XRN2 Links RNA:DNA Hybrid Resolution to Double Strand Break Repair Pathway Choice


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
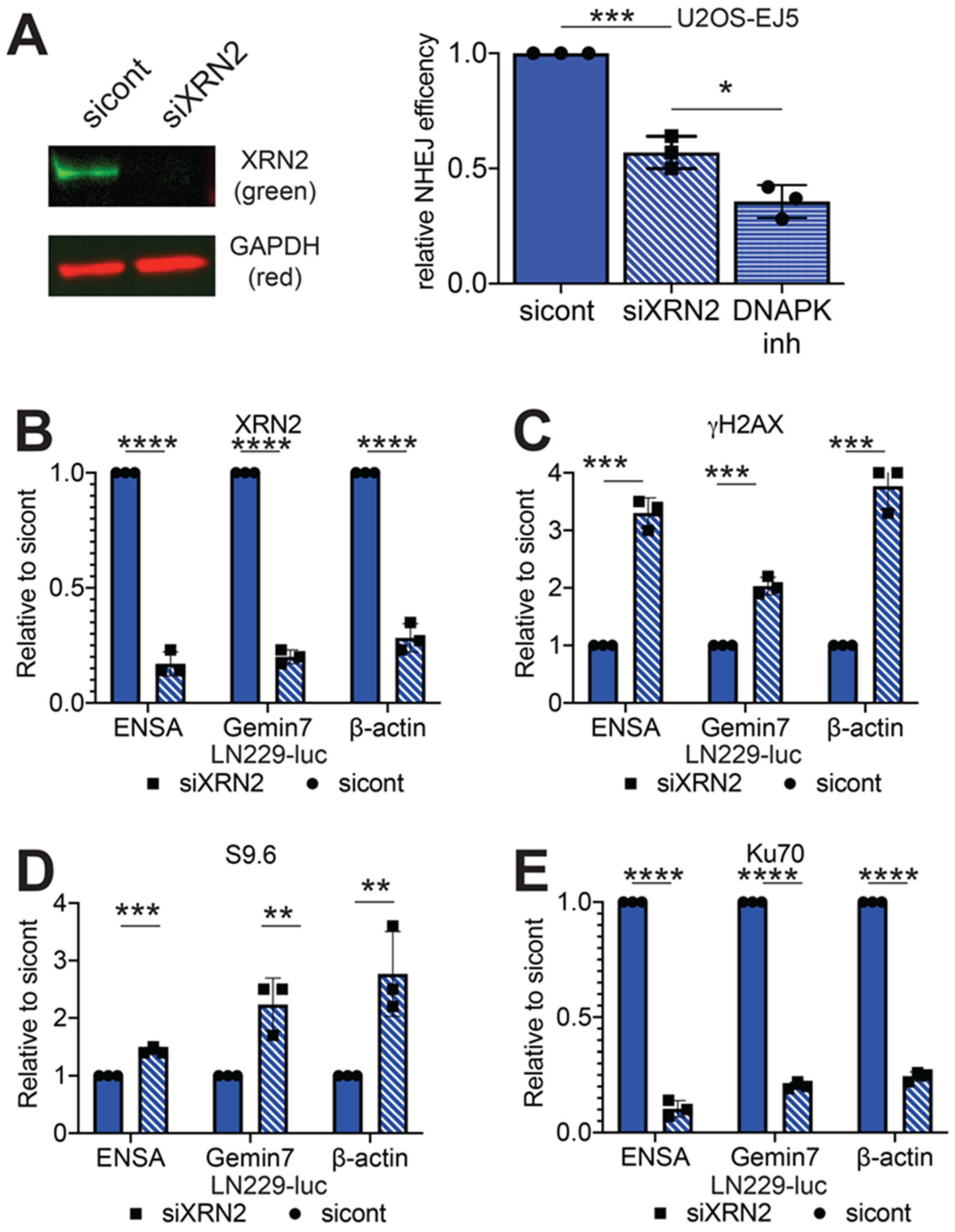
2.1. Loss of XRN2 Abrogates Ku70 Binding at the DSB Repair Site
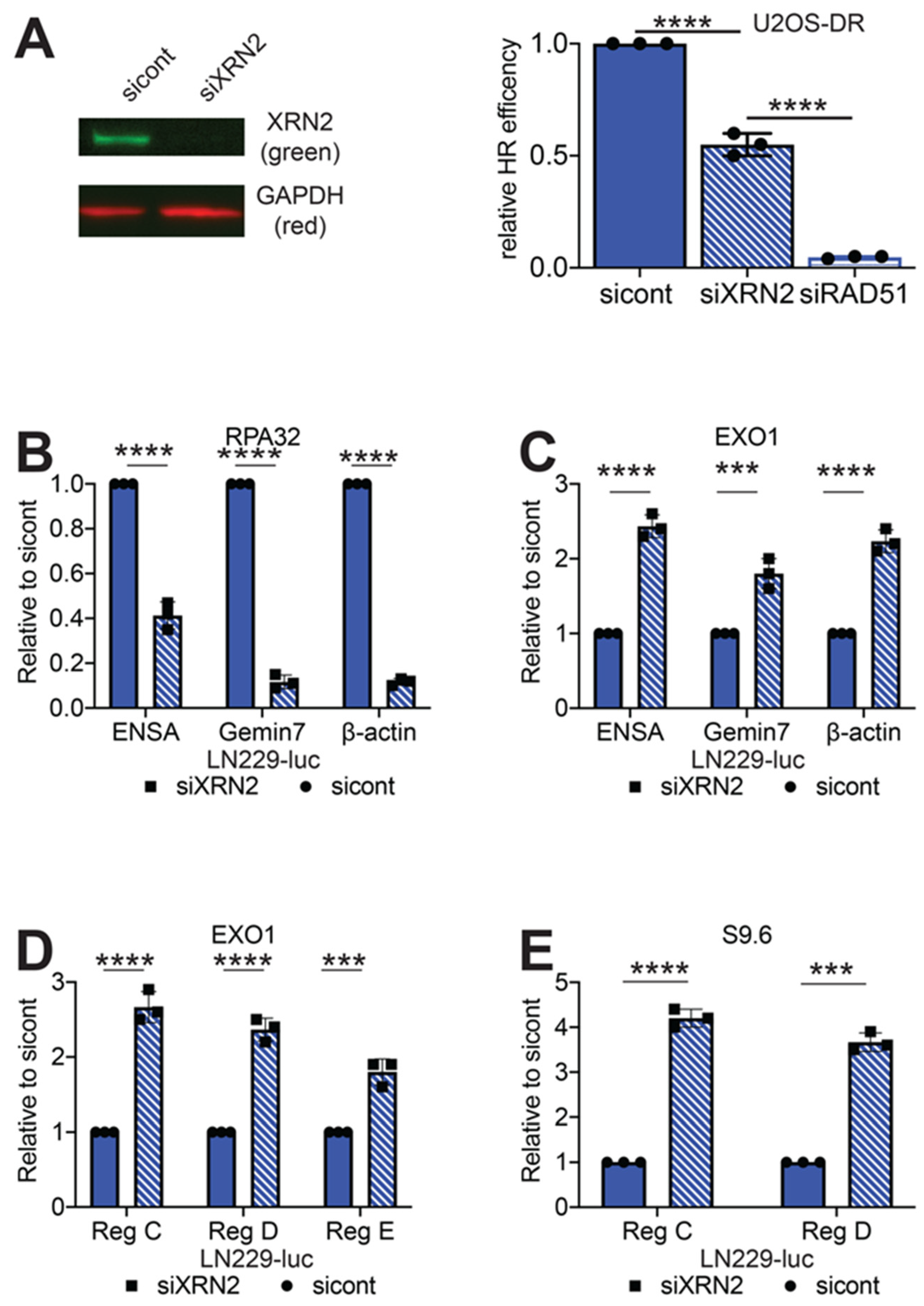
2.2. Loss of XRN2 Results in Impaired HR Repair
2.3. Loss of XRN2 Results in an Extended Association of EXO1 with Chromatin
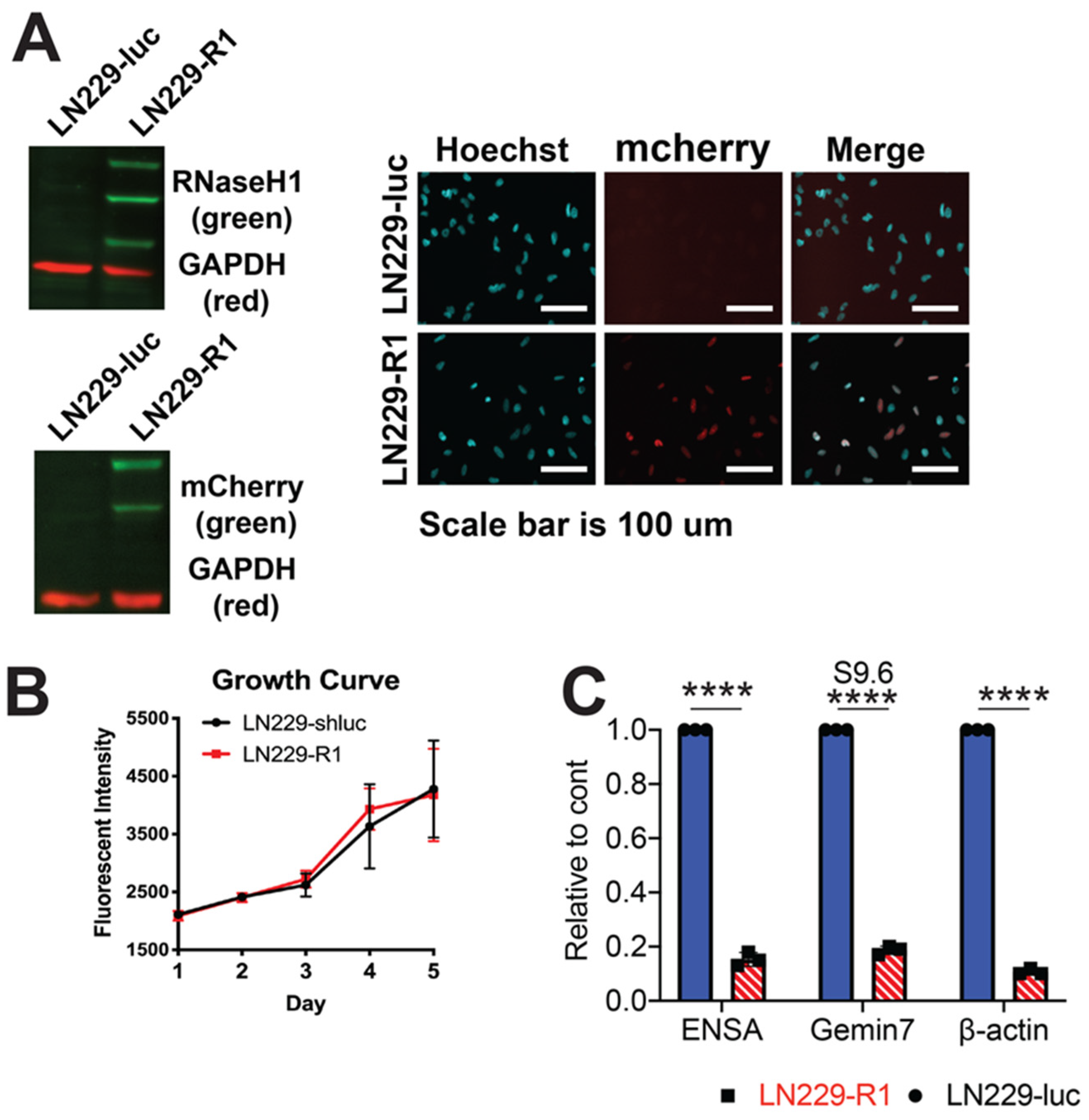
2.4. RNaseH1 Expression Restores NHEJ Repair
2.5. RNaseH1 Expression Does Not Restore HR Repair
3. Discussion
3.1. Mechanistic Function of XRN2 in NHEJ Repair
3.2. XRN2 Loss Inhibits HR Repair Due to Excessive DNA End Resection
4. Materials and Methods
4.1. Colony Forming Assays
4.2. Cell Culture
4.3. Generation of Human RNaseH1 Cells
4.4. Western Blot
4.5. Immunofluorescence (IF)
4.6. I-SecI-Based DNA Repair Assays
4.7. Imaging
4.8. Chromatin Immuno-Precipitation/qPCR (ChIP/qPCR)
4.9. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zhou, B.B.S.; Elledge, S.J. The DNA damage response: Putting checkpoints in perspective. Nature 2000, 408, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Jimeno, S.; Mejías-Navarro, F.; Prados-Carvajal, R.; Huertas, P. Chapter Four—Controlling the balance between chromosome break repair pathways. In Advances in Protein Chemistry and Structural Biology; Donev, R., Ed.; Academic Press: Cambridge, MA, USA, 2019; pp. 95–134. [Google Scholar]
- Shibata, A. Regulation of repair pathway choice at two-ended DNA double-strand breaks. Mutation Res. Fundam. Mol. Mech. Mutagenesis 2017, 803, 51–55. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, J.; Lan, L.; Zou, L. Regulation of DNA break repair by transcription and RNA. Sci. China Life Sci. 2017, 60, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Janssen, A.; Breuer, G.A.; Brinkman, E.K.; van der Meulen, A.I.; Borden, S.V.; van Steensel, B.; Bindra, R.S.; Larocque, J.R.; Karpen, G.H. A single double-strand break system reveals repair dynamics and mechanisms in heterochromatin and euchromatin. Genes Dev. 2016, 30, 1645–1657. [Google Scholar] [CrossRef]
- Aguilera, A.; Gómez-González, B. DNA–RNA hybrids: The risks of DNA breakage during transcription. Nat. Struct. Mol. Biol. 2017, 24, 439. [Google Scholar] [CrossRef]
- Li, X.; Manley, J.L. Inactivation of the SR Protein Splicing Factor ASF/SF2 Results in Genomic Instability. Cell 2005, 122, 365–378. [Google Scholar] [CrossRef]
- Morales, J.C.; Richard, P.; Patidar, P.L.; Motea, E.A.; Dang, T.T.; Manley, J.L.; Boothman, D.A. XRN2 Links Transcription Termination to DNA Damage and Replication Stress. PLoS Genet. 2016, 12, e1006107. [Google Scholar] [CrossRef]
- Chakraborty, P.; Huang, J.T.J.; Hiom, K. DHX9 helicase promotes R-loop formation in cells with impaired RNA splicing. Nat. Commun. 2018, 9, 4346. [Google Scholar] [CrossRef]
- Cohen, S.; Puget, N.; Lin, Y.; Clouaire, T.; Aguirrebengoa, M.; Rocher, V.; Pasero, P.; Canitrot, Y.; Legube, G. Senataxin resolves RNA: DNA hybrids forming at DNA double-strand breaks to prevent translocations. Nat. Commun. 2018, 9, 533. [Google Scholar] [CrossRef]
- Ohle, C.; Tesorero, R.; Schermann, G.; Dobrev, N.; Sinning, I.; Fischer, T. Transient RNA-DNA Hybrids Are Required for Efficient Double-Strand Break Repair. Cell 2016, 167, 1001–1013. [Google Scholar] [CrossRef]
- West, S.; Gromak, N.; Proudfoot, N.J. Human 5′ → 3′ exonuclease Xrn2 promotes transcription termination at co-transcriptional cleavage sites. Nature 2004, 432, 522–525. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, S.; Rozenblatt-Rosen, O.; Meyerson, M.; Manley, J.L. The multifunctional protein p54nrb/PSF recruits the exonuclease XRN2 to facilitate pre-mRNA 3′ processing and transcription termination. Genes Dev. 2007, 21, 1779–1789. [Google Scholar] [CrossRef] [PubMed]
- Bennardo, N.; Cheng, A.; Huang, N.; Stark, J.M. Alternative-NHEJ Is a Mechanistically Distinct Pathway of Mammalian Chromosome Break Repair. PLoS Genet. 2008, 4, e1000110. [Google Scholar] [CrossRef] [PubMed]
- Gunn, A.; Stark, J.M. I-SceI-Based Assays to Examine Distinct Repair Outcomes of Mammalian Chromosomal Double Strand Breaks. In DNA Repair Protocols; Bjergbæk, L., Ed.; Humana Press: Totowa, NJ, USA, 2012; pp. 379–391. [Google Scholar]
- Hollick, J.J.; Golding, B.T.; Hardcastle, I.R.; Martin, N.M.B.; Richardson, C.; Rigoreau, L.; Smith, G.C.M.; Griffin, R.J. 2,6-Disubstituted pyran-4-one and thiopyran-4-one inhibitors of DNA-Dependent protein kinase (DNA-PK). Bioorganic Med. Chem. Lett. 2003, 13, 3083–3086. [Google Scholar] [CrossRef]
- Uematsu, N.; Weterings, E.; Yano, K.; Morotomi-Yano, K.; Jakob, B.; Taucher-Scholz, G.; Mari, P.; van Gent, D.C.; Chen, B.P.C.; Chen, D.J. Autophosphorylation of DNA-PKCS regulates its dynamics at DNA double-strand breaks. J. Cell Biol. 2007, 177, 219. [Google Scholar] [CrossRef]
- Hu, Z.; Zhang, A.; Storz, G.; Gottesman, S.; Leppla, S.H. An antibody-based microarray assay for small RNA detection. Nucleic Acids Res. 2006, 34, e52. [Google Scholar] [CrossRef][Green Version]
- Mansour, W.Y.; Rhein, T.; Dahm-Daphi, J. The alternative end-joining pathway for repair of DNA double-strand breaks requires PARP1 but is not dependent upon microhomologies. Nucleic Acids Res. 2010, 38, 6065–6077. [Google Scholar] [CrossRef]
- Jones, P.; Altamura, S.; Boueres, J.K.; Ferrigno, F.; Fonsi, M.; Giomini, C.; Lamartina, S.; Monteagudo, E.; Ontoria, J.M.; Orsale, M.V.; et al. Discovery of 2-{4-[(3S)-Piperidin-3-yl]phenyl}-2H-indazole-7-carboxamide (MK-4827): A Novel Oral Poly(ADP-ribose)polymerase (PARP) Inhibitor Efficacious in BRCA-1 and -2 Mutant Tumors. J. Med. Chem. 2009, 52, 7170–7185. [Google Scholar] [CrossRef]
- Nakamoto, M.Y.; Rudolph, J.; Wuttke, D.S.; Luger, K. Nonspecific Binding of RNA to PARP1 and PARP2 Does Not Lead to Catalytic Activation. Biochemistry 2019, 58, 5107–5111. [Google Scholar] [CrossRef]
- Motea, E.A.; Fattah, F.J.; Xiao, L.; Girard, L.; Rommel, A.; Morales, J.C.; Patidar, P.; Zhou, Y.; Porter, A.; Xie, Y.; et al. Kub5-HeraRPRD1B Deficiency Promotes “BRCAness” and Vulnerability to PARP Inhibition in BRCA-proficient Breast Cancers. Clin. Cancer Res. 2018, 24, 6459. [Google Scholar] [CrossRef]
- De Lorenzo, S.; Patel, A.; Hurley, R.; Kaufmann, S. The Elephant and the Blind Men: Making Sense of PARP Inhibitors in Homologous Recombination Deficient Tumor Cells. Front. Oncol. 2013, 3. [Google Scholar] [CrossRef] [PubMed]
- Bennardo, N.; Gunn, A.; Cheng, A.; Hasty, P.; Stark, J.M. Limiting the Persistence of a Chromosome Break Diminishes Its Mutagenic Potential. PLoS Genet. 2009, 5, e1000683. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.D.; Yadav, T.; Giri, S.; Saez, B.; Graubert, T.A.; Zou, L. Functions of Replication Protein A as a Sensor of R Loops and a Regulator of RNaseH1. Mol. Cell 2017, 65, 832–847. [Google Scholar] [CrossRef] [PubMed]
- Tomimatsu, N.; Mukherjee, B.; Harris, J.L.; Boffo, F.L.; Hardebeck, M.C.; Potts, P.R.; Khanna, K.K.; Burma, S. DNA-damage-induced degradation of EXO1 exonuclease limits DNA end resection to ensure accurate DNA repair. J. Biol. Chem. 2017, 292, 10779–10790. [Google Scholar] [CrossRef]
- Hatchi, E.; Skourtistathaki, K.; Ventz, S.; Pinello, L.; Yen, A.; Kamieniarzgdula, K.; Dimitrov, S.D.; Pathania, S.; Mckinney, K.M.; Eaton, M.L.; et al. BRCA1 Recruitment to Transcriptional Pause Sites Is Required for R-Loop-Driven DNA Damage Repair. Mol. Cell 2015, 57, 636–647. [Google Scholar] [CrossRef]
- Furukawa, Y.; Nishimura, N.; Furukawa, Y.; Satoh, M.; Endo, H.; Iwase, S.; Yamada, H.; Matsuda, M.; Kano, Y.; Nakamura, M. Apaf-1 Is a Mediator of E2F-1-induced Apoptosis. J. Biol. Chem. 2002, 277, 39760–39768. [Google Scholar] [CrossRef]
- Sutheesophon, K.; Kobayashi, Y.; Takatoku, M.; Ozawa, K.; Kano, Y.; Ishii, H.; Furukawa, Y. Histone Deacetylase Inhibitor Depsipeptide (FK228) Induces Apoptosis in Leukemic Cells by Facilitating Mitochondrial Translocation of Bax, Which Is Enhanced by the Proteasome Inhibitor Bortezomib. Acta Haematol. 2006, 115, 78–90. [Google Scholar] [CrossRef]
- Bennardo, N.; Stark, J.M. ATM Limits Incorrect End Utilization during Non-Homologous End Joining of Multiple Chromosome Breaks. PLoS Genet. 2010, 6, e1001194. [Google Scholar] [CrossRef]
- Rawal, C.C.; Zardoni, L.; Di Terlizzi, M.; Galati, E.; Brambati, A.; Lazzaro, F.; Liberi, G.; Pellicioli, A. Senataxin Ortholog Sen1 Limits DNA: RNA Hybrid Accumulation at DNA Double-Strand Breaks to Control End Resection and Repair Fidelity. Cell Rep. 2020, 31, 107603. [Google Scholar] [CrossRef]
- Fell, V.L.; Schild-Poulter, C. The Ku heterodimer: Function in DNA repair and beyond. Mutat. Res. Rev. Mutat. Res. 2015, 763, 15–29. [Google Scholar] [CrossRef]
- Anisenko, A.N.; Knyazhanskaya, E.S.; Zatsepin, T.S.; Gottikh, M.B. Human Ku70 protein binds hairpin RNA and double stranded DNA through two different sites. Biochimie 2017, 132, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Horton, N.C.; Finzel, B.C. The Structure of an RNA/DNA Hybrid: A Substrate of the Ribonuclease Activity of HIV-1 Reverse Transcriptase. J. Mol. Biol. 1996, 264, 521–533. [Google Scholar] [CrossRef] [PubMed]
- Nauwelarts, K.; Lescrinier, E.; Herdewijn, P. Structure of the alpha-Homo-DNA: RNA Duplex and the Function of the Twist and Slide to Catalouge Nucleic Acid Duplex. Chemistry 2007, 13, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Collinge, M.A.; Althaus, F.R. Expression of human poly(ADP-ribose) polymerase in Saccharomyces cerevisiae. Mol. Gen. Genet. 1994, 245, 686–693. [Google Scholar] [CrossRef] [PubMed]
- Cristini, A.; Groh, M.; Kristiansen, M.S.; Gromak, N. RNA/DNA Hybrid Interactome Identifies DXH9 as a Molecular Player in Transcriptional Termination and R-Loop-Associated DNA Damage. Cell Rep. 2018, 23, 1891–1905. [Google Scholar] [CrossRef]
- Wang, I.X.; Grunseich, C.; Fox, J.; Burdick, J.T.; Zhu, Z.; Ravazian, N.; Hafner, M.; Cheung, V.G. Human proteins that interact with RNA/DNA hybrids. Genome Res. 2018, 28, 1405–1414. [Google Scholar] [CrossRef]
- Chou, D.M.; Adamson, B.S.; Dephoure, N.; Tan, X.; Nottke, A.C.; Hurov, K.E.; Gygi, S.P.; Colaiacovo, M.P.; Elledge, S.J. A chromatin localization screen reveals poly (ADP ribose)-regulated recruitment of the repressive polycomb and NuRD complexes to sites of DNA damage. Proc. Natl. Acad. Sci. USA 2010, 107, 18475. [Google Scholar] [CrossRef]
- Makharashvili, N.; Paull, T.T. CtIP: A DNA damage response protein at the intersection of DNA metabolism. DNA Repair 2015, 32, 75–81. [Google Scholar] [CrossRef]
- Zong, D.; Ray Chaudhuri, A.; Nussenzweig, A. More end resection is not merrier. Nat. Struct. Mol. Biol. 2016, 23, 699. [Google Scholar] [CrossRef]
- Pearson, G.W.; Hunter, T. Real-time imaging reveals that noninvasive mammary epithelial acini can contain motile cells. J. Cell Biol. 2007, 179, 1555–1567. [Google Scholar] [CrossRef]
- Dang, T.T.; Esparza, M.A.; Maine, E.A.; Westcott, J.M.; Pearson, G.W. ΔNp63α Promotes Breast Cancer Cell Motility through the Selective Activation of Components of the Epithelial-to-Mesenchymal Transition Program. Cancer Res. 2015, 75, 3925–3935. [Google Scholar] [CrossRef]
- Stork, C.T.; Bocek, M.J.; Crossley, M.P.; Sollier, J.; Sanz, L.A.; Chedin, F.; Swigut, T.; Cimprich, K.A. Co-transcriptional R-loops are the main cause of estrogen-induced DNA damage. eLife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; McBride, K.M.; Hensley, S.; Lu, Y.; Chedin, F.; Bedford, M.T. Arginine methylation facilitates the recruitment of TOP3B to chromatin to prevent R loop accumulation. Mol. Cell 2014, 53, 484–497. [Google Scholar] [CrossRef] [PubMed]
- Chiarle, R.; Zhang, Y.; Frock, R.L.; Lewis, S.; Molinie, B.; Ho, Y.; Myers, D.; Choi, V.W.; Compagno, M.; Malkin, D.J.; et al. Genome-wide translocation sequencing reveals mechanisms of chromosome breaks and rearrangements in B cells. Cell 2011, 147, 107–119. [Google Scholar] [CrossRef] [PubMed]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dang, T.T.; Morales, J.C. XRN2 Links RNA:DNA Hybrid Resolution to Double Strand Break Repair Pathway Choice. Cancers 2020, 12, 1821. https://doi.org/10.3390/cancers12071821
Dang TT, Morales JC. XRN2 Links RNA:DNA Hybrid Resolution to Double Strand Break Repair Pathway Choice. Cancers. 2020; 12(7):1821. https://doi.org/10.3390/cancers12071821
Chicago/Turabian StyleDang, Tuyen T., and Julio C. Morales. 2020. "XRN2 Links RNA:DNA Hybrid Resolution to Double Strand Break Repair Pathway Choice" Cancers 12, no. 7: 1821. https://doi.org/10.3390/cancers12071821
APA StyleDang, T. T., & Morales, J. C. (2020). XRN2 Links RNA:DNA Hybrid Resolution to Double Strand Break Repair Pathway Choice. Cancers, 12(7), 1821. https://doi.org/10.3390/cancers12071821