Cancer Genetics and Therapeutic Opportunities in Urologic Practice

 ,
, 
 ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
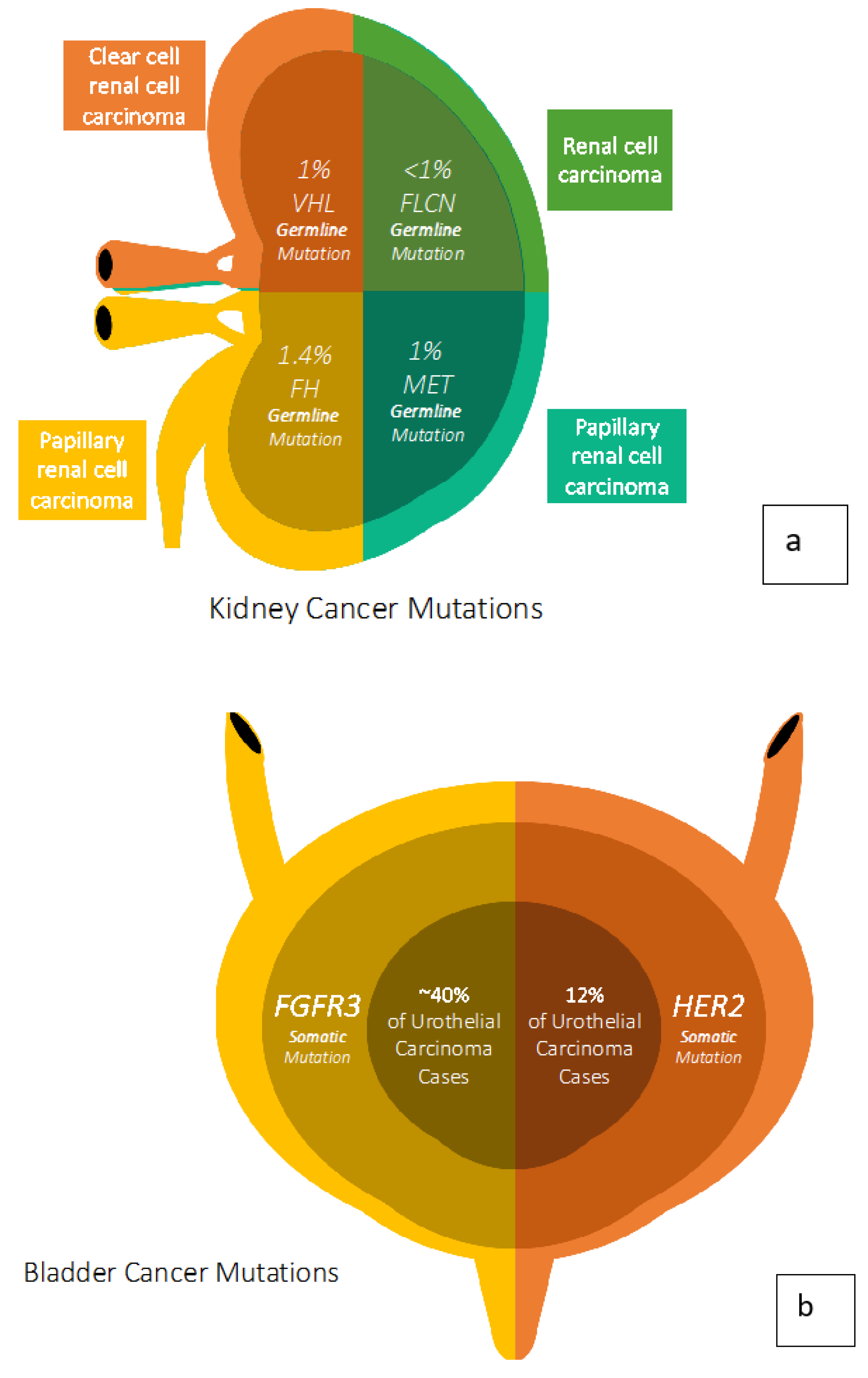
2. Kidney Cancer Genetics
3. Bladder Cancer Genetics
4. Prostate Cancer Genetics
5. Testicular Cancer Genetics
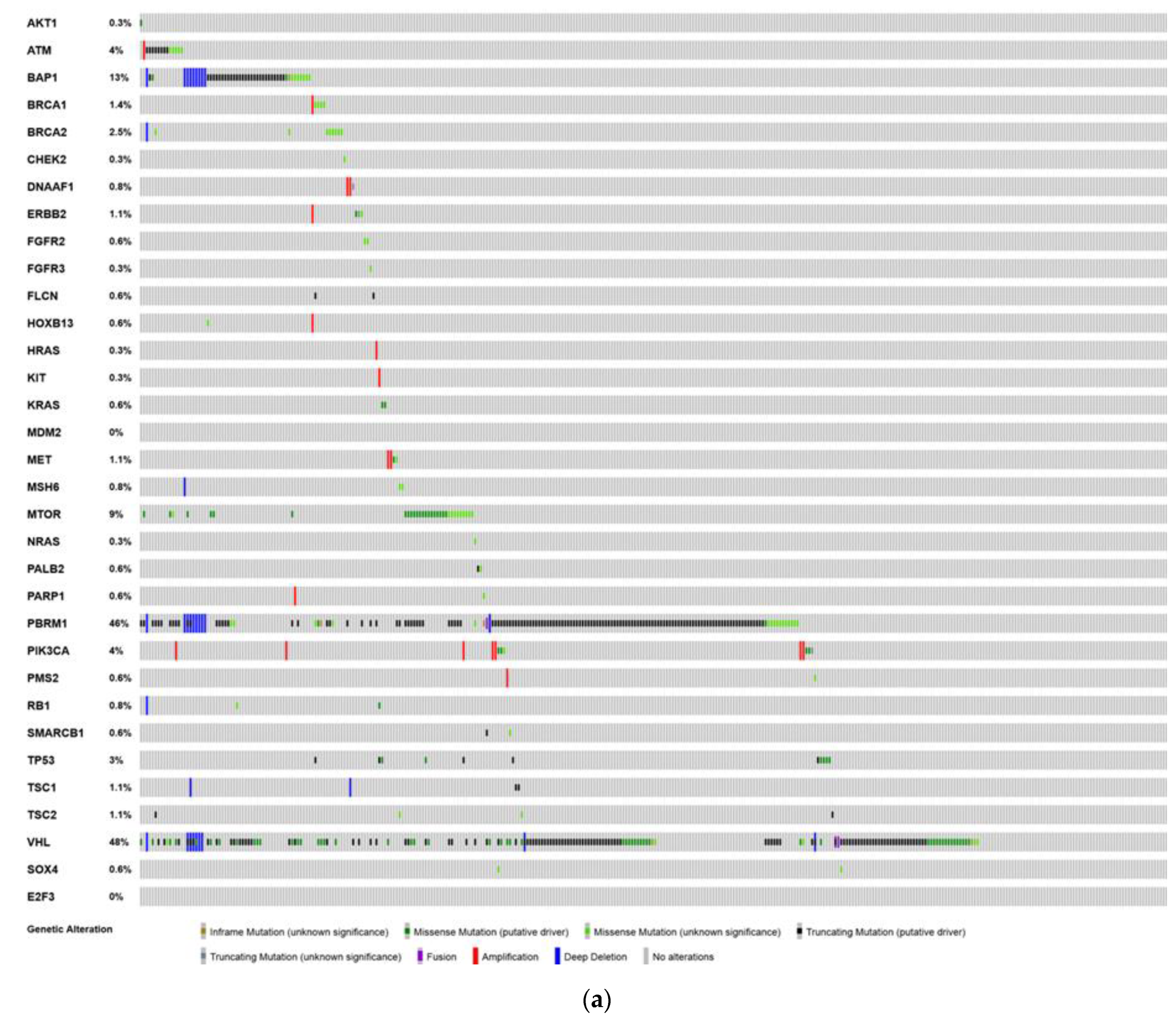
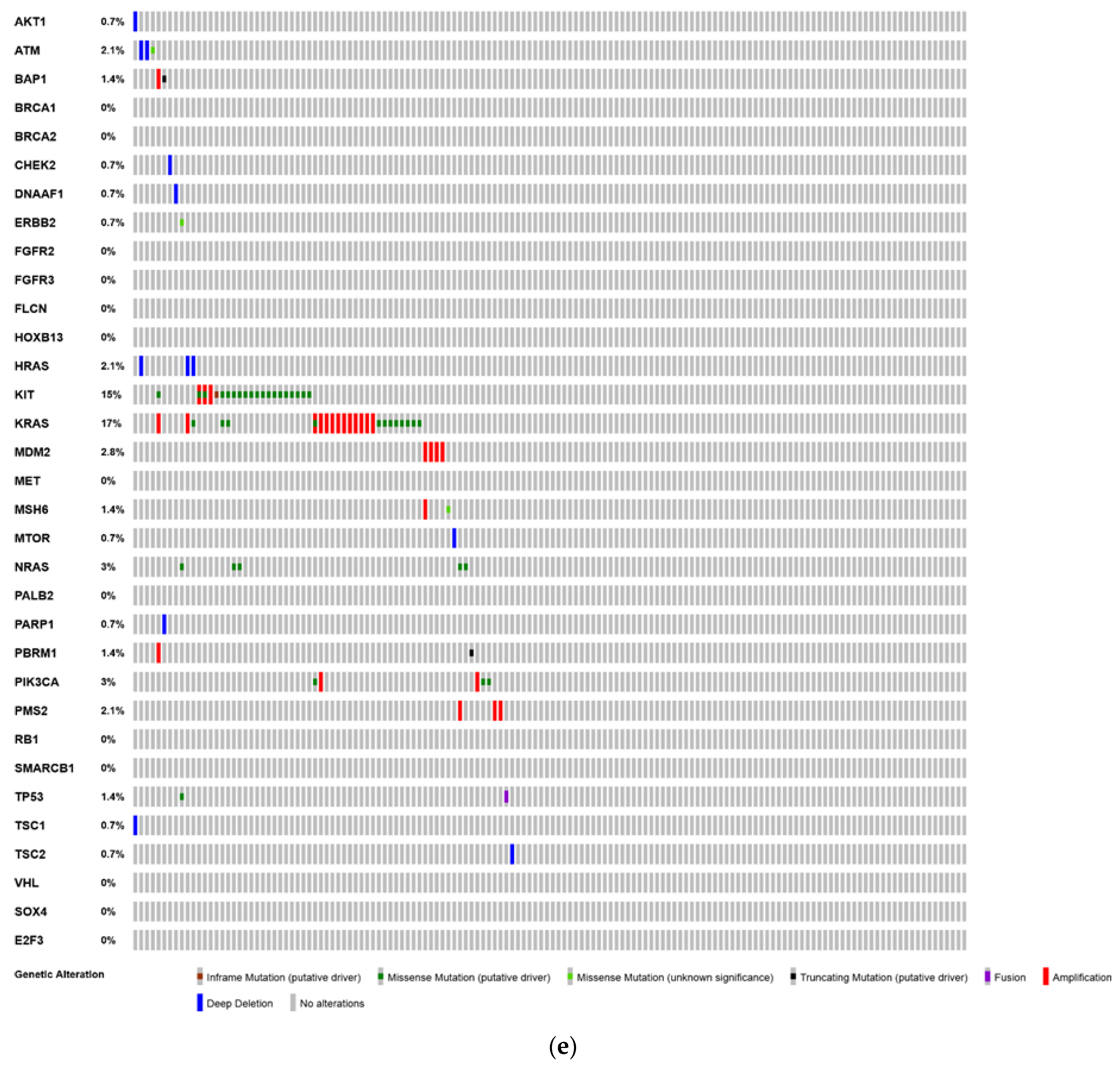
6. Analysis of The Cancer Genome Atlas (TCGA)—Last Accessed 13 March 2020
7. Genitourinary Genetic Counseling for the Practicing Physician
7.1. Genetic Counseling Overview
7.2. Genetic Testing and Prostate Cancer
7.3. Genetic Testing and Renal Cancer
7.4. Genetic Testing and Upper Tract Urothelial Carcinoma
8. Conclusions
Funding
Conflicts of Interest
References
- Haas, N.B.; Nathanson, K.L. Hereditary kidney cancer syndromes. Adv. Chronic Kidney Dis. 2014, 21, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Linehan, W.M. Genetic basis of kidney cancer: Role of genomics for the development of disease-based therapeutics. Genome Res. 2012, 22, 2089–2100. [Google Scholar] [CrossRef] [PubMed]
- Hasumi, Y.; Baba, M.; Ajima, R.; Hasumi, H.; Valera, V.A.; Klein, M.E.; Haines, D.C.; Merino, M.J.; Hong, S.B.; Yamaguchi, T.P.; et al. Homozygous loss of BHD causes early embryonic lethality and kidney tumor development with activation of mTORC1 and mTORC2. Proc. Natl. Acad. Sci. USA 2009, 106, 18722–18727. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.L.; Mashl, R.J.; Wu, Y.; Ritter, D.I.; Wang, J.; Oh, C.; Paczkowska, M.; Reynolds, S.; Wyczalkowski, M.A.; Oak, N.; et al. Pathogenic Germline Variants in 10,389 Adult Cancers. Cell 2018, 173, 355–370. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, I.; Sansom, O.J.; Leung, H.Y. Exploring molecular genetics of bladder cancer: Lessons learned from mouse models. Dis. Models Mech. 2012, 5, 323–332. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, Y. Bladder Cancer and Genetic Mutations. Cell Biochem. Biophys. 2015, 73, 65–69. [Google Scholar] [CrossRef]
- Loriot, Y.; Necchi, A.; Park, S.H.; Garcia-Donas, J.; Huddart, R.; Burgess, E.; Fleming, M.; Rezazadeh, A.; Mellado, B.; Varlamov, S.; et al. Erdafitinib in Locally Advanced or Metastatic Urothelial Carcinoma. N. Engl. J. Med. 2019, 381, 338–348. [Google Scholar] [CrossRef]
- Breast Cancer Linkage Consortium. Cancer risks in BRCA2 mutation carriers. J. Natl. Cancer Inst. 1999, 91, 1310–1316. [Google Scholar] [CrossRef]
- Thompson, D.; Easton, D.; Breast Cancer Linkage Consortium. Variation in cancer risks, by mutation position, in BRCA2 mutation carriers. Am. J. Hum. Genet. 2001, 68, 410–419. [Google Scholar] [CrossRef]
- Ostrander, E.A.; Udler, M.S. The role of the BRCA2 gene in susceptibility to prostate cancer revisited. Cancer Epidemiol. Biomark. Prev. 2008, 17, 1843–1848. [Google Scholar] [CrossRef]
- Schrader, K.A.; Cheng, D.T.; Joseph, V.; Prasad, M.; Walsh, M.; Zehir, A.; Ni, A.; Thomas, T.; Benayed, R.; Ashraf, A.; et al. Germline Variants in Targeted Tumor Sequencing Using Matched Normal DNA. JAMA Oncol. 2016, 2, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Annala, M.; Struss, W.J.; Warner, E.W.; Beja, K.; Vandekerkhove, G.; Wong, A.; Khalaf, D.; Seppala, I.L.; So, A.; Lo, G.; et al. Treatment Outcomes and Tumor Loss of Heterozygosity in Germline DNA Repair-deficient Prostate Cancer. Eur. Urol. 2017, 72, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Ewing, C.M.; Ray, A.M.; Lange, E.M.; Zuhlke, K.A.; Robbins, C.M.; Tembe, W.D.; Wiley, K.E.; Isaacs, S.D.; Johng, D.; Wang, Y.; et al. Germline mutations in HOXB13 and prostate-cancer risk. N. Engl. J. Med. 2012, 366, 141–149. [Google Scholar] [CrossRef]
- Mateo, J.; Carreira, S.; Sandhu, S.; Miranda, S.; Mossop, H.; Perez-Lopez, R.; Nava Rodrigues, D.; Robinson, D.; Omlin, A.; Tunariu, N.; et al. DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. N. Engl. J. Med. 2015, 373, 1697–1708. [Google Scholar] [CrossRef] [PubMed]
- National Comprehensive Cancer Network: NCCN Clinical Practice Guidelines in Oncology: Prostate Cancer. Available online: https://www.nccn.org/professionals/physician_gls/pdf/prostate_blocks.pdf (accessed on 6 May 2019).
- Murray, M.J.; Turnbull, C. Testicular cancer in 2017: Sequencing advances understanding. Nat. Rev. Urol. 2018, 15, 79–80. [Google Scholar] [CrossRef] [PubMed]
- Litchfield, K.; Levy, M.; Dudakia, D.; Proszek, P.; Shipley, C.; Basten, S.; Rapley, E.; Bishop, D.T.; Reid, A.; Huddart, R.; et al. Rare disruptive mutations in ciliary function genes contribute to testicular cancer susceptibility. Nat. Commun. 2016, 7, 13840. [Google Scholar] [CrossRef]
- Jonasch, E.; McCutcheon, I.E.; Gombos, D.S.; Ahrar, K.; Perrier, N.D.; Liu, D.; Robichaux, C.C.; Villarreal, M.F.; Weldon, J.A.; Woodson, A.H.; et al. Pazopanib in patients with von Hippel-Lindau disease: A single-arm, single-centre, phase 2 trial. Lancet Oncol. 2018, 19, 1351–1359. [Google Scholar] [CrossRef]
- Choueiri, T.K.; Plimack, E.; Arkenau, H.T.; Jonasch, E.; Heng, D.Y.C.; Powles, T.; Frigault, M.M.; Clark, E.A.; Handzel, A.A.; Gardner, H.; et al. Biomarker-Based Phase II Trial of Savolitinib in Patients with Advanced Papillary Renal Cell Cancer. J. Clin. Oncol. 2017, 35, 2993–3001. [Google Scholar] [CrossRef]
- Zhang, T.; Gong, J.; Maia, M.C.; Pal, S.K. Systemic Therapy for Non-Clear Cell Renal Cell Carcinoma. Am. Soc. Clin. Oncol. Educ. Book 2017, 37, 337–342. [Google Scholar] [CrossRef]
- Hartman, T.R.; Nicolas, E.; Klein-Szanto, A.; Al-Saleem, T.; Cash, T.P.; Simon, M.C.; Henske, E.P. The role of the Birt-Hogg-Dube protein in mTOR activation and renal tumorigenesis. Oncogene 2009, 28, 1594–1604. [Google Scholar] [CrossRef]
- Baba, M.; Furihata, M.; Hong, S.B.; Tessarollo, L.; Haines, D.C.; Southon, E.; Patel, V.; Igarashi, P.; Alvord, W.G.; Leighty, R.; et al. Kidney-targeted Birt-Hogg-Dube gene inactivation in a mouse model: Erk1/2 and Akt-mTOR activation, cell hyperproliferation, and polycystic kidneys. J. Natl. Cancer Inst. 2008, 100, 140–154. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, L.S.; Linehan, W.M. Clinical Features, Genetics and Potential Therapeutic Approaches for Birt-Hogg-Dube Syndrome. Expert Opin. Orphan Drugs 2015, 3, 15–29. [Google Scholar] [CrossRef] [PubMed]
- Turajlic, S.; Xu, H.; Litchfield, K.; Rowan, A.; Horswell, S.; Chambers, T.; O’Brien, T.; Lopez, J.I.; Watkins, T.B.K.; Nicol, D.; et al. Deterministic Evolutionary Trajectories Influence Primary Tumor Growth: TRACERx Renal. Cell 2018, 173, 595–610.e11. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, T.J.; Turajlic, S.; Rowan, A.; Nicol, D.; Farmery, J.H.R.; O’Brien, T.; Martincorena, I.; Tarpey, P.; Angelopoulos, N.; Yates, L.R.; et al. Timing the Landmark Events in the Evolution of Clear Cell Renal Cell Cancer: TRACERx Renal. Cell 2018, 173, 611–623. [Google Scholar] [CrossRef]
- Turajlic, S.; Xu, H.; Litchfield, K.; Rowan, A.; Chambers, T.; Lopez, J.I.; Nicol, D.; O’Brien, T.; Larkin, J.; Horswell, S.; et al. Tracking Cancer Evolution Reveals Constrained Routes to Metastases: TRACERx Renal. Cell 2018, 173, 581–594. [Google Scholar] [CrossRef]
- Ricketts, C.J.; Linehan, W.M. Multi-regional Sequencing Elucidates the Evolution of Clear Cell Renal Cell Carcinoma. Cell 2018, 173, 540–542. [Google Scholar] [CrossRef]
- Chau, C.; van Doorn, R.; van Poppelen, N.M.; van der Stoep, N.; Mensenkamp, A.R.; Sijmons, R.H.; van Paassen, B.W.; van den Ouweland, A.M.W.; Naus, N.C.; van der Hout, A.H.; et al. Families with BAP1-Tumor Predisposition Syndrome in The Netherlands: Path to Identification and a Proposal for Genetic Screening Guidelines. Cancers 2019, 11, 1114. [Google Scholar] [CrossRef]
- Walpole, S.; Pritchard, A.L.; Cebulla, C.M.; Pilarski, R.; Stautberg, M.; Davidorf, F.H.; de la Fouchardiere, A.; Cabaret, O.; Golmard, L.; Stoppa-Lyonnet, D.; et al. Comprehensive Study of the Clinical Phenotype of Germline BAP1 Variant-Carrying Families Worldwide. J. Natl. Cancer Inst. 2018, 110, 1328–1341. [Google Scholar] [CrossRef]
- Calderaro, J.; Masliah-Planchon, J.; Richer, W.; Maillot, L.; Maille, P.; Mansuy, L.; Bastien, C.; de la Taille, A.; Boussion, H.; Charpy, C.; et al. Balanced Translocations Disrupting SMARCB1 Are Hallmark Recurrent Genetic Alterations in Renal Medullary Carcinomas. Eur. Urol. 2016, 69, 1055–1061. [Google Scholar] [CrossRef]
- Alvarez, O.A. Renal Medullary Carcinoma: The Kidney Cancer That Affects Individuals With Sickle Cell Trait and Disease. J. Oncol. Pract. 2017, 13, 424–425. [Google Scholar] [CrossRef]
- Lopez-Beltran, A.; Cheng, L.; Raspollini, M.R.; Montironi, R. SMARCB1/INI1 Genetic Alterations in Renal Medullary Carcinomas. Eur. Urol. 2016, 69, 1062–1064. [Google Scholar] [CrossRef] [PubMed]
- Msaouel, P.; Tannir, N.M.; Walker, C.L. A Model Linking Sickle Cell Hemoglobinopathies and SMARCB1 Loss in Renal Medullary Carcinoma. Clin. Cancer Res. 2018, 24, 2044–2049. [Google Scholar] [CrossRef] [PubMed]
- Carugo, A.; Minelli, R.; Sapio, L.; Soeung, M.; Carbone, F.; Robinson, F.S.; Tepper, J.; Chen, Z.; Lovisa, S.; Svelto, M.; et al. p53 Is a Master Regulator of Proteostasis in SMARCB1-Deficient Malignant Rhabdoid Tumors. Cancer Cell 2019, 35, 204–220. [Google Scholar] [CrossRef] [PubMed]
- Hong, A.L.; Tseng, Y.Y.; Wala, J.A.; Kim, W.J.; Kynnap, B.D.; Doshi, M.B.; Kugener, G.; Sandoval, G.J.; Howard, T.P.; Li, J.; et al. Renal medullary carcinomas depend upon SMARCB1 loss and are sensitive to proteasome inhibition. Elife 2019, 8, e44161. [Google Scholar] [CrossRef] [PubMed]
- Linehan, W.M.; Rouault, T.A. Molecular pathways: Fumarate hydratase-deficient kidney cancer--targeting the Warburg effect in cancer. Clin. Cancer Res. 2013, 19, 3345–3352. [Google Scholar] [CrossRef] [PubMed]
- Ohe, C.; Smith, S.C.; Sirohi, D.; Divatia, M.; de Peralta-Venturina, M.; Paner, G.P.; Agaimy, A.; Amin, M.B.; Argani, P.; Chen, Y.B.; et al. Reappraisal of Morphologic Differences Between Renal Medullary Carcinoma, Collecting Duct Carcinoma, and Fumarate Hydratase-deficient Renal Cell Carcinoma. Am. J. Surg. Pathol. 2018, 42, 279–292. [Google Scholar] [CrossRef]
- Linehan, W.M.; Spellman, P.T.; Ricketts, C.J.; Creighton, C.J.; Fei, S.S.; Davis, C.; Wheeler, D.A.; Murray, B.A.; Schmidt, L.; Cancer Genome Atlas Research Network; et al. Comprehensive Molecular Characterization of Papillary Renal-Cell Carcinoma. N. Engl. J. Med. 2016, 374, 135–145. [Google Scholar] [CrossRef]
- Murta-Nascimento, C.; Silverman, D.T.; Kogevinas, M.; García-Closas, M.; Rothman, N.; Tardón, A.; García-Closas, R.; Serra, C.; Carrato, A.; Villanueva, C.; et al. Risk of Bladder Cancer Associated with Family History of Cancer: Do Low-Penetrance Polymorphisms Account for the Increase in Risk? Cancer Epidemiol. Biomark. Prev. 2007, 16, 1595–1600. [Google Scholar] [CrossRef]
- Van der Post, R.S.; Kiemeney, L.A.; Ligtenberg, M.J.L.; Witjes, J.A.; Hulsbergen-van de Kaa, C.A.; Bodmer, D.; Schaap, L.; Kets, C.M.; van Krieken, J.H.J.M.; Hoogerbrugge, N. Risk of urothelial bladder cancer in Lynch syndrome is increased, in particular among MSH2 mutation carriers. J. Med Genet. 2010, 47, 464–470. [Google Scholar] [CrossRef]
- Mancini, M.; Righetto, M.; Baggio, G. Spotlight on gender-specific disparities in bladder cancer. Urol. J. 2019. [Google Scholar] [CrossRef]
- Hurst, C.D.; Alder, O.; Platt, F.M.; Droop, A.; Stead, L.F.; Burns, J.E.; Burghel, G.J.; Jain, S.; Klimczak, L.J.; Lindsay, H.; et al. Genomic Subtypes of Non-invasive Bladder Cancer with Distinct Metabolic Profile and Female Gender Bias in KDM6A Mutation Frequency. Cancer Cell 2017, 32, 701–715. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.W.; Kim, Y.H.; Jeong, P.; Park, C.; Kim, W.T.; Ryu, D.H.; Cha, E.J.; Ha, Y.S.; Kim, T.H.; Kwon, T.G.; et al. Expression levels of FGFR3 as a prognostic marker for the progression of primary pT1 bladder cancer and its association with mutation status. Oncol. Lett. 2017, 14, 3817–3824. [Google Scholar] [CrossRef] [PubMed]
- Pichler, R.; Horninger, W.; Heidegger, I. ASCO 2018: Highlights of urothelial cancer and prostate cancer. Memo 2018, 11, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Jebar, A.H.; Hurst, C.D.; Tomlinson, D.C.; Johnston, C.; Taylor, C.F.; Knowles, M.A. FGFR3 and Ras gene mutations are mutually exclusive genetic events in urothelial cell carcinoma. Oncogene 2005, 24, 5218–5225. [Google Scholar] [CrossRef] [PubMed]
- Kompier, L.C.; Lurkin, I.; van der Aa, M.N.; van Rhijn, B.W.; van der Kwast, T.H.; Zwarthoff, E.C. FGFR3, HRAS, KRAS, NRAS and PIK3CA mutations in bladder cancer and their potential as biomarkers for surveillance and therapy. PLoS ONE 2010, 5, e13821. [Google Scholar] [CrossRef] [PubMed]
- Sjodahl, G.; Lauss, M.; Gudjonsson, S.; Liedberg, F.; Hallden, C.; Chebil, G.; Mansson, W.; Hoglund, M.; Lindgren, D. A systematic study of gene mutations in urothelial carcinoma; inactivating mutations in TSC2 and PIK3R1. PLoS ONE 2011, 6, e18583. [Google Scholar] [CrossRef]
- Yan, M.; Schwaederle, M.; Arguello, D.; Millis, S.Z.; Gatalica, Z.; Kurzrock, R. HER2 expression status in diverse cancers: Review of results from 37,992 patients. Cancer Metast. Rev. 2015, 34, 157–164. [Google Scholar] [CrossRef]
- Powles, T.; Huddart, R.A.; Elliott, T.; Sarker, S.-J.; Ackerman, C.; Jones, R.; Hussain, S.; Crabb, S.; Jagdev, S.; Chester, J.; et al. Phase III, Double-Blind, Randomized Trial That Compared Maintenance Lapatinib Versus Placebo After First-Line Chemotherapy in Patients With Human Epidermal Growth Factor Receptor 1/2–Positive Metastatic Bladder Cancer. J. Clin. Oncol. 2017, 35, 48–55. [Google Scholar] [CrossRef]
- Koshkin VS, O.D.P.; Yu, E.Y.; Grivas, P. Systematic Review: Targeting HER2 in Bladder Cancer. Bladder Cancer 2019, 5, 1–12. [Google Scholar] [CrossRef]
- Carter, B.S.; Beaty, T.H.; Steinberg, G.D.; Childs, B.; Walsh, P.C. Mendelian inheritance of familial prostate cancer. Proc. Natl. Acad. Sci. USA 1992, 89, 3367–3371. [Google Scholar] [CrossRef]
- Hjelmborg, J.B.; Scheike, T.; Holst, K.; Skytthe, A.; Penney, K.L.; Graff, R.E.; Pukkala, E.; Christensen, K.; Adami, H.O.; Holm, N.V.; et al. The heritability of prostate cancer in the Nordic Twin Study of Cancer. Cancer Epidemiol. Biomark. Prev. 2014, 23, 2303–2310. [Google Scholar] [CrossRef] [PubMed]
- Page, W.F.; Braun, M.M.; Partin, A.W.; Caporaso, N.; Walsh, P. Heredity and prostate cancer: A study of World War II veteran twins. Prostate 1997, 33, 240–245. [Google Scholar] [CrossRef]
- Pritchard, C.C.; Mateo, J.; Walsh, M.F.; De Sarkar, N.; Abida, W.; Beltran, H.; Garofalo, A.; Gulati, R.; Carreira, S.; Eeles, R.; et al. Inherited DNA-Repair Gene Mutations in Men with Metastatic Prostate Cancer. N. Engl. J. Med. 2016, 375, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Agalliu, I.; Gern, R.; Leanza, S.; Burk, R.D. Associations of high-grade prostate cancer with BRCA1 and BRCA2 founder mutations. Clin. Cancer Res. 2009, 15, 1112–1120. [Google Scholar] [CrossRef] [PubMed]
- Castro, E.; Eeles, R. The role of BRCA1 and BRCA2 in prostate cancer. Asian J. Androl. 2012, 14, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Leongamornlert, D.; Mahmud, N.; Tymrakiewicz, M.; Saunders, E.; Dadaev, T.; Castro, E.; Goh, C.; Govindasami, K.; Guy, M.; O’Brien, L.; et al. Germline BRCA1 mutations increase prostate cancer risk. Br. J. Cancer 2012, 106, 1697–1701. [Google Scholar] [CrossRef]
- Castro, E.; Goh, C.; Olmos, D.; Saunders, E.; Leongamornlert, D.; Tymrakiewicz, M.; Mahmud, N.; Dadaev, T.; Govindasami, K.; Guy, M.; et al. Germline BRCA mutations are associated with higher risk of nodal involvement, distant metastasis, and poor survival outcomes in prostate cancer. J. Clin. Oncol. 2013, 31, 1748–1757. [Google Scholar] [CrossRef]
- Castro, E.; Goh, C.; Leongamornlert, D.; Saunders, E.; Tymrakiewicz, M.; Dadaev, T.; Govindasami, K.; Guy, M.; Ellis, S.; Frost, D.; et al. Effect of BRCA Mutations on Metastatic Relapse and Cause-specific Survival After Radical Treatment for Localised Prostate Cancer. Eur. Urol. 2015, 68, 186–193. [Google Scholar] [CrossRef]
- Kim, G.; Ison, G.; McKee, A.E.; Zhang, H.; Tang, S.; Gwise, T.; Sridhara, R.; Lee, E.; Tzou, A.; Philip, R.; et al. FDA Approval Summary: Olaparib Monotherapy in Patients with Deleterious Germline BRCA-Mutated Advanced Ovarian Cancer Treated with Three or More Lines of Chemotherapy. Clin. Cancer Res. 2015, 21, 4257–4261. [Google Scholar] [CrossRef]
- Balasubramaniam, S.; Beaver, J.A.; Horton, S.; Fernandes, L.L.; Tang, S.; Horne, H.N.; Liu, J.; Liu, C.; Schrieber, S.J.; Yu, J.; et al. FDA Approval Summary: Rucaparib for the Treatment of Patients with Deleterious BRCA Mutation-Associated Advanced Ovarian Cancer. Clin. Cancer Res. 2017, 23, 7165–7170. [Google Scholar] [CrossRef]
- Hoy, S.M. Talazoparib: First Global Approval. Drugs 2018, 78, 1939–1946. [Google Scholar] [CrossRef]
- Ison, G.; Howie, L.J.; Amiri-Kordestani, L.; Zhang, L.; Tang, S.; Sridhara, R.; Pierre, V.; Charlab, R.; Ramamoorthy, A.; Song, P.; et al. FDA Approval Summary: Niraparib for the Maintenance Treatment of Patients with Recurrent Ovarian Cancer in Response to Platinum-Based Chemotherapy. Clin. Cancer Res. 2018, 24, 4066–4071. [Google Scholar] [CrossRef] [PubMed]
- Scott, L.J. Niraparib: First Global Approval. Drugs 2017, 77, 1029–1034. [Google Scholar] [CrossRef] [PubMed]
- Adashek, J.J.; Jain, R.K.; Zhang, J. Clinical Development of PARP Inhibitors in Treating Metastatic Castration-Resistant Prostate Cancer. Cells 2019, 8, 860. [Google Scholar] [CrossRef] [PubMed]
- Rimar, K.J.; Tran, P.T.; Matulewicz, R.S.; Hussain, M.; Meeks, J.J. The emerging role of homologous recombination repair and PARP inhibitors in genitourinary malignancies. Cancer 2017, 123, 1912–1924. [Google Scholar] [CrossRef]
- Geethakumari, P.R.; Schiewer, M.J.; Knudsen, K.E.; Kelly, W.K. PARP Inhibitors in Prostate Cancer. Curr. Treat. Options Oncol. 2017, 18, 37. [Google Scholar] [CrossRef]
- Cai, Q.; Wang, X.; Li, X.; Gong, R.; Guo, X.; Tang, Y.; Yang, K.; Niu, Y.; Zhao, Y. Germline HOXB13 p.Gly84Glu mutation and cancer susceptibility: A pooled analysis of 25 epidemiological studies with 145,257 participates. Oncotarget 2015, 6, 42312–42321. [Google Scholar] [CrossRef]
- Naslund-Koch, C.; Nordestgaard, B.G.; Bojesen, S.E. Increased Risk for Other Cancers in Addition to Breast Cancer for CHEK2*1100delC Heterozygotes Estimated From the Copenhagen General Population Study. J. Clin. Oncol. 2016, 34, 1208–1216. [Google Scholar] [CrossRef] [PubMed]
- Hale, V.; Weischer, M.; Park, J.Y. CHEK2 (*) 1100delC Mutation and Risk of Prostate Cancer. Prostate Cancer 2014, 2014, 294575. [Google Scholar] [CrossRef]
- Southey, M.C.; Goldgar, D.E.; Winqvist, R.; Pylkas, K.; Couch, F.; Tischkowitz, M.; Foulkes, W.D.; Dennis, J.; Michailidou, K.; van Rensburg, E.J.; et al. PALB2, CHEK2 and ATM rare variants and cancer risk: Data from COGS. J. Med. Genet. 2016, 53, 800–811. [Google Scholar] [CrossRef]
- AlDubayan, S.H.; Pyle, L.C.; Gamulin, M.; Kulis, T.; Moore, N.D.; Taylor-Weiner, A.; Hamid, A.A.; Reardon, B.; Wubbenhorst, B.; Godse, R.; et al. Association of Inherited Pathogenic Variants in Checkpoint Kinase 2 (CHEK2) With Susceptibility to Testicular Germ Cell Tumors. JAMA Oncol. 2019, 5, 514–522. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Shih, J.; Hollern, D.P.; Wang, L.; Bowlby, R.; Tickoo, S.K.; Thorsson, V.; Mungall, A.J.; Newton, Y.; Hegde, A.M.; et al. Integrated Molecular Characterization of Testicular Germ Cell Tumors. Cell Rep. 2018, 23, 3392–3406. [Google Scholar] [CrossRef] [PubMed]
- Taylor-Weiner, A.; Zack, T.; O’Donnell, E.; Guerriero, J.L.; Bernard, B.; Reddy, A.; Han, G.C.; AlDubayan, S.; Amin-Mansour, A.; Schumacher, S.E.; et al. Genomic evolution and chemoresistance in germ-cell tumours. Nature 2016, 540, 114–118. [Google Scholar] [CrossRef] [PubMed]
- Sheikine, Y.; Genega, E.; Melamed, J.; Lee, P.; Reuter, V.E.; Ye, H. Molecular genetics of testicular germ cell tumors. Am. J. Cancer Res. 2012, 2, 153–167. [Google Scholar] [PubMed]
- McGlynn, K.A.; Cook, M.B. Etiologic factors in testicular germ-cell tumors. Future Oncol. 2009, 5, 1389–1402. [Google Scholar] [CrossRef]
- Ganguly, S.; Murty, V.V.; Samaniego, F.; Reuter, V.E.; Bosl, G.J.; Chaganti, R.S. Detection of preferential NRAS mutations in human male germ cell tumors by the polymerase chain reaction. Genes Chromosomes Cancer 1990, 1, 228–232. [Google Scholar] [CrossRef]
- Honecker, F.; Wermann, H.; Mayer, F.; Gillis, A.J.; Stoop, H.; van Gurp, R.J.; Oechsle, K.; Steyerberg, E.; Hartmann, J.T.; Dinjens, W.N.; et al. Microsatellite instability, mismatch repair deficiency, and BRAF mutation in treatment-resistant germ cell tumors. J. Clin. Oncol. 2009, 27, 2129–2136. [Google Scholar] [CrossRef]
- Subbiah, V.; Meric-Bernstam, F.; Mills, G.B.; Shaw, K.R.; Bailey, A.M.; Rao, P.; Ward, J.F.; Pagliaro, L.C. Next generation sequencing analysis of platinum refractory advanced germ cell tumor sensitive to Sunitinib (Sutent(R)) a VEGFR2/PDGFRbeta/c-kit/ FLT3/RET/CSF1R inhibitor in a phase II trial. J. Hematol. Oncol. 2014, 7, 52. [Google Scholar] [CrossRef]
- Fenner, M.H.; Beutel, G.; Grunwald, V. Targeted therapies for patients with germ cell tumors. Expert Opin. Investig. Drugs 2008, 17, 511–522. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Kurzrock, R.; Sherman, S.I.; Ball, D.W.; Forastiere, A.A.; Cohen, R.B.; Mehra, R.; Pfister, D.G.; Cohen, E.E.; Janisch, L.; Nauling, F.; et al. Activity of XL184 (Cabozantinib), an oral tyrosine kinase inhibitor, in patients with medullary thyroid cancer. J. Clin. Oncol. 2011, 29, 2660–2666. [Google Scholar] [CrossRef] [PubMed]
- Braun, D.A.; Ishii, Y.; Walsh, A.M.; Van Allen, E.M.; Wu, C.J.; Shukla, S.A.; Choueiri, T.K. Clinical Validation of PBRM1 Alterations as a Marker of Immune Checkpoint Inhibitor Response in Renal Cell Carcinoma. JAMA Oncol. 2019, 5, 1631–1633. [Google Scholar] [CrossRef] [PubMed]
- Delikurt, T.; Williamson, G.R.; Anastasiadou, V.; Skirton, H. A systematic review of factors that act as barriers to patient referral to genetic services. Eur. J. Hum. Genet. 2015, 23, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Salami, S.S.; Spratt, D.E.; Kaffenberger, S.D.; Jacobs, M.F.; Morgan, T.M. Bringing Prostate Cancer Germline Genetics into Clinical Practice. J. Urol. 2019, 202, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, M.F.; Milliron, K.J. Genetic counseling and previvorship in patients with urologic malignancies. Curr. Opin. Urol. 2019, 29, 371–377. [Google Scholar] [CrossRef] [PubMed]
- Regier, D.S.; Ferreira, C.R.; Hart, S.; Hadley, D.W.; Muenke, M. Medical genetics and genomic medicine in the United States. Part 2: Reproductive genetics, newborn screening, genetic counseling, training, and registries. Mol. Genet. Genom. Med. 2017, 5, 621–630. [Google Scholar] [CrossRef] [PubMed]
- Hoskovec, J.M.; Bennett, R.L.; Carey, M.E.; DaVanzo, J.E.; Dougherty, M.; Hahn, S.E.; LeRoy, B.S.; O’Neal, S.; Richardson, J.G.; Wicklund, C.A. Projecting the Supply and Demand for Certified Genetic Counselors: A Workforce Study. J. Genet. Couns. 2018, 27, 16–20. [Google Scholar] [CrossRef]
- Colombo, N.; Huang, G.; Scambia, G.; Chalas, E.; Pignata, S.; Fiorica, J.; Van Le, L.; Ghamande, S.; Gonzalez-Santiago, S.; Bover, I.; et al. Evaluation of a Streamlined Oncologist-Led BRCA Mutation Testing and Counseling Model for Patients With Ovarian Cancer. J. Clin. Oncol. 2018, 36, 1300–1307. [Google Scholar] [CrossRef]
- Kurian, A.W.; Li, Y.; Hamilton, A.S.; Ward, K.C.; Hawley, S.T.; Morrow, M.; McLeod, M.C.; Jagsi, R.; Katz, S.J. Gaps in Incorporating Germline Genetic Testing into Treatment Decision-Making for Early-Stage Breast Cancer. J. Clin. Oncol. 2017, 35, 2232–2239. [Google Scholar] [CrossRef]
- Antoniou, A.; Pharoah, P.D.; Narod, S.; Risch, H.A.; Eyfjord, J.E.; Hopper, J.L.; Loman, N.; Olsson, H.; Johannsson, O.; Borg, A.; et al. Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case Series unselected for family history: A combined analysis of 22 studies. Am. J. Hum. Genet. 2003, 72, 1117–1130. [Google Scholar] [CrossRef]
- Domchek, S.M.; Friebel, T.M.; Singer, C.F.; Evans, D.G.; Lynch, H.T.; Isaacs, C.; Garber, J.E.; Neuhausen, S.L.; Matloff, E.; Eeles, R.; et al. Association of risk-reducing surgery in BRCA1 or BRCA2 mutation carriers with cancer risk and mortality. JAMA 2010, 304, 967–975. [Google Scholar] [CrossRef] [PubMed]
- Rebbeck, T.R.; Friebel, T.; Lynch, H.T.; Neuhausen, S.L.; van’t Veer, L.; Garber, J.E.; Evans, G.R.; Narod, S.A.; Isaacs, C.; Matloff, E.; et al. Bilateral prophylactic mastectomy reduces breast cancer risk in BRCA1 and BRCA2 mutation carriers: The PROSE Study Group. J. Clin. Oncol. 2004, 22, 1055–1062. [Google Scholar] [CrossRef] [PubMed]
- Daly, M.B. Prostate cancer genetic testing: NCCN familial high-risk assessment: Breast/ovarian. Can. J. Urol. 2019, 26, 29–30. [Google Scholar] [PubMed]
- Page, E.C.; Bancroft, E.K.; Brook, M.N.; Assel, M.; Hassan Al Battat, M.; Thomas, S.; Taylor, N.; Chamberlain, A.; Pope, J.; Raghallaigh, H.N.; et al. Interim Results from the IMPACT Study: Evidence for Prostate-specific Antigen Screening in BRCA2 Mutation Carriers. Eur. Urol. 2019, 76, 831–842. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Jonasch, E.; Michaelson, M.D.; Nandagopal, L.; Gore, J.L.; George, S.; Alva, A.; Haas, N.; Harrison, M.R.; Plimack, E.R.; et al. NCCN Guidelines Insights: Kidney Cancer, Version 2.2020. J. Natl. Compr. Cancer Netw. 2019, 17, 1278–1285. [Google Scholar] [CrossRef]
- Campbell, S.; Uzzo, R.G.; Allaf, M.E.; Bass, E.B.; Cadeddu, J.A.; Chang, A.; Clark, P.E.; Davis, B.J.; Derweesh, I.H.; Giambarresi, L.; et al. Renal Mass and Localized Renal Cancer: AUA Guideline. J. Urol. 2017, 198, 520–529. [Google Scholar] [CrossRef]
- Lui, S.T.; Shuch, B. Genetic Testing in Kidney Cancer Patients: Who, When, and How? Eur. Urol. Focus 2019, 5, 973–976. [Google Scholar] [CrossRef]
- Rossi, S.H.; Klatte, T.; Usher-Smith, J.; Stewart, G.D. Epidemiology and screening for renal cancer. World J. Urol. 2018, 36, 1341–1353. [Google Scholar] [CrossRef]
- Pradere, B.; Lotan, Y.; Roupret, M. Lynch syndrome in upper tract urothelial carcinoma: Significance, screening, and surveillance. Curr. Opin. Urol. 2017, 27, 48–55. [Google Scholar] [CrossRef]
- Roupret, M.; Babjuk, M.; Comperat, E.; Zigeuner, R.; Sylvester, R.J.; Burger, M.; Cowan, N.C.; Gontero, P.; Van Rhijn, B.W.G.; Mostafid, A.H.; et al. European Association of Urology Guidelines on Upper Urinary Tract Urothelial Carcinoma: 2017 Update. Eur. Urol. 2018, 73, 111–122. [Google Scholar] [CrossRef]
- Pinol, V.; Castells, A.; Andreu, M.; Castellvi-Bel, S.; Alenda, C.; Llor, X.; Xicola, R.M.; Rodriguez-Moranta, F.; Paya, A.; Jover, R.; et al. Accuracy of revised Bethesda guidelines, microsatellite instability, and immunohistochemistry for the identification of patients with hereditary nonpolyposis colorectal cancer. JAMA 2005, 293, 1986–1994. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Provenzale, D.; Llor, X.; Halverson, A.L.; Grady, W.; Chung, D.C.; Haraldsdottir, S.; Markowitz, A.J.; Slavin, T.P., Jr.; Hampel, H.; et al. NCCN Guidelines Insights: Genetic/Familial High-Risk Assessment: Colorectal, Version 2.2019. J. Natl. Compr. Cancer Netw. 2019, 17, 1032–1041. [Google Scholar] [CrossRef] [PubMed]
- Giardiello, F.M.; Allen, J.I.; Axilbund, J.E.; Boland, C.R.; Burke, C.A.; Burt, R.W.; Church, J.M.; Dominitz, J.A.; Johnson, D.A.; Kaltenbach, T.; et al. Guidelines on genetic evaluation and management of Lynch syndrome: A consensus statement by the US Multi-Society Task Force on colorectal cancer. Gastroenterology 2014, 147, 502–526. [Google Scholar] [CrossRef] [PubMed]
- Vasen, H.F.; Blanco, I.; Aktan-Collan, K.; Gopie, J.P.; Alonso, A.; Aretz, S.; Bernstein, I.; Bertario, L.; Burn, J.; Capella, G.; et al. Revised guidelines for the clinical management of Lynch syndrome (HNPCC): Recommendations by a group of European experts. Gut 2013, 62, 812–823. [Google Scholar] [CrossRef] [PubMed]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adashek, J.J.; Leonard, A.; Roszik, J.; Menta, A.K.; Genovese, G.; Subbiah, V.; Msaouel, P. Cancer Genetics and Therapeutic Opportunities in Urologic Practice. Cancers 2020, 12, 710. https://doi.org/10.3390/cancers12030710
Adashek JJ, Leonard A, Roszik J, Menta AK, Genovese G, Subbiah V, Msaouel P. Cancer Genetics and Therapeutic Opportunities in Urologic Practice. Cancers. 2020; 12(3):710. https://doi.org/10.3390/cancers12030710
Chicago/Turabian StyleAdashek, Jacob J., Alex Leonard, Jason Roszik, Arjun K. Menta, Giannicola Genovese, Vivek Subbiah, and Pavlos Msaouel. 2020. "Cancer Genetics and Therapeutic Opportunities in Urologic Practice" Cancers 12, no. 3: 710. https://doi.org/10.3390/cancers12030710
APA StyleAdashek, J. J., Leonard, A., Roszik, J., Menta, A. K., Genovese, G., Subbiah, V., & Msaouel, P. (2020). Cancer Genetics and Therapeutic Opportunities in Urologic Practice. Cancers, 12(3), 710. https://doi.org/10.3390/cancers12030710