
Immunotherapy Strategies for Gastrointestinal Stromal Tumor

 ,
, 
Abstract
:Simple Summary
Abstract
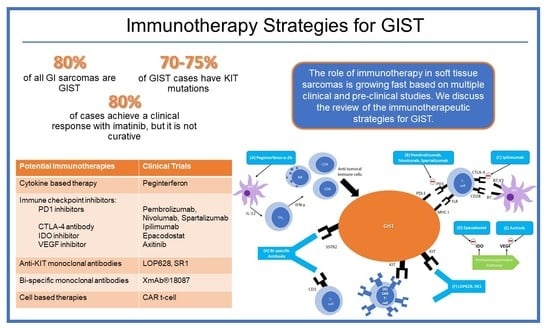
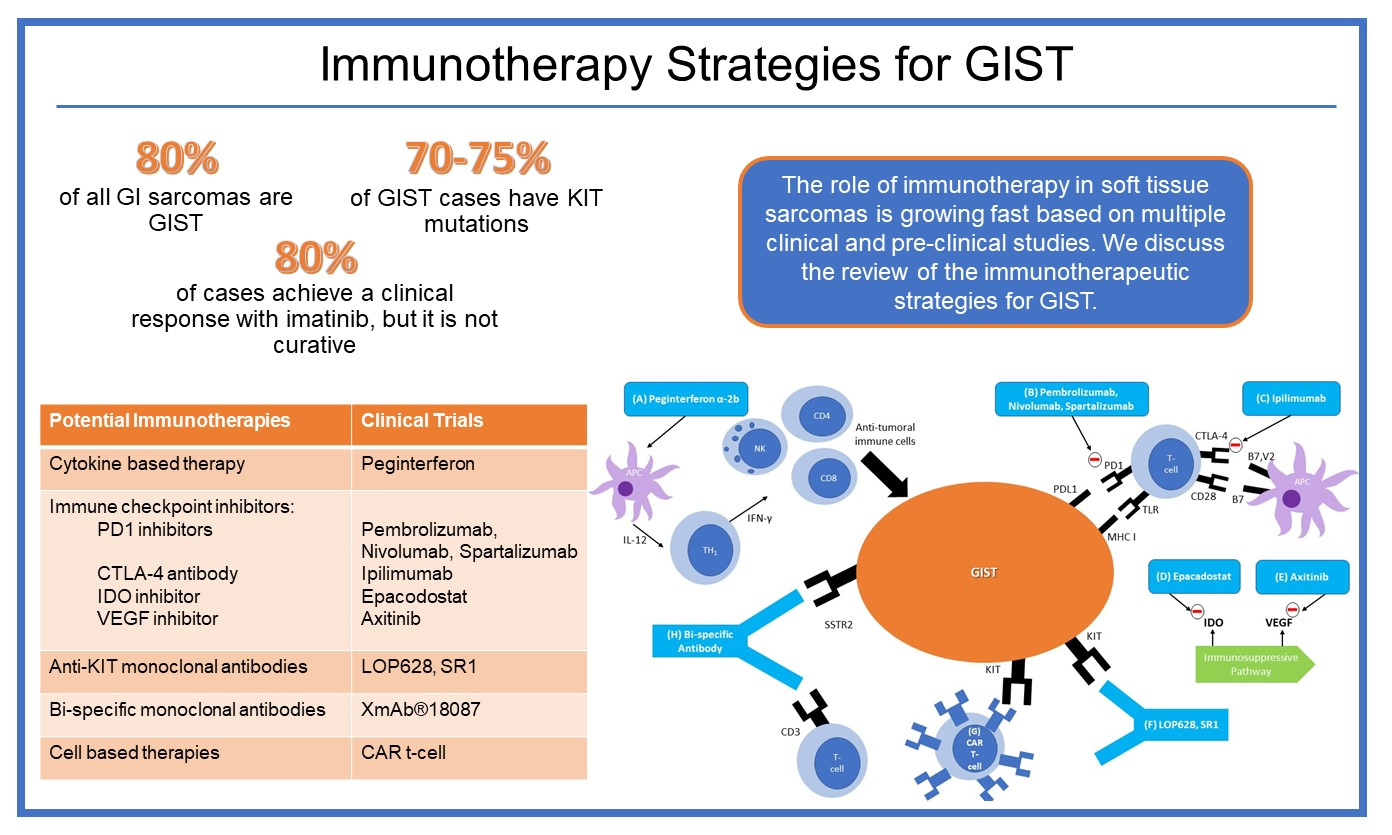
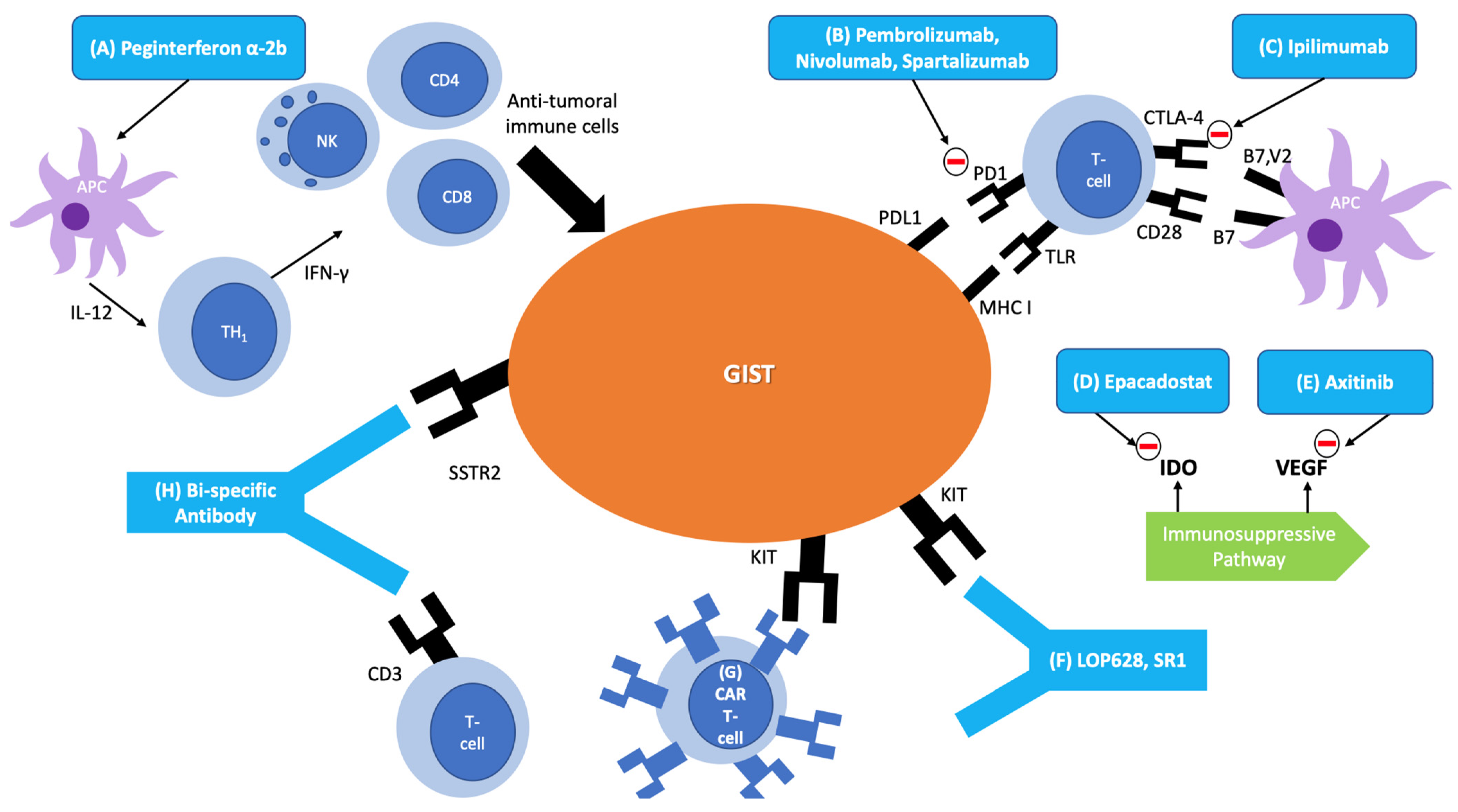
1. Introduction
2. Disease Biology
2.1. Principles of Tumor Immunology
2.2. GIST Cellular Immunology
2.3. Imatinib and the Immune System
3. Immunotherapies in GIST
3.1. Cytokine-Based Therapy
3.2. Immune Checkpoint Inhibitors
3.3. Anti-KIT Antibodies
3.4. Bi-Specific Monoclonal Antibodies
3.5. Cellular Therapy
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Burch, J.; Ahmad, I. Cancer, Gastrointestinal Stromal (GIST); StatPearls: Treasure Island, FL, USA, 2020. [Google Scholar]
- Costa, P.A.; Hana, C.K.A.; Balaji, N.C.; Skryd, A.F.; Valdes, B.N.; Minjares, R.O.; Barreto-Coelho, P.; Espejo-Freire, A.P.; Hakim, M.O.; Jonczak, E.; et al. Dose escalation of ripretinib can lead to response in advanced gastrointestinal stromal tumor patients refractory to the standard dose: A report of two cases. Gastrointestinal. Stromal Tumor 2021, 4, 1. [Google Scholar] [CrossRef]
- Nishida, T.; Blay, J.-Y.; Hirota, S.; Kitagawa, Y.; Kang, Y.-K. The standard diagnosis, treatment, and follow-up of gastrointestinal stromal tumors based on guidelines. Gastric Cancer 2016, 19, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Demetri, G.D.; Von Mehren, M.; Antonescu, C.R.; DeMatteo, R.P.; Ganjoo, K.N.; Maki, R.G.; Pisters, P.W.; Raut, C.P.; Riedel, R.F.; Schuetze, S.; et al. NCCN task force report: Update on the management of patients with gastrointestinal stromal tumors. J. Natl. Compr. Cancer Netw. 2010, 8, S1–S41, quiz S42–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaefer, I.M.; Marino-Enriquez, A.; Fletcher, J.A. What is new in gastrointestinal stromal tumor? Adv. Anat. Pathol. 2017, 24, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Trent, J.C.; Wilky, B.A.; Kerr, D.A.; Rosenberg, A.E. Current status of immunotherapy for gastrointestinal stromal tumor. Cancer Gene Ther. 2017, 24, 130–133. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, M.C.; Owzar, K.; Corless, C.L.; Hollis, D.; Borden, E.C.; Fletcher, C.D.; Ryan, C.W.; Von Mehren, M.; Blanke, C.D.; Rankin, C.; et al. Correlation of kinase genotype and clinical outcome in the North American intergroup phase III trial of imatinib mesylate for treatment of advanced gastrointestinal stromal tumor: CALGB 150105 study by cancer and leukemia group B and southwest oncology group. J. Clin. Oncol. 2008, 26, 5360–5367. [Google Scholar] [CrossRef] [Green Version]
- Nannini, M.; Biasco, G.; Astolfi, A.; Pantaleo, M.A. An overview on molecular biology of KIT/PDGFRA wild type (WT) gastrointestinal stromal tumors (GIST). J. Med. Genet. 2013, 50, 653–661. [Google Scholar] [CrossRef]
- Casali, P.G.; Zalcberg, J.; Le Cesne, A.; Reichardt, P.; Blay, J.-Y.; Lindner, L.; Judson, I.R.; Schöffski, P.; Leyvraz, S.; Italiano, A.; et al. Ten-year progression-free and overall survival in patients with unresectable or metastatic GI stromal tumors: Long-term analysis of the European Organisation for Research and Treatment of Cancer, Italian Sarcoma Group, and Australasian Gastrointestinal Trials Group intergroup phase III randomized trial on imatinib at two dose levels. J. Clin. Oncol. 2017, 35, 1713–1720. [Google Scholar] [CrossRef]
- Trent, J.C.; Subramanian, M.P. Managing GIST in the imatinib era: Optimization of adjuvant therapy. Expert Rev. Anticancer Ther. 2014, 14, 1445–1459. [Google Scholar] [CrossRef]
- Vadakara, J.; von Mehren, M. Gastrointestinal stromal tumors. Hematol. Clin. N. Am. 2013, 27, 905–920. [Google Scholar] [CrossRef] [Green Version]
- Tan, Y.; Garcia-Buitrago, M.T.; Trent, J.C.; Rosenberg, A.E. The immune system and gastrointestinal stromal tumor. Curr. Opin. Oncol. 2015, 27, 338–342. [Google Scholar] [CrossRef] [PubMed]
- Pandya, P.H.; Murray, M.E.; Pollok, K.E.; Renbarger, J.L. The immune system in cancer pathogenesis: Potential therapeutic approaches. J. Immunol. Res. 2016, 2016, 4273943. [Google Scholar] [CrossRef]
- Olszanski, A.J. Principles of immunotherapy. J. Natl. Compr. Cancer Netw. 2015, 13, 670–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Van Dongen, M.; Savage, N.D.; Jordanova, E.S.; Bruijn, I.H.B.-D.; Walburg, K.V.; Ottenhoff, T.H.; Hogendoorn, P.; Van Der Burg, S.H.; Gelderblom, H.; van Hall, T. Anti-inflammatory M2 type macrophages characterize metastasized and tyrosine kinase inhibitor-treated gastrointestinal stromal tumors. Int. J. Cancer 2010, 127, 899–909. [Google Scholar] [CrossRef]
- Cameron, S.; Gieselmann, M.; Blaschke, M.; Ramadori, G.; Fuzesi, L. Immune cells in primary and metastatic gastrointesti-nal stromal tumors (GIST). Int. J. Clin. Exp. Pathol. 2014, 7, 3563–3579. [Google Scholar]
- Rusakiewicz, S.; Semeraro, M.; Sarabi, M.; Desbois, M.; Locher, C.; Mendez, R.; Vimond, N.; Concha, A.; Garrido, F.; Isambert, N.; et al. Immune infiltrates are prognostic factors in localized gastrointestinal stromal tumors. Cancer Res. 2013, 73, 3499–3510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayers, M.; Lunceford, J.; Nebozhyn, M.; Murphy, E.; Loboda, A.; Kaufman, D.R.; Albright, A.; Cheng, J.D.; Kang, S.P.; Shankaran, V.; et al. IFN-γ–related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Investig. 2017, 127, 2930–2940. [Google Scholar] [CrossRef]
- Vitiello, G.A.; Bowler, T.G.; Liu, M.; Medina, B.D.; Zhang, J.Q.; Param, N.J.; Loo, J.K.; Goldfeder, R.L.; Chibon, F.; Rossi, F.; et al. Differential immune profiles distinguish the mutational subtypes of gastrointestinal stromal tumor. J. Clin. Investig. 2019, 129, 1863–1877. [Google Scholar] [CrossRef]
- Indio, V.; Astolfi, A.; Urbini, M.; Nannini, M.; Pantaleo, M.A. Genetics and treatment of gastrointestinal stromal tumors with immune checkpoint inhibitors: What do we know? Pharmacogenomics 2020, 21, 231–234. [Google Scholar] [CrossRef]
- Chen, L.L.; Chen, X.; Choi, H.; Sang, H.; Chen, L.C.; Zhang, H.; Gouw, L.; Andtbacka, R.H.; Chan, B.K.; Rodesch, C.K.; et al. Exploiting antitumor immunity to overcome relapse and improve remission duration. Cancer Immunol. Immunother. 2012, 61, 1113–1124. [Google Scholar] [CrossRef] [Green Version]
- Balachandran, V.P.; Cavnar, M.J.; Zeng, S.; Bamboat, Z.M.; Ocuin, L.M.; Obaid, H.; Sorenson, E.C.; Popow, R.; Ariyan, C.; Rossi, F.; et al. Imatinib potentiates antitumor T cell responses in gastrointestinal stromal tumor through the inhibition of Ido. Nat. Med. 2011, 17, 1094–1100. [Google Scholar] [CrossRef] [PubMed]
- Delahaye, N.F.; Rusakiewicz, S.; Martins, I.; Ménard, C.; Roux, S.; Lyonnet, L.; Paul, P.; Sarabi, M.; Chaput, N.; Semeraro, M.; et al. Alternatively spliced NKp30 isoforms affect the prognosis of gastrointestinal stromal tumors. Nat. Med. 2011, 17, 700–707. [Google Scholar] [CrossRef] [PubMed]
- Florou, V.; Rosenberg, A.E.; Wieder, E.; Komanduri, K.V.; Kolonias, D.; Uduman, M.; Castle, J.C.; Buell, J.S.; Trent, J.C.; Wilky, B.A. Angiosarcoma patients treated with immune checkpoint inhibitors: A case series of seven patients from a single institution. J. Immunother. Cancer 2019, 7, 213. [Google Scholar] [CrossRef] [Green Version]
- Farag, S.; Verschoor, A.J.; Bosma, J.W.; Gelderblom, H.; Kerst, J.M.; Sleijfer, S.; Steeghs, N. Imatinib-induced agranulocytosis in patients with gastrointestinal stromal tumors. J. Clin. Pharmacol. 2015, 55, 920–925. [Google Scholar] [CrossRef]
- Bertucci, F.; Finetti, P.; Mamessier, E.; Pantaleo, M.A.; Astolfi, A.; Ostrowski, J.; Birnbaum, D. PDL1 expression is an independent prognostic factor in localized GIST. OncoImmunology 2015, 4, e1002729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seifert, A.M.; Zeng, S.; Zhang, J.Q.; Kim, T.; Cohen, N.; Beckman, M.J.; Medina, B.D.; Maltbaek, J.H.; Loo, J.K.; Crawley, M.H.; et al. PD-1/PD-L1 blockade enhances T-cell activity and antitumor efficacy of imatinib in gastrointestinal stromal tumors. Clin. Cancer Res. 2017, 23, 454–465. [Google Scholar] [CrossRef] [Green Version]
- PDR001 Plus Imatinib for Metastatic or Unresectable GIST. Available online: https://clinicaltrials.gov/ct2/show/NCT03609424 (accessed on 19 May 2021).
- D’Angelo, S.P.; Shoushtari, A.N.; Keohan, M.L.; Dickson, M.A.; Gounder, M.M.; Chi, P.; Loo, J.K.; Gaffney, L.; Schneider, L.; Patel, Z.; et al. combined kit and ctla-4 blockade in patients with refractory gist and other advanced sarcomas: A phase Ib study of dasatinib plus ipilimumab. Clin. Cancer Res. 2017, 23, 2972–2980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Epacadostat and Pembrolizumab in Patients With GIST. Available online: https://clinicaltrials.gov/ct2/show/NCT03291054 (accessed on 19 May 2021).
- Singh, A.S.; Chmielowski, B.; Hecht, J.R.; Rosen, L.S.; Chow, W.A.; Wang, X.; Brackert, S.; Adame, C.; Bovill, J.; Schink, E.; et al. A randomized phase II study of nivolumab monotherapy versus nivolumab combined with ipilimumab in advanced gastrointestinal stromal tumor (GIST). J. Clin. Oncol. 2019, 37, 11017. [Google Scholar] [CrossRef]
- Edris, B.; Willingham, S.B.; Weiskopf, K.; Volkmer, A.K.; Volkmer, J.-P.; Mühlenberg, T.; Montgomery, K.D.; Contreras-Trujillo, H.; Czechowicz, A.; Fletcher, J.A.; et al. Anti-KIT monoclonal antibody inhibits imatinib-resistant gastrointestinal stromal tumor growth. Proc. Natl. Acad. Sci. USA 2013, 110, 3501–3506. [Google Scholar] [CrossRef] [Green Version]
- Abrams, T.J.; Connor, A.; Fanton, C.P.; Cohen, S.B.; Huber, T.; Miller, K.; Hong, E.E.; Niu, X.; Kline, J.; Ison-Dugenny, M.; et al. preclinical antitumor activity of a novel anti-c-KIT antibody-drug conjugate against mutant and wild-type c-KIT-positive solid tumors. Clin. Cancer Res. 2018, 24, 4297–4308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, W.Y.; Zhuang, C.; Xu, J.; Wang, M.; Zhang, Z.Z.; Tu, L.; Wang, C.J.; Ling, T.L.; Cao, H.; Zhang, Z.G. Somatostatin re-ceptors in gastrointestinal stromal tumors: New prognostic biomarker and potential therapeutic strategy. Am. J. Transl. Res. 2014, 6, 831–840. [Google Scholar]
- A Study of XmAb®18087 in Subjects with NET and GIST. Available online: https://clinicaltrials.gov/ct2/show/NCT03411915 (accessed on 19 May 2021).
- Katz, S.C.; Burga, R.A.; Naheed, S.; Licata, L.A.; Thorn, M.; Osgood, D.; Nguyen, C.T.; Espat, N.J.; Fletcher, J.A.; Junghans, R.P. Anti-KIT designer T cells for the treatment of gastrointestinal stromal tumor. J. Transl. Med. 2013, 11, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siozopoulou, V.; Domen, A.; Zwaenepoel, K.; Van Beeck, A.; Smits, E.; Pauwels, P.; Marcq, E. Immune checkpoint inhibitory therapy in sarcomas: Is there light at the end of the tunnel? Cancers 2021, 13, 360. [Google Scholar] [CrossRef]
- Arshad, J.; Roberts, A.; Ahmed, J.; Cotta, J.; Pico, B.; Kwon, D.; Trent, J. Utility of circulating tumor DNA in the management of patients with GI stromal tumors: Analysis of 243 patients. J. Clin. Oncol. Precis. Oncol. 2020, 4, 66–73. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Trial | Phase | Drug | GIST Population | Results |
|---|---|---|---|---|
| NCT00585221 | Phase 2 | Peginterferon alfa-2b plus Imatinib | Stage III/IV | 7 PR and 1 CR (100%). 2 (25%) patients progressed during the 3.6 m follow-up. |
| NCT01643278 | Phase 1 | Dasatinib plus Ipilimumab | Advanced/unresectable refractory to imatinib and sunitinib | 7 PR (53%), 3 SD (23%), and 3 PD (23%), with a median duration of response of 82 days |
| NCT02636725 | Phase 2 | Axitinib plus Pembrolizumab | Unresectable and refractory to first-line therapy | No response seen in GIST patients (n = 3) |
| NCT03609424 | Phase 1b/2 | Spartalizumab (PDR001) | Metastatic or unresectable | Ongoing |
| NCT03411915 | Phase 1 | XmAb®18087 | Advanced or metastatic refractory to all FDA-approved therapies | Ongoing |
| NCT03291054 | Phase 2 | Epacadostat plus Pembrolizumab | Unresectable or metastatic refractory to imatinib and another TKI | Ongoing |
| NCT02880020 | Phase 2 | Nivolumab Monotherapy versus Nivolumab Combined with Ipilimumab | Metastatic or Unresectable | Ongoing |
| NCT03475953 | Phase 1/2 | Regorafenib with Avelumab | Metastatic or Unresectable | Ongoing |
| NCT02406781 | Phase 2 | Pembrolizumab with metronomic cyclophosphamide | Metastatic or Unresectable after imatinib and sunitinib. | Ongoing |
| NCT04258956 | Phase 2 | Avelumab with Axitinib | KIT or PDGFRA positive advanced or metastatic, with no more than 3 previous therapies, which must include imatinib and sunitinib | Ongoing |
| NCT02834013 | Phase 2 | Nivolumab and Ipilimumab | Progressed in at least one line, and no further approved standard therapy that prolongs survival | Ongoing |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arshad, J.; Costa, P.A.; Barreto-Coelho, P.; Valdes, B.N.; Trent, J.C. Immunotherapy Strategies for Gastrointestinal Stromal Tumor. Cancers 2021, 13, 3525. https://doi.org/10.3390/cancers13143525
Arshad J, Costa PA, Barreto-Coelho P, Valdes BN, Trent JC. Immunotherapy Strategies for Gastrointestinal Stromal Tumor. Cancers. 2021; 13(14):3525. https://doi.org/10.3390/cancers13143525
Chicago/Turabian StyleArshad, Junaid, Philippos A. Costa, Priscila Barreto-Coelho, Brianna Nicole Valdes, and Jonathan C. Trent. 2021. "Immunotherapy Strategies for Gastrointestinal Stromal Tumor" Cancers 13, no. 14: 3525. https://doi.org/10.3390/cancers13143525
APA StyleArshad, J., Costa, P. A., Barreto-Coelho, P., Valdes, B. N., & Trent, J. C. (2021). Immunotherapy Strategies for Gastrointestinal Stromal Tumor. Cancers, 13(14), 3525. https://doi.org/10.3390/cancers13143525