Therapeutic Strategies and Biomarkers to Modulate PARP Activity for Targeted Cancer Therapy

 ,
,
Abstract
1. Introduction
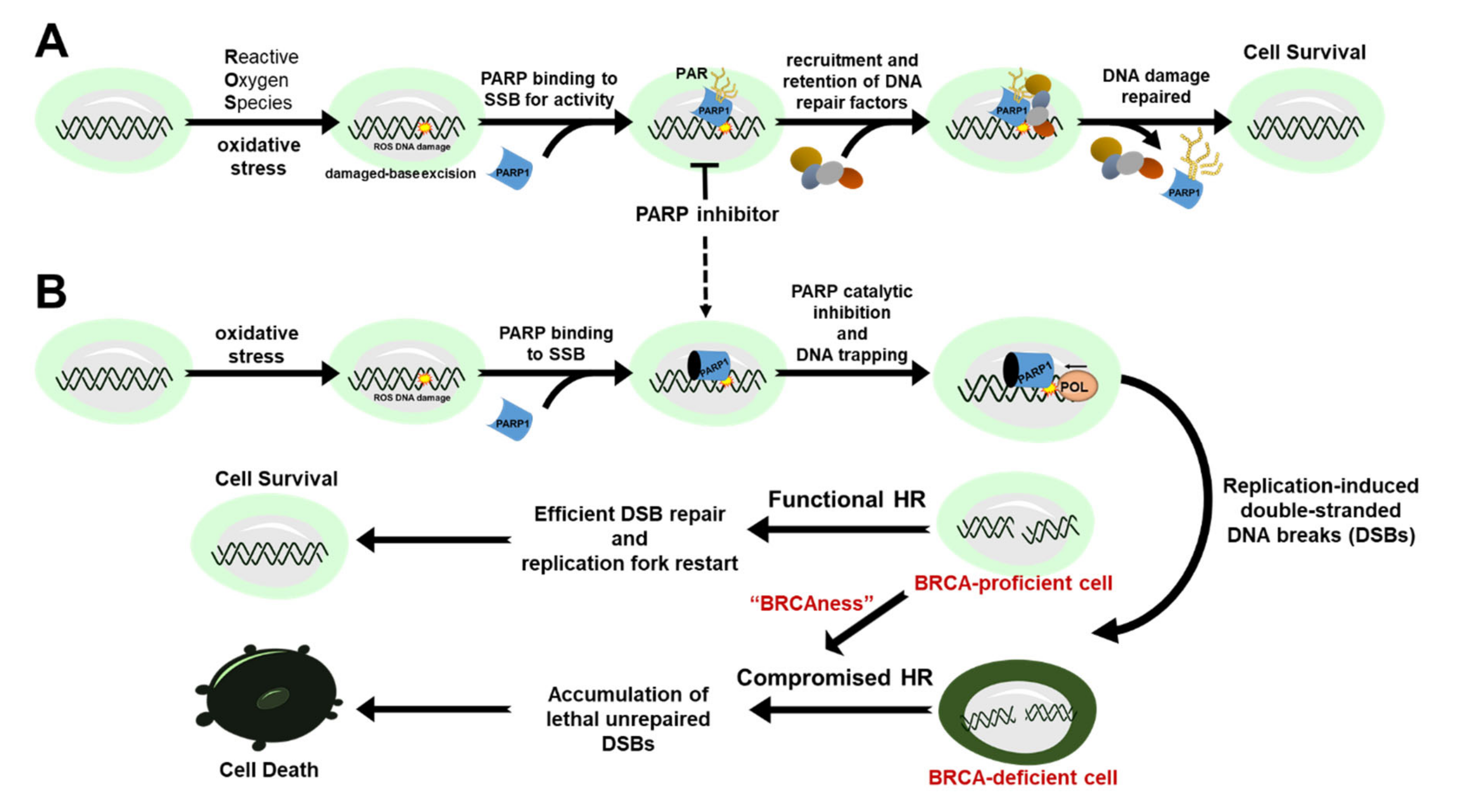
2. Mechanism of Action for PARP Inhibitors
3. Approved PARP Inhibitors for Targeted Cancer Therapy
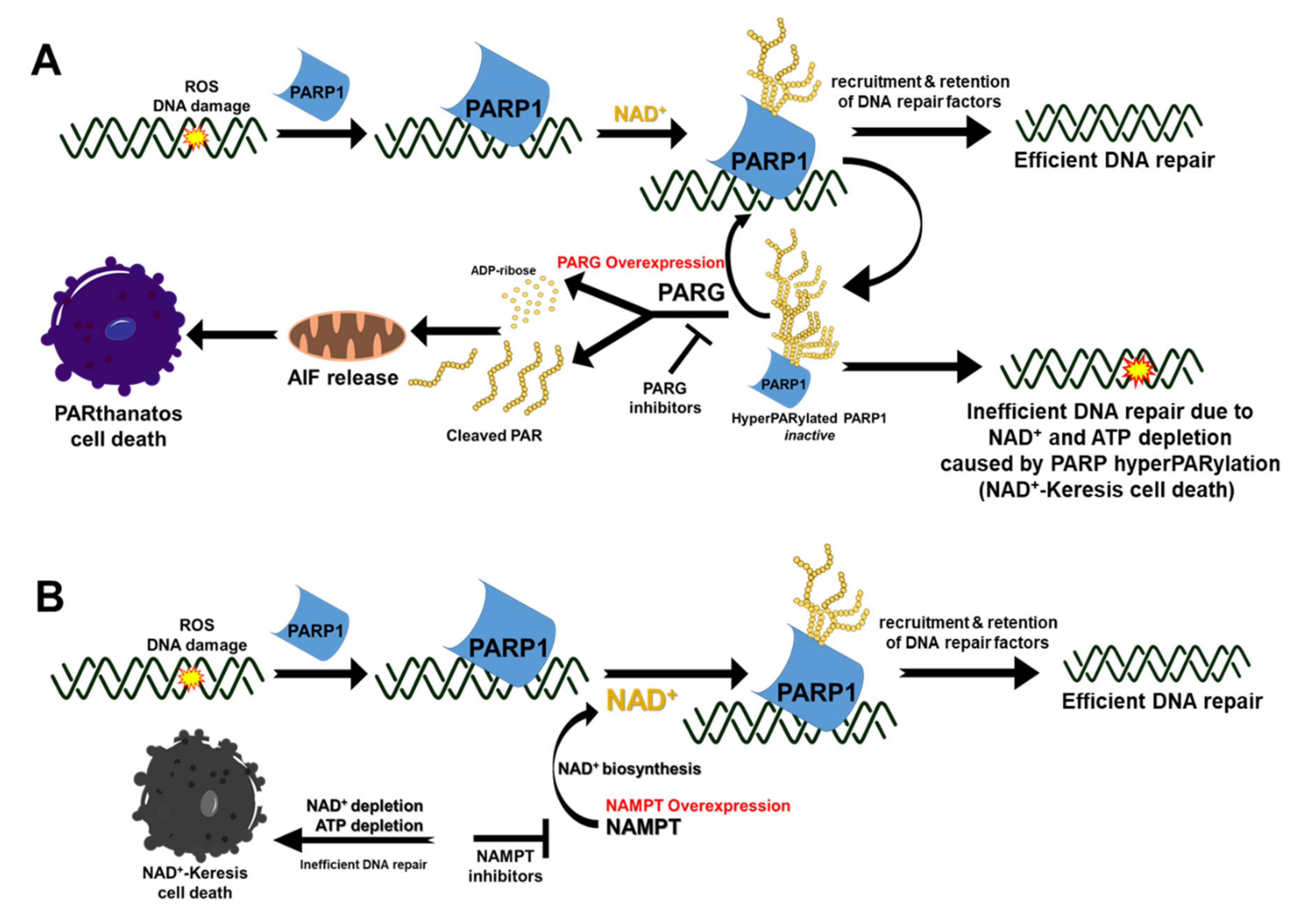
4. Harnessing the Power of PAR and PARG Inhibition for Cancer Therapy
5. Targeting NAD+ for Cancer Therapy
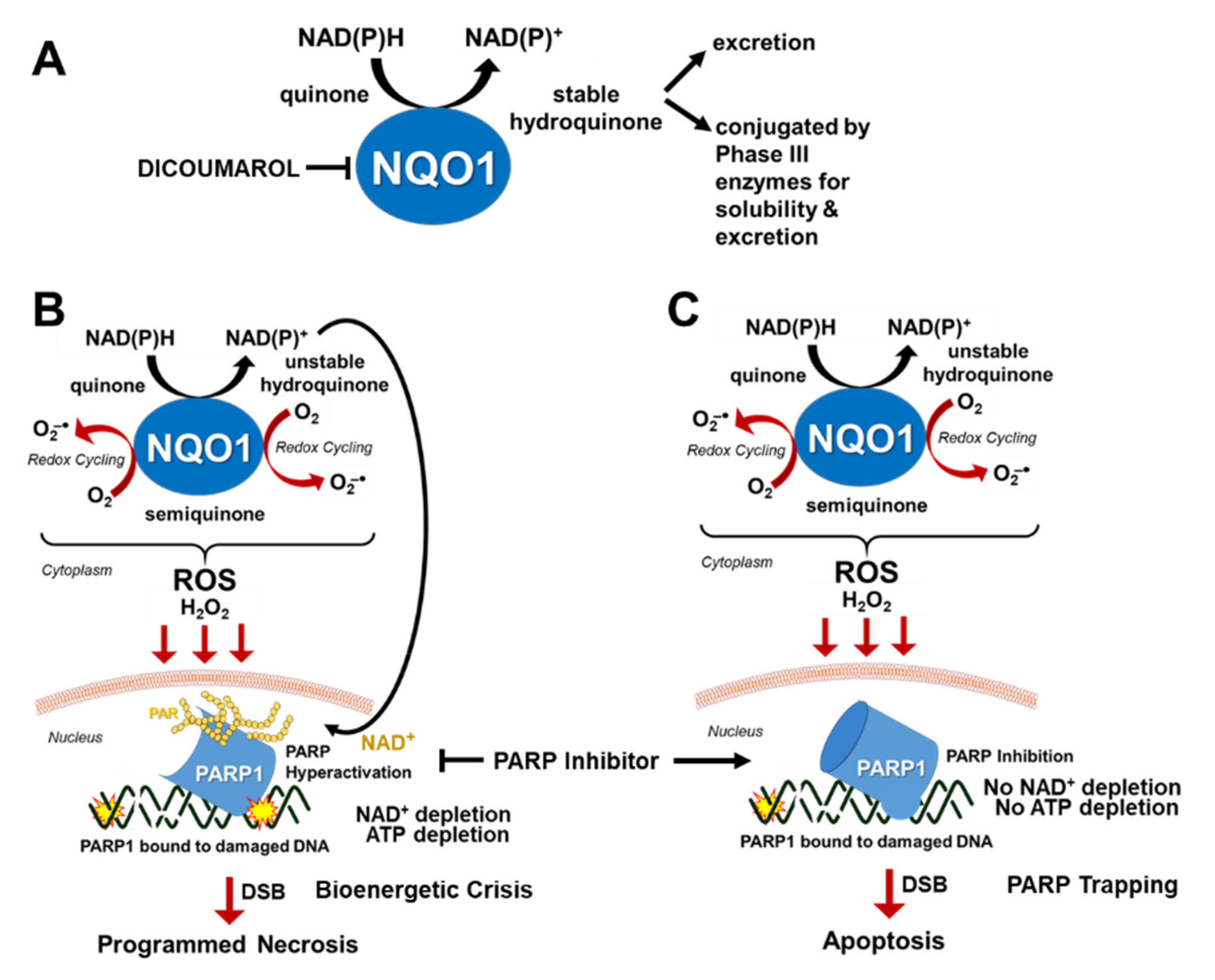
6. Leveraging NQO1 as a Biomarker for Tumor-Selective Use of PARP Inhibitors with NQO1-Bioactivatable Drugs
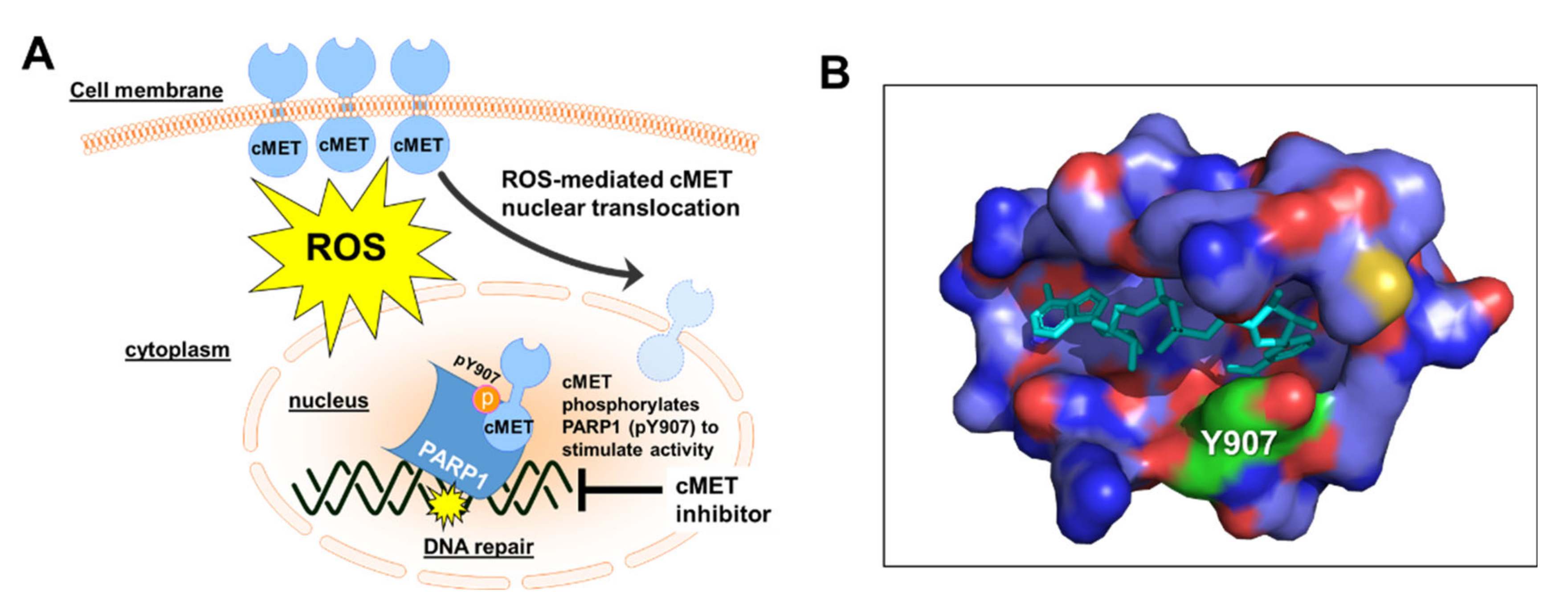
7. Targeting cMET to Attenuate PARP1 Activity
8. Targeting of PARP Activity in Non-Oncological Events
9. Perspectives and Conclusions

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein Name | Primary Function/Activity | Association with “BRCAness” | Reference |
|---|---|---|---|
| CDK1 | Cell cycle regulation | Loss of expression or activity inhibition compromises phosphorylation of BRCA1 for proper HR function | [30] |
| CDK12/13 | Phosphorylates RNAPII CTD | Loss of expression and activity inhibition suppresses expression of specific HR proteins such as RAD51 and BRCA1 | [147] |
| AXL | A receptor tyrosine kinase associated with metastasis, invasion and migration in many cancers | Loss of expression or activity inhibition decreases expression of specific HR genes and proteins | [148] |
| Kub5-Hera, RPRD1B, CREPT | Transcription termination factor | Loss of expression compromises HR by decreasing CDK1 expression | [25] |
| WEE1 | Involved in the terminal phosphorylation and inactivation of CDK1-bound cyclin B | Activity inhibition with AZD1775 indirectly inhibits BRCA2 | [149] |
| UCHL3 | Deubiquitinase | Activity inhibition with perifosine promotes ubiquitination of RAD51 and blocks the binding of RAD51 with BRCA2 | [150] |
| BET | Transcriptional regulators | Activity inhibition with JQ1 decreases expression of RAD51 and Ku80 | [151] |
| PI3K | Kinase involve in cell growth, proliferation, differentiation, motility, survival and intracellular trafficking | Inhibition of activity impairs BRCA1/2 expression | [152] |
| Cyclin D1 | Regulator of CDKs (cyclin dependent kinases), required for cell cycle G1/S transition | Loss of expression impairs recruitment of RAD51 | [153,154] |
| AURKA | Play important role in mitosis/ regulation of cell cycle progression | Activity inhibition or loss of expression decreases expression of BRCA1 and BRCA2 | [155,156] |
| HKMT | Regulation of histone methylation | Inhibition of activity abolishes retention of BRCA1/BARD1 complexes at sites of DSB | [157,158] |
| CCDC6 | Tumor suppressor | Loss of expression compromises BRCC3 and DNA damage checkpoints in response to DNA damage. | [158,159] |
| MEK | Kinase that phosphorylates and activates MAPK | Activity inhibition or loss of expression downregulates BRCA2 | [160] |
| HDAC | Removes acetyl groups from an amino acid on a histone | Activity inhibition with SAHA reduces BRCA1 protein levels by targeting the UHRF1/BRCA1 protein complex | [161] |
| PAK1 | Regulates cytoskeleton remodeling, phenotypic signaling and gene expression | Reduced activity and loss of expression downregulates the expression of genes involved in FA/BRCA pathway | [162] |
| Androgen receptor | DNA-binding transcription factor that regulates gene expression | Activity inhibition or loss of expression suppresses the expression of HR genes, thus creating HR deficiency and BRCAness | [163] |
| TGFβ | Involved in embryonic development, cell proliferation, motility and apoptosis, extracellular matrix production, and immunomodulation | Overexpression suppresses BRCA1, ATM, and MSH2 | [164] |
Author Contributions
Funding
Conflicts of Interest
References
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [PubMed]
- Hottiger, M.O.; Hassa, P.O.; Luscher, B.; Schuler, H.; Koch-Nolte, F. Toward a unified nomenclature for mammalian ADP-ribosyltransferases. Trends Biochem. Sci. 2010, 35, 208–219. [Google Scholar] [CrossRef] [PubMed]
- D’Amours, D.; Desnoyers, S.; D’Silva, I.; Poirier, G.G. Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem. J. 1999, 342 Pt 2, 249–268. [Google Scholar] [CrossRef]
- Gibson, B.A.; Kraus, W.L. New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat. Rev. Mol. Cell Biol. 2012, 13, 411–424. [Google Scholar] [CrossRef] [PubMed]
- Hottiger, M.O. Nuclear ADP-Ribosylation and Its Role in Chromatin Plasticity, Cell Differentiation, and Epigenetics. Annu. Rev. Biochem. 2015, 84, 227–263. [Google Scholar] [CrossRef] [PubMed]
- Pascal, J.M. The comings and goings of PARP-1 in response to DNA damage. DNA Repair (Amst) 2018, 71, 177–182. [Google Scholar] [CrossRef]
- Wei, H.; Yu, X. Functions of PARylation in DNA Damage Repair Pathways. Genomics Proteomics Bioinforma. 2016, 14, 131–139. [Google Scholar] [CrossRef]
- Hossain, M.A.; Lin, Y.; Yan, S. Single-Strand Break End Resection in Genome Integrity: Mechanism and Regulation by APE2. Int. J. Mol. Sci. 2018, 19, 2389. [Google Scholar] [CrossRef]
- Panier, S.; Durocher, D. Push back to respond better: Regulatory inhibition of the DNA double-strand break response. Nat. Rev. Mol. Cell Biol. 2013, 14, 661–672. [Google Scholar] [CrossRef]
- Lieber, M.R. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem. 2010, 79, 181–211. [Google Scholar] [CrossRef]
- Deriano, L.; Roth, D.B. Modernizing the nonhomologous end-joining repertoire: Alternative and classical NHEJ share the stage. Annu. Rev. Genet. 2013, 47, 433–455. [Google Scholar] [CrossRef] [PubMed]
- Sfeir, A.; Symington, L.S. Microhomology-Mediated End Joining: A Back-up Survival Mechanism or Dedicated Pathway? Trends Biochem. Sci. 2015, 40, 701–714. [Google Scholar] [CrossRef] [PubMed]
- Chae, Y.K.; Anker, J.F.; Carneiro, B.A.; Chandra, S.; Kaplan, J.; Kalyan, A.; Santa-Maria, C.A.; Platanias, L.C.; Giles, F.J. Genomic landscape of DNA repair genes in cancer. Oncotarget 2016, 7, 23312–23321. [Google Scholar] [CrossRef] [PubMed]
- Miki, Y.; Swensen, J.; Shattuck-Eidens, D.; Futreal, P.A.; Harshman, K.; Tavtigian, S.; Liu, Q.; Cochran, C.; Bennett, L.M.; Ding, W.; et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 1994, 266, 66–71. [Google Scholar] [CrossRef]
- Wooster, R.; Bignell, G.; Lancaster, J.; Swift, S.; Seal, S.; Mangion, J.; Collins, N.; Gregory, S.; Gumbs, C.; Micklem, G. Identification of the breast cancer susceptibility gene BRCA2. Nature 1995, 378, 789–792. [Google Scholar] [CrossRef]
- Gottipati, P.; Vischioni, B.; Schultz, N.; Solomons, J.; Bryant, H.E.; Djureinovic, T.; Issaeva, N.; Sleeth, K.; Sharma, R.A.; Helleday, T. Poly(ADP-ribose) polymerase is hyperactivated in homologous recombination-defective cells. Cancer Res. 2010, 70, 5389–5398. [Google Scholar] [CrossRef]
- Helleday, T. The underlying mechanism for the PARP and BRCA synthetic lethality: Clearing up the misunderstandings. Mol. Oncol. 2011, 5, 387–393. [Google Scholar] [CrossRef]
- Gelmon, K.A.; Tischkowitz, M.; Mackay, H.; Swenerton, K.; Robidoux, A.; Tonkin, K.; Hirte, H.; Huntsman, D.; Clemons, M.; Gilks, B.; et al. Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: A phase 2, multicentre, open-label, non-randomised study. Lancet Oncol. 2011, 12, 852–861. [Google Scholar] [CrossRef]
- Evans, T.; Matulonis, U. PARP inhibitors in ovarian cancer: Evidence, experience and clinical potential. Ther. Adv. Med. Oncol. 2017, 9, 253–267. [Google Scholar] [CrossRef]
- Ledermann, J.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.L.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: A preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol. 2014, 15, 852–861. [Google Scholar] [CrossRef]
- Scott, C.L.; Swisher, E.M.; Kaufmann, S.H. Poly (ADP-ribose) polymerase inhibitors: Recent advances and future development. J. Clin. Oncol. 2015, 33, 1397–1406. [Google Scholar] [CrossRef] [PubMed]
- Murai, J.; Huang, S.Y.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y.; O’Connor, M.J.; de Bono, J. Laying a trap to kill cancer cells: PARP inhibitors and their mechanisms of action. Sci. Transl. Med. 2016, 8, 362ps17. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, T.A.; Shi, Y.; Rodriguez, L.E.; Solomon, L.R.; Donawho, C.K.; DiGiammarino, E.L.; Panchal, S.C.; Wilsbacher, J.L.; Gao, W.; Olson, A.M.; et al. Mechanistic Dissection of PARP1 Trapping and the Impact on In Vivo Tolerability and Efficacy of PARP Inhibitors. Mol. Cancer Res. 2015, 13, 1465–1477. [Google Scholar] [CrossRef]
- Motea, E.A.; Fattah, F.J.; Xiao, L.; Girard, L.; Rommel, A.; Morales, J.C.; Patidar, P.; Zhou, Y.; Porter, A.; Xie, Y.; et al. Kub5-Hera (RPRD1B) Deficiency Promotes “BRCAness” and Vulnerability to PARP Inhibition in BRCA-proficient Breast Cancers. Clin. Cancer Res. 2018, 24, 6459–6470. [Google Scholar] [CrossRef]
- Fong, P.C.; Boss, D.S.; Yap, T.A.; Tutt, A.; Wu, P.; Mergui-Roelvink, M.; Mortimer, P.; Swaisland, H.; Lau, A.; O’Connor, M.J.; et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N. Engl. J. Med. 2009, 361, 123–134. [Google Scholar] [CrossRef]
- Kaufman, B.; Shapira-Frommer, R.; Schmutzler, R.K.; Audeh, M.W.; Friedlander, M.; Balmana, J.; Mitchell, G.; Fried, G.; Stemmer, S.M.; Hubert, A.; et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J. Clin. Oncol. 2015, 33, 244–250. [Google Scholar] [CrossRef]
- Yang, L.; Zhang, Y.; Shan, W.; Hu, Z.; Yuan, J.; Pi, J.; Wang, Y.; Fan, L.; Tang, Z.; Li, C.; et al. Repression of BET activity sensitizes homologous recombination-proficient cancers to PARP inhibition. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef]
- Mateo, J.; Carreira, S.; Sandhu, S.; Miranda, S.; Mossop, H.; Perez-Lopez, R.; Nava Rodrigues, D.; Robinson, D.; Omlin, A.; Tunariu, N.; et al. DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. N. Engl. J. Med. 2015, 373, 1697–1708. [Google Scholar] [CrossRef]
- Johnson, N.; Li, Y.C.; Walton, Z.E.; Cheng, K.A.; Li, D.; Rodig, S.J.; Moreau, L.A.; Unitt, C.; Bronson, R.T.; Thomas, H.D.; et al. Compromised CDK1 activity sensitizes BRCA-proficient cancers to PARP inhibition. Nat. Med. 2011, 17, 875–882. [Google Scholar] [CrossRef]
- Conrad, L.B.; Lin, K.Y.; Nandu, T.; Gibson, B.A.; Lea, J.S.; Kraus, W.L. ADP-Ribosylation Levels and Patterns Correlate with Gene Expression and Clinical Outcomes in Ovarian Cancers. Mol. Cancer Ther. 2020, 19, 282–291. [Google Scholar] [CrossRef]
- Ledermann, J.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer. N. Engl. J. Med. 2012, 366, 1382–1392. [Google Scholar] [CrossRef] [PubMed]
- Pujade-Lauraine, E.; Ledermann, J.A.; Selle, F.; Gebski, V.; Penson, R.T.; Oza, A.M.; Korach, J.; Huzarski, T.; Poveda, A.; Pignata, S.; et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): A double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2017, 18, 1274–1284. [Google Scholar] [CrossRef]
- Coleman, R.L.; Oza, A.M.; Lorusso, D.; Aghajanian, C.; Oaknin, A.; Dean, A.; Colombo, N.; Weberpals, J.I.; Clamp, A.; Scambia, G.; et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 390, 1949–1961. [Google Scholar] [CrossRef]
- Mirza, M.R.; Monk, B.J.; Herrstedt, J.; Oza, A.M.; Mahner, S.; Redondo, A.; Fabbro, M.; Ledermann, J.A.; Lorusso, D.; Vergote, I.; et al. Niraparib Maintenance Therapy in Platinum-Sensitive, Recurrent Ovarian Cancer. N. Engl. J. Med. 2016, 375, 2154–2164. [Google Scholar] [CrossRef]
- Mittica, G.; Ghisoni, E.; Giannone, G.; Genta, S.; Aglietta, M.; Sapino, A.; Valabrega, G. PARP Inhibitors in Ovarian Cancer. Recent Pat. Anticancer Drug Discov. 2018, 13, 392–410. [Google Scholar] [CrossRef]
- Weng, C.S.; Wu, C.C.; Chen, T.C.; Chen, J.R.; Huang, C.Y.; Chang, C.L. Retrospective Analysis Of Comparative Outcomes In Recurrent Platinum-Sensitive Ovarian Cancer Treated With Pegylated Liposomal Doxorubicin (Lipo-Dox) And Carboplatin Versus Paclitaxel And Carboplatin. Cancer Manag. Res. 2019, 11, 9899–9905. [Google Scholar] [CrossRef]
- Fong, P.C.; Yap, T.A.; Boss, D.S.; Carden, C.P.; Mergui-Roelvink, M.; Gourley, C.; De Greve, J.; Lubinski, J.; Shanley, S.; Messiou, C.; et al. Poly(ADP)-ribose polymerase inhibition: Frequent durable responses in BRCA carrier ovarian cancer correlating with platinum-free interval. J. Clin. Oncol. 2010, 28, 2512–2519. [Google Scholar] [CrossRef]
- Tutt, A.; Robson, M.; Garber, J.E.; Domchek, S.M.; Audeh, M.W.; Weitzel, J.N.; Friedlander, M.; Arun, B.; Loman, N.; Schmutzler, R.K.; et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: A proof-of-concept trial. Lancet 2010, 376, 235–244. [Google Scholar] [CrossRef]
- Audeh, M.W.; Carmichael, J.; Penson, R.T.; Friedlander, M.; Powell, B.; Bell-McGuinn, K.M.; Scott, C.; Weitzel, J.N.; Oaknin, A.; Loman, N.; et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: A proof-of-concept trial. Lancet 2010, 376, 245–251. [Google Scholar] [CrossRef]
- Olaparib Keeps Hereditary Breast Tumors in Check. Cancer Discov. 2017, 7, OF10. [CrossRef] [PubMed][Green Version]
- Robson, M.; Im, S.A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N. Engl. J. Med. 2017, 377, 523–533. [Google Scholar] [CrossRef] [PubMed]
- Domchek, S.M.; Aghajanian, C.; Shapira-Frommer, R.; Schmutzler, R.K.; Audeh, M.W.; Friedlander, M.; Balmana, J.; Mitchell, G.; Fried, G.; Stemmer, S.M.; et al. Efficacy and safety of olaparib monotherapy in germline BRCA1/2 mutation carriers with advanced ovarian cancer and three or more lines of prior therapy. Gynecol. Oncol. 2016, 140, 199–203. [Google Scholar] [CrossRef] [PubMed]
- Swisher, E.M.; Lin, K.K.; Oza, A.M.; Scott, C.L.; Giordano, H.; Sun, J.; Konecny, G.E.; Coleman, R.L.; Tinker, A.V.; O’Malley, D.M.; et al. Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): An international, multicentre, open-label, phase 2 trial. Lancet Oncol. 2017, 18, 75–87. [Google Scholar] [CrossRef]
- Kristeleit, R.; Shapiro, G.I.; Burris, H.A.; Oza, A.M.; LoRusso, P.; Patel, M.R.; Domchek, S.M.; Balmana, J.; Drew, Y.; Chen, L.M.; et al. A Phase I-II Study of the Oral PARP Inhibitor Rucaparib in Patients with Germline BRCA1/2-Mutated Ovarian Carcinoma or Other Solid Tumors. Clin. Cancer Res. 2017, 23, 4095–4106. [Google Scholar] [CrossRef]
- Moore, K.N.; Secord, A.A.; Geller, M.A.; Miller, D.S.; Cloven, N.; Fleming, G.F.; Wahner Hendrickson, A.E.; Azodi, M.; DiSilvestro, P.; Oza, A.M.; et al. Niraparib monotherapy for late-line treatment of ovarian cancer (QUADRA): A multicentre, open-label, single-arm, phase 2 trial. Lancet Oncol. 2019, 20, 636–648. [Google Scholar] [CrossRef]
- Guney Eskiler, G. Talazoparib to treat BRCA-positive breast cancer. Drugs Today (Barc) 2019, 55, 459–467. [Google Scholar] [CrossRef]
- Okayama, H.; Edson, C.M.; Fukushima, M.; Ueda, K.; Hayaishi, O. Purification and properties of poly(adenosine diphosphate ribose) synthetase. J. Biol. Chem. 1977, 252, 7000–7005. [Google Scholar]
- Brochu, G.; Duchaine, C.; Thibeault, L.; Lagueux, J.; Shah, G.M.; Poirier, G.G. Mode of action of poly(ADP-ribose) glycohydrolase. Biochim. Biophys. Acta (BBA) Gene Struct. Expr. 1994, 1219, 342–350. [Google Scholar] [CrossRef]
- Hassa, P.O.; Haenni, S.S.; Elser, M.; Hottiger, M.O. Nuclear ADP-ribosylation reactions in mammalian cells: Where are we today and where are we going? Microbiol. Mol. Biol. Rev. 2006, 70, 789–829. [Google Scholar] [CrossRef]
- Hassa, P.O.; Hottiger, M.O. The diverse biological roles of mammalian PARPS, a small but powerful family of poly-ADP-ribose polymerases. Front. Biosci. 2008, 13, 3046–3082. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, K.; Toth, A.; Deres, P.; Kalai, T.; Hideg, K.; Gallyas, F., Jr.; Sumegi, B. Critical role of PI3-kinase/Akt activation in the PARP inhibitor induced heart function recovery during ischemia-reperfusion. Biochem. Pharmacol. 2006, 71, 441–452. [Google Scholar] [CrossRef] [PubMed]
- Juarez-Salinas, H.; Sims, J.L.; Jacobson, M.K. Poly(ADP-ribose) levels in carcinogen-treated cells. Nature 1979, 282, 740–741. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, J.; Tedim Ferreira, M.; Gagne, J.P.; Sharma, A.K.; Hendzel, M.J.; Masson, J.Y.; Poirier, G.G. Emerging roles of eraser enzymes in the dynamic control of protein ADP-ribosylation. Nat. Commun. 2019, 10, 1182. [Google Scholar] [CrossRef] [PubMed]
- Botta, D.; Jacobson, M.K. Identification of a regulatory segment of poly(ADP-ribose) glycohydrolase. Biochemistry 2010, 49, 7674–7682. [Google Scholar] [CrossRef] [PubMed]
- Meyer-Ficca, M.L.; Meyer, R.G.; Coyle, D.L.; Jacobson, E.L.; Jacobson, M.K. Human poly(ADP-ribose) glycohydrolase is expressed in alternative splice variants yielding isoforms that localize to different cell compartments. Exp. Cell Res. 2004, 297, 521–532. [Google Scholar] [CrossRef] [PubMed]
- Braun, S.A.; Panzeter, P.L.; Collinge, M.A.; Althaus, F.R. Endoglycosidic cleavage of branched polymers by poly(ADP-ribose) glycohydrolase. Eur. J. Biochem. 1994, 220, 369–375. [Google Scholar] [CrossRef]
- David, K.K.; Andrabi, S.A.; Dawson, T.M.; Dawson, V.L. Parthanatos, a messenger of death. Front. Biosci. (Landmark Ed) 2009, 14, 1116–1128. [Google Scholar] [CrossRef]
- Andrabi, S.A.; Kim, N.S.; Yu, S.W.; Wang, H.; Koh, D.W.; Sasaki, M.; Klaus, J.A.; Otsuka, T.; Zhang, Z.; Koehler, R.C.; et al. Poly(ADP-ribose) (PAR) polymer is a death signal. Proc. Natl. Acad. Sci. USA 2006, 103, 18308–18313. [Google Scholar] [CrossRef]
- Wang, Y.; Kim, N.S.; Haince, J.F.; Kang, H.C.; David, K.K.; Andrabi, S.A.; Poirier, G.G.; Dawson, V.L.; Dawson, T.M. Poly(ADP-ribose) (PAR) binding to apoptosis-inducing factor is critical for PAR polymerase-1-dependent cell death (parthanatos). Sci. Signal. 2011, 4, ra20. [Google Scholar] [CrossRef]
- Yu, S.W.; Wang, H.; Poitras, M.F.; Coombs, C.; Bowers, W.J.; Federoff, H.J.; Poirier, G.G.; Dawson, T.M.; Dawson, V.L. Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science 2002, 297, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Niere, M.; Mashimo, M.; Agledal, L.; Dolle, C.; Kasamatsu, A.; Kato, J.; Moss, J.; Ziegler, M. ADP-ribosylhydrolase 3 (ARH3), not poly(ADP-ribose) glycohydrolase (PARG) isoforms, is responsible for degradation of mitochondrial matrix-associated poly(ADP-ribose). J. Biol. Chem. 2012, 287, 16088–16102. [Google Scholar] [CrossRef] [PubMed]
- Marques, M.; Jangal, M.; Wang, L.C.; Kazanets, A.; da Silva, S.D.; Zhao, T.; Lovato, A.; Yu, H.; Jie, S.; Del Rincon, S.; et al. Oncogenic activity of poly (ADP-ribose) glycohydrolase. Oncogene 2019, 38, 2177–2191. [Google Scholar] [CrossRef] [PubMed]
- Tavassoli, M.; Tavassoli, M.H.; Shall, S. Effect of DNA intercalators on poly(ADP-ribose) glycohydrolase activity. Biochim. Biophys. Acta 1985, 827, 228–234. [Google Scholar] [CrossRef]
- Li, Q.; Li, M.; Wang, Y.L.; Fauzee, N.J.; Yang, Y.; Pan, J.; Yang, L.; Lazar, A. RNA interference of PARG could inhibit the metastatic potency of colon carcinoma cells via PI3-kinase/Akt pathway. Cell. Physiol. Biochem. 2012, 29, 361–372. [Google Scholar] [CrossRef]
- Putt, K.S.; Hergenrother, P.J. A nonradiometric, high-throughput assay for poly(ADP-ribose) glycohydrolase (PARG): Application to inhibitor identification and evaluation. Anal. Biochem. 2004, 333, 256–264. [Google Scholar] [CrossRef]
- Aoki, T.; Kojima, M.; Adachi, J.; Okano, M. Effect of short-term egg exclusion diet on infantile atopic dermatitis and its relation to egg allergy: A single-blind test. Acta Derm. Venereol. Suppl. (Stockh) 1992, 176, 99–102. [Google Scholar]
- Tsai, Y.J.; Aoki, T.; Maruta, H.; Abe, H.; Sakagami, H.; Hatano, T.; Okuda, T.; Tanuma, S. Mouse mammary tumor virus gene expression is suppressed by oligomeric ellagitannins, novel inhibitors of poly(ADP-ribose) glycohydrolase. J. Biol. Chem. 1992, 267, 14436–14442. [Google Scholar]
- Formentini, L.; Arapistas, P.; Pittelli, M.; Jacomelli, M.; Pitozzi, V.; Menichetti, S.; Romani, A.; Giovannelli, L.; Moroni, F.; Chiarugi, A. Mono-galloyl glucose derivatives are potent poly(ADP-ribose) glycohydrolase (PARG) inhibitors and partially reduce PARP-1-dependent cell death. Br. J. Pharmacol. 2008, 155, 1235–1249. [Google Scholar] [CrossRef]
- Finch, K.E.; Knezevic, C.E.; Nottbohm, A.C.; Partlow, K.C.; Hergenrother, P.J. Selective Small Molecule Inhibition of Poly(ADP-Ribose) Glycohydrolase (PARG). ACS Chem. Biol. 2012, 7, 563–570. [Google Scholar] [CrossRef]
- Pillay, N.; Tighe, A.; Nelson, L.; Littler, S.; Coulson-Gilmer, C.; Bah, N.; Golder, A.; Bakker, B.; Spierings, D.C.J.; James, D.I.; et al. DNA Replication Vulnerabilities Render Ovarian Cancer Cells Sensitive to Poly(ADP-Ribose) Glycohydrolase Inhibitors. Cancer Cell 2019, 35, 519–533. [Google Scholar] [CrossRef] [PubMed]
- Fujihara, H.; Ogino, H.; Maeda, D.; Shirai, H.; Nozaki, T.; Kamada, N.; Jishage, K.; Tanuma, S.; Takato, T.; Ochiya, T.; et al. Poly(ADP-ribose) Glycohydrolase deficiency sensitizes mouse ES cells to DNA damaging agents. Curr. Cancer Drug Targets 2009, 9, 953–962. [Google Scholar] [CrossRef] [PubMed]
- Shirai, H.; Poetsch, A.R.; Gunji, A.; Maeda, D.; Fujimori, H.; Fujihara, H.; Yoshida, T.; Ogino, H.; Masutani, M. PARG dysfunction enhances DNA double strand break formation in S-phase after alkylation DNA damage and augments different cell death pathways. Cell Death Dis. 2013, 4, e656. [Google Scholar] [CrossRef] [PubMed]
- Murphy, J.P.; Giacomantonio, M.A.; Paulo, J.A.; Everley, R.A.; Kennedy, B.E.; Pathak, G.P.; Clements, D.R.; Kim, Y.; Dai, C.; Sharif, T.; et al. The NAD(+) Salvage Pathway Supports PHGDH-Driven Serine Biosynthesis. Cell Rep. 2018, 24, 2381–2391. [Google Scholar] [CrossRef]
- Verdin, E. NAD(+) in aging, metabolism, and neurodegeneration. Science 2015, 350, 1208–1213. [Google Scholar] [CrossRef]
- Garrido, A.; Djouder, N. NAD(+) Deficits in Age-Related Diseases and Cancer. Trends Cancer 2017, 3, 593–610. [Google Scholar] [CrossRef]
- Yaku, K.; Okabe, K.; Nakagawa, T. NAD metabolism: Implications in aging and longevity. Ageing Res. Rev. 2018, 47, 1–17. [Google Scholar] [CrossRef]
- Yamamoto, M.; Inohara, H.; Nakagawa, T. Targeting metabolic pathways for head and neck cancers therapeutics. Cancer Metastasis Rev. 2017, 36, 503–514. [Google Scholar] [CrossRef]
- Tan, B.; Young, D.A.; Lu, Z.H.; Wang, T.; Meier, T.I.; Shepard, R.L.; Roth, K.; Zhai, Y.; Huss, K.; Kuo, M.S.; et al. Pharmacological inhibition of nicotinamide phosphoribosyltransferase (NAMPT), an enzyme essential for NAD+ biosynthesis, in human cancer cells: Metabolic basis and potential clinical implications. J. Biol. Chem. 2013, 288, 3500–3511. [Google Scholar] [CrossRef]
- Cerna, D.; Li, H.; Flaherty, S.; Takebe, N.; Coleman, C.N.; Yoo, S.S. Inhibition of nicotinamide phosphoribosyltransferase (NAMPT) activity by small molecule GMX1778 regulates reactive oxygen species (ROS)-mediated cytotoxicity in a p53- and nicotinic acid phosphoribosyltransferase1 (NAPRT1)-dependent manner. J. Biol. Chem. 2012, 287, 22408–22417. [Google Scholar] [CrossRef]
- Kato, H.; Ito, E.; Shi, W.; Alajez, N.M.; Yue, S.; Lee, C.; Chan, N.; Bhogal, N.; Coackley, C.L.; Vines, D.; et al. Efficacy of combining GMX1777 with radiation therapy for human head and neck carcinoma. Clin. Cancer Res. 2010, 16, 898–911. [Google Scholar] [CrossRef] [PubMed]
- Moore, Z.; Chakrabarti, G.; Luo, X.; Ali, A.; Hu, Z.; Fattah, F.J.; Vemireddy, R.; DeBerardinis, R.J.; Brekken, R.A.; Boothman, D.A. NAMPT inhibition sensitizes pancreatic adenocarcinoma cells to tumor-selective, PAR-independent metabolic catastrophe and cell death induced by beta-lapachone. Cell Death Dis. 2015, 6, e1599. [Google Scholar] [CrossRef] [PubMed]
- Zerp, S.F.; Vens, C.; Floot, B.; Verheij, M.; van Triest, B. NAD(+) depletion by APO866 in combination with radiation in a prostate cancer model, results from an in vitro and in vivo study. Radiother. Oncol. 2014, 110, 348–354. [Google Scholar] [CrossRef] [PubMed]
- Ju, H.Q.; Zhuang, Z.N.; Li, H.; Tian, T.; Lu, Y.X.; Fan, X.Q.; Zhou, H.J.; Mo, H.Y.; Sheng, H.; Chiao, P.J.; et al. Regulation of the Nampt-mediated NAD salvage pathway and its therapeutic implications in pancreatic cancer. Cancer Lett. 2016, 379, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Lucena-Cacace, A.; Otero-Albiol, D.; Jimenez-Garcia, M.P.; Munoz-Galvan, S.; Carnero, A. NAMPT Is a Potent Oncogene in Colon Cancer Progression that Modulates Cancer Stem Cell Properties and Resistance to Therapy through Sirt1 and PARP. Clin. Cancer Res. 2018, 24, 1202–1215. [Google Scholar] [CrossRef]
- Lucena-Cacace, A.; Otero-Albiol, D.; Jiménez-García, M.P.; Peinado-Serrano, J.; Carnero, A. NAMPT overexpression induces cancer stemness and defines a novel tumor signature for glioma prognosis. Oncotarget 2017, 8, 99514–99530. [Google Scholar] [CrossRef]
- Zhu, Y.; Guo, M.; Zhang, L.; Xu, T.; Wang, L.; Xu, G. Biomarker triplet NAMPT/VEGF/HER2 as a de novo detection panel for the diagnosis and prognosis of human breast cancer. Oncol. Rep. 2016, 35, 454–462. [Google Scholar] [CrossRef]
- Wang, B.; Hasan, M.K.; Alvarado, E.; Yuan, H.; Wu, H.; Chen, W.Y. NAMPT overexpression in prostate cancer and its contribution to tumor cell survival and stress response. Oncogene 2011, 30, 907–921. [Google Scholar] [CrossRef]
- Shackelford, R.E.; Bui, M.M.; Coppola, D.; Hakam, A. Over-expression of nicotinamide phosphoribosyltransferase in ovarian cancers. Int. J. Clin. Exp. Pathol. 2010, 3, 522–527. [Google Scholar]
- Gehrke, I.; Bouchard, E.D.; Beiggi, S.; Poeppl, A.G.; Johnston, J.B.; Gibson, S.B.; Banerji, V. On-target effect of FK866, a nicotinamide phosphoribosyl transferase inhibitor, by apoptosis-mediated death in chronic lymphocytic leukemia cells. Clin. Cancer Res. 2014, 20, 4861–4872. [Google Scholar] [CrossRef]
- Del Nagro, C.; Xiao, Y.; Rangell, L.; Reichelt, M.; O’Brien, T. Depletion of the central metabolite NAD leads to oncosis-mediated cell death. J. Biol. Chem. 2014, 289, 35182–35192. [Google Scholar] [CrossRef] [PubMed]
- Hasmann, M.; Schemainda, I. FK866, a highly specific noncompetitive inhibitor of nicotinamide phosphoribosyltransferase, represents a novel mechanism for induction of tumor cell apoptosis. Cancer Res. 2003, 63, 7436–7442. [Google Scholar] [PubMed]
- Olesen, U.H.; Christensen, M.K.; Björkling, F.; Jäättelä, M.; Jensen, P.B.; Sehested, M.; Nielsen, S.J. Anticancer agent CHS-828 inhibits cellular synthesis of NAD. Biochem. Biophys. Res. Commun. 2008, 367, 799–804. [Google Scholar] [CrossRef] [PubMed]
- Watson, M.; Roulston, A.; Belec, L.; Billot, X.; Marcellus, R.; Bedard, D.; Bernier, C.; Branchaud, S.; Chan, H.; Dairi, K.; et al. The small molecule GMX1778 is a potent inhibitor of NAD+ biosynthesis: Strategy for enhanced therapy in nicotinic acid phosphoribosyltransferase 1-deficient tumors. Mol. Cell. Biol. 2009, 29, 5872–5888. [Google Scholar] [CrossRef]
- Chan, D.A.; Sutphin, P.D.; Nguyen, P.; Turcotte, S.; Lai, E.W.; Banh, A.; Reynolds, G.E.; Chi, J.T.; Wu, J.; Solow-Cordero, D.E.; et al. Targeting GLUT1 and the Warburg effect in renal cell carcinoma by chemical synthetic lethality. Sci. Transl. Med. 2011, 3, 94ra70. [Google Scholar] [CrossRef]
- Espindola-Netto, J.M.; Chini, C.C.S.; Tarrago, M.; Wang, E.; Dutta, S.; Pal, K.; Mukhopadhyay, D.; Sola-Penna, M.; Chini, E.N. Preclinical efficacy of the novel competitive NAMPT inhibitor STF-118804 in pancreatic cancer. Oncotarget 2017, 8, 85054–85067. [Google Scholar] [CrossRef]
- Zhao, G.; Green, C.F.; Hui, Y.H.; Prieto, L.; Shepard, R.; Dong, S.; Wang, T.; Tan, B.; Gong, X.; Kays, L.; et al. Discovery of a Highly Selective NAMPT Inhibitor That Demonstrates Robust Efficacy and Improved Retinal Toxicity with Nicotinic Acid Coadministration. Mol. Cancer Ther. 2017, 16, 2677–2688. [Google Scholar] [CrossRef]
- Abu Aboud, O.; Chen, C.H.; Senapedis, W.; Baloglu, E.; Argueta, C.; Weiss, R.H. Dual and Specific Inhibition of NAMPT and PAK4 By KPT-9274 Decreases Kidney Cancer Growth. Mol. Cancer Ther. 2016, 15, 2119–2129. [Google Scholar] [CrossRef]
- Von Heideman, A.; Berglund, A.; Larsson, R.; Nygren, P. Safety and efficacy of NAD depleting cancer drugs: Results of a phase I clinical trial of CHS 828 and overview of published data. Cancer Chemother. Pharmacol. 2010, 65, 1165–1172. [Google Scholar] [CrossRef]
- Goldinger, S.M.; Gobbi Bischof, S.; Fink-Puches, R.; Klemke, C.D.; Dreno, B.; Bagot, M.; Dummer, R. Efficacy and Safety of APO866 in Patients With Refractory or Relapsed Cutaneous T-Cell Lymphoma: A Phase 2 Clinical Trial. JAMA Dermatol. 2016, 152, 837–839. [Google Scholar] [CrossRef]
- Holen, K.; Saltz, L.B.; Hollywood, E.; Burk, K.; Hanauske, A.R. The pharmacokinetics, toxicities, and biologic effects of FK866, a nicotinamide adenine dinucleotide biosynthesis inhibitor. Investig. New Drugs 2008, 26, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Ravaud, A.; Cerny, T.; Terret, C.; Wanders, J.; Bui, B.N.; Hess, D.; Droz, J.P.; Fumoleau, P.; Twelves, C. Phase I study and pharmacokinetic of CHS-828, a guanidino-containing compound, administered orally as a single dose every 3 weeks in solid tumours: An ECSG/EORTC study. Eur. J. Cancer 2005, 41, 702–707. [Google Scholar] [CrossRef] [PubMed]
- Hovstadius, P.; Larsson, R.; Jonsson, E.; Skov, T.; Kissmeyer, A.M.; Krasilnikoff, K.; Bergh, J.; Karlsson, M.O.; Lonnebo, A.; Ahlgren, J. A Phase I study of CHS 828 in patients with solid tumor malignancy. Clin. Cancer Res. 2002, 8, 2843–2850. [Google Scholar] [PubMed]
- Liu, H.Y.; Li, Q.R.; Cheng, X.F.; Wang, G.J.; Hao, H.P. NAMPT inhibition synergizes with NQO1-targeting agents in inducing apoptotic cell death in non-small cell lung cancer cells. Chin. J. Nat. Med. 2016, 14, 582–589. [Google Scholar] [CrossRef]
- Zhang, K.; Chen, D.; Ma, K.; Wu, X.; Hao, H.; Jiang, S. NAD(P)H:Quinone Oxidoreductase 1 (NQO1) as a Therapeutic and Diagnostic Target in Cancer. J. Med. Chem. 2018, 61, 6983–7003. [Google Scholar] [CrossRef]
- Pardee, A.B.; Li, Y.Z.; Li, C.J. Cancer therapy with beta-lapachone. Curr. Cancer Drug Targets 2002, 2, 227–242. [Google Scholar] [CrossRef]
- Beg, M.S.; Huang, X.; Silvers, M.A.; Gerber, D.E.; Bolluyt, J.; Sarode, V.; Fattah, F.; Deberardinis, R.J.; Merritt, M.E.; Xie, X.J.; et al. Using a novel NQO1 bioactivatable drug, beta-lapachone (ARQ761), to enhance chemotherapeutic effects by metabolic modulation in pancreatic cancer. J. Surg. Oncol. 2017, 116, 83–88. [Google Scholar] [CrossRef]
- Bey, E.A.; Bentle, M.S.; Reinicke, K.E.; Dong, Y.; Yang, C.R.; Girard, L.; Minna, J.D.; Bornmann, W.G.; Gao, J.; Boothman, D.A. An NQO1- and PARP-1-mediated cell death pathway induced in non-small-cell lung cancer cells by beta-lapachone. Proc. Natl. Acad. Sci. USA 2007, 104, 11832–11837. [Google Scholar] [CrossRef]
- Huang, X.; Motea, E.A.; Moore, Z.R.; Yao, J.; Dong, Y.; Chakrabarti, G.; Kilgore, J.A.; Silvers, M.A.; Patidar, P.L.; Cholka, A.; et al. Leveraging an NQO1 Bioactivatable Drug for Tumor-Selective Use of Poly(ADP-ribose) Polymerase Inhibitors. Cancer Cell 2016, 30, 940–952. [Google Scholar] [CrossRef]
- Motea, E.A.; Huang, X.; Singh, N.; Kilgore, J.A.; Williams, N.S.; Xie, X.J.; Gerber, D.E.; Beg, M.S.; Bey, E.A.; Boothman, D.A. NQO1-dependent, Tumor-selective Radiosensitization of Non-small Cell Lung Cancers. Clin. Cancer Res. 2019, 25, 2601–2609. [Google Scholar] [CrossRef]
- Blanco, E.; Bey, E.A.; Khemtong, C.; Yang, S.G.; Setti-Guthi, J.; Chen, H.; Kessinger, C.W.; Carnevale, K.A.; Bornmann, W.G.; Boothman, D.A.; et al. Beta-lapachone micellar nanotherapeutics for non-small cell lung cancer therapy. Cancer Res. 2010, 70, 3896–3904. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Bey, E.A.; Li, L.S.; Kabbani, W.; Yan, J.; Xie, X.J.; Hsieh, J.T.; Gao, J.; Boothman, D.A. Prostate cancer radiosensitization through poly(ADP-Ribose) polymerase-1 hyperactivation. Cancer Res. 2010, 70, 8088–8096. [Google Scholar] [CrossRef] [PubMed]
- Reinicke, K.E.; Bey, E.A.; Bentle, M.S.; Pink, J.J.; Ingalls, S.T.; Hoppel, C.L.; Misico, R.I.; Arzac, G.M.; Burton, G.; Bornmann, W.G.; et al. Development of beta-lapachone prodrugs for therapy against human cancer cells with elevated NAD(P)H:quinone oxidoreductase 1 levels. Clin. Cancer Res. 2005, 11, 3055–3064. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Silvers, M.A.; Deja, S.; Singh, N.; Egnatchik, R.A.; Sudderth, J.; Luo, X.; Beg, M.S.; Burgess, S.C.; DeBerardinis, R.J.; Boothman, D.A.; et al. The NQO1 bioactivatable drug, beta-lapachone, alters the redox state of NQO1+ pancreatic cancer cells, causing perturbation in central carbon metabolism. J. Biol. Chem. 2017, 292, 18203–18216. [Google Scholar] [CrossRef]
- Pink, J.J.; Planchon, S.M.; Tagliarino, C.; Varnes, M.E.; Siegel, D.; Boothman, D.A. NAD(P)H:Quinone oxidoreductase activity is the principal determinant of beta-lapachone cytotoxicity. J. Biol. Chem. 2000, 275, 5416–5424. [Google Scholar] [CrossRef]
- Imlay, J.A.; Chin, S.M.; Linn, S. Toxic DNA damage by hydrogen peroxide through the Fenton reaction in vivo and in vitro. Science 1988, 240, 640–642. [Google Scholar] [CrossRef]
- Andrabi, S.A.; Umanah, G.K.; Chang, C.; Stevens, D.A.; Karuppagounder, S.S.; Gagne, J.P.; Poirier, G.G.; Dawson, V.L.; Dawson, T.M. Poly(ADP-ribose) polymerase-dependent energy depletion occurs through inhibition of glycolysis. Proc. Natl. Acad. Sci. USA 2014, 111, 10209–10214. [Google Scholar] [CrossRef]
- Kim, S.; Lee, S.; Cho, J.Y.; Yoon, S.H.; Jang, I.J.; Yu, K.S. Pharmacokinetics and tolerability of MB12066, a beta-lapachone derivative targeting NAD(P)H: Quinone oxidoreductase 1: Two independent, double-blind, placebo-controlled, combined single and multiple ascending dose first-in-human clinical trials. Drug Des. Dev. Ther. 2017, 11, 3187–3195. [Google Scholar] [CrossRef]
- Hartner, L.P.; Rosen, L.; Hensley, M.; Mendelson, D.; Staddon, A.P.; Chow, W.; Kovalyov, O.; Ruka, W.; Skladowski, K.; Jagiello-Gruszfeld, A.; et al. Phase 2 dose multi-center, open-label study of ARQ 501, a checkpoint activator, in adult patients with persistent, recurrent or metastatic leiomyosarcoma (LMS). J. Clin. Oncol. 2007, 25 (Suppl. 18), 20521. [Google Scholar] [CrossRef]
- Buranrat, B.; Chau-in, S.; Prawan, A.; Puapairoj, A.; Zeekpudsa, P.; Kukongviriyapan, V. NQO1 expression correlates with cholangiocarcinoma prognosis. Asian Pac. J. Cancer Prev. 2012, 13, 131–136. [Google Scholar]
- Cui, X.; Jin, T.; Wang, X.; Jin, G.; Li, Z.; Lin, L. NAD(P)H:quinone oxidoreductase-1 overexpression predicts poor prognosis in small cell lung cancer. Oncol. Rep. 2014, 32, 2589–2595. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Li, L.; Yan, G.; Meng, K.; Lin, Z.; Nan, Y.; Jin, G.; Li, C. High expression of NQO1 is associated with poor prognosis in serous ovarian carcinoma. BMC Cancer 2015, 15, 244. [Google Scholar] [CrossRef] [PubMed]
- Kolesar, J.M.; Dahlberg, S.E.; Marsh, S.; McLeod, H.L.; Johnson, D.H.; Keller, S.M.; Schiller, J.H. The NQO1*2/*2 polymorphism is associated with poor overall survival in patients following resection of stages II and IIIa non-small cell lung cancer. Oncol. Rep. 2011, 25, 1765–1772. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhang, Y.; Jin, T.; Men, J.; Lin, Z.; Qi, P.; Piao, Y.; Yan, G. NQO1 protein expression predicts poor prognosis of non-small cell lung cancers. BMC Cancer 2015, 15, 207. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Qin, Y.; Jin, T.; Liu, S.; Zhang, S.; Shen, X.; Lin, Z. Significance of NQO1 overexpression for prognostic evaluation of gastric adenocarcinoma. Exp. Mol. Pathol. 2014, 96, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhang, Y.; Wu, Q.; Cui, X.; Lin, Z.; Liu, S.; Chen, L. Clinical implications of high NQO1 expression in breast cancers. J. Exp. Clin. Cancer Res. 2014, 33, 14. [Google Scholar] [CrossRef]
- Morales, J.; Li, L.; Fattah, F.J.; Dong, Y.; Bey, E.A.; Patel, M.; Gao, J.; Boothman, D.A. Review of poly (ADP-ribose) polymerase (PARP) mechanisms of action and rationale for targeting in cancer and other diseases. Crit. Rev. Eukaryot. Gene Expr. 2014, 24, 15–28. [Google Scholar] [CrossRef]
- Du, Y.; Yamaguchi, H.; Wei, Y.; Hsu, J.L.; Wang, H.-L.; Hsu, Y.-H.; Lin, W.-C.; Yu, W.-H.; Leonard, P.G.; Lee IV, G.R. Blocking c-Met–mediated PARP1 phosphorylation enhances anti-tumor effects of PARP inhibitors. Nat. Med. 2016, 22, 194. [Google Scholar] [CrossRef]
- Zhang, Y.; Xia, M.; Jin, K.; Wang, S.; Wei, H.; Fan, C.; Wu, Y.; Li, X.; Li, X.; Li, G. Function of the c-Met receptor tyrosine kinase in carcinogenesis and associated therapeutic opportunities. Mol. Cancer 2018, 17, 45. [Google Scholar] [CrossRef]
- Chiarugi, P.; Cirri, P. Redox regulation of protein tyrosine phosphatases during receptor tyrosine kinase signal transduction. Trends Biochem. Sci. 2003, 28, 509–514. [Google Scholar] [CrossRef]
- Chen, M.-K.; Du, Y.; Sun, L.; Hsu, J.L.; Wang, Y.-H.; Gao, Y.; Huang, J.; Hung, M.-C. H2O2 induces nuclear transport of the receptor tyrosine kinase c-MET in breast cancer cells via a membrane-bound retrograde trafficking mechanism. J. Biol. Chem. 2019, 294, 8516–8528. [Google Scholar] [CrossRef] [PubMed]
- Dong, Q.; Du, Y.; Li, H.; Liu, C.; Wei, Y.; Chen, M.-K.; Zhao, X.; Chu, Y.-Y.; Qiu, Y.; Qin, L. EGFR and c-MET cooperate to enhance resistance to PARP inhibitors in hepatocellular carcinoma. Cancer Res. 2019, 79, 819–829. [Google Scholar] [CrossRef] [PubMed]
- Crawford, R.S.; Albadawi, H.; Atkins, M.D.; Jones, J.E.; Yoo, H.J.; Conrad, M.F.; Austen, W.G., Jr.; Watkins, M.T. Postischemic poly (ADP-ribose) polymerase (PARP) inhibition reduces ischemia reperfusion injury in a hind-limb ischemia model. Surgery 2010, 148, 110–118. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zingarelli, B.; Hake, P.W.; O’Connor, M.; Denenberg, A.; Kong, S.; Aronow, B.J. Absence of poly(ADP-ribose)polymerase-1 alters nuclear factor-kappa B activation and gene expression of apoptosis regulators after reperfusion injury. Mol. Med. 2003, 9, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Blaser, H.; Dostert, C.; Mak, T.W.; Brenner, D. TNF and ROS Crosstalk in Inflammation. Trends Cell Biol. 2016, 26, 249–261. [Google Scholar] [CrossRef]
- Johnson, P.M.; Sagerman, R.H.; Dombrowski, C.S. Ischemia of the lung due to ionizing radiation: Quantitative studies. J. Nucl. Med. 1970, 11, 491–495. [Google Scholar]
- Zhou, Z.B.; Meng, L.; Gelb, A.W.; Lee, R.; Huang, W.Q. Cerebral ischemia during surgery: An overview. J. Biomed. Res. 2016, 30, 83–87. [Google Scholar]
- Wang, S.; Han, L.; Han, J.; Li, P.; Ding, Q.; Zhang, Q.J.; Liu, Z.P.; Chen, C.; Yu, Y. Uncoupling of PARP1 trapping and inhibition using selective PARP1 degradation. Nat. Chem. Biol. 2019, 15, 1223–1231. [Google Scholar] [CrossRef]
- Hatachi, G.; Tsuchiya, T.; Miyazaki, T.; Matsumoto, K.; Yamasaki, N.; Okita, N.; Nanashima, A.; Higami, Y.; Nagayasu, T. The poly(adenosine diphosphate-ribose) polymerase inhibitor PJ34 reduces pulmonary ischemia-reperfusion injury in rats. Transplantation 2014, 98, 618–624. [Google Scholar] [CrossRef]
- Xu, J.C.; Fan, J.; Wang, X.; Eacker, S.M.; Kam, T.I.; Chen, L.; Yin, X.; Zhu, J.; Chi, Z.; Jiang, H.; et al. Cultured networks of excitatory projection neurons and inhibitory interneurons for studying human cortical neurotoxicity. Sci. Transl. Med. 2016, 8, 333ra48. [Google Scholar] [CrossRef]
- Chatterjee, P.K.; Chatterjee, B.E.; Pedersen, H.; Sivarajah, A.; McDonald, M.C.; Mota-Filipe, H.; Brown, P.A.; Stewart, K.N.; Cuzzocrea, S.; Threadgill, M.D.; et al. 5-Aminoisoquinolinone reduces renal injury and dysfunction caused by experimental ischemia/reperfusion. Kidney Int. 2004, 65, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Mouchiroud, L.; Houtkooper, R.H.; Moullan, N.; Katsyuba, E.; Ryu, D.; Canto, C.; Mottis, A.; Jo, Y.S.; Viswanathan, M.; Schoonjans, K.; et al. The NAD(+)/Sirtuin Pathway Modulates Longevity through Activation of Mitochondrial UPR and FOXO Signaling. Cell 2013, 154, 430–441. [Google Scholar] [CrossRef] [PubMed]
- Mota, R.; Sanchez-Bueno, F.; Berenguer-Pina, J.J.; Hernandez-Espinosa, D.; Parrilla, P.; Yelamos, J. Therapeutic treatment with poly(ADP-ribose) polymerase inhibitors attenuates the severity of acute pancreatitis and associated liver and lung injury. Br. J. Pharmacol. 2007, 151, 998–1005. [Google Scholar] [CrossRef] [PubMed]
- Montoni, A.; Robu, M.; Pouliot, E.; Shah, G.M. Resistance to PARP-Inhibitors in Cancer Therapy. Front. Pharmacol. 2013, 4, 18. [Google Scholar] [CrossRef]
- Prensner, J.R.; Chen, W.; Iyer, M.K.; Cao, Q.; Ma, T.; Han, S.; Sahu, A.; Malik, R.; Wilder-Romans, K.; Navone, N.; et al. PCAT-1, a long noncoding RNA, regulates BRCA2 and controls homologous recombination in cancer. Cancer Res. 2014, 74, 1651–1660. [Google Scholar] [CrossRef]
- Livraghi, L.; Garber, J.E. PARP inhibitors in the management of breast cancer: Current data and future prospects. BMC Med. 2015, 13, 188. [Google Scholar] [CrossRef]
- Quereda, V.; Bayle, S.; Vena, F.; Frydman, S.M.; Monastyrskyi, A.; Roush, W.R.; Duckett, D.R. Therapeutic targeting of CDK12/CDK13 in triple-negative breast cancer. Cancer Cell 2019, 36, 545–558. [Google Scholar] [CrossRef]
- Balaji, K.; Vijayaraghavan, S.; Diao, L.; Tong, P.; Fan, Y.; Carey, J.P.; Bui, T.N.; Warner, S.; Heymach, J.V.; Hunt, K.K. AXL inhibition suppresses the DNA damage response and sensitizes cells to PARP inhibition in multiple cancers. Mol. Cancer Res. 2017, 15, 45–58. [Google Scholar] [CrossRef]
- Garcia, T.B.; Snedeker, J.C.; Baturin, D.; Gardner, L.; Fosmire, S.P.; Zhou, C.; Jordan, C.T.; Venkataraman, S.; Vibhakar, R.; Porter, C.C. A small-molecule inhibitor of WEE1, AZD1775, synergizes with olaparib by impairing homologous recombination and enhancing DNA damage and apoptosis in acute leukemia. Mol. Cancer Ther. 2017, 16, 2058–2068. [Google Scholar] [CrossRef]
- Song, Z.; Tu, X.; Zhou, Q.; Huang, J.; Chen, Y.; Liu, J.; Lee, S.; Kim, W.; Nowsheen, S.; Luo, K. A novel UCHL 3 inhibitor, perifosine, enhances PARP inhibitor cytotoxicity through inhibition of homologous recombination-mediated DNA double strand break repair. Cell Death & Disease 2019, 10, 398. [Google Scholar]
- Miller, A.L.; Fehling, S.C.; Garcia, P.L.; Gamblin, T.L.; Council, L.N.; van Waardenburg, R.C.; Yang, E.S.; Bradner, J.E.; Yoon, K.J. The BET inhibitor JQ1 attenuates double-strand break repair and sensitizes models of pancreatic ductal adenocarcinoma to PARP inhibitors. EBioMedicine 2019, 44, 419–430. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, Y.H.; García-García, C.; Serra, V.; He, L.; Torres-Lockhart, K.; Prat, A.; Anton, P.; Cozar, P.; Guzmán, M.; Grueso, J. PI3K inhibition impairs BRCA1/2 expression and sensitizes BRCA-proficient triple-negative breast cancer to PARP inhibition. Cancer Discov. 2012, 2, 1036–1047. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Q.; Hu, Z.; Li, Q.; Yi, T.; Li, J.; Yang, H. Cyclin D1 silencing impairs DNA double strand break repair, sensitizes BRCA1 wildtype ovarian cancer cells to olaparib. Gynecol. Oncol. 2019, 152, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Jirawatnotai, S.; Hu, Y.; Michowski, W.; Elias, J.E.; Becks, L.; Bienvenu, F.; Zagozdzon, A.; Goswami, T.; Wang, Y.E.; Clark, A.B.; et al. A function for cyclin D1 in DNA repair uncovered by protein interactome analyses in human cancers. Nature 2011, 474, 230–234. [Google Scholar] [CrossRef]
- Hirst, J.; Godwin, A.K. AURKA inhibition mimics BRCAness. Aging (Albany NY) 2017, 9, 1945. [Google Scholar] [CrossRef]
- Byrum, A.K.; Vindigni, A.; Mosammaparast, N. Defining and Modulating ‘BRCAness’. Trends Cell Biol. 2019. [Google Scholar] [CrossRef]
- Wu, W.; Nishikawa, H.; Fukuda, T.; Vittal, V.; Asano, M.; Miyoshi, Y.; Klevit, R.E.; Ohta, T. Interaction of BARD1 and HP1 is required for BRCA1 retention at sites of DNA damage. Cancer Res. 2015, 75, 1311–1321. [Google Scholar] [CrossRef]
- Criscuolo, D.; Morra, F.; Giannella, R.; Visconti, R.; Cerrato, A.; Celetti, A. New combinatorial strategies to improve the PARP inhibitors efficacy in the urothelial bladder Cancer treatment. J. Exp. Clin. Cancer Res. 2019, 38, 91. [Google Scholar] [CrossRef]
- Cerrato, A.; Merolla, F.; Morra, F.; Celetti, A. CCDC6: The identity of a protein known to be partner in fusion. Int. J. Cancer 2018, 142, 1300–1308. [Google Scholar] [CrossRef]
- Vena, F.; Jia, R.; Esfandiari, A.; Garcia-Gomez, J.J.; Rodriguez-Justo, M.; Ma, J.; Syed, S.; Crowley, L.; Elenbaas, B.; Goodstal, S. MEK inhibition leads to BRCA2 downregulation and sensitization to DNA damaging agents in pancreas and ovarian cancer models. Oncotarget 2018, 9, 11592. [Google Scholar] [CrossRef]
- Yin, L.; Liu, Y.; Peng, Y.; Peng, Y.; Yu, X.; Gao, Y.; Yuan, B.; Zhu, Q.; Cao, T.; He, L. PARP inhibitor veliparib and HDAC inhibitor SAHA synergistically co-target the UHRF1/BRCA1 DNA damage repair complex in prostate cancer cells. J. Exp. Clin. Cancer Res. 2018, 37, 153. [Google Scholar] [CrossRef] [PubMed]
- Cruz, O.V.; Prudnikova, T.Y.; Araiza-Olivera, D.; Perez-Plasencia, C.; Johnson, N.; Bernhardy, A.J.; Slifker, M.; Renner, C.; Chernoff, J.; Arias, L.E. Reduced PAK1 activity sensitizes FA/BRCA-proficient breast cancer cells to PARP inhibition. Oncotarget 2016, 7, 76590. [Google Scholar]
- Li, L.; Karanika, S.; Yang, G.; Wang, J.; Park, S.; Broom, B.; Manyam, G.C.; Wu, W.; Luo, Y.; Basourakos, S. Enzalutamide-induced “BRCAness” and PARP inhibition is a synthetic lethal therapy for castration-resistant prostate cancer. Sci. Signal. 2017, 10. [Google Scholar] [CrossRef]
- Liu, L.; Zhou, W.; Cheng, C.-T.; Ren, X.; Somlo, G.; Fong, M.Y.; Chin, A.R.; Li, H.; Yu, Y.; Xu, Y. TGFβ induces “BRCAness” and sensitivity to PARP inhibition in breast cancer by regulating DNA-repair genes. Mol. Cancer Res. 2014, 12, 1597–1609. [Google Scholar] [CrossRef] [PubMed]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, N.; Pay, S.L.; Bhandare, S.B.; Arimpur, U.; Motea, E.A. Therapeutic Strategies and Biomarkers to Modulate PARP Activity for Targeted Cancer Therapy. Cancers 2020, 12, 972. https://doi.org/10.3390/cancers12040972
Singh N, Pay SL, Bhandare SB, Arimpur U, Motea EA. Therapeutic Strategies and Biomarkers to Modulate PARP Activity for Targeted Cancer Therapy. Cancers. 2020; 12(4):972. https://doi.org/10.3390/cancers12040972
Chicago/Turabian StyleSingh, Naveen, S. Louise Pay, Snehal B. Bhandare, Udhaya Arimpur, and Edward A. Motea. 2020. "Therapeutic Strategies and Biomarkers to Modulate PARP Activity for Targeted Cancer Therapy" Cancers 12, no. 4: 972. https://doi.org/10.3390/cancers12040972
APA StyleSingh, N., Pay, S. L., Bhandare, S. B., Arimpur, U., & Motea, E. A. (2020). Therapeutic Strategies and Biomarkers to Modulate PARP Activity for Targeted Cancer Therapy. Cancers, 12(4), 972. https://doi.org/10.3390/cancers12040972