
Focal Adhesion Kinase Fine Tunes Multifaced Signals toward Breast Cancer Progression


 and
and 
Simple Summary
Abstract
1. Introduction
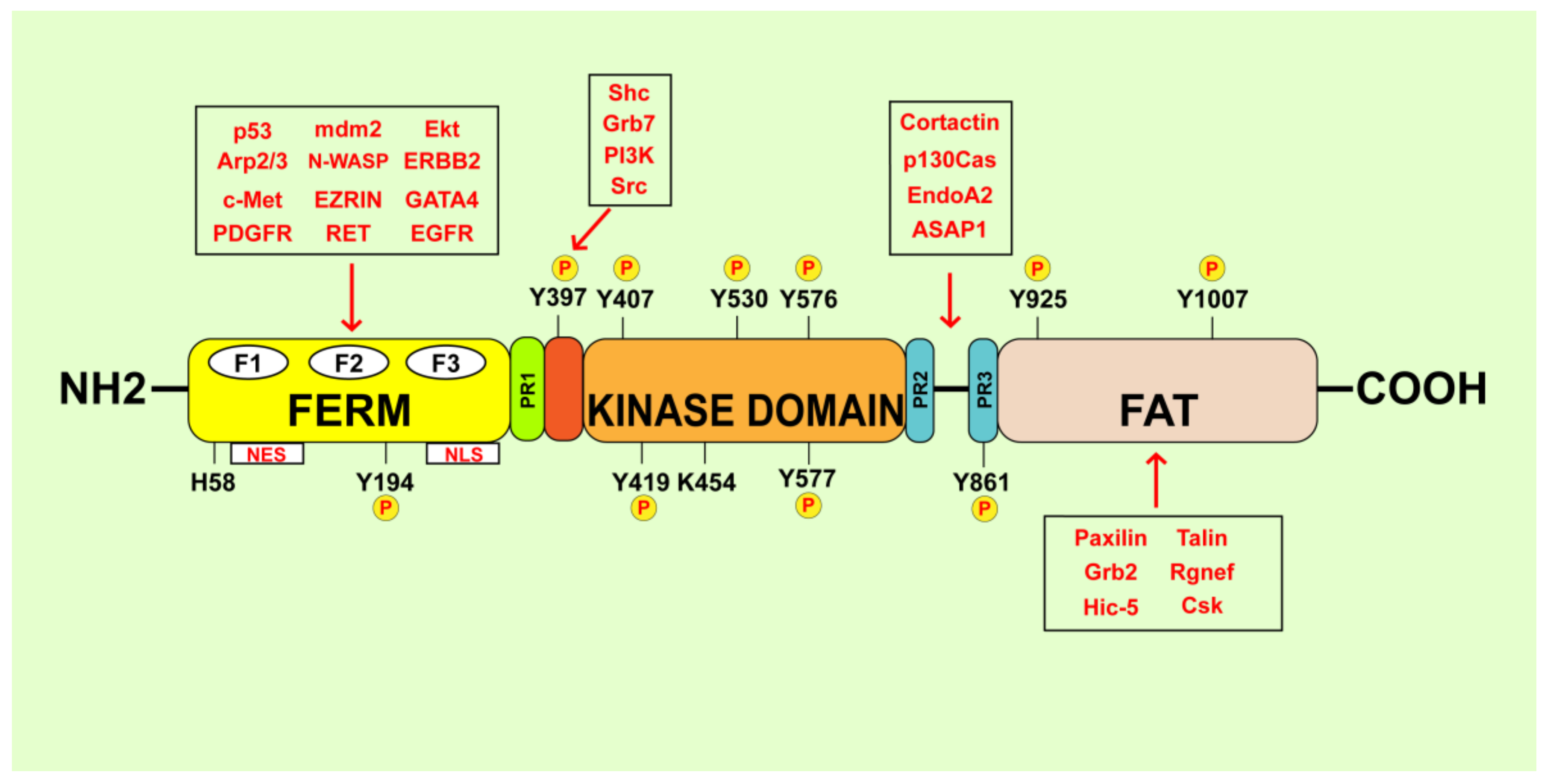
2. FAK Structural Organization and Activation
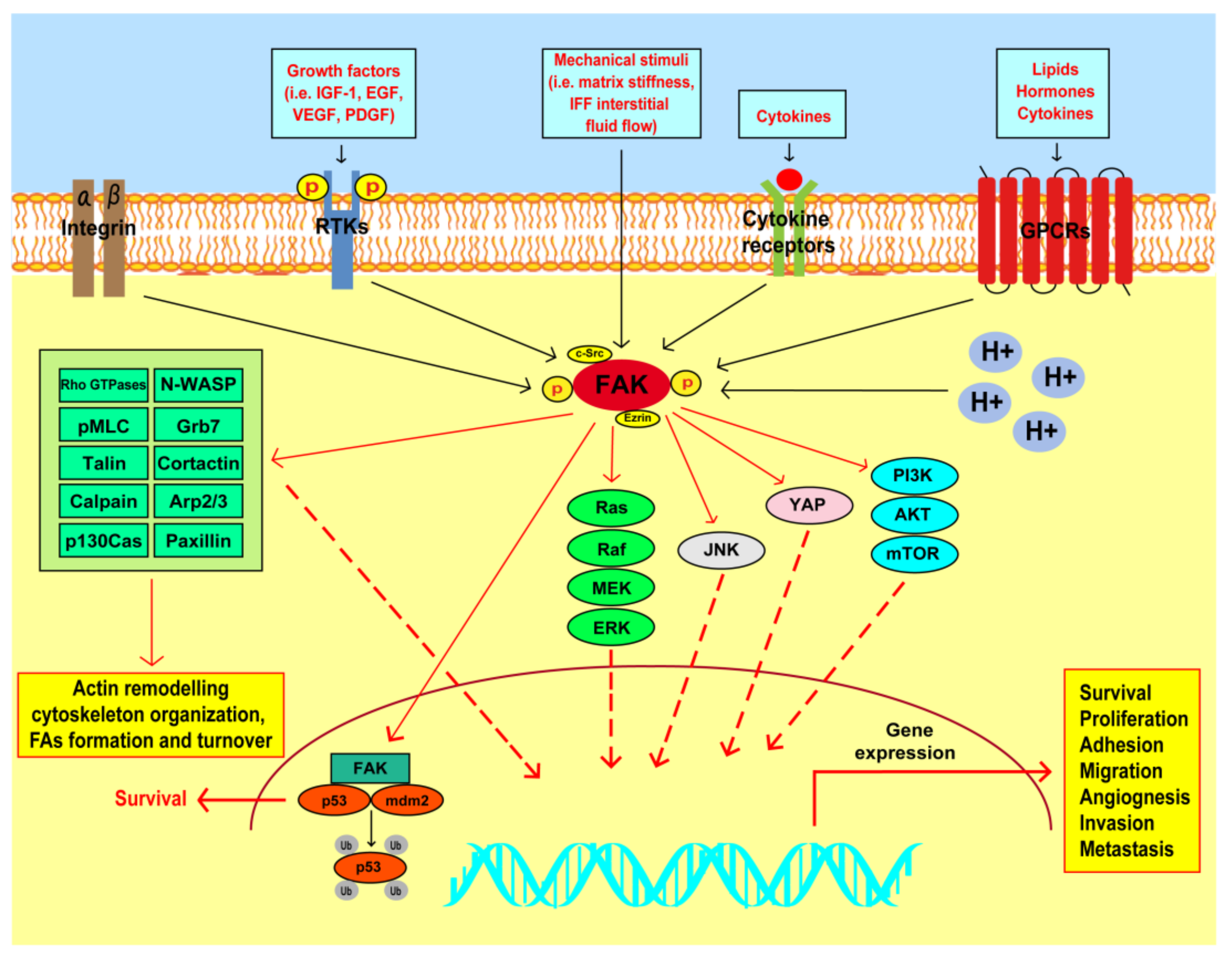
3. FAK Involvement in the Survival and Proliferation of Breast Cancer Cells
4. FAK Action in the Invasive and Metastatic Features of Breast Tumors
5. FAK as a Mechanotransducer in Breast Cancer
6. Role of FAK in CSC Self-Renewal and Maintenance
7. FAK and the Breast TME
8. FAK as a Predictive and Prognostic Determinant in Breast Cancer
9. Clinical Usefulness of FAK Inhibitors
10. Conclusions and Perspectives
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 4-1BB | Tumor necrosis factor receptor superfamily member 9 |
| ACM | adipose tissue conditioned media |
| AGTR1 | angiotensin II type I receptor |
| AKR1B10 | aldo-keto reductase 1B10 |
| Akt | Protein kinase B |
| AMPK | AMP-activated protein kinase |
| AR | Androgen Receptor |
| ANGPTL4 | Angiopoietin Like 4 |
| APCs | antigen-presenting cells |
| Arp2/3 | Actin Related Protein 2/3 |
| Bad | BCL2 associated agonist of cell death |
| CAA | cancer-associated adipocytes |
| CAFs | cancer-associated fibroblasts |
| Ccl11 | C–C Motif Chemokine Ligand 11 |
| Ccl12 | C–C Motif Chemokine Ligand 12 |
| Ccl6 | C–C Motif Chemokine Ligand 6 |
| cdc42 | cell division cycle 42 |
| CM | conditioned-medium |
| c-Myc | c-Myc Proto-Oncogene |
| CSCs | Cancer Stem Cells |
| c-Src | proto-oncogene tyrosine-protein kinase Src |
| CXCR4 | C-X-C Motif Chemokine Receptor 4 |
| DCIS | mammary ductal carcinoma in situ |
| DFG | Asp-Phe-Gly |
| DFS | disease-free survival |
| DMP1 | cyclin D binding protein 1 |
| ECM | extracellular matrix |
| ECs | endothelial cells |
| EGCG | epigallocatechin-3-gallate |
| EGFR | epidermal growth factor receptor |
| EMT | Epithelial-Mesenchymal Transition |
| ER | estrogen receptor |
| ERK | Extracellular Signal-Regulated Kinase |
| ERM | Ezrin–radixin–moesin |
| FADD | Fas-associated death domain |
| FAK | focal adhesion kinase |
| FAK-CD | C-terminal domain of FAK |
| FAs | focal adhesions |
| FAT | C-terminal focal adhesion-targeting domain |
| FERM | band 4.1-ezrin-radixin-moesin domain |
| FISH | fluorescence in situ hybridization |
| FRNK | FAK-related non-kinase |
| FUT8 | fucosyltranferase-8 |
| GPCRs | G-protein coupled receptors |
| GPER | G protein estrogen receptor |
| Gpx1 | glutathione peroxidase-1 |
| Grb2 | growth factor receptor bound protein 2 |
| Grb7 | Growth factor receptor-bound protein 7 |
| GSK-3β | Glycogen Synthase Kinase 3 Beta |
| HER2 | human epidermal growth factor receptor 2 |
| Hippo | Salvador-Warts-Hippo SWH |
| HPIP/PBXIP1 | hematopoietic PBX-interacting protein |
| HSP90β | heat shock protein 90β |
| ICAM-1 | Intercellular Adhesion Molecule 1 |
| ICAM-1 | Intercellular Adhesion Molecule 1 |
| IF | interstitial flow |
| IFF | interstitial fluid flow |
| IGF-I | insulin-like growth factor-1 |
| IGF-IR | insulin-like growth factor-1 receptor |
| IL-1β | interleukin-1β |
| ILK | integrin-linked kinase |
| JNK | c-Jun amino terminal kinase |
| KD | kinase-dead |
| KLF8 | Krüppel-like factor 8 |
| KO | knock out |
| Lox | Lysyl oxidase |
| LOXL2 | lysyl oxidase-like 2 |
| MaCSCs | mammary cancer stem cells |
| MAPK | mitogen-activated protein kinase |
| MDSCs | myeloid-derived suppressor cells |
| METABRIC | Molecular Taxonomy of Breast Cancer International Consortium |
| MLC | Myosin light-chain |
| MLCK | myosin light chain-kinase |
| MMP14 | Matrix Metallopeptidase 14 |
| MMP2 | Matrix Metallopeptidase 2 |
| MMP9 | Matrix Metallopeptidase 9 |
| MMPs | matrix metalloproteinases |
| MMTV | mouse mammary virus tumor |
| MMTV-PyMT | Mammary Tumor Virus-Polyoma Middle T-Antigen |
| MPS | mononuclear phagocytes |
| mrcPCR | modified real competitive PCR |
| MT1-MMP | Membrane type 1-matrix metalloproteinase |
| mTORC1 | mammalian target of rapamycin complex 1 |
| MUCL1 | Mucin-like 1 |
| NAD | Nicotinamide Adenine Dinucleotide |
| NANOG | Nanog Homeobox |
| NES | nuclear export sequence |
| NGS | next generation sequencing |
| NK | Natural Killer |
| NLS | nuclear localization sequence |
| N-WASP | Neural Wiskott-Aldrich syndrome protein |
| OA | Oroxylin A |
| OS | overall survival |
| OX-40 | Tumor necrosis factor receptor superfamily, member 4 |
| PAK1 | P21 (RAC1) Activated Kinase 1 |
| PARP | poly (ADP-ribose) polymerase |
| PD-L1 | Programmed death-ligand 1 |
| PI (4,5)-P2 | phosphatidylinositol 4,5-bisphosphate |
| PI3K | Phosphatidylinositol 3-Kinase |
| PPAR | Peroxisome Proliferator Activated Receptor |
| PR | progesterone receptor |
| PRMT7 | protein arginine methyltransferase 7 |
| PTK2 | Protein Tyrosine Kinase 2 |
| PTX-S-LA | ROS-responsive thioether-linked paclitaxel-linoleic acid conjugates |
| Pyk2 | FAK-related proline-rich tyrosine kinase 2 |
| Rac1 | Rac family small GTPase 1 |
| RFS | relapse-free survival |
| RhoA | Ras Homolog Family Member A |
| RIP | receptor-interacting protein |
| ROCK1 | Rho-associated coiled-coil kinase 1 |
| ROCK2 | Rho-associated coiled-coil kinase 2 |
| ROS | reactive oxygen species |
| RTKs | receptor tyrosine kinases |
| SEPT9_i1 | SEPT9 isoform 1 protein |
| SH2 | Src Homology 2 |
| SH3 | Src-homology 3 |
| SHANK2 | SH3 and Multiple Ankyrin Repeat Domains 2 |
| SIPA1 | signal-induced proliferation-associated protein 1 |
| SIRT3 | sirtuin 3 |
| Slug | Snail Family Transcriptional Repressor 2 |
| Snail | Snail Family Transcriptional Repressor |
| Sox9 | SRY-Box Transcription Factor 9 |
| SRC-3Δ4 | SRC-3 oncogene/c-Src signaling adaptor molecule |
| ST8SIA1 | GD3 synthase ST8 Alpha-N-Acetyl-Neuraminide Alpha-2,8-Sialyltransferase 1 |
| STAT3 | Signal Transducer and Activator of Transcription 3 |
| TAMs | tumor-associated macrophages |
| TCGA | The Cancer Genome Atlas Program |
| Tet-ON | tetracycline-inducible |
| TGF-β | Transforming Growth Factor Beta |
| THBS1 | thrombospondin 1 |
| Tinagl-1 | tubulo interstitial nephritis antigen-like 1 |
| TME | tumor microenvironment |
| TNBC | triple-negative breast cancer |
| TNFα | Tumor Necrosis Factor α |
| TKIs | tyrosine kinase inhibitors |
| TRAF2 | TNF receptor–associated factor 2 |
| TWIST | Helix-Loop-Helix Twist Family BHLH Transcription Factor 1 |
| uPA | urokinase plasminogen activator |
| VEC | vascular endothelial cadherin |
| VEGF | Vascular-Endothelial Growth Factor |
| VEGFR2 | Vascular endothelial growth factor receptor-2 |
| VEGFR-3 | Vascular Endothelial Growth Factor Receptor-3 |
| WNT | Wingless-type MMTV integration site family |
| Wnt3a | Wnt Family Member 3A |
| WT | wild-type |
| YAP | Yes-associated protein |
References
- Torre, A.L.; Islami, F.; Siegel, R.L.; Ward, E.M.; Jemal, A. Global Cancer in Women: Burden and Trends. Cancer Epidemiol. Biomark. Prev. 2017, 26, 444–457. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Li, T.; Bai, Z.; Yang, Y.; Liu, X.; Zhan, J.; Shi, B. Breast cancer intrinsic subtype classification, clinical use and future trends. Am. J. Cancer Res. 2015, 5, 2929–2943. [Google Scholar]
- Waks, A.G.; Winer, E.P. Breast Cancer Treatment: A Review. JAMA 2019, 321, 288–300. [Google Scholar] [CrossRef]
- Kumar, P.; Aggarwal, R. An overview of triple-negative breast cancer. Arch. Gynecol. Obstet. 2016, 293, 247–269. [Google Scholar] [CrossRef]
- Marti, J.L.G.; Hyder, T.; Nasrazadani, A.; Brufsky, A.M. The Evolving Landscape of HER2-Directed Breast Cancer Therapy. Curr. Treat Options Oncol. 2020, 21, 82. [Google Scholar] [CrossRef]
- Szostakowska, M.; Trębińska-Stryjewska, A.; Grzybowska, E.A.; Fabisiewicz, A. Resistance to endocrine therapy in breast cancer: Molecular mechanisms and future goals. Breast Cancer Res. Treat. 2019, 173, 489–497. [Google Scholar] [CrossRef] [PubMed]
- Gupta, G.K.; Collier, A.L.; Lee, D.; Hoefer, R.A.; Zheleva, V.; Siewertsz van Reesema, L.L.; Tang-Tan, A.M.; Guye, M.L.; Chang, D.Z.; Winston, J.S.; et al. Perspectives on Triple-Negative Breast Cancer: Current Treatment Strategies, Unmet Needs, and Potential Targets for Future Therapies. Cancers 2020, 12, 2392. [Google Scholar] [CrossRef] [PubMed]
- Van Hoeck, A.; Tjoonk, N.H.; van Boxtel, R.; Cuppen, E. Portrait of a cancer: Mutational signature analyses for cancer diagnostics. BMC Cancer 2019, 19, 457. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, J.L.; Cardoso Nunes, N.C.; Izetti, P.; de Mesquita, G.G.; de Melo, A.C. Triple negative breast cancer: A thorough review of biomarkers. Crit. Rev. Oncol. Hematol. 2020, 145, 102855. [Google Scholar] [CrossRef]
- Mertins, P.; Mani, D.R.; Ruggles, K.V.; Gillette, M.A.; Clauser, K.R.; Wang, P.; Wang, X.; Qiao, J.W.; Cao, S.; Petralia, F.; et al. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature 2016, 534, 55–62. [Google Scholar] [CrossRef]
- Murphy, J.M.; Rodriguez, Y.A.R.; Jeong, K.; Ahn, E.E.; Lim, S.S. Targeting focal adhesion kinase in cancer cells and the tumor microenvironment. Exp. Mol. Med. 2020, 52, 877–886. [Google Scholar] [CrossRef]
- Parsons, J.T. Focal adhesion kinase: The first ten years. J. Cell Sci. 2003, 116, 1409–1416. [Google Scholar] [CrossRef]
- Sulzmaier, F.J.; Jean, C.; Schlaepfer, D.D. FAK in cancer: Mechanistic findings and clinical applications. Nat. Rev. Cancer 2014, 14, 598–610. [Google Scholar] [CrossRef]
- Owens, L.V.; Xu, L.; Craven, R.J.; Dent, G.A.; Weiner, T.M.; Kornberg, L.; Liu, E.T.; Cance, W.G. Overexpression of the focal adhesion kinase (p125FAK) in invasive human tumors. Cancer Res. 1995, 55, 2752–2755. [Google Scholar] [PubMed]
- Lark, A.L.; Livasy, C.A.; Dressler, L.; Moore, D.T.; Millikan, R.C.; Geradts, J.; Iacocca, M.; Cowan, D.; Little, D.; Craven, R.J. High focal adhesion kinase expression in invasive breast carcinomas is associated with an aggressive phenotype. Mod. Pathol. 2005, 18, 1289–1294. [Google Scholar] [CrossRef] [PubMed]
- Cance, W.G.; Harris, J.E.; Iacocca, M.V.; Roche, E.; Yang, X.; Chang, J.; Simkins, S.; Xu, L. Immunohistochemical analyses of focal adhesion kinase expression in benign and malignant human breast and colon tissues: Correlation with preinvasive and invasive phenotypes. Clin. Cancer Res. 2000, 6, 2417–2423. [Google Scholar] [PubMed]
- Qiao, W.; Wang, W.; Liu, H.; Guo, W.; Li, P.; Deng, M. Prognostic and clinical significance of focal adhesion kinase expression in breast cancer: A systematic review and meta-analysis. Transl. Oncol. 2020, 13, 100835. [Google Scholar] [CrossRef]
- Lahlou, H.; Sanguin-Gendreau, V.; Zuo, D.; Cardiff, R.D.; McLean, G.W.; Frame, M.C.; Muller, W.J. Mammary epithelial-specific disruption of the focal adhesion kinase blocks mammary tumor progression. Proc. Natl. Acad. Sci. USA 2007, 104, 20302–20307. [Google Scholar] [CrossRef]
- Katoh, K. FAK-Dependent Cell Motility and Cell Elongation. Cells 2020, 9, 192. [Google Scholar] [CrossRef] [PubMed]
- Brandão-Costa, R.M.; Helal-Neto, E.; Vieira, A.M.; Barcellos-de-Souza, P.; Morgado-Diaz, J.; Barja-Fidalgo, C. Extracellular Matrix Derived from High Metastatic Human Breast Cancer Triggers Epithelial-Mesenchymal Transition in Epithelial Breast Cancer Cells through αvβ3 Integrin. Int. J. Mol. Sci. 2020, 21, 2995. [Google Scholar] [CrossRef] [PubMed]
- Carragher, N.O.; Frame, M.C. Focal adhesion and actin dynamics: A place where kinases and proteases meet to promote invasion. Trends Cell Biol. 2004, 14, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Cooper, J.; Giancotti, F.G. Integrin Signaling in Cancer: Mechanotransduction, Stemness, Epithelial Plasticity, and Therapeutic Resistance. Cancer Cell 2019, 35, 347–367. [Google Scholar] [CrossRef] [PubMed]
- Lechertier, T.; Hodivala-Dilke, K. Focal adhesion kinase and tumour angiogenesis. J. Pathol. 2012, 226, 404–412. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.L. Integrin signaling through FAK in the regulation of mammary stem cells and breast cancer. IUBMB Life 2010, 62, 268–276. [Google Scholar] [CrossRef]
- Kobayashi, H.; Azumi, M.; Kimura, Y.; Sato, K.; Aoki, N.; Kimura, S.; Honma, M.; Iizuka, H.; Tateno, M.; Celiset, E. Focal adhesion kinase as an immunotherapeutic target. Cancer Immunol. Immunother. 2009, 58, 931–940. [Google Scholar] [CrossRef][Green Version]
- Demircioglu, F.; Wang, J.; Candido, J.; Costa, A.S.H.; Casado, P.; de Luxan Delgado, B.; Reynolds, L.E.; Gomez-Escudero, J.; Newport, E.; Rajeeve, V.; et al. Cancer associated fibroblast FAK regulates malignant cell metabolism. Nat. Commun. 2020, 11, 1290. [Google Scholar] [CrossRef]
- Jeong, K.; Murphy, J.M.; Rodriguez, Y.A.R.; Kim, J.S.; Ahn, E.Y.E.; Lim, S.T.S. FAK inhibition reduces metastasis of α4 integrin-expressing melanoma to lymph nodes by targeting lymphatic VCAM-1 expression. Biochem. Biophys. Res. Commun. 2019, 509, 1034–1040. [Google Scholar] [CrossRef]
- Kanteti, R.; Mirzapoiazova, T.; Riehm, J.J.; Dhanasingh, I.; Mambetsariev, B.; Wang, J.; Kulkarni, P.; Kaushik, G.; Seshacharyulu, P.; Ponnusamy, M.P.; et al. Focal adhesion kinase a potential therapeutic target for pancreatic cancer and malignant pleural mesotelioma. Cancer Biol. Ther. 2018, 19, 316–327. [Google Scholar] [CrossRef]
- Feng, X.; Arang, N.; Rigiracciolo, D.C.; Lee, J.S.; Yeerna, H.; Wang, Z.; Lubrano, S.; Kishore, A.; Pachter, J.A.; König, G.M.; et al. A Platform of Synthetic Lethal Gene Interaction Networks Reveals that the GNAQ Uveal Melanoma Oncogene Controls the Hippo Pathway through FAK. Cancer Cell 2019, 35, 457–472. [Google Scholar] [CrossRef]
- Tanjoni, I.; Walsh, C.; Uryu, S.; Tomar, A.; Nam, J.O.; Mielgo, A.; Lim, S.T.; Liang, C.; Koenig, M.; Sun, C.; et al. PND-1186 FAK inhibitor selectively promotes tumor cell apoptosis in three-dimensional environments. Cancer Biol. Ther. 2010, 9, 764–777. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.L.; Shalloway, D. Regulation of focal adhesion-associated protein tyrosine kinase by both cellular adhesion and oncogenic transformation. Nature 1992, 358, 690–692. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Guan, J.L. Signal transduction by focal adhesion kinase in cancer. Cancer Metastasis Rev. 2009, 28, 35–49. [Google Scholar] [CrossRef]
- Kleinschmidt, E.G.; Schlaepfer, D.D. Focal adhesion kinase signaling in unexpected places. Curr. Opin. Cell Biol. 2017, 45, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Ceccarelli, D.F.J.; Song, H.K.; Poy, F.; Schaller, M.D.; Eck, M.J. Crystal structure of the FERM domain of focal adhesion kinase. J. Biol. Chem. 2006, 281, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Martínez, P.T.; Navajas, P.L.; Lietha, D. FAK Structure and Regulation by Membrane Interactions and Force in Focal Adhesions. Biomolecules 2020, 10, 179. [Google Scholar] [CrossRef]
- Dunty, J.M.; Gabarra-Niecko, V.; King, M.L.; Ceccarelli, D.F.J.; Eck, M.J.; Schaller, M.D. FERM domain interaction promotes FAK signaling. Mol. Cell Biol. 2004, 24, 5353–5368. [Google Scholar] [CrossRef]
- Frame, M.C.; Patel, H.; Serrels, B.; Lietha, D.; Eck, M.J. The FERM domain: Organizing the structure and function of FAK. Nat. Rev. Mol. Cell Biol. 2010, 11, 802–814. [Google Scholar] [CrossRef]
- Sieg, D.J.; Hauck, C.R.; Ilic, D.; Klingbeil, C.K.; Schaefer, E.; Damsky, C.H.; Schlaepfer, D.D. FAK integrates growth-factor and integrin signals to promote cell migration. Nat. Cell Biol. 2000, 2, 249–256. [Google Scholar] [CrossRef]
- Stahl, E.; Nott, R.; Koessel, K.; Cance, W.; Marlowe, T. Computational-based discovery of FAK FERM domain chemical probes that inhibit HER2-FAK cancer signaling. Chem. Biol. Drug Des. 2020, 95, 584–599. [Google Scholar] [CrossRef]
- Plaza-Menacho, I.; Morandi, A.; Mologni, L.; Boender, P.; Gambacorti-Passerini, C.; Magee, A.I.; Hofstra, R.M.W.; Knowles, P.; McDonald, N.Q.; Isacke, C.M. Focal adhesion kinase (FAK) binds RET kinase via its FERM domain, priming a direct and reciprocal RET-FAK transactivation mechanism. J. Biol. Chem. 2011, 286, 17292–17302. [Google Scholar] [CrossRef]
- Poullet, P.; Gautreau, A.; Kadaré, G.; Girault, J.A.; Louvard, D.; Arpin, M. Ezrin interacts with focal adhesion kinase and induces its activation independently of cell-matrix adhesion. J. Biol. Chem. 2001, 276, 37686–37691. [Google Scholar] [CrossRef]
- Lim, S.T.; Chen, X.L.; Lim, Y.; Hanson, D.A.; Vo, T.T.; Howerton, K.; Larocque, N.; Fisher, S.J.; Schlaepfer, D.D.; Ilic, D. Nuclear FAK promotes cell proliferation and survival through FERM-enhanced p53 degradation. Mol. Cell. 2008, 29, 9–22. [Google Scholar] [CrossRef]
- Serrels, B.; McGivern, N.; Canel, M.; Byron, A.; Johnson, S.C.; McSorley, H.J.; Quinn, N.; Taggart, D.; Von Kreigsheim, A.; Anderton, S.M.; et al. IL-33 and ST2 mediate FAK-dependent antitumor immune evasion through transcriptional networks. Sci. Signal. 2017, 10, eaan8355. [Google Scholar] [CrossRef]
- Mei, L.; Xiong, W. FAK interaction with MBD2: A link from cell adhesion to nuclear chromatin remodeling? Cell Adhes. Migr. 2010, 4, 77–80. [Google Scholar] [CrossRef][Green Version]
- Lim, S.; Miller, N.L.G.; Chen, X.L.; Tancioni, I.; Walsh, C.T.; Lawson, C.; Uryu, S.; Weis, S.M.; Cheresh, D.A.; Schlaepfer, D.D. Nuclear-localized focal adhesion kinase regulates inflammatory VCAM-1 expression. J. Cell Biol. 2012, 197, 907–919. [Google Scholar] [CrossRef]
- Papusheva, E.; de Queiroz, F.M.; Dalous, J.; Han, Y.; Esposito, A.; Jares-Erijmanxa, E.A.; Jovin, T.M.; Bunt, G. Dynamic conformational changes in the FERM domain of FAK are involved in focal-adhesion behavior during cell spreading and motility. J. Cell Sci. 2009, 122, 656–666. [Google Scholar] [CrossRef] [PubMed]
- Lietha, D.; Cai, X.; Ceccarelli, D.F.J.; Li, Y.; Schaller, M.D.; Eck, M.J. Structural basis for the autoinhibition of focal adhesion kinase. Cell 2007, 129, 1177–1187. [Google Scholar] [CrossRef]
- Tomar, A.; Lim, S.; Lim, Y.; Schlaepfer, D.D. A FAK-p120RasGAP-p190RhoGAP complex regulates polarity in migrating cells. J. Cell Sci. 2009, 122, 1852–1862. [Google Scholar] [CrossRef] [PubMed]
- Kasahara, T.; Koguchi, E.; Funakoshi, M.; Aizu-Yokota, E.; Sonoda, Y. Antiapoptotic action of focal adhesion kinase (FAK) against ionizing radiation. Antioxid Redox Signal. 2002, 4, 491–499. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Gelman, I.H.; Katsuta, E.; Liang, Y.; Wang, X.; Li, J.; Qu, J.; Yan, L.; Takabe, K.; Hochwald, S.N. Glucose Drives Growth Factor-Independent Esophageal Cancer Proliferation via Phosphohistidine-Focal Adhesion Kinase Signaling. Cell. Mol. Gastroenterol. Hepatol. 2019, 8, 37–60. [Google Scholar] [CrossRef]
- Hayashi, I.; Vuori, K.; Liddington, R.C. The focal adhesion targeting (FAT) region of focal adhesion kinase is a four-helix bundle that binds paxillin. Nat. Struct. Biol. 2002, 9, 101–106. [Google Scholar] [CrossRef]
- Bertolucci, C.M.; Guibao, C.D.; Zheng, J. Structural features of the focal adhesion kinase-paxillin complex give insight into the dynamics of focal adhesion assembly. Protein Sci. 2005, 14, 644–652. [Google Scholar] [CrossRef]
- Arold, S.T.; Hoellerer, M.K.; Noble, M.E.M. The structural basis of localization and signaling by the focal adhesion targeting domain. Structure 2002, 10, 319–327. [Google Scholar] [CrossRef]
- Schlaepfer, D.D.; Hanks, S.K.; Hunter, T.; van der Geer, P. Integrin-mediated signal transduction linked to Ras pathway by GRB2 binding to focal adhesion kinase. Nature 1994, 372, 786–791. [Google Scholar] [CrossRef]
- Garces, C.A.; Kurenova, E.V.; Golubovskaya, V.M.; Cance, W.G. Vascular endothelial growth factor receptor-3 and focal adhesion kinase bind and suppress apoptosis in breast cancer cells. Cancer Res. 2006, 66, 1446–1454. [Google Scholar] [CrossRef] [PubMed]
- Acebrón, I.; Righetto, R.D.; Schoenherr, C.; de Buhr, S.; Redondo, P.; Culley, J.; Rodríguez, C.F.; Daday, C.; Biyani, N.; Llorca, O.; et al. Structural basis of Focal Adhesion Kinase activation on lipid membranes. EMBO J. 2020, 39, e104743. [Google Scholar] [CrossRef] [PubMed]
- Brod, F.; Fässler, R. A FAK conundrum is solved: Activation and organization of focal adhesion kinase at the plasma membrane. EMBO J. 2020, 39, e106234. [Google Scholar] [CrossRef] [PubMed]
- Pylayeva, Y.; Gillen, K.M.; Gerald, W.; Beggs, H.E.; Reichardt, L.F.; Giancotti, F.G. Ras- and PI3K-dependent breast tumorigenesis in mice and humans requires focal adhesion kinase signaling. J. Clin. Investig. 2009, 119, 252–266. [Google Scholar] [CrossRef] [PubMed]
- Luey, B.C.; May, F.E.B. Insulin-like growth factors are essential to prevent anoikis in oestrogen-responsive breast cancer cells: Importance of the type I IGF receptor and PI3-kinase/Akt pathway. Mol. Cancer. 2016, 15, 8. [Google Scholar] [CrossRef]
- Paul, R.; Luo, M.; Mo, X.; Lu, J.; Yeo, S.K.; Guan, J.L. FAK activates AKT-mTOR signaling to promote the growth and progression of MMTV-Wnt1-driven basal-like mammary tumors. Breast Cancer Res. 2020, 22, 59. [Google Scholar] [CrossRef]
- Golubovskaya, V.; Beviglia, L.; Xu, L.H.; Earp 3rd, H.S.; Craven, R.; Cance, W. Dual inhibition of focal adhesion kinase and epidermal growth factor receptor pathways cooperatively induces death receptor-mediated apoptosis in human breast cancer cells. J. Biol. Chem. 2002, 277, 38978–38987. [Google Scholar] [CrossRef]
- da Silva, S.D.; Xu, B.; Maschietto, M.; Marchi, F.A.; Alkailani, M.I.; Bijian, K.; Xiao, D.; Alaoui-Jamali, M.A. TRAF2 Cooperates with Focal Adhesion Signaling to Regulate Cancer Cell Susceptibility to Anoikis. Mol. Cancer Ther. 2019, 18, 139–146. [Google Scholar] [CrossRef]
- Xu, L.H.; Yang, X.; Bradham, C.A.; Brenner, D.A.; Baldwin Jr, A.S.; Craven, R.J.; Cance, W.G. The focal adhesion kinase suppresses transformation-associated, anchorage-independent apoptosis in human breast cancer cells. Involvement of death receptor-related signaling pathways. J Biol. Chem. 2000, 275, 30597–30604. [Google Scholar] [CrossRef] [PubMed]
- Kurenova, E.; Xu, L.H.; Yang, X.; Baldwin Jr, A.S.; Craven, R.J.; Hanks, S.K.; Liu, Z.G.; Cance, W.G. Focal adhesion kinase suppresses apoptosis by binding to the death domain of receptor-interacting protein. Mol. Cell Biol. 2004, 24, 4361–4371. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Pestell, R.; Guan, J.L. Transcriptional activation of cyclin D1 promoter by FAK contributes to cell cycle progression. Mol. Biol. Cell. 2001, 12, 4066–4077. [Google Scholar] [CrossRef] [PubMed]
- Conley, S.J.; Bosco, E.E.; Tice, D.A.; Hollingsworth, R.E.; Herbst, R.; Xiao, Z. HER2 drives Mucin-like 1 to control proliferation in breast cancer cells. Oncogene 2016, 35, 4225–4234. [Google Scholar] [CrossRef] [PubMed]
- Rigiracciolo, D.C.; Nohata, N.; Lappano, R.; Cirillo, F.; Talia, M.; Scordamaglia, D.; Gutkind, J.S.; Maggiolini, M. IGF-1/IGF-1R/FAK/YAP Transduction Signaling Prompts Growth Effects in Triple-Negative Breast Cancer (TNBC) Cells. Cells 2020, 9, 2619. [Google Scholar] [CrossRef]
- Cao, W.; Zheng, W.; Chen, T. Ruthenium polypyridyl complex inhibits growth and metastasis of breast cancer cells by suppressing FAK signaling with enhancement of TRAIL-induced apoptosis. Sci. Rep. 2015, 5, 9157. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Chen, Y.; Zhao, X.; Yu, S.; Yang, B.; Wu, T.; Guo, J.; Hao, C.; Zhao, D.; Cheng, M. Design, synthesis and biological evaluation of novel 7H-pyrrolo[2,3-d]pyrimidine derivatives as potential FAK inhibitors and anticancer agents. Eur. J. Med. Chem. 2019, 183, 111716. [Google Scholar] [CrossRef]
- Kandil, S.B.; Jones, S.R.; Smith, S.; Hiscox, S.E.; Westwell, A.D. Structure-Based Virtual Screening, Synthesis and Biological Evaluation of Potential FAK-FAT Domain Inhibitors for Treatment of Metastatic Cancer. Molecules 2020, 25, 3488. [Google Scholar] [CrossRef]
- Alanko, J.; Mai, A.; Jacquemet, G.; Schauer, K.; Kaukonen, R.; Saari, M.; Goud, B.; Ivaska, J. Integrin endosomal signalling suppresses anoikis. Nat. Cell Biol. 2015, 17, 1412–1421. [Google Scholar] [CrossRef] [PubMed]
- Tiede, S.; Meyer-Schaller, N.; Kalathur, R.K.R.; Ivanek, R.; Fagiani, E.; Schmassmann, P.; Stillhard, P.; Häfliger, S.; Kraut, N.; Schweifer, N.; et al. The FAK inhibitor BI 853520 exerts anti-tumor effects in breast cancer. Oncogenesis 2018, 7, 73. [Google Scholar] [CrossRef] [PubMed]
- Behmoaram, E.; Bijian, K.; Jie, S.; Xu, Y.; Darnel, A.; Bismar, T.A.; Alaoui-Jamali, M.A. Focal adhesion kinase-related proline-rich tyrosine kinase 2 and focal adhesion kinase are co-overexpressed in early-stage and invasive ErbB-2-positive breast cancer and cooperate for breast cancer cell tumorigenesis and invasiveness. Am. J. Pathol. 2008, 173, 1540–1550. [Google Scholar] [CrossRef]
- Tai, Y.L.; Chu, P.Y.; Lai, I.R.; Wang, M.Y.; Tseng, H.Y.; Guan, J.L.; Liou, J.Y.; Shen, T.L. An EGFR/Src-dependent β4 integrin/FAK complex contributes to malignancy of breast cancer. Sci. Rep. 2015, 5, 16408. [Google Scholar] [CrossRef]
- Luo, X.; Guo, L.; Zhang, L.; Hu, Y.; Shang, D.; Ji, D. Bioinformatics analysis of microarray profiling identifies the mechanism of focal adhesion kinase signalling pathway in proliferation and apoptosis of breast cancer cells modulated by green tea polyphenol epigallocatechin 3-gallate. J. Pharm. Pharmacol. 2018, 70, 1606–1618. [Google Scholar] [CrossRef]
- van Miltenburg, M.H.A.M.; van Nimwegen, M.J.; Tijdens, I.; Lalai, R.; Kuiper, R.; Klarenbeek, S.; Schouten, P.C.; de Vries, A.; Jonkers, J.; van de Water, B. Mammary gland-specific ablation of focal adhesion kinase reduces the incidence of p53-mediated mammary tumour formation. Br. J. Cancer 2014, 110, 2747–2755. [Google Scholar] [CrossRef]
- Golubovskaya, V.M.; Zheng, M.; Zhang, L.; Li, J.L.; Cance, W.G. The direct effect of focal adhesion kinase (FAK), dominant-negative FAK, FAK-CD and FAK siRNA on gene expression and human MCF-7 breast cancer cell tumorigenesis. BMC Cancer 2009, 9, 280. [Google Scholar] [CrossRef]
- Inoue, K.; Mallakin, A.; Frazier, D.P. Dmp1 and tumor suppression. Oncogene 2007, 26, 4329–4335. [Google Scholar] [CrossRef]
- Provenzano, P.P.; Inman, D.R.; Eliceiri, K.W.; Beggs, H.E.; Keely, P.J. Mammary epithelial-specific disruption of focal adhesion kinase retards tumor formation and metastasis in a transgenic mouse model of human breast cancer. Am. J. Pathol. 2008, 173, 1551–1565. [Google Scholar] [CrossRef]
- Nagy, T.; Wei, H.; Shen, T.L.; Peng, X.; Liang, C.C.; Gan, B.; Guan, J.L. Mammary epithelial-specific deletion of the focal adhesion kinase gene leads to severe lobulo-alveolar hypoplasia and secretory immaturity of the murine mammary gland. J. Biol. Chem. 2007, 282, 31766–31776. [Google Scholar] [CrossRef]
- San Juan, B.P.; Garcia-Leon, M.J.; Rangel, L.; Goetz, J.G.; Chaffer, C.L. The Complexities of Metastasis. Cancers 2019, 11, 1575. [Google Scholar] [CrossRef]
- Welch, D.R.; Hurst, D.R. Defining the Hallmarks of Metastasis. Cancer Res. 2019, 79, 3011–3027. [Google Scholar] [CrossRef]
- Vicente-Manzanares, M.; Webb, D.J.; Horwitz, A.R. Cell migration at a glance. J. Cell Sci. 2005, 118, 4917–4919. [Google Scholar] [CrossRef] [PubMed]
- Hall, J.E.; Fu, W.; Schaller, M.D. Focal adhesion kinase: Exploring Fak structure to gain insight into function. Int. Rev. Cell Mol. Biol. 2011, 288, 185–225. [Google Scholar] [CrossRef] [PubMed]
- Schaller, M.D. Cellular functions of FAK kinases: Insight into molecular mechanisms and novel functions. J. Cell Sci. 2010, 123, 1007–1013. [Google Scholar] [CrossRef]
- Luo, M.; Guan, J.L. Focal adhesion kinase: A prominent determinant in breast cancer initiation, progression and metastasis. Cancer Lett. 2010, 289, 127–139. [Google Scholar] [CrossRef]
- Huveneers, S.; Danen, E.H.J. Adhesion signaling–crosstalk between integrins, Src and Rho. J. Cell Sci. 2009, 122, 1059–1069. [Google Scholar] [CrossRef]
- Chodniewicz, D.; Klemke, R.L. Regulation of integrin-mediated cellular responses through assembly of a CAS/Crk scaffold. Biochim. Biophys. Acta. 2004, 1692, 63–76. [Google Scholar] [CrossRef]
- Chen, C.; Tao, T.; Wen, C.; He, W.Q.; Qiao, Y.N.; Gao, Y.Q.; Chen, X.; Wang, P.; Chen, C.P.; Zhao, W.; et al. Myosin Light Chain Kinase (MLCK) Regulates Cell Migration in a Myosin Regulatory Light Chain Phosphorylation-independent Mechanism. J. Biol. Chem. 2014, 289, 28478–28488. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.L.; Lu, S.; Szeto, K.W.; Sun, J.; Wang, Y.; Lasheras, J.C.; Chien, S. FAK and paxillin dynamics at focal adhesions in the protrusions of migrating cells. Sci. Rep. 2014, 4, 6024. [Google Scholar] [CrossRef]
- Carragher, N.O.; Westhoff, M.A.; Fincham, V.J.; Schaller, M.D.; Frame, M.C. A novel role for FAK as a protease-targeting adaptor protein: Regulation by p42 ERK and Src. Curr. Biol. 2003, 13, 1442–1450. [Google Scholar] [CrossRef]
- Han, D.C.; Guan, J.L. Association of focal adhesion kinase with Grb7 and its role in cell migration. J. Biol. Chem. 1999, 274, 24425–24430. [Google Scholar] [CrossRef] [PubMed]
- Tomar, A.; Schlaepfer, D.D. Focal adhesion kinase: Switching between GAPs and GEFs in the regulation of cell motility. Curr. Opin. Cell Biol. 2009, 21, 676–683. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Suetsugu, S.; Cooper, L.A.; Takenawa, T.; Guan, J.L. Focal adhesion kinase regulation of N-WASP subcellular localization and function. J. Biol. Chem. 2004, 279, 9565–9576. [Google Scholar] [CrossRef]
- Swaminathan, V.; Fischer, R.S.; Waterman, C.M. The FAK-Arp2/3 interaction promotes leading edge advance and haptosensing by coupling nascent adhesions to lamellipodia actin. Mol. Biol. Cell. 2016, 27, 1085–1100. [Google Scholar] [CrossRef]
- Gilkes, D.M.; Xiang, L.; Lee, S.J.; Chaturvedi, P.; Hubbi, M.E.; Wirtz, D.; Semenza, G.L. Hypoxia-inducible factors mediate coordinated RhoA-ROCK1 expression and signaling in breast cancer cells. Proc. Natl. Acad. Sci. USA 2014, 111, E384–E393. [Google Scholar] [CrossRef]
- Zeng, Y.; Cao, Y.; Liu, L.; Zhao, J.; Zhang, T.; Xiao, L.; Jia, M.; Tian, Q.; Yu, H.; Chen, S.; et al. SEPT9_i1 regulates human breast cancer cell motility through cytoskeletal and RhoA/FAK signaling pathway regulation. Cell Death Dis. 2019, 10, 720. [Google Scholar] [CrossRef]
- Ma, Y.; Xia, Z.; Ye, C.; Lu, C.; Zhou, S.; Pan, J.; Liu, C.; Zhang, J.; Liu, T.; Hu, T.; et al. AGTR1 promotes lymph node metastasis in breast cancer by upregulating CXCR4/SDF-1α and inducing cell migration and invasion. Aging 2019, 11, 3969–3992. [Google Scholar] [CrossRef]
- Huang, C.; Verhulst, S.; Shen, Y.; Bu, Y.; Cao, Y.; He, Y.; Wang, Y.; Huang, D.; Cai, C.; Rao, K.; et al. AKR1B10 promotes breast cancer metastasis through integrin α5/δ-catenin mediated FAK/Src/Rac1 signaling pathway. Oncotarget 2016, 7, 43779–43791. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.J.; van de Ven, R.A.H.; Zaganjor, E.; Ng, M.R.; Barakat, A.; Demmers, J.J.P.G.; Finley, L.W.S.; Gonzalez Herrera, K.N.; Hung, Y.P.; Harris, I.S.; et al. Inhibition of epithelial cell migration and Src/FAK signaling by SIRT3. Proc. Natl. Acad. Sci. USA 2018, 115, 7057–7062. [Google Scholar] [CrossRef]
- Taliaferro-Smith, L.; Oberlick, E.; Liu, T.; McGlothen, T.; Alcaide, T.; Tobin, R.; Donnelly, S.; Commander, R.; Kline, E.; Nagaraju, G.P.; et al. FAK activation is required for IGF1R-mediated regulation of EMT, migration, and invasion in mesenchymal triple negative breast cancer cells. Oncotarget 2015, 6, 4757–4772. [Google Scholar] [CrossRef]
- Long, W.; Yi, P.; Amazit, L.; LaMarca, H.L.; Ashcroft, F.; Kumar, R.; Mancini, M.A.; Tsai, S.Y.; Tsai, M.J.; O’Malley, B.W. SRC-3Delta4 mediates the interaction of EGFR with FAK to promote cell migration. Mol. Cell. 2010, 37, 321–332. [Google Scholar] [CrossRef]
- Bugide, S.; David, D.; Nair, A.; Kannan, N.; Samanthapudi, V.S.K.; Prabhakar, J.; Manavathi, B. Hematopoietic PBX-interacting protein (HPIP) is over expressed in breast infiltrative ductal carcinoma and regulates cell adhesion and migration through modulation of focal adhesion dynamics. Oncogene 2015, 34, 4601–4612. [Google Scholar] [CrossRef][Green Version]
- Lee, E.; Choi, A.; Jun, Y.; Kim, N.; Yook, J.I.; Kim, S.Y.; Lee, S.; Kang, S.W. Glutathione peroxidase-1 regulates adhesion and metastasis of triple-negative breast cancer cells via FAK signaling. Redox Biol. 2020, 29, 101391. [Google Scholar] [CrossRef] [PubMed]
- Yue, L.; Han, C.; Li, Z.; Li, X.; Liu, D.; Liu, S.; Yu, H. Fucosyltransferase 8 expression in breast cancer patients: A high throughput tissue microarray analysis. Histol. Histopathol. 2016, 31, 547–555. [Google Scholar] [CrossRef]
- Guo, D.; Guo, J.; Li, X.; Guan, F. Enhanced motility and proliferation by miR-10b/FUT8/p-AKT axis in breast cancer cells. Oncol. Lett. 2018, 16, 2097–2104. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Gao, Z.; Yue, L. Fucosyltransferase 8 deficiency suppresses breast cancer cell migration by interference of the FAK/integrin pathway. Cancer Biomark. 2019, 25, 303–311. [Google Scholar] [CrossRef]
- Shen, J.; Cao, B.; Wang, Y.; Ma, C.; Zeng, Z.; Liu, L.; Li, X.; Tao, D.; Gong, J.; Xie, D. Hippo component YAP promotes focal adhesion and tumour aggressiveness via transcriptionally activating THBS1/FAK signalling in breast cancer. J. Exp. Clin. Cancer Res. 2018, 37, 175. [Google Scholar] [CrossRef]
- Rigiracciolo, D.C.; Santolla, M.F.; Lappano, R.; Vivacqua, A.; Cirillo, F.; Galli, G.R.; Talia, M.; Muglia, L.; Pellegrino, M.; Nohata, N.; et al. Focal adhesion kinase (FAK) activation by estrogens involves GPER in triple-negative breast cancer cells. J. Exp. Clin. Cancer Res. 2019, 38, 58. [Google Scholar] [CrossRef] [PubMed]
- Nagaharu, K.; Zhang, X.; Yoshida, T.; Katoh, D.; Hanamura, N.; Kozuka, Y.; Ogawa, T.; Shiraishi, T.; Imanaka-Yoshida, K. Tenascin C induces epithelial-mesenchymal transition-like change accompanied by SRC activation and focal adhesion kinase phosphorylation in human breast cancer cells. Am. J. Pathol. 2011, 178, 754–763. [Google Scholar] [CrossRef]
- Smith, B.N.; Bhowmick, N.A. Role of EMT in Metastasis and Therapy Resistance. J. Clin. Med. 2016, 5, 17. [Google Scholar] [CrossRef]
- Wang, Y.; Mc Niven, A. Invasive matrix degradation at focal adhesions occurs via protease recruitment by a FAK–p130Cas complex. J. Cell Biol. 2012, 196, 375–385. [Google Scholar] [CrossRef]
- Mitra, S.K.; Lim, S.T.; Chi, A.; Schlaepfer, D.D. Intrinsic focal adhesion kinase activity controls orthotopic breast carcinoma metastasis via the regulation of urokinase plasminogen activator expression in a syngeneic tumor model. Oncogene 2006, 25, 4429–4440. [Google Scholar] [CrossRef]
- Lu, H.; Hu, L.; Yu, L.; Wang, X.; Urvalek, A.M.; Li, T.; Shen, C.; Mukherjee, D.; Lahiri, S.K.; Wason, M.S.; et al. KLF8 and FAK cooperatively enrich the active MMP14 on the cell surface required for the metastatic progression of breast cancer. Oncogene 2014, 33, 2909–2917. [Google Scholar] [CrossRef]
- Lappano, R.; Talia, M.; Cirillo, F.; Rigiracciolo, D.C.; Scordamaglia, D.; Guzzi, R.; Miglietta, A.M.; De Francesco, E.M.; Belfiore, A.; Sims, A.H.; et al. The IL1β-IL1R signaling is involved in the stimulatory effects triggered by hypoxia in breast cancer cells and cancer-associated fibroblasts (CAFs). J. Exp. Clin. Cancer Res. 2020, 39, 153. [Google Scholar] [CrossRef]
- Mon, N.N.; Senga, T.; Ito, S. Interleukin-1β activates focal adhesion kinase and Src to induce matrix metalloproteinase-9 production and invasion of MCF-7 breast cancer cells. Oncol. Lett. 2017, 13, 955–960. [Google Scholar] [CrossRef]
- Zhang, Y.; Gong, Y.; Hu, D.; Zhu, P.; Wang, N.; Zhang, Q.; Wang, M.; Aldeewan, A.; Xia, H.; Qu, X.; et al. Nuclear SIPA1 activates integrin β1 promoter and promotes invasion of breast cancer cells. Oncogene 2015, 34, 1451–1462. [Google Scholar] [CrossRef]
- Yang, J.; Hou, Y.; Zhou, M.; Wen, S.; Zhou, J.; Xu, L.; Tang, X.; Du, Y.E.; Hu, P.; Liu, M. Twist induces epithelial-mesenchymal transition and cell motility in breast cancer via ITGB1-FAK/ILK signaling axis and its associated downstream network. Int. J. Biochem. Cell. Biol. 2016, 71, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.; Zhao, X.; Sun, S.; Luo, M.; Guan, J.L. Function of focal adhesion kinase scaffolding to mediate endophilin A2 phosphorylation promotes epithelial-mesenchymal transition and mammary cancer stem cell activities in vivo. J. Biol. Chem. 2013, 288, 3322–3333. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Gan, B.; Yoo, Y.; Guan, J.L. FAK-mediated src phosphorylation of endophilin A2 inhibits endocytosis of MT1-MMP and promotes ECM degradation. Dev. Cell. 2005, 9, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Xiong, X.; Wang, Y.; Liu, C.; Lu, Q.; Liu, T.; Chen, G.; Rao, H.; Luo, S. Heat shock protein 90β stabilizes focal adhesion kinase and enhances cell migration and invasion in breast cancer cells. Exp. Cell Res. 2014, 326, 78–89. [Google Scholar] [CrossRef] [PubMed]
- Hoskin, V.; Szeto, A.; Ghaffari, A.; Greer, P.A.; Côté, G.P.; Elliott, B.E. Ezrin regulates focal adhesion and invadopodia dynamics by altering calpain activity to promote breast cancer cell invasion. Mol. Biol. Cell. 2015, 26, 3464–3479. [Google Scholar] [CrossRef] [PubMed]
- Van Nimwegen, M.J.; Verkoeijen, S.; van Buren, L.; Burg, D.; van de Water, B. Requirement for focal adhesion kinase in the early phase of mammary adenocarcinoma lung metastasis formation. Cancer Res. 2005, 65, 4698–4706. [Google Scholar] [CrossRef]
- Shen, M.; Jiang, Y.Z.; Wei, Y.; Ell, B.; Sheng, X.; Esposito, M.; Kang, J.; Hang, X.; Zheng, H.; Rowicki, M.; et al. Tinagl1 Suppresses Triple-Negative Breast Cancer Progression and Metastasis by Simultaneously Inhibiting Integrin/FAK and EGFR Signaling. Cancer Cell 2019, 35, 64–80. [Google Scholar] [CrossRef]
- Wang, K.; Ye, H.; Zhang, X.; Wang, X.; Yang, B.; Luo, C.; Zhao, Z.; Zhao, J.; Lu, Q.; Zhang, H.; et al. An exosome-like programmable-bioactivating paclitaxel prodrug nanoplatform for enhanced breast cancer metastasis inhibition. Biomaterials 2020, 257, 120224. [Google Scholar] [CrossRef]
- Liu, Y.; Li, L.; Liu, X.; Wang, Y.; Liu, L.; Peng, L.; Liu, J.; Zhang, L.; Wang, G.; Li, H.; et al. Arginine methylation of SHANK2 by PRMT7 promotes human breast cancer metastasis through activating endosomal FAK signalling. Elife 2020, 9, e57617. [Google Scholar] [CrossRef]
- Pratt, S.J.P.; Lee, R.M.; Martin, S.S. The Mechanical Microenvironment in Breast Cancer. Cancers 2020, 12, 1452. [Google Scholar] [CrossRef]
- Boyd, N.F.; Li, Q.; Melnichouk, O.; Huszti, E.; Martin, L.J.; Gunasekara, A.; Mawdsley, G.; Yaffe, M.J.; Minkin, S. Evidence that breast tissue stiffness is associated with risk of breast cancer. PLoS ONE 2014, 9, e100937. [Google Scholar] [CrossRef]
- Plotnikov, S.V.; Pasapera, A.M.; Sabass, B.; Waterman, C.M. Force fluctuations within focal adhesions mediate ECM-rigidity sensing to guide directed cell migration. Cell 2012, 151, 1513–1527. [Google Scholar] [CrossRef]
- Peng, Y.; Chen, Z.; Chen, Y.; Li, S.; Jiang, Y.; Yang, H.; Wu, C.; You, F.; Zheng, C.; Zhu, J.; et al. ROCK isoforms differentially modulate cancer cell motility by mechanosensing the substrate stiffness. Acta Biomater. 2019, 88, 86–101. [Google Scholar] [CrossRef]
- Allen, S.C.; Widman, J.A.; Datta, A.; Suggs, L.J. Dynamic extracellular matrix stiffening induces a phenotypic transformation and a migratory shift in epithelial cells. Integr. Biol. 2020, 12, 161–174. [Google Scholar] [CrossRef]
- Dupont, S.; Morsut, L.; Aragona, M.; Enzo, E.; Giulitti, S.; Cordenonsi, M.; Zanconato, F.; Digabel, J.L.; Forcato, M.; Bicciato, S.; et al. Role of YAP/TAZ in mechanotransduction. Nature 2011, 474, 179–183. [Google Scholar] [CrossRef]
- Lachowski, D.; Cortes, E.; Robinson, B.; Rice, A.; Rombouts, K.; Del Río Hernández, A.E. FAK controls the mechanical activation of YAP, a transcriptional regulator required for durotaxis. FASEB J. 2018, 32, 1099–1107. [Google Scholar] [CrossRef]
- Kim, N.G.; Gumbiner, B.M. Adhesion to fibronectin regulates Hippo signaling via the FAK-Src-PI3K pathway. J. Cell Biol. 2015, 210, 503–515. [Google Scholar] [CrossRef] [PubMed]
- Cox, T.R.; Erler, J.T. Remodeling and homeostasis of the extracellular matrix: Implications for fibrotic diseases and cancer. Dis. Model Mech. 2011, 4, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Wullkopf, L.; West, A.K.V.; Leijnse, N.; Cox, T.R.; Madsen, C.D.; Oddershede, L.B.; Erler, J.T. Cancer cells’ ability to mechanically adjust to extracellular matrix stiffness correlates with their invasive potential. Mol. Biol. Cell. 2018, 29, 2378–2385. [Google Scholar] [CrossRef] [PubMed]
- Gehler, S.; Compere, F.V.; Miller, A.M. Semaphorin 3A Increases FAK Phosphorylation at Focal Adhesions to Modulate MDA-MB-231 Cell Migration and Spreading on Different Substratum Concentrations. Int. J. Breast Cancer 2017, 2017, 9619734. [Google Scholar] [CrossRef]
- Provenzano, P.P.; Inman, D.R.; Eliceiri, K.W.; Keely, P.J. Matrix density-induced mechanoregulation of breast cell phenotype, signaling and gene expression through a FAK-ERK linkage. Oncogene 2009, 28, 4326–4343. [Google Scholar] [CrossRef]
- Levental, K.R.; Yu, H.; Kass, L.; Lakins, J.N.; Egeblad, M.; Erler, J.T.; Fong, S.F.T.; Csiszar, K.; Giaccia, A.; Weninger, W.; et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell 2009, 139, 891–906. [Google Scholar] [CrossRef]
- Shields, J.D.; Fleury, M.E.; Yong, C.; Tomei, A.A.; Randolph, G.J.; Swartz, M.A. Autologous chemotaxis as a mechanism of tumor cell homing to lymphatics via interstitial flow and autocrine CCR7 signaling. Cancer Cell 2007, 11, 526–538. [Google Scholar] [CrossRef]
- Polacheck, W.J.; German, A.E.; Mammoto, A.; Ingber, D.E.; Kamm, R.D. Mechanotransduction of fluid stresses governs 3D cell migration. Proc. Natl. Acad. Sci. USA 2014, 111, 2447–2452. [Google Scholar] [CrossRef]
- Steele, H.E.; Guo, Y.; Li, B.Y.; Na, S. Mechanotransduction of mitochondrial AMPK and its distinct role in flow-induced breast cancer cell migration. Biochem. Biophys. Res. Commun. 2019, 514, 524–529. [Google Scholar] [CrossRef]
- Guo, Y.; Steele, H.E.; Li, B.Y.; Na, S. Fluid flow-induced activation of subcellular AMPK and its interaction with FAK and Src. Arch Biochem. Biophys. 2020, 679, 108208. [Google Scholar] [CrossRef]
- Pan, M.P.; Hou, M.F.; Ou-Yang, F.; Wu, C.C.; Chang, S.J.; Hung, W.C.; Yip, H.K.; Luo, C.W. FAK is Required for Tumor Metastasis-Related Fluid Microenvironment in Triple-Negative Breast Cancer. J. Clin. Med. 2019, 8, 38. [Google Scholar] [CrossRef]
- Bauer, M.S.; Baumann, F.; Daday, C.; Redondo, P.; Durner, E.; Jobst, M.A.; Milles, L.F.; Mercadante, D.; Pippig, D.A.; Gaub, H.E.; et al. Structural and mechanistic insights into mechanoactivation of focal adhesion kinase. Proc. Natl. Acad. Sci. USA 2019, 116, 6766–6774. [Google Scholar] [CrossRef]
- Velasco-Velázquez, M.A.; Popov, V.M.; Lisanti, M.P.; Pestell, R.G. The Role of Breast Cancer Stem Cells in Metastasis and Therapeutic Implications. Am. J. Pathol. 2011, 179, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Powell, K.; Li, L. Breast Cancer Stem Cells: Biomarkers, Identification and Isolation Methods, Regulating Mechanisms, Cellular Origin, and Beyond. Cancers 2020, 12, 3765. [Google Scholar] [CrossRef]
- Casarsa, C.; Saro, O.; Coradini, D. The Controversial Clinicobiological Role of Breast Cancer Stem Cells. J. Oncol. 2008, 2008, 492643. [Google Scholar] [CrossRef]
- Iqbal, J.; Chong, P.Y.; Tan, P.H. Breast cancer stem cells: An update. J. Clin. Pathol. 2013, 66, 485–490. [Google Scholar] [CrossRef] [PubMed]
- Stingl, J.; Caldas, C. Molecular heterogeneity of breast carcinomas and the cancer stem cell hypothesis. Nat. Rev. Cancer. 2007, 7, 791–799. [Google Scholar] [CrossRef]
- Osterman, C.J.D.; Ozmadenci, D.; Kleinschmidt, E.G.; Taylor, K.N.; Barrie, A.M.; Jiang, S.; Bean, L.M.; Sulzmaier, F.J.; Jean, C.; Tancioni, I.; et al. FAK activity sustains intrinsic and acquired ovarian cancer resistance to platinum chemotherapy. Elife 2019, 8, e47327. [Google Scholar] [CrossRef]
- Luo, M.; Zhao, X.; Chen, S.; Liu, S.; Wicha, M.S.; Guan, J.L. Distinct FAK activities determine progenitor and mammary stem cell characteristics. Cancer Res. 2013, 73, 5591–5602. [Google Scholar] [CrossRef]
- Luo, M.; Fan, H.; Nagy, T.; Wei, H.; Wang, C.; Liu, S.; Wicha, M.S.; Guan, J.L. Mammary epithelial-specific ablation of the focal adhesion kinase suppresses mammary tumorigenesis by affecting mammary cancer stem/progenitor cells. Cancer Res. 2009, 69, 466–474. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, K.; Yan, Y.; Yuan, B.; Dasgupta, A.; Sun, J.; Mu, H.; Do, K.A.; Ueno, N.T.; Andreeff, M.; Lokesh Battula, V. ST8SIA1 Regulates Tumor Growth and Metastasis in TNBC by Activating the FAK-AKT-mTOR Signaling Pathway. Mol. Cancer Ther. 2018, 17, 2689–2701. [Google Scholar] [CrossRef] [PubMed]
- Barnawi, R.; Al-Khaldi, S.; Bakheet, T.; Fallatah, M.; Alaiya, A.; Ghebeh, H.; Al-Alwan, M. Fascin Activates β-Catenin Signaling and Promotes Breast Cancer Stem Cell Function Mainly Through Focal Adhesion Kinase (FAK): Relation With Disease Progression. Front. Oncol. 2020, 10, 440. [Google Scholar] [CrossRef]
- Thiagarajan, P.S.; Sinyuk, M.; Turaga, S.M.; Mulkearns-Hubert, E.E.; Hale, J.S.; Rao, V.; Demelash, A.; Saygin, C.; China, A.; Alban, T.J.; et al. Cx26 drives self-renewal in triple-negative breast cancer via interaction with NANOG and focal adhesion kinase. Nat. Commun. 2018, 9, 578. [Google Scholar] [CrossRef] [PubMed]
- Katakam, S.K.; Tria, V.; Sim, W.C.; Yip, G.W.; Molgora, S.; Karnavas, T.; Elghonaimy, E.A.; Pelucchi, P.; Piscitelli, E.; Ibrahim, S.A.; et al. The heparan sulfate proteoglycan syndecan-1 regulates colon cancer stem cell function via a focal adhesion kinase-Wnt signaling axis. FEBS J. 2020. [Google Scholar] [CrossRef]
- Kolev, V.N.; Tam, W.F.; Wright, Q.G.; McDermott, S.P.; Vidal, C.M.; Shapiro, I.M.; Xu, Q.; Wicha, M.S.; Pachter, J.A.; Weaver, D.T. Inhibition of FAK kinase activity preferentially targets cancer stem cells. Oncotarget 2017, 8, 51733–51747. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.E.; Bundred, N.J.; Landberg, G.; Clarke, R.B.; Farnie, G. Focal adhesion kinase and Wnt signaling regulate human ductal carcinoma in situ stem cell activity and response to radiotherapy. Stem Cells 2015, 33, 327–341. [Google Scholar] [CrossRef]
- Tancioni, I.; Miller, N.L.G.; Uryu, S.; Lawson, C.; Jean, C.; Chen, X.L.; Kleinschmidt, E.G.; Schlaepfer, D.D. FAK activity protects nucleostemin in facilitating breast cancer spheroid and tumor growth. Breast Cancer Res. 2015, 17, 47. [Google Scholar] [CrossRef]
- Baghban, R.; Roshangar, L.; Jahanban-Esfahlan, R.; Seidi, K.; Ebrahimi-Kalan, A.; Jaymand, M.; Kolahian, S.; Javaheri, T.; Zare, P. Tumor microenvironment complexity and therapeutic implications at a glance. Cell Commun Signal. 2020, 18, 59. [Google Scholar] [CrossRef] [PubMed]
- Saman, H.; Raza, S.S.; Uddin, S.; Rasul, K. Inducing Angiogenesis, a Key Step in Cancer Vascularization, and Treatment Approaches. Cancers 2020, 12, 1172. [Google Scholar] [CrossRef]
- Pedrosa, A.R.; Bodrug, N.; Gomez-Escudero, J.; Carter, E.P.; Reynolds, L.E.; Georgiou, P.N.; Fernandez, I.; Lees, D.M.; Kostourou, V.; Alexopoulou, A.N.; et al. Tumor Angiogenesis Is Differentially Regulated by Phosphorylation of Endothelial Cell Focal Adhesion Kinase Tyrosines-397 and -861. Cancer Res. 2019, 79, 4371–4386. [Google Scholar] [CrossRef]
- Sun, S.; Wu, H.J.; Guan, J.L. Nuclear FAK and its kinase activity regulate VEGFR2 transcription in angiogenesis of adult mice. Sci. Rep. 2018, 8, 2550. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.K.; Naidansuren, P.; Lee, S.W.; Kim, R.W.; Lee, S.J.; Lee, S.K.; Hong, Y.K.; Joe, Y.A. Urokinase-derived peptide UP-7 suppresses tumor angiogenesis and metastasis through inhibition of FAK activation. Oncotarget 2018, 9, 9951–9962. [Google Scholar] [CrossRef] [PubMed]
- Tavora, B.; Batista, S.; Reynolds, L.E.; Jadeja, S.; Robinson, S.; Kostourou, V.; Hart, I.; Fruttiger, M.; Parsons, M.; Hodivala-Dilke, K.M. Endothelial FAK is required for tumour angiogenesis. EMBO Mol. Med. 2010, 2, 516–528. [Google Scholar] [CrossRef] [PubMed]
- Tavora, B.; Reynolds, L.E.; Batista, S.; Demircioglu, F.; Fernandez, I.; Lechertier, T.; Lees, D.M.; Wong, P.P.; Alexopoulou, A.; Elia, G.; et al. Endothelial-cell FAK targeting sensitizes tumours to DNA-damaging therapy. Nature 2014, 514, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Lechertier, T.; Reynolds, L.E.; Kim, H.; Pedrosa, A.R.; Gómez-Escudero, J.; Muñoz-Félix, J.M.; Batista, S.; Dukinfield, M.; Demircioglu, F.; Wong, P.P.; et al. Pericyte FAK negatively regulates Gas6/Axl signalling to suppress tumour angiogenesis and tumour growth. Nat. Commun. 2020, 11, 2810. [Google Scholar] [CrossRef]
- Alexopoulou, A.N.; Ho-Yen, C.M.; Papalazarou, V.; Elia, G.; Jones, J.L.; Hodivala-Dilke, K. Tumour-associated endothelial-FAK correlated with molecular sub-type and prognostic factors in invasive breast cancer. BMC Cancer 2014, 14, 237. [Google Scholar] [CrossRef]
- Jean, C.; Chen, X.L.; Nam, J.O.; Tancioni, I.; Uryu, S.; Lawson, C.; Ward, K.K.; Walsh, C.T.; Miller, N.L.G.; Ghassemian, M.; et al. Inhibition of endothelial FAK activity prevents tumor metastasis by enhancing barrier function. J. Cell Biol. 2014, 204, 247–263. [Google Scholar] [CrossRef]
- Mitra, S.K.; Mikolon, D.; Molina, J.E.; Hsia, D.A.; Hanson, D.A.; Chi, A.; Lim, S.T.; Bernard-Trifilo, J.A.; Ilic, D.; Stupack, D.G.; et al. Intrinsic FAK activity and Y925 phosphorylation facilitate an angiogenic switch in tumors. Oncogene 2006, 25, 5969–5984. [Google Scholar] [CrossRef] [PubMed]
- Sp, N.; Kang, D.Y.; Joung, Y.H.; Park, J.H.; Kim, W.S.; Lee, H.K.; Song, K.D.; Park, Y.M.; Yang, Y.M. Nobiletin Inhibits Angiogenesis by Regulating Src/FAK/STAT3-Mediated Signaling through PXN in ER⁺ Breast Cancer Cells. Int. J. Mol. Sci. 2017, 18, 935. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Khan, S.; Shukla, S.; Lakra, A.D.; Kumar, S.; Das, G.; Maurya, R.; Meeran, S.M. Cucurbitacin B inhibits breast cancer metastasis and angiogenesis through VEGF-mediated suppression of FAK/MMP-9 signaling axis. Int. J. Biochem. Cell Biol. 2016, 77, 41–56. [Google Scholar] [CrossRef]
- Louault, K.; Li, R.R.; DeClerck, Y.A. Cancer-Associated Fibroblasts: Understanding Their Heterogeneity. Cancers 2020, 12, 3108. [Google Scholar] [CrossRef]
- Wu, H.J.; Hao, M.; Yeo, S.K.; Guan, J.L. FAK signaling in cancer-associated fibroblasts promotes breast cancer cell migration and metastasis by exosomal miRNAs-mediated intercellular communication. Oncogene 2020, 39, 2539–2549. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Liu, X.; Knolhoff, B.L.; Hegde, S.; Lee, K.B.; Jiang, H.; Fields, R.C.; Pachter, J.A.; Lim, K.H.; DeNardo, D.G. Development of resistance to FAK inhibition in pancreatic cancer is linked to stromal depletion. Gut 2020, 69, 122–132. [Google Scholar] [CrossRef]
- Min, A.; Zhu, C.; Wang, J.; Peng, S.; Shuai, C.; Gao, S.; Tang, Z.; Su, T. Focal adhesion kinase knockdown in carcinoma-associated fibroblasts inhibits oral squamous cell carcinoma metastasis via downregulating MCP-1/CCL2 expression. J. Biochem. Mol. Toxicol. 2015, 29, 70–76. [Google Scholar] [CrossRef]
- Wang, X.; Zhou, Q.; Yu, Z.; Wu, X.; Chen, X.; Li, J.; Li, C.; Yan, M.; Zhu, Z.; Liu, B.; et al. Cancer-associated fibroblast-derived Lumican promotes gastric cancer progression via the integrin β1-FAK signaling pathway. Int. J. Cancer. 2017, 141, 998–1010. [Google Scholar] [CrossRef]
- Barker, H.E.; Bird, D.; Lang, G.; Erler, J.T. Tumor-secreted LOXL2 activates fibroblasts through FAK signaling. Mol. Cancer Res. 2013, 11, 1425–1436. [Google Scholar] [CrossRef]
- Cao, Y.; Cao, W.; Qiu, Y.; Zhou, Y.; Guo, Q.; Gao, Y.; Lu, N. Oroxylin A suppresses ACTN1 expression to inactivate cancer-associated fibroblasts and restrain breast cancer metastasis. Pharmacol. Res. 2020, 159, 104981. [Google Scholar] [CrossRef] [PubMed]
- Goreczny, G.J.; Ouderkirk-Pecone, J.L.; Olson, E.C.; Krendel, M.; Turner, C.E. Hic-5 remodeling of the stromal matrix promotes breast tumor progression. Oncogene 2017, 36, 2693–2703. [Google Scholar] [CrossRef]
- Yu, T.; Yang, G.; Hou, Y.; Tang, X.; Wu, C.; Wu, X.A.; Guo, L.; Zhu, Q.; Luo, H.; Du, Y.E.; et al. Cytoplasmic GPER translocation in cancer-associated fibroblasts mediates cAMP/PKA/CREB/glycolytic axis to confer tumor cells with multidrug resistance. Oncogene 2017, 36, 2131–2145. [Google Scholar] [CrossRef]
- D’Esposito, V.; Ambrosio, M.R.; Giuliano, M.; Cabaro, S.; Miele, C.; Beguinot, F.; Formisano, P. Mammary Adipose Tissue Control of Breast Cancer Progression: Impact of Obesity and Diabetes. Front. Oncol. 2020, 10, 1554. [Google Scholar] [CrossRef] [PubMed]
- Chu, D.T.; Phuong, T.N.T.; Tien, N.L.B.; Tran, D.K.; Nguyen, T.T.; Thanh, V.V.; Quang, T.L.; Minh, L.B.; Pham, V.H.; Ngoc, V.T.N.; et al. The Effects of Adipocytes on the Regulation of Breast Cancer in the Tumor Microenvironment: An Update. Cells 2019, 8, 857. [Google Scholar] [CrossRef]
- Gyamfi, J.; Lee, Y.H.; Min, B.S.; Choi, J. Niclosamide reverses adipocyte induced epithelial-mesenchymal transition in breast cancer cells via suppression of the interleukin-6/STAT3 signalling axis. Sci. Rep. 2019, 9, 11336. [Google Scholar] [CrossRef]
- Blücher, C.; Iberl, S.; Schwagarus, N.; Müller, S.; Liebisch, G.; Höring, M.; Hidrobo, M.S.; Ecker, J.; Spindler, N.; Dietrich, A.; et al. Secreted Factors from Adipose Tissue Reprogram Tumor Lipid Metabolism and Induce Motility by Modulating PPARα/ANGPTL4 and FAK. Mol. Cancer Res. 2020, 18, 1849–1862. [Google Scholar] [CrossRef]
- Wu, Q.; Li, B.; Li, Z.; Li, J.; Sun, S.; Sun, S. Cancer-associated adipocytes: Key players in breast cancer progression. J. Hematol. Oncol. 2019, 12, 95. [Google Scholar] [CrossRef]
- Olea-Flores, M.; Zuñiga-Eulogio, M.; Tacuba-Saavedra, A.; Bueno-Salgado, M.; Sánchez-Carvajal, A.; Vargas-Santiago, Y.; Mendoza-Catalán, M.A.; Salazar, E.P.; García-Hernández, A.; Padilla-Benavides, T.; et al. Leptin Promotes Expression of EMT-Related Transcription Factors and Invasion in a Src and FAK-Dependent Pathway in MCF10A Mammary Epithelial Cells. Cells 2019, 8, 1133. [Google Scholar] [CrossRef] [PubMed]
- Juárez-Cruz, J.C.; Zuñiga-Eulogio, M.D.; Olea-Flores, M.; Castañeda-Saucedo, E.; Mendoza-Catalán, M.A.; Ortuño-Pineda, C.; Moreno-Godínez, M.E.; Villegas-Comonfort, S.; Padilla-Benavides, T.; Navarro-Tito, N. Leptin induces cell migration and invasion in a FAK-Src-dependent manner in breast cancer cells. Endocr. Connect. 2019, 8, 1539–1552. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.F.; Chen, J.H.; Wu, C.T.; Chang, P.C.; Wang, S.L.; Yeh, W.L. Induction of osteoclast-like cell formation by leptin-induced soluble intercellular adhesion molecule secreted from cancer cells. Ther. Adv. Med. Oncol. 2019, 11, 1758835919846806. [Google Scholar] [CrossRef]
- Engblom, C.; Pfirschke, C.; Pittet, M.J. The role of myeloid cells in cancer therapies. Nat. Rev. Cancer. 2016, 16, 447–462. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, D.; Cang, H.; Guo, B. Crosstalk between cancer and immune cells: Role of tumor-associated macrophages in the tumor microenvironment. Cancer Med. 2019, 8, 4709–4721. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Liu, H.; Zhang, X.; Zhang, W.; Liu, L.; Yu, Y.; Ren, S.; Yang, Q.; Liu, B.; Li, J.; et al. Tumor Cells Interleukin-22 Expression Associates with Elevated Tumor-Associated Macrophages Infiltrating and Poor Prognosis in Patients with Breast Cancer. Cancer Biother. Radiopharm. 2020. [Google Scholar] [CrossRef] [PubMed]
- Osipov, A.; Saung, M.T.; Zheng, L.; Murphy, A.G. Small molecule immunomodulation: The tumor microenvironment and overcoming immune escape. J. Immunother. Cancer 2019, 7, 224. [Google Scholar] [CrossRef]
- Walsh, C.; Tanjoni, I.; Uryu, S.; Tomar, A.; Nam, J.O.; Luo, H.; Phillips, A.; Patel, N.; Kwok, C.; McMahon, G.; et al. Oral delivery of PND-1186 FAK inhibitor decreases tumor growth and spontaneous breast to lung metastasis in pre-clinical models. Cancer Biol. Ther. 2010, 9, 778–790. [Google Scholar] [CrossRef]
- Wendt, M.K.; Schiemann, W.P. Therapeutic targeting of the focal adhesion complex prevents oncogenic TGF-beta signaling and metastasis. Breast Cancer Res. 2009, 11, R68. [Google Scholar] [CrossRef]
- Llewellyn, R.A.; Gutknecht, M.F.; Thomas, K.S.; Conaway, M.R.; Bouton, A.H. Focal adhesion kinase (FAK) deficiency in mononuclear phagocytes alters murine breast tumor progression. Am. J. Cancer Res. 2018, 8, 675–687. [Google Scholar] [PubMed]
- Canel, M.; Taggart, D.; Sims, A.H.; Lonergan, D.W.; Waizenegger, I.C.; Serrels, A. T-cell co-stimulation in combination with targeting FAK drives enhanced anti-tumor immunity. Elife 2020, 9, e48092. [Google Scholar] [CrossRef]
- Mohan, N.; Hosain, S.; Zhao, J.; Shen, Y.; Luo, X.; Jiang, J.; Endo, Y.; Wu, W.J. Atezolizumab potentiates Tcell-mediated cytotoxicity and coordinates with FAK to suppress cell invasion and motility in PD-L1 + triple negative breast cancer cells. Oncoimmunology 2019, 8, e1624128. [Google Scholar] [CrossRef]
- Corsi, J.M.; Rouer, E.; Girault, J.A.; Enslen, H. Organization and post-transcriptional processing of focal adhesion kinase gene. BMC Genom. 2006, 7, 198. [Google Scholar] [CrossRef] [PubMed]
- Yom, C.K.; Noh, D.Y.; Kim, W.H.; Kim, H.S. Clinical significance of high focal adhesion kinase gene copy number and overexpression in invasive breast cancer. Breast Cancer Res. Treat. 2011, 128, 647–655. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef]
- Kim, H.K.; Hwang, H.L.; Park, S.Y.; Lee, K.M.; Park, W.C.; Kim, H.S.; Um, T.H.; Hong, Y.J.; Lee, J.K.; Joo, S.Y.; et al. Simple and versatile molecular method of copy-number measurement using cloned competitors. PLoS ONE 2013, 8, e69414. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.H.; Kim, H.K.; Kim, H.Y.; Gawk, H.R.; Bae, S.H.; Sim, H.W.; Kang, E.K.; Seoh, J.Y.; Jang, H.; Hong, K.M. FAK-Copy-Gain Is a Predictive Marker for Sensitivity to FAK Inhibition in Breast Cancer. Cancers 2019, 11, 1288. [Google Scholar] [CrossRef]
- Yao, L.; Li, K.; Peng, W.; Lin, Q.; Li, S.; Hu, X.; Zheng, X.; Shao, Z. An aberrant spliced transcript of focal adhesion kinase is exclusively expressed in human breast cancer. J. Transl. Med. 2014, 12, 136. [Google Scholar] [CrossRef]
- Fang, X.Q.; Liu, X.F.; Yao, L.; Chen, C.Q.; Gu, Z.D.; Ni, P.H.; Zheng, X.M.; Fan, Q.S. Somatic mutational analysis of FAK in breast cancer: A novel gain-of-function mutation due to deletion of exon 33. Biochem. Biophys. Res. Commun. 2014, 443, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Sethuraman, A.; Brown, M.; Seagroves, T.N.; Wu, Z.H.; Pfeffer, L.M.; Fan, M. SMARCE1 regulates metastatic potential of breast cancer cells through the HIF1A/PTK2 pathway. Breast Cancer Res. 2016, 18, 81. [Google Scholar] [CrossRef] [PubMed]
- Andisha, N.M.; McMillan, D.C.; Gujam, F.J.A.; Roseweir, A.; Edwards, J. The relationship between phosphorylation status of focal adhesion kinases, molecular subtypes, tumour microenvironment and survival in patients with primary operable ductal breast cancer. Cell Signal. 2019, 60, 91–99. [Google Scholar] [CrossRef]
- Almstedt, K.; Sicking, I.; Battista, M.J.; Huangfu, S.; Heimes, A.S.; Weyer-Elberich, V.; Hasenburg, A.; Schmidt, M. Prognostic Significance of Focal Adhesion Kinase in Node-Negative Breast Cancer. Breast Care 2017, 12, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, K.J.; Grabellus, F.; Callies, R.; Otterbach, F.; Wohlschlaeger, J.; Levkau, B.; Kimmig, R.; Schmid, K.W.; Baba, H.B. High expression of focal adhesion kinase (p125FAK) in node-negative breast cancer is related to overexpression of HER-2/neu and activated Akt kinase but does not predict outcome. Breast Cancer Res. 2005, 7, R194–R203. [Google Scholar] [CrossRef]
- Lv, P.C.; Jiang, A.Q.; Zhang, W.M.; Zhu, H.L. FAK inhibitors in Cancer, a patent review. Expert Opin. Ther. Pat. 2018, 28, 139–145. [Google Scholar] [CrossRef]
- Shimizu, T.; Fukuoka, K.; Takeda, M.; Iwasa, T.; Yoshida, T.; Horobin, J.; Keegan, M.; Vaickus, L.; Chavan, A.; Padval, M.; et al. A first-in-Asian phase 1 study to evaluate safety, pharmacokinetics and clinical activity of VS-6063, a focal adhesion kinase (FAK) inhibitor in Japanese patients with advanced solid tumors. Cancer Chemother Pharmacol. 2016, 77, 997–1003. [Google Scholar] [CrossRef] [PubMed]
- Infante, J.R.; Camidge, D.R.; Mileshkin, L.R.; Chen, E.X.; Hicks, R.J.; Rischin, D.; Fingert, H.; Pierce, K.J.; Xu, H.; Roberts, W.G.; et al. Safety, pharmacokinetic, and pharmacodynamic phase I dose-escalation trial of PF-00562271, an inhibitor of focal adhesion kinase, in advanced solid tumors. J. Clin. Oncol. 2012, 30, 1527–1533. [Google Scholar] [CrossRef]
- Liu, T.J.; LaFortune, T.; Honda, T.; Ohmori, O.; Hatakeyama, S.; Meyer, T.; Jackson, D.; de Groot, J.; Yung, W.K.A. Inhibition of both focal adhesion kinase and insulin-like growth factor-I receptor kinase suppresses glioma proliferation in vitro and in vivo. Mol. Cancer Ther. 2007, 6, 1357–1367. [Google Scholar] [CrossRef]
- Fukami, S.; Tomioka, D.; Murakami, Y.; Honda, T.; Hatakeyama, S. Pharmacological profiling of a dual FAK/IGF-1R kinase inhibitor TAE226 in cellular and in vivo tumor models. BMC Res. Notes 2019, 12, 347. [Google Scholar] [CrossRef] [PubMed]
- Kurio, N.; Shimo, T.; Fukazawa, T.; Takaoka, M.; Okui, T.; Hassan, N.M.M.; Honami, T.; Hatakeyama, S.; Ikeda, M.; Naomoto, Y.; et al. Anti-tumor effect in human breast cancer by TAE226, a dual inhibitor for FAK and IGF-IR in vitro and in vivo. Exp. Cell Res. 2011, 317, 1134–1146. [Google Scholar] [CrossRef] [PubMed]
- Slack-Davis, J.K.; Martin, K.H.; Tilghman, R.W.; Iwanicki, M.; Ung, E.J.; Autry, C.; Luzzio, M.J.; Cooper, B.; Kath, J.C.; Roberts, W.G.; et al. Cellular characterization of a novel focal adhesion kinase inhibitor. J. Biol. Chem. 2007, 282, 14845–14852. [Google Scholar] [CrossRef]
- Hiscox, S.; Barnfather, P.; Hayes, E.; Bramble, P.; Christensen, J.; Nicholson, R.I.; Barrett-Lee, P. Inhibition of focal adhesion kinase suppresses the adverse phenotype of endocrine-resistant breast cancer cells and improves endocrine response in endocrine-sensitive cells. Breast Cancer Res. Treat. 2011, 125, 659–669. [Google Scholar] [CrossRef] [PubMed]
- Stokes, J.B.; Adair, S.J.; Slack-Davis, J.K.; Walters, D.M.; Tilghman, R.W.; Hershey, E.D.; Lowrey, B.; Thomas, K.S.; Bouton, A.H.; Hwang, R.F.; et al. Inhibition of focal adhesion kinase by PF-562,271 inhibits the growth and metastasis of pancreatic cancer concomitant with altering the tumor microenvironment. Mol. Cancer Ther. 2011, 10, 2135–2145. [Google Scholar] [CrossRef] [PubMed]
- Wiemer, A.J.; Wernimont, S.A.; Cung, T.D.; Bennin, D.A.; Beggs, H.E.; Huttenlocher, A. The focal adhesion kinase inhibitor PF-562,271 impairs primary CD4+ T cell activation. Biochem. Pharmacol. 2013, 86, 770–781. [Google Scholar] [CrossRef]
- Jones, S.F.; Siu, L.L.; Bendell, J.C.; Cleary, J.M.; Razak, A.R.A.; Infante, J.R.; Pandya, S.S.; Bedard, P.L.; Pierce, K.J.; Houk, B.; et al. A phase I study of VS-6063, a second-generation focal adhesion kinase inhibitor, in patients with advanced solid tumors. Investig. New Drugs 2015, 33, 1100–1107. [Google Scholar] [CrossRef] [PubMed]
- Schultze, A.; Fiedler, W. Clinical importance and potential use of small molecule inhibitors of focal adhesion kinase. Anticancer Agents Med. Chem. 2011, 11, 593–599. [Google Scholar] [CrossRef] [PubMed]
- Soria, J.C.; Gan, H.K.; Blagden, S.P.; Plummer, R.; Arkenau, H.T.; Ranson, M.; Evans, T.R.J.; Zalcman, G.; Bahleda, R.; Hollebecque, A.; et al. A phase I, pharmacokinetic and pharmacodynamic study of GSK2256098, a focal adhesion kinase inhibitor, in patients with advanced solid tumors. Ann. Oncol. 2016, 27, 2268–2274. [Google Scholar] [CrossRef] [PubMed]
- Mak, G.; Soria, J.C.; Blagden, S.P.; Plummer, R.; Fleming, R.A.; Nebot, N.; Zhang, J.; Mazumdar, J.; Rogan, D.; Gazzah, A.; et al. A phase Ib dose-finding, pharmacokinetic study of the focal adhesion kinase inhibitor GSK2256098 and trametinib in patients with advanced solid tumours. Br. J. Cancer 2019, 120, 975–981. [Google Scholar] [CrossRef]
- Laszlo, V.; Valko, Z.; Ozsvar, J.; Kovacs, I.; Garay, T.; Hoda, M.A.; Klikovits, T.; Stockhammer, P.; Aigner, C.; Gröger, M.; et al. The FAK inhibitor BI 853520 inhibits spheroid formation and orthotopic tumor growth in malignant pleural mesotelioma. J. Mol. Med. 2019, 97, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Wörthmüller, J.; Curzio Rüegg, C. The Crosstalk between FAK and Wnt Signaling Pathways in Cancer and Its Therapeutic Implication. Int. J. Mol. Sci. 2020, 21, 9107. [Google Scholar] [CrossRef] [PubMed]
- Lazaro, G.; Smith, C.; Goddard, L.; Jordan, N.; McClelland, R.; Barrett-Lee, P.; Nicholson, R.I.; Hiscox, S. Targeting focal adhesion kinase in ER+/HER2+ breast cancer improves trastuzumab response. Endocr. Relat. Cancer 2013, 20, 691–704. [Google Scholar] [CrossRef] [PubMed]
- Thakur, R.; Trivedi, R.; Rastogi, N.; Singh, M.; Mishra, D.P. Inhibition of STAT3, FAK and Src mediated signaling reduces cancer stem cell load, tumorigenic potential and metastasis in breast cancer. Sci. Rep. 2015, 5, 10194. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Wu, D.; Yuan, S. Tyrosine Kinase Inhibitors in the Combination Therapy of HER2 Positive Breast Cancer. Technol. Cancer Res. Treat. 2020, 19, 1533033820962140. [Google Scholar] [CrossRef]
- Fremd, C.; Jaeger, D.; Schneeweiss, A. Targeted and immuno-biology driven treatment strategies for triple-negative breast cancer: Current knowledge and future perspectives. Expert Rev. Anticancer Ther. 2019, 19, 29–42. [Google Scholar] [CrossRef]
- Kang, C.; Syed, Y.Y. Atezolizumab (in Combination with Nab-Paclitaxel): A Review in Advanced Triple-Negative Breast Cancer. Drugs 2020, 80, 601–607. [Google Scholar] [CrossRef] [PubMed]
- Papadimitriou, M.; Mountzios, G.; Papadimitriou, C.A. The role of PARP inhibition in triple-negative breast cancer: Unraveling the wide spectrum of synthetic lethality. Cancer Treat. Rev. 2018, 67, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Sukumar, J.; Gast, K.; Quiroga, D.; Lustberg, M.; Williams, N. Triple-negative breast cancer: Promising prognostic biomarkers currently in development. Expert Rev. Anticancer Ther. 2020. [Google Scholar] [CrossRef] [PubMed]
- Mohanty, A.; Pharaon, R.R.; Nam, A.; Salgia, S.; Kulkarni, P.; Massarelli, E. FAK-targeted and combination therapies for the treatment of cancer: An overview of phase I and II clinical trials. Expert Opin. Investig. Drugs 2020, 29, 399–409. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| FAK Inhibitors | Type | Target (s) | In Vitro Studies | In Vivo Studies |
|---|---|---|---|---|
| NVPTAE-226 | ATP-competitive inhibitor | FAK IGF-1R | + | + |
| GSK-2256098 | ATP-competitive inhibitor | FAK | − | − |
| PF-573228 | ATP-competitive inhibitor | FAK | + | + |
| PF-03814735 | ATP-competitive inhibitor | FAK Aurora1/2 | + | + |
| PF-431396 | ATP-competitive inhibitor | FAK Pyk2 | − | − |
| VS-6062 | ATP-competitive inhibitor | FAK Pyk2 | + | + |
| VS-6063 | ATP-competitive inhibitor | FAK Pyk2 | + | + |
| VS-4718 | ATP-competitive inhibitor | FAK | + | + |
| BI-853520 | ATP-competitive scaffold FAK inhibitor | FAK | + | + |
| C4 | FAK scaffold inhibitor | FAK-VEGFR3 interaction | + | + |
| R2 | FAK scaffold inhibitor | FAK-p53 interaction | + | − |
| Y11 | FAK scaffold inhibitor | FAK | + | − |
| Y15 | FAK scaffold inhibitor | FAK | + | − |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rigiracciolo, D.C.; Cirillo, F.; Talia, M.; Muglia, L.; Gutkind, J.S.; Maggiolini, M.; Lappano, R. Focal Adhesion Kinase Fine Tunes Multifaced Signals toward Breast Cancer Progression. Cancers 2021, 13, 645. https://doi.org/10.3390/cancers13040645
Rigiracciolo DC, Cirillo F, Talia M, Muglia L, Gutkind JS, Maggiolini M, Lappano R. Focal Adhesion Kinase Fine Tunes Multifaced Signals toward Breast Cancer Progression. Cancers. 2021; 13(4):645. https://doi.org/10.3390/cancers13040645
Chicago/Turabian StyleRigiracciolo, Damiano Cosimo, Francesca Cirillo, Marianna Talia, Lucia Muglia, Jorge Silvio Gutkind, Marcello Maggiolini, and Rosamaria Lappano. 2021. "Focal Adhesion Kinase Fine Tunes Multifaced Signals toward Breast Cancer Progression" Cancers 13, no. 4: 645. https://doi.org/10.3390/cancers13040645
APA StyleRigiracciolo, D. C., Cirillo, F., Talia, M., Muglia, L., Gutkind, J. S., Maggiolini, M., & Lappano, R. (2021). Focal Adhesion Kinase Fine Tunes Multifaced Signals toward Breast Cancer Progression. Cancers, 13(4), 645. https://doi.org/10.3390/cancers13040645