Insights into the New Cancer Therapy through Redox Homeostasis and Metabolic Shifts


Abstract
{kind=link}
{kind=link}
{kind=link}
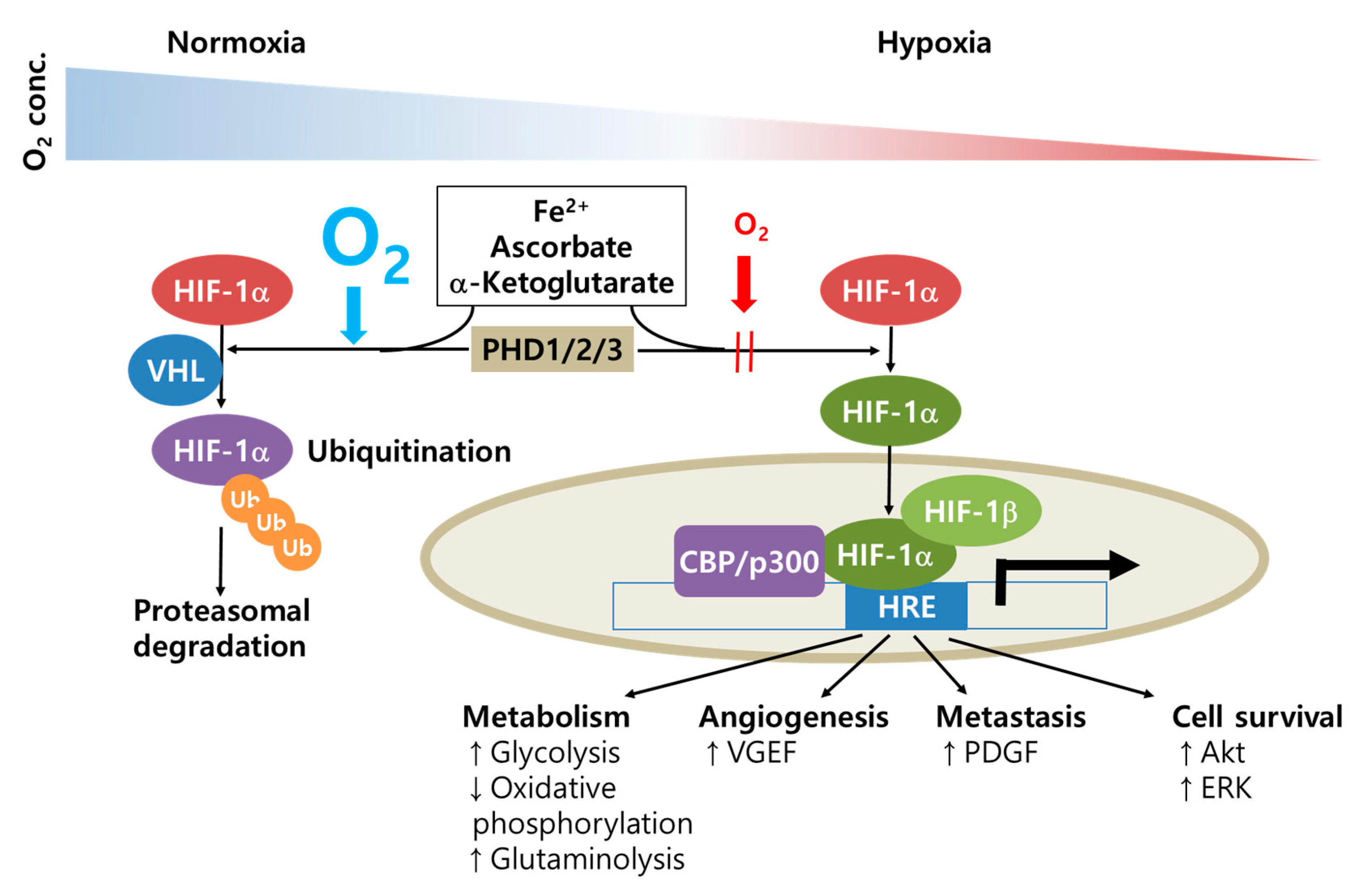{kind=link}
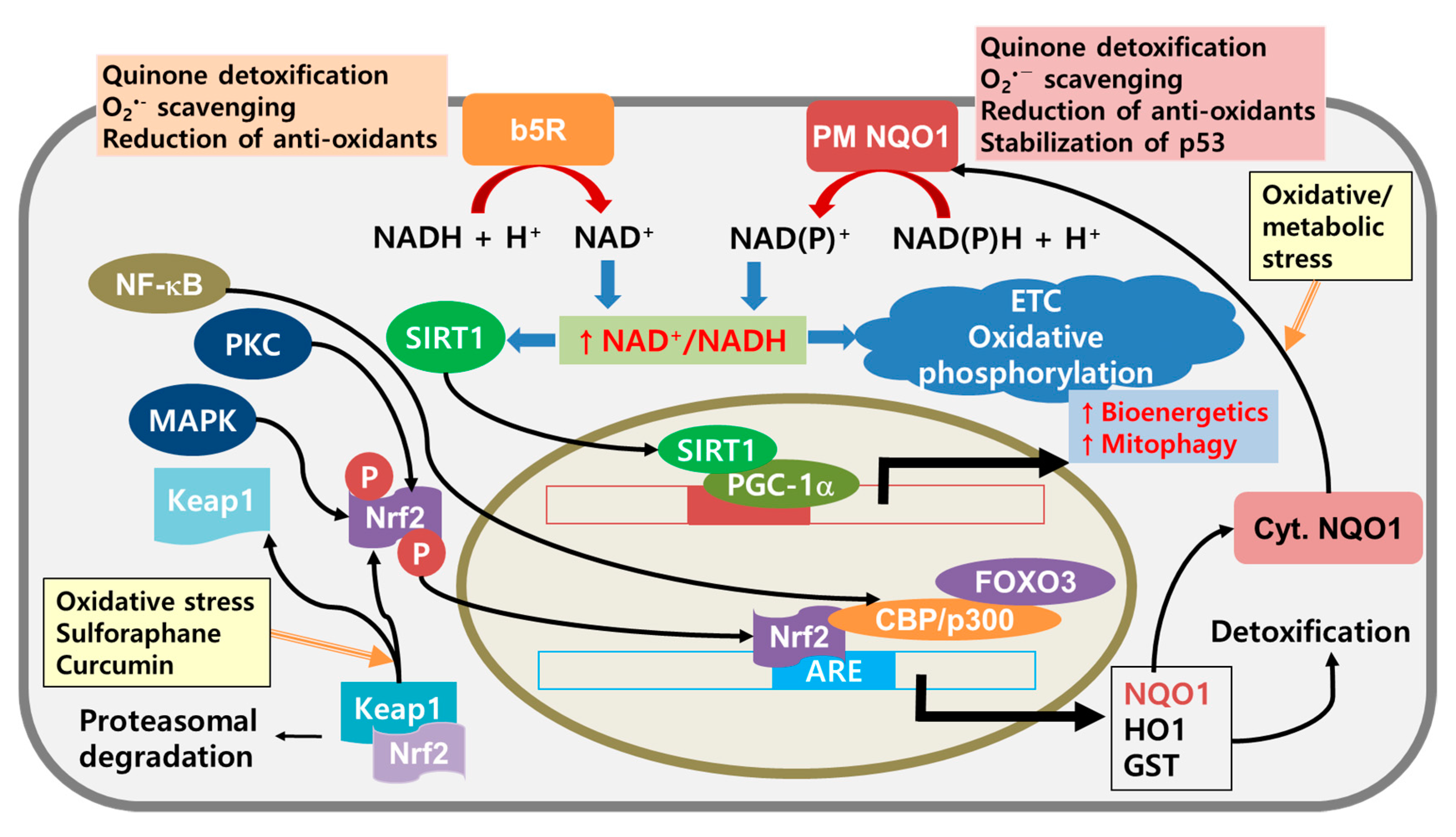{kind=link}
1. Introduction
2. Oxidative Stress and the Antioxidant Defense System
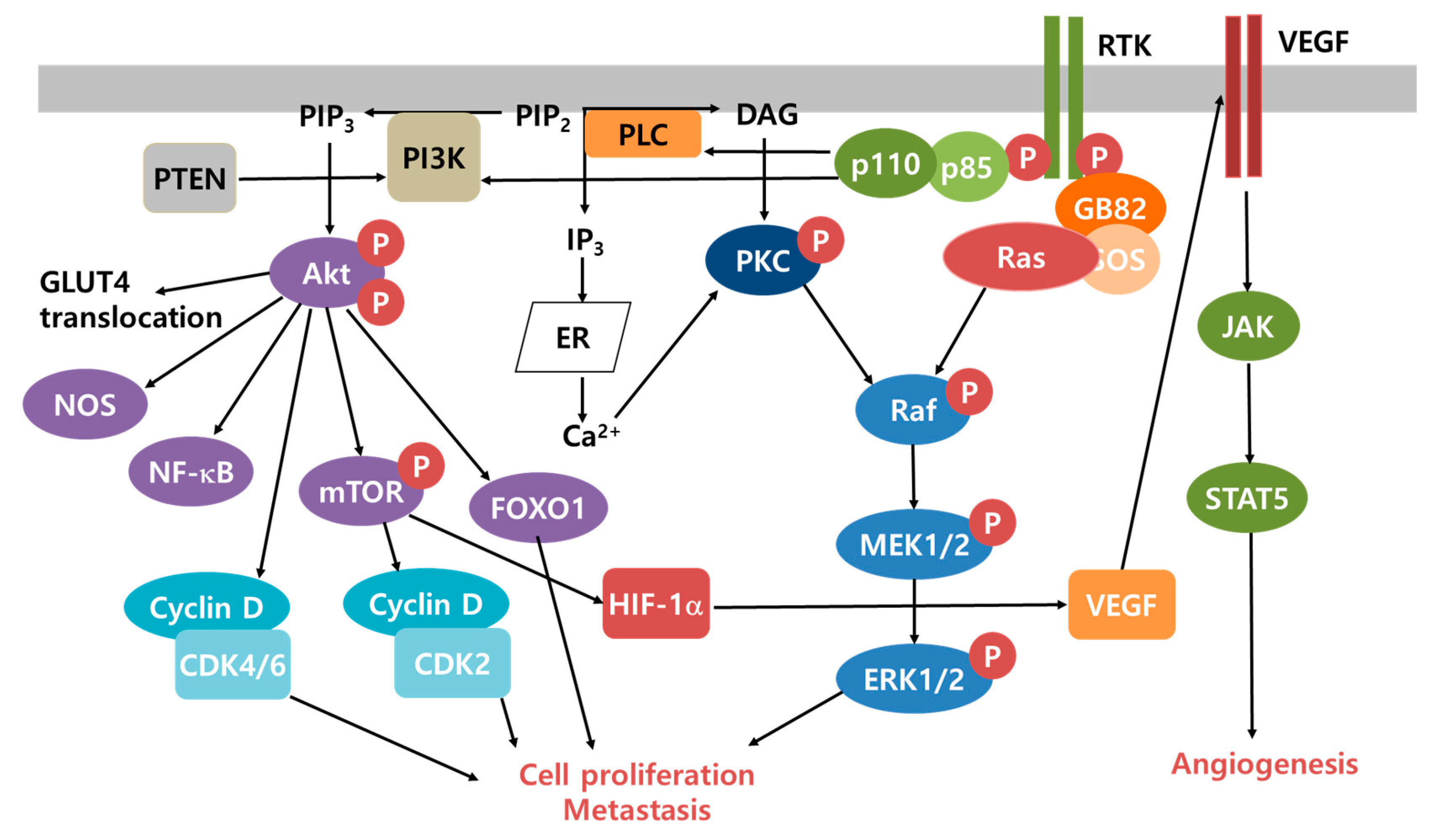
3. Cancer Microenvironment and Survival Mechanisms
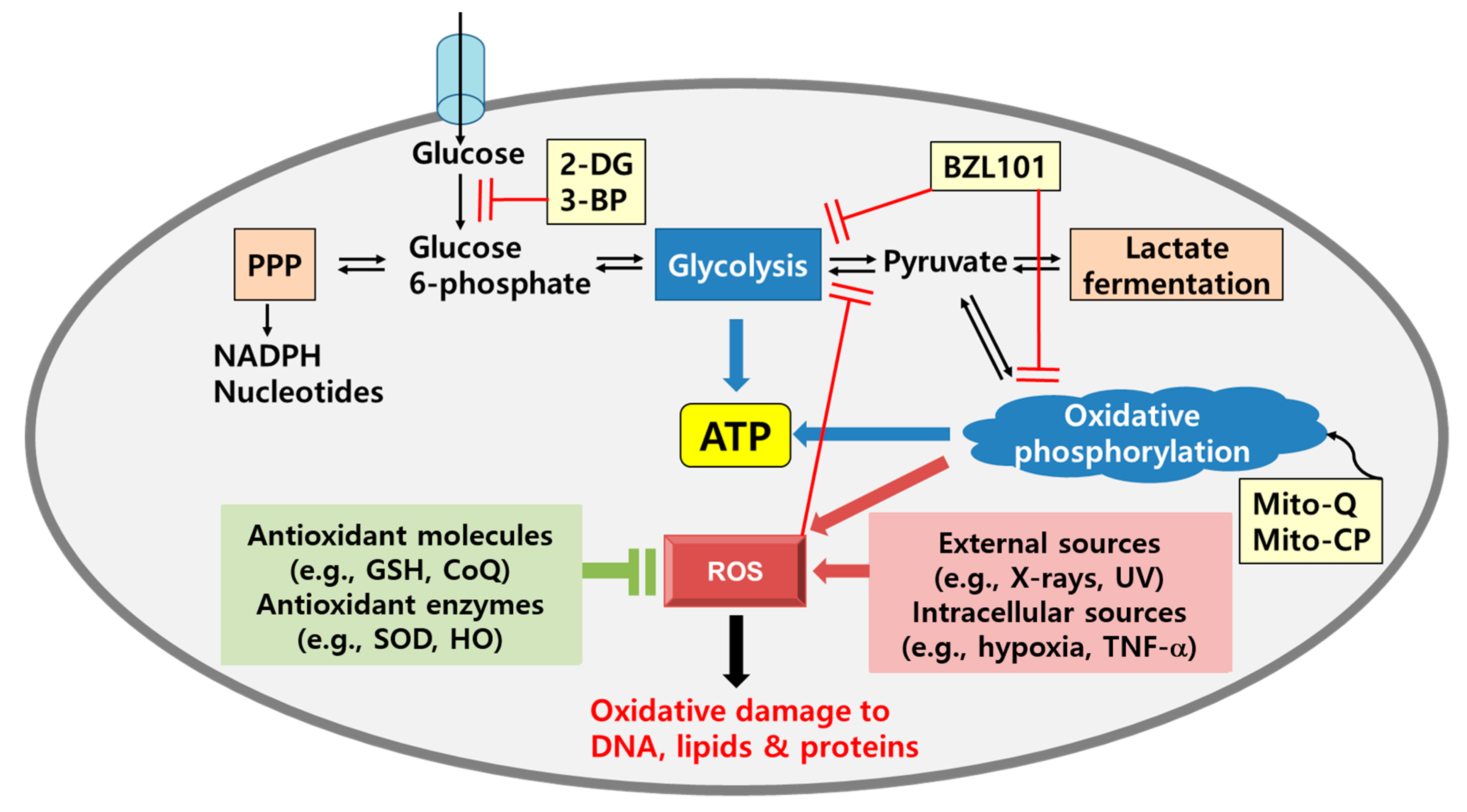
4. Metabolic Shifts in Cancer: Glycolysis and Oxidative Phosphorylation
5. Redox Homeostasis as a Cancer Therapeutic Target
6. Improvement of Mitochondrial Functions as a Cancer Therapeutic Target
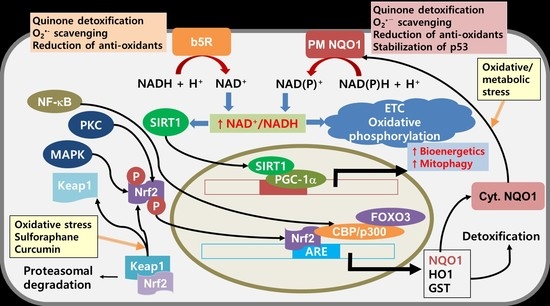
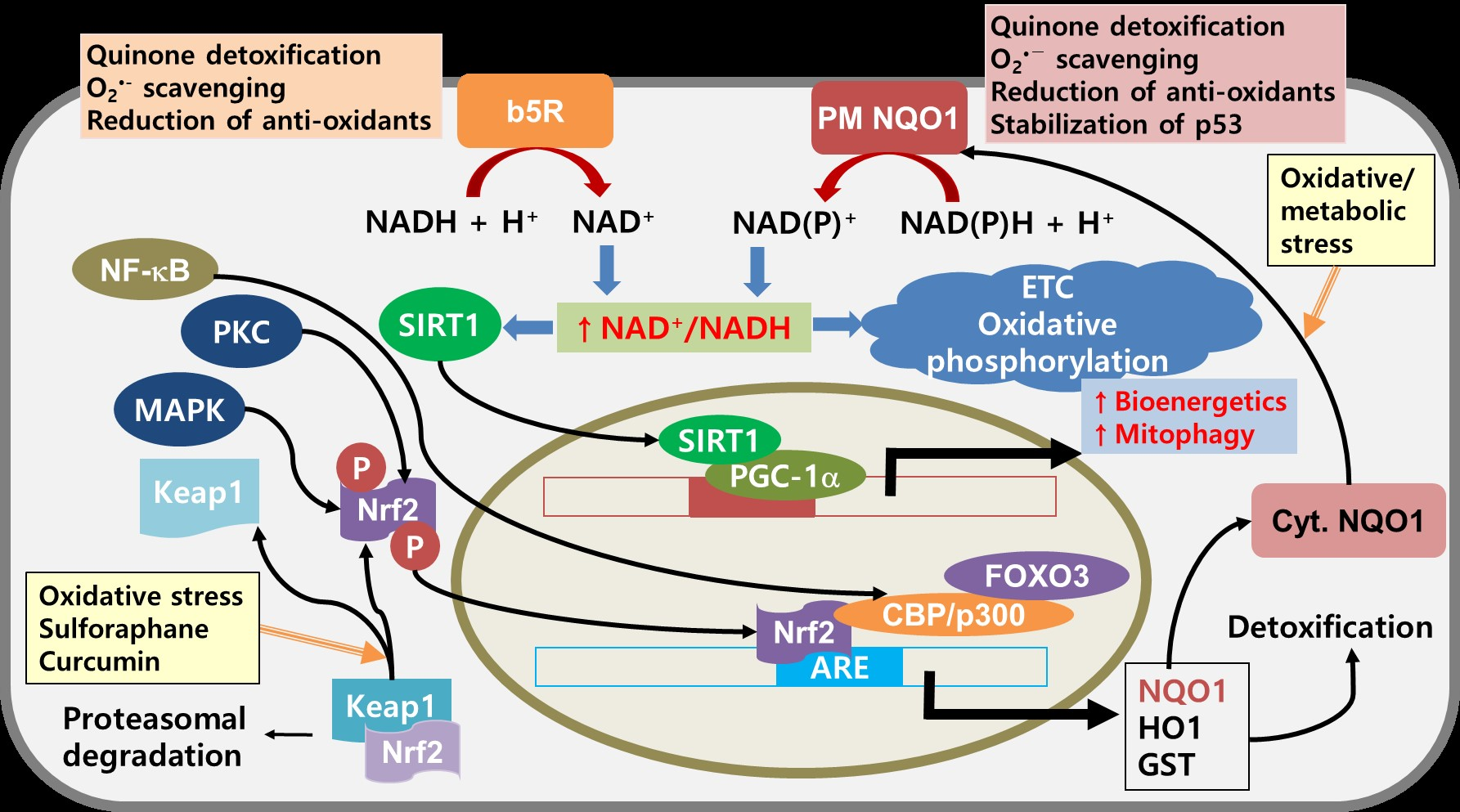
7. NQO1 as a Potential Target for Cancer Therapy
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Melton, C.; Reuter, J.A.; Spacek, D.V.; Snyder, M. Recurrent somatic mutations in regulatory regions of human cancer genomes. Nat. Genet. 2015, 47, 710–716. [Google Scholar] [CrossRef]
- Jose, C.; Bellance, N.; Rossignol, R. Choosing between glycolysis and oxidative phosphorylation: A tumor’s dilemma? Biochim. Biophys. Acta (BBA) Bioenerg. 2011, 1807, 552–561. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Suo, C.; Li, S.T.; Zhang, H.; Gao, P. Metabolic reprogramming for cancer cells and their microenvironment: Beyond the Warburg Effect. Biochim. Biophys. Acta (BBA) Rev. Cancer 2018, 1870, 51–66. [Google Scholar] [CrossRef] [PubMed]
- Gough, D.J.; Corlett, A.; Schlessinger, K.; Wegrzyn, J.; Larner, A.C.; Levy, D.E. Mitochondrial STAT3 supports Ras-dependent oncogenic transformation. Science 2009, 324, 1713–1716. [Google Scholar] [CrossRef]
- Kruiswijk, F.; Labuschagne, C.F.; Vousden, K.H. p53 in survival, death and metabolic health: A lifeguard with a licence to kill. Nat. Rev. Mol. Cell Biol. 2015, 16, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Urra, F.A.; Munoz, F.; Lovy, A.; Cardenas, C. The Mitochondrial complex(I)ty of cancer. Front. Oncol. 2017, 7, 118. [Google Scholar] [CrossRef]
- Birsoy, K.; Possemato, R.; Lorbeer, F.K.; Bayraktar, E.C.; Thiru, P.; Yucel, B.; Wang, T.; Chen, W.W.; Clish, C.B.; Sabatini, D.M. Metabolic determinants of cancer cell sensitivity to glucose limitation and biguanides. Nature 2014, 508, 108–112. [Google Scholar] [CrossRef]
- Cheng, G.; Zielonka, J.; Dranka, B.P.; McAllister, D.; Mackinnon, A.C., Jr.; Joseph, J.; Kalyanaraman, B. Mitochondria-targeted drugs synergize with 2-deoxyglucose to trigger breast cancer cell death. Cancer Res. 2012, 72, 2634–2644. [Google Scholar] [CrossRef]
- Van der Bliek, A.M.; Sedensky, M.M.; Morgan, P.G. Cell biology of the mitochondrion. Genetics 2017, 207, 843–871. [Google Scholar] [CrossRef]
- Liochev, S.I. Reactive oxygen species and the free radical theory of aging. Free Radic. Biol. Med. 2013, 60, 1–4. [Google Scholar] [CrossRef]
- Halliwell, B. The chemistry of free radicals. Toxicol. Ind. Health 1993, 9, 1–21. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Radi, R. Oxygen radicals, nitric oxide, and peroxynitrite: Redox pathways in molecular medicine. Proc. Natl. Acad. Sci. USA 2018, 115, 5839–5848. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B. Free radicals and antioxidants: Updating a personal view. Nutr. Rev. 2012, 70, 257–265. [Google Scholar] [CrossRef]
- Kondo, T.; Ohshima, T.; Ishida, Y. Age-dependent expression of 8-hydroxy-2′-deoxyguanosine in human pituitary gland. Histochem. J. 2001, 33, 647–651. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Takeshita, H.; Komatsu, M.; Xu, B.; Aoyama, K.; Takeuchi, T. Generation of 8-hydroxydeoxyguanosine from DNA using rat liver homogenates. Cancer Sci. 2005, 96, 13–18. [Google Scholar] [CrossRef]
- Choi, I.Y.; Lim, J.H.; Kim, C.; Song, H.Y.; Ju, C.; Kim, W.K. 4-hydroxy-2(E)-Nonenal facilitates NMDA-induced neurotoxicity via triggering mitochondrial permeability transition pore opening and mitochondrial calcium overload. Exp. Neurobiol. 2013, 22, 200–207. [Google Scholar] [CrossRef][Green Version]
- Raess, B.U.; Keenan, C.E.; McConnell, E.J. Effects of 4-OH-2,3-trans-nonenal on human erythrocyte plasma membrane Ca2+ pump and passive Ca2+ permeability. Biochem. Biophys. Res. Commun. 1997, 235, 451–454. [Google Scholar] [CrossRef]
- Chevion, M.; Berenshtein, E.; Stadtman, E.R. Human studies related to protein oxidation: Protein carbonyl content as a marker of damage. Free Radic. Res. 2000, 33, S99–S108. [Google Scholar]
- Jomova, K.; Vondrakova, D.; Lawson, M.; Valko, M. Metals, oxidative stress and neurodegenerative disorders. Mol. Cell Biochem. 2010, 345, 91–104. [Google Scholar] [CrossRef]
- Vaziri, N.D.; Ni, Z.; Oveisi, F.; Liang, K.; Pandian, R. Enhanced nitric oxide inactivation and protein nitration by reactive oxygen species in renal insufficiency. Hypertension 2002, 39, 135–141. [Google Scholar] [CrossRef]
- Weiss, W.P. A 100-year review: From ascorbic acid to zinc-mineral and vitamin nutrition of dairy cows. J. Dairy Sci. 2017, 100, 10045–10060. [Google Scholar] [CrossRef]
- Liguori, I.; Russo, G.; Curcio, F.; Bulli, G.; Aran, L.; Della-Morte, D.; Gargiulo, G.; Testa, G.; Cacciatore, F.; Bonaduce, D.; et al. Oxidative stress, aging, and diseases. Clin. Interv. Aging 2018, 13, 757–772. [Google Scholar] [CrossRef] [PubMed]
- Vona, R.; Gambardella, L.; Cittadini, C.; Straface, E.; Pietraforte, D. Biomarkers of oxidative stress in metabolic syndrome and associated diseases. Oxid. Med. Cell. Longev. 2019, 2019, 8267234. [Google Scholar] [CrossRef]
- Liu, C.Y.; Lee, C.F.; Wei, Y.H. Role of reactive oxygen species-elicited apoptosis in the pathophysiology of mitochondrial and neurodegenerative diseases associated with mitochondrial DNA mutations. J. Formos. Med. Assoc. 2009, 108, 599–611. [Google Scholar] [CrossRef]
- Mawrin, C.; Kirches, E.; Krause, G.; Schneider-Stock, R.; Bogerts, B.; Vorwerk, C.K.; Dietzmann, K. Region-specific analysis of mitochondrial DNA deletions in neurodegenerative disorders in humans. Neurosci. Lett. 2004, 357, 111–114. [Google Scholar] [CrossRef] [PubMed]
- Moran, M.; Moreno-Lastres, D.; Marin-Buera, L.; Arenas, J.; Martin, M.A.; Ugalde, C. Mitochondrial respiratory chain dysfunction: Implications in neurodegeneration. Free Radic. Biol. Med. 2012, 53, 595–609. [Google Scholar] [CrossRef]
- Reddy, P.H.; Reddy, T.P. Mitochondria as a therapeutic target for aging and neurodegenerative diseases. Curr. Alzheimer Res. 2011, 8, 393–409. [Google Scholar] [CrossRef]
- Wen, Y.; Li, W.; Poteet, E.C.; Xie, L.; Tan, C.; Yan, L.J.; Ju, X.; Liu, R.; Qian, H.; Marvin, M.A.; et al. Alternative mitochondrial electron transfer as a novel strategy for neuroprotection. J. Biol. Chem. 2011, 286, 16504–16515. [Google Scholar] [CrossRef]
- Keogh, M.J.; Chinnery, P.F. Mitochondrial DNA mutations in neurodegeneration. Biochim. Biophys. Acta 2015, 1847, 1401–1411. [Google Scholar] [CrossRef]
- Maruszak, A.; Gaweda-Walerych, K.; Soltyszewski, I.; Zekanowski, C. Mitochondrial DNA in pathogenesis of Alzheimer’s and Parkinson’s diseases. Acta Neurobiol. Exp. 2006, 66, 153–176. [Google Scholar]
- Ghiasi, P.; Hosseinkhani, S.; Noori, A.; Nafissi, S.; Khajeh, K. Mitochondrial complex I deficiency and ATP/ADP ratio in lymphocytes of amyotrophic lateral sclerosis patients. Neurol. Res. 2012, 34, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Zhang, Q.; Chen, S.; Fang, B.; Yang, Q.; Chen, C.; Miele, L.; Sarkar, F.H.; Xia, J.; Wang, Z. Mitochondrial dysfunction promotes breast cancer cell migration and invasion through HIF1alpha accumulation via increased production of reactive oxygen species. PLoS ONE 2013, 8, e69485. [Google Scholar]
- Onyango, I.G.; Khan, S.M.; Bennett, J.P., Jr. Mitochondria in the pathophysiology of Alzheimer’s and Parkinson’s diseases. Front. Biosci. (Landmark Ed) 2017, 22, 854–872. [Google Scholar] [CrossRef] [PubMed]
- Menzies, F.M.; Cookson, M.R.; Taylor, R.W.; Turnbull, D.M.; Chrzanowska-Lightowlers, Z.M.; Dong, L.; Figlewicz, D.A.; Shaw, P.J. Mitochondrial dysfunction in a cell culture model of familial amyotrophic lateral sclerosis. Brain 2002, 125, 1522–1533. [Google Scholar] [CrossRef]
- Lesnefsky, E.J.; Gudz, T.I.; Moghaddas, S.; Migita, C.T.; Ikeda-Saito, M.; Turkaly, P.J.; Hoppel, C.L. Aging decreases electron transport complex III activity in heart interfibrillar mitochondria by alteration of the cytochrome c binding site. J. Mol. Cell. Cardiol. 2001, 33, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Kudryavtseva, A.V.; Krasnov, G.S.; Dmitriev, A.A.; Alekseev, B.Y.; Kardymon, O.L.; Sadritdinova, A.F.; Fedorova, M.S.; Pokrovsky, A.V.; Melnikova, N.V.; Kaprin, A.D.; et al. Mitochondrial dysfunction and oxidative stress in aging and cancer. Oncotarget 2016, 7, 44879–44905. [Google Scholar] [CrossRef]
- Mancuso, M.; Coppede, F.; Migliore, L.; Siciliano, G.; Murri, L. Mitochondrial dysfunction, oxidative stress and neurodegeneration. J. Alzheimers Dis. 2006, 10, 59–73. [Google Scholar] [CrossRef]
- Hinshaw, D.C.; Shevde, L.A. The tumor microenvironment innately modulates cancer progression. Cancer Res. 2019, 79, 4557–4566. [Google Scholar] [CrossRef] [PubMed]
- Roma-Rodrigues, C.; Mendes, R.; Baptista, P.V.; Fernandes, A.R. Targeting tumor microenvironment for cancer therapy. Int. J. Mol. Sci. 2019, 20, 840. [Google Scholar] [CrossRef]
- Hanahan, D.; Coussens, L.M. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef]
- Rasanen, K.; Vaheri, A. Activation of fibroblasts in cancer stroma. Exp. Cell Res. 2010, 316, 2713–2722. [Google Scholar] [CrossRef] [PubMed]
- Viallard, C.; Larrivee, B. Tumor angiogenesis and vascular normalization: Alternative therapeutic targets. Angiogenesis 2017, 20, 409–426. [Google Scholar] [CrossRef] [PubMed]
- Gavalas, N.G.; Liontos, M.; Trachana, S.P.; Bagratuni, T.; Arapinis, C.; Liacos, C.; Dimopoulos, M.A.; Bamias, A. Angiogenesis-related pathways in the pathogenesis of ovarian cancer. Int. J. Mol. Sci. 2013, 14, 15885–15909. [Google Scholar] [CrossRef] [PubMed]
- Ushio-Fukai, M.; Nakamura, Y. Reactive oxygen species and angiogenesis: NADPH oxidase as target for cancer therapy. Cancer Lett. 2008, 266, 37–52. [Google Scholar] [CrossRef]
- Dong, G.; Lin, X.H.; Liu, H.H.; Gao, D.M.; Cui, J.F.; Ren, Z.G.; Chen, R.X. Intermittent hypoxia alleviates increased VEGF and pro-angiogenic potential in liver cancer cells. Oncol. Lett. 2019, 18, 1831–1839. [Google Scholar] [CrossRef]
- Triner, D.; Shah, Y.M. Hypoxia-inducible factors: A central link between inflammation and cancer. J. Clin. Investig. 2016, 126, 3689–3698. [Google Scholar] [CrossRef]
- Blanchetot, C.; Boonstra, J. The ROS-NOX connection in cancer and angiogenesis. Crit. Rev. Eukaryot Gene Expr. 2008, 18, 35–45. [Google Scholar] [CrossRef]
- Pittayapruek, P.; Meephansan, J.; Prapapan, O.; Komine, M.; Ohtsuki, M. Role of matrix metalloproteinases in photoaging and photocarcinogenesis. Int. J. Mol. Sci. 2016, 17, 868. [Google Scholar] [CrossRef]
- Anderson, K.G.; Stromnes, I.M.; Greenberg, P.D. Obstacles posed by the tumor microenvironment to T cell activity: A case for synergistic therapies. Cancer Cell 2017, 31, 311–325. [Google Scholar] [CrossRef]
- Marsh, T.; Pietras, K.; McAllister, S.S. Fibroblasts as architects of cancer pathogenesis. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2013, 1832, 1070–1078. [Google Scholar] [CrossRef] [PubMed]
- Stover, D.G.; Bierie, B.; Moses, H.L. A delicate balance: TGF-beta and the tumor microenvironment. J. Cell. Biochem. 2007, 101, 851–861. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Li, Y.; Zamyatnin, A.A., Jr.; Werner, J.; Bazhin, A.V. Reactive oxygen species and colorectal cancer. J. Cell. Physiol. 2018, 233, 5119–5132. [Google Scholar] [CrossRef]
- Bahrambeigi, S.; Shafiei-Irannejad, V. Immune-mediated anti-tumor effects of metformin; targeting metabolic reprogramming of T cells as a new possible mechanism for anti-cancer effects of metformin. Biochem. Pharmacol. 2020, 174, 113787. [Google Scholar] [CrossRef] [PubMed]
- Gibbons, D.L.; Byers, L.A.; Kurie, J.M. Smoking, p53 mutation, and lung cancer. Mol. Cancer Res. 2014, 12, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Soussi, T.; Lozano, G. p53 mutation heterogeneity in cancer. Biochem. Biophys. Res. Commun. 2005, 331, 834–842. [Google Scholar] [CrossRef]
- Guo, G.; Yu, M.; Xiao, W.; Celis, E.; Cui, Y. Local activation of p53 in the tumor microenvironment overcomes immune suppression and enhances antitumor immunity. Cancer Res. 2017, 77, 2292–2305. [Google Scholar] [CrossRef]
- Lu, P.; Weaver, V.M.; Werb, Z. The extracellular matrix: A dynamic niche in cancer progression. J. Cell Biol. 2012, 196, 395–406. [Google Scholar] [CrossRef]
- Nasser, N.J. Heparanase involvement in physiology and disease. Cell. Mol. Life Sci. 2008, 65, 1706–1715. [Google Scholar] [CrossRef]
- Wielenga, V.J.; van der Voort, R.; Taher, T.E.; Smit, L.; Beuling, E.A.; van Krimpen, C.; Spaargaren, M.; Pals, S.T. Expression of c-Met and heparan-sulfate proteoglycan forms of CD44 in colorectal cancer. Am. J. Pathol. 2000, 157, 1563–1573. [Google Scholar] [CrossRef]
- Herishanu, Y.; Gibellini, F.; Njuguna, N.; Hazan-Halevy, I.; Farooqui, M.; Bern, S.; Keyvanfar, K.; Lee, E.; Wilson, W.; Wiestner, A. Activation of CD44, a receptor for extracellular matrix components, protects chronic lymphocytic leukemia cells from spontaneous and drug induced apoptosis through MCL-1. Leuk. Lymphoma 2011, 52, 1758–1769. [Google Scholar] [CrossRef] [PubMed]
- Barker, H.E.; Chang, J.; Cox, T.R.; Lang, G.; Bird, D.; Nicolau, M.; Evans, H.R.; Gartland, A.; Erler, J.T. LOXL2-mediated matrix remodeling in metastasis and mammary gland involution. Cancer Res. 2011, 71, 1561–1572. [Google Scholar] [CrossRef] [PubMed]
- Cox, T.R.; Erler, J.T. Lysyl oxidase in colorectal cancer. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 305, G659–G666. [Google Scholar] [CrossRef]
- Levental, K.R.; Yu, H.; Kass, L.; Lakins, J.N.; Egeblad, M.; Erler, J.T.; Fong, S.F.; Csiszar, K.; Giaccia, A.; Weninger, W.; et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell 2009, 139, 891–906. [Google Scholar] [CrossRef]
- Erler, J.T.; Bennewith, K.L.; Nicolau, M.; Dornhofer, N.; Kong, C.; Le, Q.T.; Chi, J.T.; Jeffrey, S.S.; Giaccia, A.J. Lysyl oxidase is essential for hypoxia-induced metastasis. Nature 2006, 440, 1222–1226. [Google Scholar] [CrossRef] [PubMed]
- Gong, R.; Lin, W.; Gao, A.; Liu, Y.; Li, J.; Sun, M.; Chen, X.; Han, S.; Men, C.; Sun, Y.; et al. Forkhead box C1 promotes metastasis and invasion of non-small cell lung cancer by binding directly to the lysyl oxidase promoter. Cancer Sci. 2019, 110, 3663–3676. [Google Scholar] [CrossRef]
- Moloney, J.N.; Cotter, T.G. ROS signalling in the biology of cancer. Semin. Cell Dev. Biol. 2018, 80, 50–64. [Google Scholar] [CrossRef]
- Du, Z.; Lovly, C.M. Mechanisms of receptor tyrosine kinase activation in cancer. Mol. Cancer 2018, 17, 58. [Google Scholar] [CrossRef]
- Kidger, A.M.; Sipthorp, J.; Cook, S.J. ERK1/2 inhibitors: New weapons to inhibit the RAS-regulated RAF-MEK1/2-ERK1/2 pathway. Pharmacol. Ther. 2018, 187, 45–60. [Google Scholar] [CrossRef]
- DiDonato, J.A.; Mercurio, F.; Karin, M. NF-kappaB and the link between inflammation and cancer. Immunol. Rev. 2012, 246, 379–400. [Google Scholar] [CrossRef]
- Fresno Vara, J.A.; Casado, E.; de Castro, J.; Cejas, P.; Belda-Iniesta, C.; Gonzalez-Baron, M. PI3K/Akt signalling pathway and cancer. Cancer Treat. Rev. 2004, 30, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P. VEGF as a key mediator of angiogenesis in cancer. Oncology 2005, 69, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Fagiani, E.; Christofori, G. Angiopoietins in angiogenesis. Cancer Lett. 2013, 328, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Basseres, D.S.; Ebbs, A.; Levantini, E.; Baldwin, A.S. Requirement of the NF-kappaB subunit p65/RelA for K-Ras-induced lung tumorigenesis. Cancer Res. 2010, 70, 3537–3546. [Google Scholar] [CrossRef]
- Taguchi, K.; Yamamoto, M. The KEAP1-NRF2 system in cancer. Front. Oncol. 2017, 7, 85. [Google Scholar] [CrossRef] [PubMed]
- Wagner, E.F.; Nebreda, A.R. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat. Rev. Cancer 2009, 9, 537–549. [Google Scholar] [CrossRef]
- Caino, M.C.; Altieri, D.C. Molecular Pathways: Mitochondrial Reprogramming in Tumor Progression and Therapy. Clin. Cancer Res. 2016, 22, 540–545. [Google Scholar] [CrossRef]
- Ritt, D.A.; Abreu-Blanco, M.T.; Bindu, L.; Durrant, D.E.; Zhou, M.; Specht, S.I.; Stephen, A.G.; Holderfield, M.; Morrison, D.K. Inhibition of Ras/Raf/MEK/ERK pathway signaling by a stress-induced phospho-regulatory circuit. Mol. Cell 2016, 64, 875–887. [Google Scholar] [CrossRef]
- Bos, J.L. Ras oncogenes in human cancer: A review. Cancer Res. 1989, 49, 4682–4689. [Google Scholar]
- Imamura, Y.; Morikawa, T.; Liao, X.; Lochhead, P.; Kuchiba, A.; Yamauchi, M.; Qian, Z.R.; Nishihara, R.; Meyerhardt, J.A.; Haigis, K.M.; et al. Specific mutations in KRAS codons 12 and 13, and patient prognosis in 1075 BRAF wild-type colorectal cancers. Clin. Cancer Res. 2012, 18, 4753–4763. [Google Scholar] [CrossRef] [PubMed]
- Omholt, K.; Karsberg, S.; Platz, A.; Kanter, L.; Ringborg, U.; Hansson, J. Screening of N-ras codon 61 mutations in paired primary and metastatic cutaneous melanomas: Mutations occur early and persist throughout tumor progression. Clin. Cancer Res. 2002, 8, 3468–3474. [Google Scholar] [PubMed]
- Quilliam, L.A.; Castro, A.F.; Rogers-Graham, K.S.; Martin, C.B.; Der, C.J.; Bi, C. M-Ras/R-Ras3, a transforming ras protein regulated by Sos1, GRF1, and p120 Ras GTPase-activating protein, interacts with the putative Ras effector AF6. J. Biol. Chem. 1999, 274, 23850–23857. [Google Scholar] [CrossRef]
- Saez, R.; Chan, A.M.; Miki, T.; Aaronson, S.A. Oncogenic activation of human R-ras by point mutations analogous to those of prototype H-ras oncogenes. Oncogene 1994, 9, 2977–2982. [Google Scholar]
- Weyemi, U.; Lagente-Chevallier, O.; Boufraqech, M.; Prenois, F.; Courtin, F.; Caillou, B.; Talbot, M.; Dardalhon, M.; Al Ghuzlan, A.; Bidart, J.M.; et al. ROS-generating NADPH oxidase NOX4 is a critical mediator in oncogenic H-Ras-induced DNA damage and subsequent senescence. Oncogene 2012, 31, 1117–1129. [Google Scholar] [CrossRef] [PubMed]
- Indran, I.R.; Hande, M.P.; Pervaiz, S. Tumor cell redox state and mitochondria at the center of the non-canonical activity of telomerase reverse transcriptase. Mol. Aspects Med. 2010, 31, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Pan, G.; Chen, Y.; Yang, Q.; Hao, T.; Zhao, L.; Zhao, L.; Cong, Y.; Diao, A.; Yu, P. Inhibitor of the human telomerase reverse trancriptase (hTERT) gene promoter induces cell apoptosis via a mitochondrial-dependent pathway. Eur. J. Med. Chem. 2018, 145, 370–378. [Google Scholar] [CrossRef]
- Whibley, C.; Pharoah, P.D.; Hollstein, M. p53 polymorphisms: Cancer implications. Nat. Rev. Cancer 2009, 9, 95–107. [Google Scholar] [CrossRef]
- Liberti, M.V.; Locasale, J.W. The warburg effect: How does it benefit cancer cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef]
- Schwartz, L.; Supuran, C.T.; Alfarouk, K.O. The Warburg Effect and the Hallmarks of Cancer. Anticancer Agents Med. Chem. 2017, 17, 164–170. [Google Scholar] [CrossRef]
- Witney, T.H.; Lewis, D.Y. Imaging Cancer Metabolism with Positron Emission Tomography (PET). Methods Mol. Biol. 2019, 1928, 29–44. [Google Scholar] [PubMed]
- Heurling, K.; Leuzy, A.; Jonasson, M.; Frick, A.; Zimmer, E.R.; Nordberg, A.; Lubberink, M. Quantitative positron emission tomography in brain research. Brain Res. 2017, 1670, 220–234. [Google Scholar] [CrossRef] [PubMed]
- Abbaszadeh, Z.; Cesmeli, S.; Biray Avci, C. Crucial players in glycolysis: Cancer progress. Gene 2020, 726, 144158. [Google Scholar] [CrossRef] [PubMed]
- Ralph, S.J.; Rodriguez-Enriquez, S.; Neuzil, J.; Saavedra, E.; Moreno-Sanchez, R. The causes of cancer revisited: “Mitochondrial malignancy” and ROS-induced oncogenic transformation—Why mitochondria are targets for cancer therapy. Mol. Aspects Med. 2010, 31, 145–170. [Google Scholar] [CrossRef] [PubMed]
- Bertout, J.A.; Patel, S.A.; Simon, M.C. The impact of O2 availability on human cancer. Nat. Rev. Cancer 2008, 8, 967–975. [Google Scholar] [CrossRef] [PubMed]
- Schito, L.; Semenza, G.L. Hypoxia-inducible factors: Master regulators of cancer progression. Trends Cancer 2016, 2, 758–770. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Zhou, Y. Crucial role of the pentose phosphate pathway in malignant tumors. Oncol. Lett. 2019, 17, 4213–4221. [Google Scholar] [CrossRef]
- Patra, K.C.; Hay, N. The pentose phosphate pathway and cancer. Trends Biochem. Sci. 2014, 39, 347–354. [Google Scholar] [CrossRef]
- Chihara, N.; Amo, T.; Tokunaga, A.; Yuzuriha, R.; Wolf, A.M.; Asoh, S.; Suzuki, H.; Uchida, E.; Ohta, S. Mitochondrial DNA alterations in colorectal cancer cell lines. J. Nippon Med. Sch. 2011, 78, 13–21. [Google Scholar] [CrossRef][Green Version]
- Hopkins, J.F.; Sabelnykova, V.Y.; Weischenfeldt, J.; Simon, R.; Aguiar, J.A.; Alkallas, R.; Heisler, L.E.; Zhang, J.; Watson, J.D.; Chua, M.L.K.; et al. Mitochondrial mutations drive prostate cancer aggression. Nat. Commun. 2017, 8, 656. [Google Scholar] [CrossRef]
- Lenaz, G.; Baracca, A.; Carelli, V.; D’Aurelio, M.; Sgarbi, G.; Solaini, G. Bioenergetics of mitochondrial diseases associated with mtDNA mutations. Biochim. Biophys. Acta (BBA) Bioenerg. 2004, 1658, 89–94. [Google Scholar] [CrossRef]
- Ishikawa, K.; Takenaga, K.; Akimoto, M.; Koshikawa, N.; Yamaguchi, A.; Imanishi, H.; Nakada, K.; Honma, Y.; Hayashi, J. ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science 2008, 320, 661–664. [Google Scholar] [CrossRef] [PubMed]
- Guppy, M.; Leedman, P.; Zu, X.; Russell, V. Contribution by different fuels and metabolic pathways to the total ATP turnover of proliferating MCF-7 breast cancer cells. Biochem. J. 2002, 364, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Hervouet, E.; Cizkova, A.; Demont, J.; Vojtiskova, A.; Pecina, P.; Franssen-van Hal, N.L.; Keijer, J.; Simonnet, H.; Ivanek, R.; Kmoch, S.; et al. HIF and reactive oxygen species regulate oxidative phosphorylation in cancer. Carcinogenesis 2008, 29, 1528–1537. [Google Scholar] [CrossRef] [PubMed]
- Selak, M.A.; Armour, S.M.; MacKenzie, E.D.; Boulahbel, H.; Watson, D.G.; Mansfield, K.D.; Pan, Y.; Simon, M.C.; Thompson, C.B.; Gottlieb, E. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell 2005, 7, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Torrens-Mas, M.; Hernandez-Lopez, R.; Pons, D.G.; Roca, P.; Oliver, J.; Sastre-Serra, J. Sirtuin 3 silencing impairs mitochondrial biogenesis and metabolism in colon cancer cells. Am. J. Physiol. Cell Physiol. 2019, 317, C398–C404. [Google Scholar] [CrossRef] [PubMed]
- Gunter, J.; Ruiz-Serrano, A.; Pickel, C.; Wenger, R.H.; Scholz, C.C. The functional interplay between the HIF pathway and the ubiquitin system—More than a one-way road. Exp. Cell Res. 2017, 356, 152–159. [Google Scholar] [CrossRef]
- Alasiri, G.; Fan, L.Y.; Zona, S.; Goldsbrough, I.G.; Ke, H.L.; Auner, H.W.; Lam, E.W. ER stress and cancer: The FOXO forkhead transcription factor link. Mol. Cell. Endocrinol. 2018, 462, 67–81. [Google Scholar] [CrossRef] [PubMed]
- Denko, N.C. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat. Rev. Cancer 2008, 8, 705–713. [Google Scholar] [CrossRef]
- Hyun, D.H.; Hunt, N.D.; Emerson, S.S.; Hernandez, J.O.; Mattson, M.P.; de Cabo, R. Up-regulation of plasma membrane-associated redox activities in neuronal cells lacking functional mitochondria. J. Neurochem. 2007, 100, 1364–1374. [Google Scholar] [CrossRef] [PubMed]
- Lebedeva, M.A.; Eaton, J.S.; Shadel, G.S. Loss of p53 causes mitochondrial DNA depletion and altered mitochondrial reactive oxygen species homeostasis. Biochim. Biophys. Acta (BBA) Bioenerg. 2009, 1787, 328–334. [Google Scholar] [CrossRef]
- Burns, D.M.; Richter, J.D. CPEB regulation of human cellular senescence, energy metabolism, and p53 mRNA translation. Genes Dev. 2008, 22, 3449–3460. [Google Scholar] [CrossRef] [PubMed]
- Guerra, F.; Arbini, A.A.; Moro, L. Mitochondria and cancer chemoresistance. Biochim. Biophys. Acta (BBA) Bioenerg. 2017, 1858, 686–699. [Google Scholar] [CrossRef] [PubMed]
- Pustylnikov, S.; Costabile, F.; Beghi, S.; Facciabene, A. Targeting mitochondria in cancer: Current concepts and immunotherapy approaches. Transl. Res. 2018, 202, 35–51. [Google Scholar] [CrossRef] [PubMed]
- Stefano, G.B.; Kream, R.M. Cancer: Mitochondrial origins. Med. Sci. Monit. 2015, 21, 3736–3739. [Google Scholar] [CrossRef]
- Dei Cas, M.; Ghidoni, R. Cancer Prevention and therapy with polyphenols: Sphingolipid-mediated mechanisms. Nutrients 2018, 10, 940. [Google Scholar] [CrossRef]
- Saso, L.; Firuzi, O. Pharmacological applications of antioxidants: Lights and shadows. Curr. Drug Targets 2014, 15, 1177–1199. [Google Scholar] [CrossRef]
- Cuadrado, A.; Rojo, A.I.; Wells, G.; Hayes, J.D.; Cousin, S.P.; Rumsey, W.L.; Attucks, O.C.; Franklin, S.; Levonen, A.L.; Kensler, T.W.; et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat. Rev. Drug Discov. 2019, 18, 295–317. [Google Scholar] [CrossRef]
- Biersack, B.; Schobert, R. Current state of metal-based drugs for the efficient therapy of lung cancers and lung metastases. Adv. Exp. Med. Biol. 2016, 893, 211–224. [Google Scholar]
- Galadari, S.; Rahman, A.; Pallichankandy, S.; Thayyullathil, F. Reactive oxygen species and cancer paradox: To promote or to suppress? Free Radic. Biol. Med. 2017, 104, 144–164. [Google Scholar] [CrossRef]
- Afzal, S.; Jensen, S.A.; Sorensen, J.B.; Henriksen, T.; Weimann, A.; Poulsen, H.E. Oxidative damage to guanine nucleosides following combination chemotherapy with 5-fluorouracil and oxaliplatin. Cancer Chemother. Pharmacol. 2012, 69, 301–307. [Google Scholar] [CrossRef]
- Alexandre, J.; Hu, Y.; Lu, W.; Pelicano, H.; Huang, P. Novel action of paclitaxel against cancer cells: Bystander effect mediated by reactive oxygen species. Cancer Res. 2007, 67, 3512–3517. [Google Scholar] [CrossRef] [PubMed]
- Ramanathan, B.; Jan, K.Y.; Chen, C.H.; Hour, T.C.; Yu, H.J.; Pu, Y.S. Resistance to paclitaxel is proportional to cellular total antioxidant capacity. Cancer Res. 2005, 65, 8455–8460. [Google Scholar] [CrossRef] [PubMed]
- Fong, S.; Shoemaker, M.; Cadaoas, J.; Lo, A.; Liao, W.; Tagliaferri, M.; Cohen, I.; Shtivelman, E. Molecular mechanisms underlying selective cytotoxic activity of BZL101, an extract of Scutellaria barbata, towards breast cancer cells. Cancer Biol. Ther. 2008, 7, 577–586. [Google Scholar] [CrossRef] [PubMed]
- Zong, W.X.; Ditsworth, D.; Bauer, D.E.; Wang, Z.Q.; Thompson, C.B. Alkylating DNA damage stimulates a regulated form of necrotic cell death. Genes Dev. 2004, 18, 1272–1282. [Google Scholar] [CrossRef] [PubMed]
- Kondoh, H.; Lleonart, M.E.; Nakashima, Y.; Yokode, M.; Tanaka, M.; Bernard, D.; Gil, J.; Beach, D. A high glycolytic flux supports the proliferative potential of murine embryonic stem cells. Antioxid. Redox Signal. 2007, 9, 293–299. [Google Scholar] [CrossRef]
- Geschwind, J.F.; Ko, Y.H.; Torbenson, M.S.; Magee, C.; Pedersen, P.L. Novel therapy for liver cancer: Direct intraarterial injection of a potent inhibitor of ATP production. Cancer Res. 2002, 62, 3909–3913. [Google Scholar]
- Pichla, M.; Sroka, J.; Pienkowska, N.; Piwowarczyk, K.; Madeja, Z.; Bartosz, G.; Sadowska-Bartosz, I. Metastatic prostate cancer cells are highly sensitive to 3-bromopyruvic acid. Life Sci. 2019, 227, 212–223. [Google Scholar] [CrossRef]
- Xian, S.L.; Cao, W.; Zhang, X.D.; Lu, Y.F. 3-Bromopyruvate inhibits human gastric cancer tumor growth in nude mice via the inhibition of glycolysis. Oncol. Lett. 2016, 12, 5377. [Google Scholar] [CrossRef]
- Chen, V.; Staub, R.E.; Fong, S.; Tagliaferri, M.; Cohen, I.; Shtivelman, E. Bezielle selectively targets mitochondria of cancer cells to inhibit glycolysis and OXPHOS. PLoS ONE 2012, 7, e30300. [Google Scholar] [CrossRef]
- Kalyanaraman, B.; Cheng, G.; Hardy, M.; Ouari, O.; Lopez, M.; Joseph, J.; Zielonka, J.; Dwinell, M.B. A review of the basics of mitochondrial bioenergetics, metabolism, and related signaling pathways in cancer cells: Therapeutic targeting of tumor mitochondria with lipophilic cationic compounds. Redox Biol. 2018, 14, 316–327. [Google Scholar] [CrossRef]
- Cheng, G.; Zielonka, J.; McAllister, D.; Tsai, S.; Dwinell, M.B.; Kalyanaraman, B. Profiling and targeting of cellular bioenergetics: Inhibition of pancreatic cancer cell proliferation. Br. J. Cancer 2014, 111, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Glasauer, A.; Chandel, N.S. Targeting antioxidants for cancer therapy. Biochem. Pharmacol. 2014, 92, 90–101. [Google Scholar] [CrossRef] [PubMed]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.T.; Gu, W. SIRT1: Regulator of p53 Deacetylation. Genes Cancer 2013, 4, 112–117. [Google Scholar] [CrossRef]
- Lin, S.J.; Ford, E.; Haigis, M.; Liszt, G.; Guarente, L. Calorie restriction extends yeast life span by lowering the level of NADH. Genes Dev. 2004, 18, 12–16. [Google Scholar] [CrossRef] [PubMed]
- Merker, M.P.; Bongard, R.D.; Kettenhofen, N.J.; Okamoto, Y.; Dawson, C.A. Intracellular redox status affects transplasma membrane electron transport in pulmonary arterial endothelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002, 282, L36–L43. [Google Scholar] [CrossRef]
- Larm, J.A.; Vaillant, F.; Linnane, A.W.; Lawen, A. Up-regulation of the plasma membrane oxidoreductase as a prerequisite for the viability of human Namalwa rho 0 cells. J. Biol. Chem. 1994, 269, 30097–30100. [Google Scholar]
- Scarlett, D.J.; Herst, P.; Tan, A.; Prata, C.; Berridge, M. Mitochondrial gene-knockout (rho0) cells: A versatile model for exploring the secrets of trans-plasma membrane electron transport. Biofactors 2004, 20, 199–206. [Google Scholar] [CrossRef]
- Crane, F.L.; Sun, I.L.; Barr, R.; Low, H. Electron and proton transport across the plasma membrane. J. Bioenerg. Biomembr. 1991, 23, 773–803. [Google Scholar] [CrossRef]
- Hyun, D.H.; Lee, G.H. Cytochrome b5 reductase, a plasma membrane redox enzyme, protects neuronal cells against metabolic and oxidative stress through maintaining redox state and bioenergetics. Age 2015, 37, 122. [Google Scholar] [CrossRef]
- Kim, J.; Kim, S.K.; Kim, H.K.; Mattson, M.P.; Hyun, D.H. Mitochondrial function in human neuroblastoma cells is up-regulated and protected by NQO1, a plasma membrane redox enzyme. PLoS ONE 2013, 8, e69030. [Google Scholar] [CrossRef] [PubMed]
- Martin-Montalvo, A.; Sun, Y.; Diaz-Ruiz, A.; Ali, A.; Gutierrez, V.; Palacios, H.H.; Curtis, J.; Siendones, E.; Ariza, J.; Abulwerdi, G.A.; et al. Cytochrome b5 reductase and the control of lipid metabolism and healthspan. NPJ Aging Mech. Dis. 2016, 2, 16006. [Google Scholar] [CrossRef]
- Xiao, X.; Zhao, W.; Tian, F.; Zhou, X.; Zhang, J.; Huang, T.; Hou, B.; Du, C.; Wang, S.; Mo, Y.; et al. Cytochrome b5 reductase 2 is a novel candidate tumor suppressor gene frequently inactivated by promoter hypermethylation in human nasopharyngeal carcinoma. Tumour Biol. 2014, 35, 3755–3763. [Google Scholar] [CrossRef]
- Kurian, J.R.; Chin, N.A.; Longlais, B.J.; Hayes, K.L.; Trepanier, L.A. Reductive detoxification of arylhydroxylamine carcinogens by human NADH cytochrome b5 reductase and cytochrome b5. Chem. Res. Toxicol. 2006, 19, 1366–1373. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tong, Y.H.; Zhang, B.; Yan, Y.Y.; Fan, Y.; Yu, J.W.; Kong, S.S.; Zhang, D.; Fang, L.; Su, D.; Lin, N.M. Dual-negative expression of Nrf2 and NQO1 predicts superior outcomes in patients with non-small cell lung cancer. Oncotarget 2017, 8, 45750–45758. [Google Scholar] [CrossRef] [PubMed]
- Begleiter, A.; Robotham, E.; Leith, M.K. Role of NAD(P)H:(quinone acceptor) oxidoreductase (DT-diaphorase) in activation of mitomycin C under hypoxia. Mol. Pharmacol. 1992, 41, 677–682. [Google Scholar] [PubMed]
- Cui, X.; Jin, T.; Wang, X.; Jin, G.; Li, Z.; Lin, L. NAD(P)H:quinone oxidoreductase-1 overexpression predicts poor prognosis in small cell lung cancer. Oncol Rep. 2014, 32, 2589–2595. [Google Scholar] [CrossRef]
- Wakai, T.; Shirai, Y.; Sakata, J.; Matsuda, Y.; Korita, P.V.; Takamura, M.; Ajioka, Y.; Hatakeyama, K. Prognostic significance of NQO1 expression in intrahepatic cholangiocarcinoma. Int. J. Clin. Exp. Pathol. 2011, 4, 363–370. [Google Scholar]
- Siegel, D.; Yan, C.; Ross, D. NAD(P)H:quinone oxidoreductase 1 (NQO1) in the sensitivity and resistance to antitumor quinones. Biochem. Pharmacol. 2012, 83, 1033–1040. [Google Scholar] [CrossRef]
- Park, E.J.; Choi, K.S.; Kwon, T.K. beta-Lapachone-induced reactive oxygen species (ROS) generation mediates autophagic cell death in glioma U87 MG cells. Chem. Biol. Interact. 2011, 189, 37–44. [Google Scholar] [CrossRef]
- Dong, G.Z.; Youn, H.; Park, M.T.; Oh, E.T.; Park, K.H.; Song, C.W.; Choi, E.K.; Park, H.J. Heat shock increases expression of NAD(P)H:quinone oxidoreductase (NQO1), mediator of beta-lapachone cytotoxicity, by increasing NQO1 gene activity and via Hsp70-mediated stabilisation of NQO1 protein. Int. J. Hyperth. 2009, 25, 477–487. [Google Scholar] [CrossRef] [PubMed]
- Park, M.T.; Song, M.J.; Lee, H.; Oh, E.T.; Choi, B.H.; Jeong, S.Y.; Choi, E.K.; Park, H.J. Beta-lapachone significantly increases the effect of ionizing radiation to cause mitochondrial apoptosis via JNK activation in cancer cells. PLoS ONE 2011, 6, e25976. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.K.; Terai, K.; Ji, I.M.; Kook, Y.H.; Park, K.H.; Oh, E.T.; Griffin, R.J.; Lim, B.U.; Kim, J.S.; Lee, D.S.; et al. Upregulation of NAD(P)H:quinone oxidoreductase by radiation potentiates the effect of bioreductive beta-lapachone on cancer cells. Neoplasia 2007, 9, 634–642. [Google Scholar] [CrossRef] [PubMed]
- Park, H.J.; Ahn, K.J.; Ahn, S.D.; Choi, E.; Lee, S.W.; Williams, B.; Kim, E.J.; Griffin, R.; Bey, E.A.; Bornmann, W.G.; et al. Susceptibility of cancer cells to beta-lapachone is enhanced by ionizing radiation. Int. J. Radiat. Oncol. Biol. Phys. 2005, 61, 212–219. [Google Scholar] [CrossRef]
- Beaver, S.K.; Mesa-Torres, N.; Pey, A.L.; Timson, D.J. NQO1: A target for the treatment of cancer and neurological diseases, and a model to understand loss of function disease mechanisms. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2019, 1867, 663–676. [Google Scholar] [CrossRef]
- Peng, Q.; Lu, Y.; Lao, X.; Chen, Z.; Li, R.; Sui, J.; Qin, X.; Li, S. The NQO1 Pro187Ser polymorphism and breast cancer susceptibility: Evidence from an updated meta-analysis. Diagn Pathol 2014, 9, 100. [Google Scholar] [CrossRef]
- Renaud, C.O.; Ziros, P.G.; Chartoumpekis, D.V.; Bongiovanni, M.; Sykiotis, G.P. Keap1/Nrf2 Signaling: A new player in thyroid pathophysiology and thyroid cancer. Front. Endocrinol. 2019, 10, 510. [Google Scholar] [CrossRef]
- Yanling, H.; Yuhong, Z.; Wenwu, H.; Lei, X.; Mingwu, C. NQO1 C609T polymorphism and esophageal cancer risk: A HuGE review and meta-analysis. BMC Med. Genet. 2013, 14, 31. [Google Scholar] [CrossRef]
- Siegel, D.; Anwar, A.; Winski, S.L.; Kepa, J.K.; Zolman, K.L.; Ross, D. Rapid polyubiquitination and proteasomal degradation of a mutant form of NAD(P)H:quinone oxidoreductase 1. Mol. Pharmacol. 2001, 59, 263–268. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T.; Talalay, P. NAD(P)H:quinone acceptor oxidoreductase 1 (NQO1), a multifunctional antioxidant enzyme and exceptionally versatile cytoprotector. Arch. Biochem. Biophys. 2010, 501, 116–123. [Google Scholar] [CrossRef]
- Rothman, N.; Smith, M.T.; Hayes, R.B.; Traver, R.D.; Hoener, B.; Campleman, S.; Li, G.L.; Dosemeci, M.; Linet, M.; Zhang, L.; et al. Benzene poisoning, a risk factor for hematological malignancy, is associated with the NQO1 609C-->T mutation and rapid fractional excretion of chlorzoxazone. Cancer Res. 1997, 57, 2839–2842. [Google Scholar] [PubMed]
- Chiu, M.M.; Ko, Y.J.; Tsou, A.P.; Chau, G.Y.; Chau, Y.P. Analysis of NQO1 polymorphisms and p53 protein expression in patients with hepatocellular carcinoma. Histol. Histopathol. 2009, 24, 1223–1232. [Google Scholar]
- Gong, X.; Kole, L.; Iskander, K.; Jaiswal, A.K. NRH:quinone oxidoreductase 2 and NAD(P)H:quinone oxidoreductase 1 protect tumor suppressor p53 against 20 s proteasomal degradation leading to stabilization and activation of p53. Cancer Res. 2007, 67, 5380–5388. [Google Scholar] [CrossRef]
- Anwar, A.; Dehn, D.; Siegel, D.; Kepa, J.K.; Tang, L.J.; Pietenpol, J.A.; Ross, D. Interaction of human NAD(P)H:quinone oxidoreductase 1 (NQO1) with the tumor suppressor protein p53 in cells and cell-free systems. J. Biol. Chem. 2003, 278, 10368–10373. [Google Scholar] [CrossRef] [PubMed]
- Asher, G.; Lotem, J.; Kama, R.; Sachs, L.; Shaul, Y. NQO1 stabilizes p53 through a distinct pathway. Proc. Natl. Acad. Sci. USA 2002, 99, 3099–3104. [Google Scholar] [CrossRef] [PubMed]
- Jaber, S.; Polster, B.M. Idebenone and neuroprotection: Antioxidant, pro-oxidant, or electron carrier? J. Bioenerg. Biomembr. 2015, 47, 111–118. [Google Scholar] [CrossRef]
- Gan, F.F.; Ling, H.; Ang, X.; Reddy, S.A.; Lee, S.S.; Yang, H.; Tan, S.H.; Hayes, J.D.; Chui, W.K.; Chew, E.H. A novel shogaol analog suppresses cancer cell invasion and inflammation, and displays cytoprotective effects through modulation of NF-kappaB and Nrf2-Keap1 signaling pathways. Toxicol. Appl. Pharmacol. 2013, 272, 852–862. [Google Scholar] [CrossRef] [PubMed]
- Bauman, J.E.; Zang, Y.; Sen, M.; Li, C.; Wang, L.; Egner, P.A.; Fahey, J.W.; Normolle, D.P.; Grandis, J.R.; Kensler, T.W.; et al. Prevention of carcinogen-induced oral cancer by sulforaphane. Cancer Prev. Res. 2016, 9, 547–557. [Google Scholar] [CrossRef]
- Leone, A.; Diorio, G.; Sexton, W.; Schell, M.; Alexandrow, M.; Fahey, J.W.; Kumar, N.B. Sulforaphane for the chemoprevention of bladder cancer: Molecular mechanism targeted approach. Oncotarget 2017, 8, 35412–35424. [Google Scholar] [CrossRef]
- Kan, S.F.; Wang, J.; Sun, G.X. Sulforaphane regulates apoptosis- and proliferationrelated signaling pathways and synergizes with cisplatin to suppress human ovarian cancer. Int. J. Mol. Med. 2018, 42, 2447–2458. [Google Scholar]
- Aumeeruddy, M.Z.; Mahomoodally, M.F. Combating breast cancer using combination therapy with 3 phytochemicals: Piperine, sulforaphane, and thymoquinone. Cancer 2019, 125, 1600–1611. [Google Scholar] [CrossRef]
- Deguchi, A. Curcumin targets in inflammation and cancer. Endocr. Metab. Immune Disord. Drug Targets 2015, 15, 88–96. [Google Scholar] [CrossRef]
- Giordano, A.; Tommonaro, G. Curcumin and cancer. Nutrients 2019, 11, 2376. [Google Scholar] [CrossRef]
- Bettum, I.J.; Gorad, S.S.; Barkovskaya, A.; Pettersen, S.; Moestue, S.A.; Vasiliauskaite, K.; Tenstad, E.; Oyjord, T.; Risa, O.; Nygaard, V.; et al. Metabolic reprogramming supports the invasive phenotype in malignant melanoma. Cancer Lett. 2015, 366, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Hyun, D.H. Plasma membrane redox enzymes: New therapeutic targets for neurodegenerative diseases. Arch. Pharm. Res. 2019, 42, 436–445. [Google Scholar] [CrossRef] [PubMed]
- Hyun, D.H.; Hernandez, J.O.; Mattson, M.P.; de Cabo, R. The plasma membrane redox system in aging. Ageing Res. Rev. 2006, 5, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Sajadimajd, S.; Khazaei, M. Oxidative stress and cancer: The role of Nrf2. Curr. Cancer Drug Targets 2018, 18, 538–557. [Google Scholar] [CrossRef]




© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hyun, D.-H. Insights into the New Cancer Therapy through Redox Homeostasis and Metabolic Shifts. Cancers 2020, 12, 1822. https://doi.org/10.3390/cancers12071822
Hyun D-H. Insights into the New Cancer Therapy through Redox Homeostasis and Metabolic Shifts. Cancers. 2020; 12(7):1822. https://doi.org/10.3390/cancers12071822
Chicago/Turabian StyleHyun, Dong-Hoon. 2020. "Insights into the New Cancer Therapy through Redox Homeostasis and Metabolic Shifts" Cancers 12, no. 7: 1822. https://doi.org/10.3390/cancers12071822
APA StyleHyun, D.-H. (2020). Insights into the New Cancer Therapy through Redox Homeostasis and Metabolic Shifts. Cancers, 12(7), 1822. https://doi.org/10.3390/cancers12071822