
Remodeling of Cancer-Specific Metabolism under Hypoxia with Lactate Calcium Salt in Human Colorectal Cancer Cells


Abstract
Simple Summary
Abstract
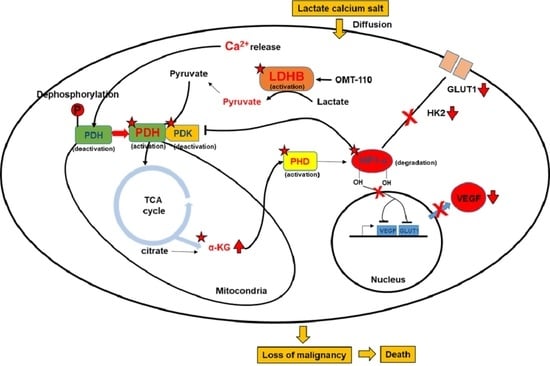
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Culture Conditions
2.2. Reagents
2.3. Measurement of Intracellular Calcium and Lactate
2.4. Quantification of Enzyme Levels
2.5. Immunocytochemistry
2.6. Western Blotting
2.7. Metabolite Assay
2.8. Small Interfering RNA (siRNA) Transfection
2.9. RNA Extraction and Quantitative Real-Time Polymerase Chain Reaction (PCR)
2.10. Cell Viability Assay
2.11. Tube Formation Assay
2.12. Wound-Healing Assay
2.13. Xenograft Animal Model
2.14. Mouse Fluorine-18-Fluorodeoxyglucose Positron Emission Tomography/Computed Tomography (18F-FDG-PET/CT) Scanning
2.15. Immunohistochemistry
2.16. Immunofluorescence
2.17. Statistical Analysis
3. Results
3.1. The Direct Effect of Lactate Calcium Salt (CaLac) Influx into Colorectal Cancer (CRC) Cells on Anaerobic Glycolysis
3.2. Restoration of the Tricarboxylic Acid (TCA) Cycle under Hypoxia Subsequent to Increased Intracellular Calcium and Pyruvate Levels
3.3. Suppression of Hypoxia-Inducible Factor (HIF)-1α Transcriptional Activation by TCA Cycle Restoration
3.4. Anti-Cancer Effect on CRC and Suppressing Oncogene Expression Characterized by Hypoxia
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jeong, K.-Y. Cancer-specific metabolism: Promising approaches for colorectal cancer treatment. World J. Gastrointest. Oncol. 2019, 11, 768–772. [Google Scholar] [CrossRef] [PubMed]
- Al Tameemi, W.; Dale, T.P.; Al-Jumaily, R.M.K.; Forsyth, N.R. Hypoxia-Modified Cancer Cell Metabolism. Front. Cell Dev. Biol. 2019, 7, 4. [Google Scholar] [CrossRef] [PubMed]
- Ziehler, K. Whole body glucose metabolism. Am. J. Physiol. 1999, 276, E409–E426. [Google Scholar]
- Burns, J.S.; Manda, G. Metabolic Pathways of the Warburg Effect in Health and Disease: Perspectives of Choice, Chain or Chance. Int. J. Mol. Sci. 2017, 18, 2755. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J. Energy metabolism of cancer: Glycolysis versus oxidative phosphorylation (Review). Oncol. Lett. 2012, 4, 1151–1157. [Google Scholar] [CrossRef]
- Brooks, G.A. The Science and Translation of Lactate Shuttle Theory. Cell Metab. 2018, 27, 757–785. [Google Scholar] [CrossRef] [PubMed]
- San-Millán, I.; Brooks, G.A. Reexamining cancer metabolism: Lactate production for carcinogenesis could be the purpose and explanation of the Warburg Effect. Carcinogen 2016, 38, 119–133. [Google Scholar] [CrossRef]
- Granchi, C.; Minutolo, F. Anticancer Agents That Counteract Tumor Glycolysis. ChemMedChem 2012, 7, 1318–1350. [Google Scholar] [CrossRef]
- Miskimins, W.K.; Ahn, H.J.; Kim, J.Y.; Ryu, S.; Jung, Y.-S.; Choi, J.Y. Synergistic Anti-Cancer Effect of Phenformin and Oxamate. PLoS ONE 2014, 9, e85576. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Hou, L.; Li, L.; Li, L.; Zhu, L.; Wang, Y.; Huang, X.; Hou, Y.; Zhu, D.; Zou, H.; et al. Dichloroacetate restores colorectal cancer chemosensitivity through the p53/miR-149-3p/PDK2-mediated glucose metabolic pathway. Oncogene 2020, 39, 469–485. [Google Scholar] [CrossRef] [PubMed]
- Stacpoole, P.W.; Henderson, G.N.; Yan, Z.; O James, M. Clinical pharmacology and toxicology of dichloroacetate. Environ. Health Perspect. 1998, 106, 989–994. [Google Scholar] [CrossRef]
- Stacpoole, P.W. The Dichloroacetate Dilemma: Environmental Hazard versus Therapeutic Goldmine—Both or Neither? Environ. Health Perspect. 2011, 119, 155–158. [Google Scholar] [CrossRef] [PubMed]
- Mishra, D.; Banerjee, D. Lactate Dehydrogenases as Metabolic Links between Tumor and Stroma in the Tumor Microenvironment. Cancers 2019, 11, 750. [Google Scholar] [CrossRef] [PubMed]
- Eales, K.L.; Hollinshead, K.E.R.; Tennant, D.A. Hypoxia and metabolic adaptation of cancer cells. Oncogene 2016, 5, e190. [Google Scholar] [CrossRef]
- Davies, C.W. 397. The extent of dissociation of salts in water. Part VIII. An equation for the mean ionic activity coefficient of an electrolyte in water, and a revision of the dissociation constants of some sulphates. J. Chem. Soc. 1938, 2093–2098. [Google Scholar] [CrossRef]
- Hay, N. Reprogramming glucose metabolism in cancer: Can it be exploited for cancer therapy? Nat. Rev. Cancer 2016, 16, 635–649. [Google Scholar] [CrossRef]
- Saunier, E.; Benelli, C.; Bortoli, S. The pyruvate dehydrogenase complex in cancer: An old metabolic gatekeeper regulated by new pathways and pharmacological agents. Int. J. Cancer 2015, 138, 809–817. [Google Scholar] [CrossRef]
- Golias, T.; Papandreou, I.; Sun, R.; Kumar, B.; Brown, N.V.; Swanson, B.J.; Pai, R.; Jaitin, D.; Le, Q.-T.; Teknos, T.N.; et al. Hypoxic repression of pyruvate dehydrogenase activity is necessary for metabolic reprogramming and growth of model tumours. Sci. Rep. 2016, 6, 31146. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Jeon, J.-H.; Min, B.-K.; Ha, C.-M.; Thoudam, T.; Park, B.-Y.; Lee, I.-K. Role of the Pyruvate Dehydrogenase Complex in Metabolic Remodeling: Differential Pyruvate Dehydrogenase Complex Functions in Metabolism. Diabetes Metab. J. 2018, 42, 270–281. [Google Scholar] [CrossRef]
- Strowitzki, M.J.; Cummins, E.P.; Taylor, C.T. Protein Hydroxylation by Hypoxia-Inducible Factor (HIF) Hydroxylases: Unique or Ubiquitous? Cells 2019, 8, 384. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.; Kim, S.S.; Lee, J. Cancer cell metabolism: Implications for therapeutic targets. Exp. Mol. Med. 2013, 45, e45. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.; Chandel, N.S.; Simon, M.C. Cellular adaptation to hypoxia through hypoxia inducible factors and beyond. Nat. Rev. Mol. Cell Biol. 2020, 21, 268–283. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Liu, L.; Fu, T.; Zhou, Q.; Zhou, D.; Xiao, L.; Liu, J.; Kong, Y.; Xie, H.; Yi, F.; et al. Exercise Inducible Lactate Dehydrogenase B Regulates Mitochondrial Function in Skeletal Muscle. J. Biol. Chem. 2016, 291, 25306–25318. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Saunier, E.; Antonio, S.; Regazzetti, A.; Auzeil, N.; Laprévote, O.; Shay, J.W.; Coumoul, X.; Barouki, R.; Benelli, C.; Huc-Lemarié, L.; et al. Resveratrol reverses the Warburg effect by targeting the pyruvate dehydrogenase complex in colon cancer cells. Sci. Rep. 2017, 7, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Reyes, I.; Chandel, N.S. Mitochondrial TCA cycle metabolites control physiology and disease. Nat. Commun. 2020, 11, 1–11. [Google Scholar] [CrossRef]
- Fendt, S.-M.; Bell, E.L.; Keibler, M.A.; Olenchock, B.A.; Mayers, J.R.; Wasylenko, T.M.; Vokes, N.I.; Guarente, L.; Heiden, M.G.V.; Stephanopoulos, G. Reductive glutamine metabolism is a function of the α-ketoglutarate to citrate ratio in cells. Nat. Commun. 2013, 4, 1–11. [Google Scholar] [CrossRef]
- Cluntun, A.A.; Lukey, M.J.; Cerione, R.A.; Locasale, J.W. Glutamine Metabolism in Cancer: Understanding the Heterogeneity. Trends Cancer 2017, 3, 169–180. [Google Scholar] [CrossRef]
- Pollard, P.J.; Yang, M.; Su, H.; Soga, T.; Kranc, K.R. Prolyl hydroxylase domain enzymes: Important regulators of cancer metabolism. Hypoxia 2014, 2, 127–142. [Google Scholar] [CrossRef] [PubMed]
- Koh, M.Y.; Darnay, B.G.; Powis, G. Hypoxia-Associated Factor, a Novel E3-Ubiquitin Ligase, Binds and Ubiquitinates Hypoxia-Inducible Factor 1α, Leading to Its Oxygen-Independent Degradation. Mol. Cell. Biol. 2008, 28, 7081–7095. [Google Scholar] [CrossRef]
- Shi, Y.-H.; Fang, W.-G. Hypoxia-inducible factor-1 in tumour angiogenesis. World J. Gastroenterol. 2004, 10, 1082–1087. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.A.; Jung, Y.D.; Liub, W.; Reinmuthb, N.; Parikha, A.; Stoeltzingb, O.; Fanb, F.; Ellis, L.M. The role of the microenvironment and intercellular cross-talk in tumor angiogenesis. Semin. Cancer Biol. 2002, 12, 105–112. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Direction | Primer Sequences |
|---|---|---|
| Actin | Sense | 5′-AACTGGAACGGTGAAGGT-3′ |
| Anti-sense | 5′-CCTGTAACAACGCATCTCAT-3′ | |
| VEGF | Sense | 5′-ACATCTTCCAGGAGTACCC-3′ |
| Anti-sense | 5′-CTTGGTGAGGTTTGATCCG-3′ | |
| PDK-1 | Sense | 5′-TGTAGGTGGTATCATTCTCTTTC-3′ |
| Anti-sense | 5′-GGATAACTAACAACACAGTCTCT-3′ | |
| HK2 | Sense | 5′-CAAAGTGACAGTGGGTGTGG-3′ |
| Anti-sense | 5′-GCCAGGTCCTTCACTGTCTC-3′ | |
| GLUT1 | Sense | 5′-ACCACCTCACTCCTGTTA-3′ |
| Anti-sense | 5′-CCACTTACTTCTGTCTCACTC-3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jeong, K.-Y.; Sim, J.-J.; Park, M.H.; Kim, H.M. Remodeling of Cancer-Specific Metabolism under Hypoxia with Lactate Calcium Salt in Human Colorectal Cancer Cells. Cancers 2021, 13, 1518. https://doi.org/10.3390/cancers13071518
Jeong K-Y, Sim J-J, Park MH, Kim HM. Remodeling of Cancer-Specific Metabolism under Hypoxia with Lactate Calcium Salt in Human Colorectal Cancer Cells. Cancers. 2021; 13(7):1518. https://doi.org/10.3390/cancers13071518
Chicago/Turabian StyleJeong, Keun-Yeong, Jae-Jun Sim, Min Hee Park, and Hwan Mook Kim. 2021. "Remodeling of Cancer-Specific Metabolism under Hypoxia with Lactate Calcium Salt in Human Colorectal Cancer Cells" Cancers 13, no. 7: 1518. https://doi.org/10.3390/cancers13071518
APA StyleJeong, K.-Y., Sim, J.-J., Park, M. H., & Kim, H. M. (2021). Remodeling of Cancer-Specific Metabolism under Hypoxia with Lactate Calcium Salt in Human Colorectal Cancer Cells. Cancers, 13(7), 1518. https://doi.org/10.3390/cancers13071518