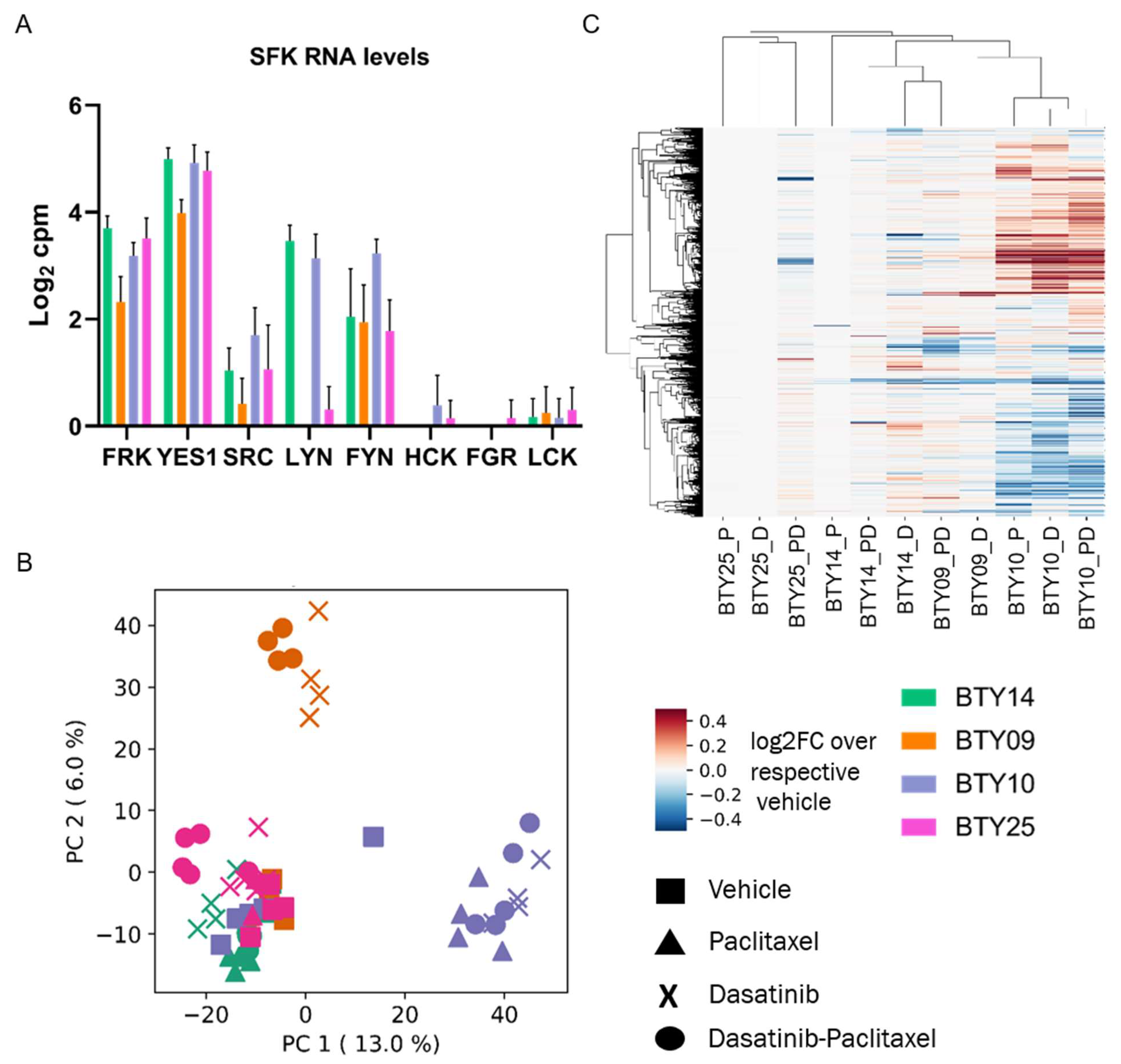
3.5. Gene Expression Profiles Are Not Correlated with Dasatinib Sensitivity
3.6. High SFK Signature in TNBC Patients
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Anders, C.K.; Carey, L.A. Biology, metastatic patterns, and treatment of patients with triple-negative breast cancer. Clin. Breast Cancer 2009, 9, S73. [Google Scholar] [CrossRef] [PubMed]
- DeSantis, C.E.; Ma, J.; Gaudet, M.M.; Newman, L.A.; Miller, K.D.; Sauer, A.G.; Jemal, A.; Siegel, R.L. Breast cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 438–451. [Google Scholar] [CrossRef] [PubMed]
- Cortazar, P.; Zhang, L.; Untch, M.; Mehta, K.; Costantino, J.P.; Wolmark, N.; Bonnefoi, H.; Cameron, D.; Gianni, L.; Valagussa, P.; et al. Pathological complete response and long-term clinical benefit in breast cancer: The CTNeoBC pooled analysis. Lancet 2014, 384, 164–172. [Google Scholar] [CrossRef]
- Albain, K.; Anderson, S.; Arriagada, R.; Barlow, W.; Bergh, J.; Bliss, J.; Buyse, M.; Cameron, D.; Carrasco, E.; Clarke, M.; et al. Comparisons between different polychemotherapy regimens for early breast cancer: Meta-analyses of long-term outcome among 100,000 women in 123 randomised trials. Lancet 2012, 379, 432–444. [Google Scholar] [CrossRef]
- Yau, C.; Osdoit, M.; van der Noordaa, M.; Shad, S.; Wei, J.; de Croze, D.; Hamy, A.S.; Laé, M.; Reyal, F.; Sonke, G.S.; et al. Residual cancer burden after neoadjuvant chemotherapy and long-term survival outcomes in breast cancer: A multicentre pooled analysis of 5161 patients. Lancet Oncol. 2022, 23, 149–160. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef]
- Herold, C.I.; Chadaram, V.; Peterson, B.L.; Marcom, P.K.; Hopkins, J.; Kimmick, G.G.; Favaro, J.; Hamilton, E.; Welch, R.A.; Bacus, S.; et al. Phase II trial of dasatinib in patients with metastatic breast cancer using real-time pharmacodynamic tissue biomarkers of Src inhibition to escalate dosing. Clin. Cancer Res. 2011, 17, 6061–6070. [Google Scholar] [CrossRef]
- Finn, R.S.; Bengala, C.; Ibrahim, N.; Roche, H.; Sparano, J.; Strauss, L.C.; Fairchild, J.; Sy, O.; Goldstein, L.J. Dasatinib as a single agent in triple-negative breast cancer: Results of an open-label phase 2 study. Clin. Cancer Res. 2011, 17, 6905–6913. [Google Scholar] [CrossRef] [Green Version]
- Baselga, J.; Gómez, P.; Greil, R.; Braga, S.; Climent, M.A.; Wardley, A.M.; Kaufman, B.; Stemmer, S.M.; Pêgo, A.; Chan, A.; et al. Randomized phase II study of the anti–epidermal growth factor receptor monoclonal antibody cetuximab with cisplatin versus cisplatin alone in patients with metastatic triple-negative breast cancer. J. Clin. Oncol. 2013, 31, 2586. [Google Scholar] [CrossRef] [PubMed]
- Carey, L.A.; Rugo, H.S.; Marcom, P.K.; Mayer, E.L.; Esteva, F.J.; Ma, C.X.; Liu, M.C.; Storniolo, A.M.; Rimawi, M.F.; Forero-Torres, A.; et al. TBCRC 001: Randomized phase II study of cetuximab in combination with carboplatin in stage IV triple-negative breast cancer. J. Clin. Oncol. 2012, 30, 2615–2623. [Google Scholar] [CrossRef]
- Goetz, M.P.; Kalari, K.R.; Suman, V.J.; Moyer, A.M.; Yu, J.; Visscher, D.W.; Dockter, T.J.; Vedell, P.T.; Sinnwell, J.P.; Tang, X.; et al. Tumor sequencing and patient-derived xenografts in the neoadjuvant treatment of breast cancer. JNCI J. Natl. Cancer Inst. 2017, 109, djw306. [Google Scholar] [CrossRef]
- Derose, Y.S.; Wang, G.; Lin, Y.C.; Bernard, P.S.; Buys, S.S.; Ebbert, M.T.W.; Factor, R.; Matsen, C.; Milash, B.A.; Nelson, E.; et al. Tumor grafts derived from women with breast cancer authentically reflect tumor pathology, growth, metastasis and disease outcomes. Nat. Med. 2011, 17, 1514. [Google Scholar] [CrossRef]
- Hidalgo, M.; Amant, F.; Biankin, A.V.; Budinská, E.; Byrne, A.T.; Caldas, C.; Clarke, R.B.; de Jong, S.; Jonkers, J.; Mælandsmo, G.M.; et al. Patient derived xenograft models: An emerging platform for translational cancer research. Cancer Discov. 2014, 4, 998. [Google Scholar] [CrossRef]
- Yu, J.; Qin, B.; Moyer, A.M.; Sinnwell, J.P.; Thompson, K.J.; Copland, J.A., III; Marlow, L.A.; Miller, J.L.; Yin, P.; Gao, B.; et al. Establishing and characterizing patient-derived xenografts using pre-chemotherapy percutaneous biopsy and post-chemotherapy surgical samples from a prospective neoadjuvant breast cancer study. Breast Cancer Res. 2017, 19, 1–13. [Google Scholar] [CrossRef]
- Kohale, I.N.; Burgenske, D.M.; Mladek, A.C.; Bakken, K.K.; Kuang, J.; Boughey, J.C.; Wang, L.; Carter, J.M.; Haura, E.B.; Goetz, M.P.; et al. Quantitative analysis of tyrosine phosphorylation from FFPE tissues reveals patient-specific signaling networks. Cancer Res. 2021, 81, 3930–3941. [Google Scholar] [CrossRef]
- Emdal, K.B.; Dittmann, A.; Reddy, R.J.; Lescarbeau, R.S.; Moores, S.L.; Laquerre, S.; White, F.M. Characterization of in vivo resistance to osimertinib and JNJ-61186372, an EGFR/Met bispecific antibody, reveals unique and consensus mechanisms of resistance. Mol. Cancer Ther. 2017, 16, 2572–2585. [Google Scholar] [CrossRef]
- Taus, T.; Köcher, T.; Pichler, P.; Paschke, C.; Schmidt, A.; Henrich, C.; Mechtler, K. Universal and confident phosphorylation site localization using phosphoRS. J. Proteome Res. 2011, 10, 5354–5362. [Google Scholar] [CrossRef]
- Nichols, A.M.; White, F.M. Manual validation of peptide sequence and sites of tyrosine phosphorylation from MS/MS spectra. Methods Mol. Biol. 2009, 492, 143–160. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Jensen, L.J.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2018, 47, 607–613. [Google Scholar] [CrossRef] [PubMed]
- Lachmann, A.; Ma’ayan, A. KEA: Kinase enrichment analysis. Bioinformatics 2009, 25, 684–686. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Reich, M.; Liefeld, T.; Gould, J.; Lerner, J.; Tamayo, P.; Mesirov, J.P. GenePattern 2.0. Nat. Genet. 2006, 38, 500–501. [Google Scholar] [CrossRef] [PubMed]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef]
- Soneson, C.; Love, M.I.; Robinson, M.D. Differential analyses for RNA-seq: Transcript-level estimates improve gene-level inferences. F1000Research 2016, 4, 1521. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- McCarthy, D.J.; Chen, Y.; Smyth, G.K. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012, 40, 4288. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D450. [Google Scholar] [CrossRef] [PubMed]
- Lindauer, M.; Hochhaus, A. Dasatinib. Recent Results Cancer Res. 2015, 201, 27–65. [Google Scholar] [CrossRef]
- Kothiwale, S.; Borza, C.M.; Lowe, W.; Pozzi, A.; Meiler, J. Discoidin domain receptor 1 (DDR1) kinase as target for structure-based drug discovery. Drug Discov. Today 2015, 20, 255. [Google Scholar] [CrossRef] [PubMed]
- Kajiyama, H.; Shibata, K.; Terauchi, M.; Yamashita, M.; Ino, K.; Nawa, A.; Kikkawa, F. Chemoresistance to paclitaxel induces epithelial-mesenchymal transition and enhances metastatic potential for epithelial ovarian carcinoma cells. Int. J. Oncol. 2007, 31, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Gajadhar, A.S.; Johnson, H.; Slebos, R.J.C.; Shaddox, K.; Wiles, K.; Washington, M.K.; Herline, A.J.; Levine, D.A.; Liebler, D.C.; White, F.M. Phosphotyrosine signaling analysis in human tumors is confounded by systemic ischemia-driven artifacts and intra-specimen heterogeneity. Cancer Res. 2015, 75, 1495–1503. [Google Scholar] [CrossRef]
- Mertins, P.; Yang, F.; Liu, T.; Mani, D.R.; Petyuk, V.A.; Gillette, M.A.; Clauser, K.R.; Qiao, J.W.; Gritsenko, M.A.; Moore, R.J.; et al. Ischemia in tumors induces early and sustained phosphorylation changes in stress kinase pathways but does not affect global protein levels. Mol. Cell. Proteom. 2014, 13, 1690–1704. [Google Scholar] [CrossRef]
- Lowell, C.A. Src-family kinases: Rheostats of immune cell signaling. Mol. Immunol. 2004, 41, 631–643. [Google Scholar] [CrossRef]
- Stopfer, L.E.; Flower, C.T.; Gajadhar, A.S.; Patel, B.; Gallien, S.; Lopez-Ferrer, D.; White, F.M. High-density, targeted monitoring of tyrosine phosphorylation reveals activated signaling networks in human tumors. Cancer Res. 2021, 81, 2495–2509. [Google Scholar] [CrossRef]
- McVeigh, T.P.; Kerin, M.J. Clinical use of the oncotype DX genomic test to guide treatment decisions for patients with invasive breast cancer. Breast Cancer 2017, 9, 393. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kohale, I.N.; Yu, J.; Zhuang, Y.; Fan, X.; Reddy, R.J.; Sinnwell, J.; Kalari, K.R.; Boughey, J.C.; Carter, J.M.; Goetz, M.P.; et al. Identification of Src Family Kinases as Potential Therapeutic Targets for Chemotherapy-Resistant Triple Negative Breast Cancer. Cancers 2022, 14, 4220. https://doi.org/10.3390/cancers14174220
Kohale IN, Yu J, Zhuang Y, Fan X, Reddy RJ, Sinnwell J, Kalari KR, Boughey JC, Carter JM, Goetz MP, et al. Identification of Src Family Kinases as Potential Therapeutic Targets for Chemotherapy-Resistant Triple Negative Breast Cancer. Cancers. 2022; 14(17):4220. https://doi.org/10.3390/cancers14174220
Chicago/Turabian StyleKohale, Ishwar N., Jia Yu, Yongxian Zhuang, Xiaoyang Fan, Raven J. Reddy, Jason Sinnwell, Krishna R. Kalari, Judy C. Boughey, Jodi M. Carter, Matthew P. Goetz, and et al. 2022. "Identification of Src Family Kinases as Potential Therapeutic Targets for Chemotherapy-Resistant Triple Negative Breast Cancer" Cancers 14, no. 17: 4220. https://doi.org/10.3390/cancers14174220
APA StyleKohale, I. N., Yu, J., Zhuang, Y., Fan, X., Reddy, R. J., Sinnwell, J., Kalari, K. R., Boughey, J. C., Carter, J. M., Goetz, M. P., Wang, L., & White, F. M. (2022). Identification of Src Family Kinases as Potential Therapeutic Targets for Chemotherapy-Resistant Triple Negative Breast Cancer. Cancers, 14(17), 4220. https://doi.org/10.3390/cancers14174220