Cytoskeletal Proteins in Cancer and Intracellular Stress: A Therapeutic Perspective

 ,
, 
 and
and
Abstract
1. Introduction
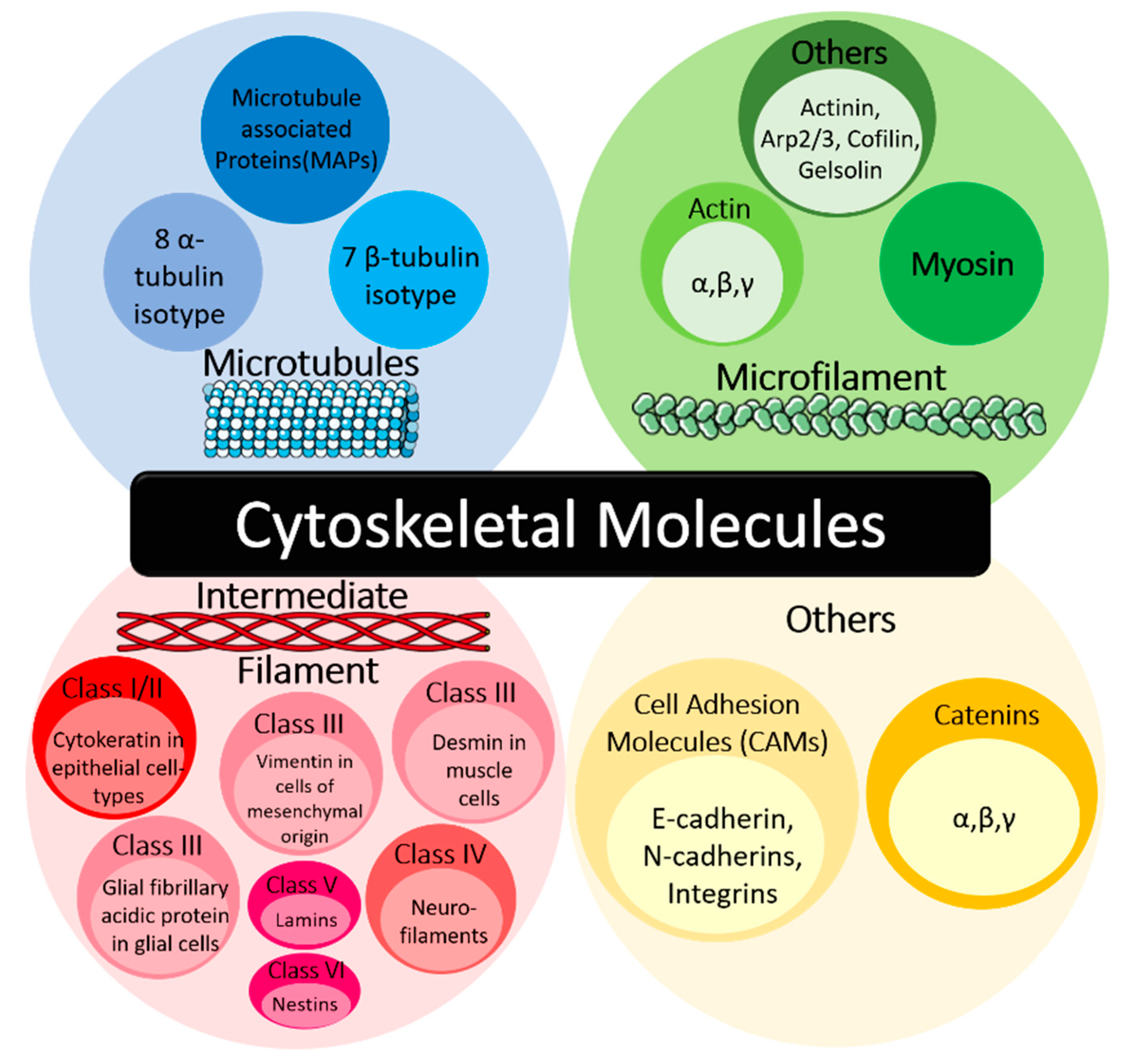
1.1. Cytoskeletal Molecules in Cancer
1.2. Intracellular Stress in Cancer
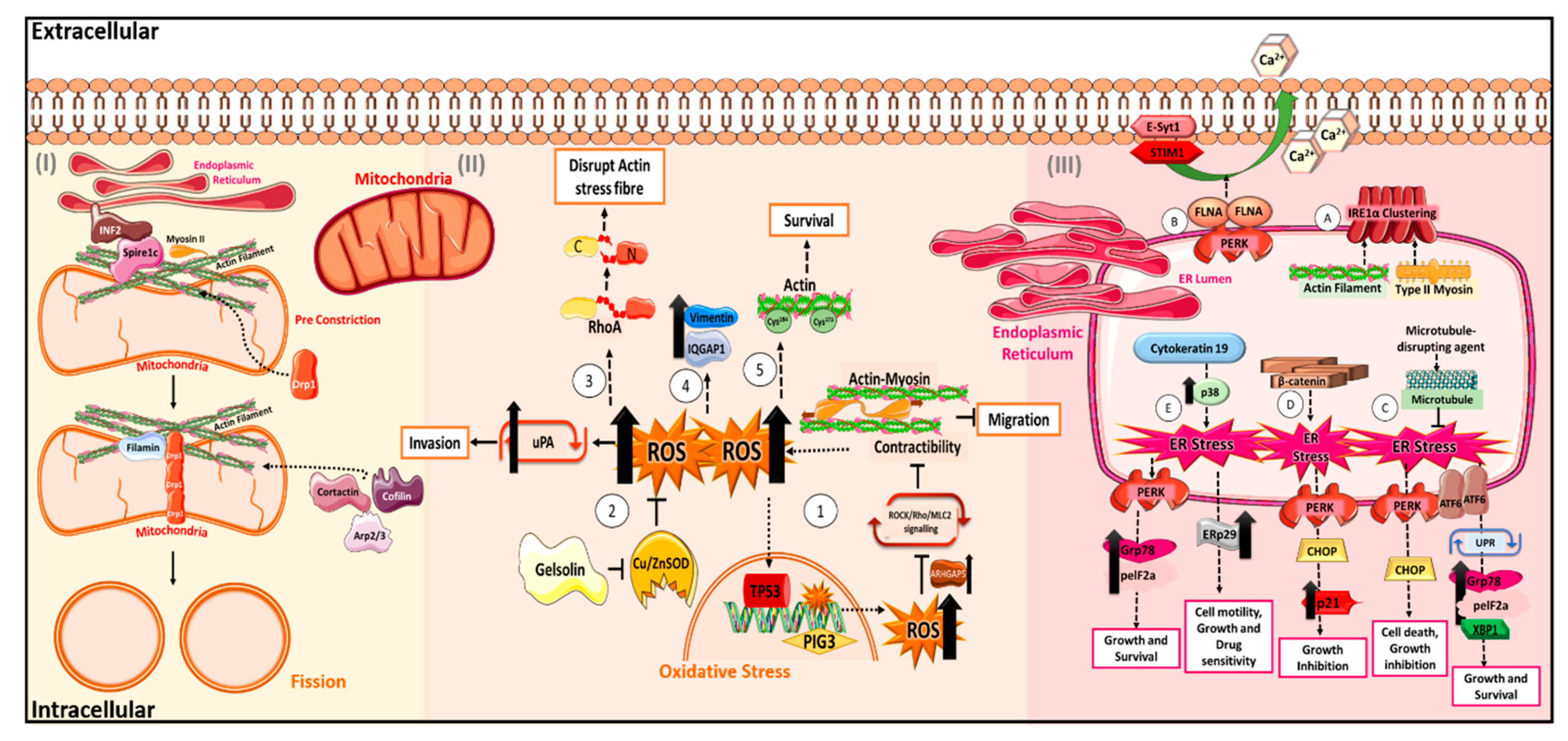
2. The “Cytoskeletons” in Oxidative Stress
2.1. Actin and Its Regulators in Oxidative Stress
2.2. Other Cytoskeletal Proteins in Oxidative Stress
3. The “Cytoskeletons” in Mitochondrial Stress
3.1. Drp1 and Actin in Mitochondrial Fission and Fusion
3.2. Regulation of VDAC by Cytoskeleton
4. The “Cytoskeletons” in Endoplasmic Reticulum Stress
4.1. Cytoskeleton in ER Sensors
4.2. Regulation of ER Signalling Cascade by Cytoskeleton
5. Effects of Cellular Stress on Chemotherapeutic Response
6. Targeting the Cytoskeleton as Potential Therapeutics
6.1. Targeting Microtubules in Cancer
6.2. Unexplored Therapeutic Targets—Actin Microfilaments
6.3. Unexplored Therapeutic Targets—Intermediate Filaments
6.4. Unexplored Therapeutic Targets—Cytoskeletal-Associated Proteins
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Fletcher, D.A.; Mullins, R.D. Cell mechanics and the cytoskeleton. Nature 2010, 463, 485–492. [Google Scholar] [CrossRef] [PubMed]
- Pawlak, G.; Helfman, D.M. Cytoskeletal changes in cell transformation and tumorigenesis. Curr. Opin. Genet. Dev. 2001, 11, 41–47. [Google Scholar] [CrossRef]
- Parker, A.L.; Kavallaris, M.; McCarroll, J.A. Microtubules and their role in cellular stress in cancer. Front. Oncol. 2014, 4, 153. [Google Scholar] [CrossRef] [PubMed]
- Don, S.; Verrills, N.M.; Liaw, T.Y.E.; Liu, M.L.M.; Norris, M.D.; Haber, M.; Kavallaris, M. Neuronal-associated microtubule proteins class III B-tubulins and MAP2c in neuroblastoma: Role in resistance to microtubule-targeted drugs. Mol. Cancer Ther. 2004, 3, 1137–1146. [Google Scholar]
- Kavallaris, M. Microtubules and resistance to tubulin-binding agents. Nat. Rev. Cancer 2010, 10, 194–204. [Google Scholar] [CrossRef]
- Rao, J.; Li, N. Microfilament Actin Remodeling as a Potential Target for Cancer Drug Development. Curr. Cancer Drug Targets 2004, 4, 345–354. [Google Scholar] [CrossRef]
- Qiao, Y.; Chen, J.; Lim, Y.B.; Finch-Edmondson, M.L.; Seshachalam, V.P.; Qin, L.; Jiang, T.; Low, B.C.; Singh, H.; Lim, C.T. YAP regulates actin dynamics through ARHGAP29 and promotes metastasis. Cell Rep. 2017, 19, 1495–1502. [Google Scholar] [CrossRef]
- Tojkander, S.; Gateva, G.; Lappalainen, P. Actin stress fibers—Assembly, dynamics and biological roles. J. Cell Sci. 2012, 125, 1855–1864. [Google Scholar] [CrossRef]
- Daly, N.; Meleady, P.; Walsh, D.; Clynes, M. Regulation of keratin and integrin gene expression in cancer and drug resistance. Cytotechnology 1998, 27, 321. [Google Scholar] [CrossRef]
- Gomes, F.; Paulin, D.; Moura Neto, V. Glial fibrillary acidic protein (GFAP): Modulation by growth factors and its implication in astrocyte differentiation. Braz. J. Med Biol. Res. 1999, 32, 619–631. [Google Scholar] [CrossRef]
- Ben-Ze’ev, A. The cytoskeleton in cancer cells. Biochim. Biophys. Acta 1985, 780, 197–212. [Google Scholar] [CrossRef]
- Hendrix, M.J.; Seftor, E.A.; Chu, Y.W.; Trevor, K.T.; Seftor, R.E. Role of intermediate filaments in migration, invasion and metastasis. Cancer Metastasis Rev. 1996, 15, 507–525. [Google Scholar] [CrossRef] [PubMed]
- Holle, A.W.; Kalafat, M.; Ramos, A.S.; Seufferlein, T.; Kemkemer, R.; Spatz, J.P. Intermediate filament reorganization dynamically influences cancer cell alignment and migration. Sci. Rep. 2017, 7, 45152. [Google Scholar] [CrossRef]
- Satelli, A.; Li, S. Vimentin in cancer and its potential as a molecular target for cancer therapy. Cell. Mol. Life Sci. CMLS 2011, 68, 3033–3046. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Sadacharan, S.; Su, S.; Belldegrun, A.; Persad, S.; Singh, G. Overexpression of vimentin: Role in the invasive phenotype in an androgen-independent model of prostate cancer. Cancer Res. 2003, 63, 2306–2311. [Google Scholar] [PubMed]
- Kidd, M.E.; Shumaker, D.K.; Ridge, K.M. The role of vimentin intermediate filaments in the progression of lung cancer. Am. J. Respir. Cell Mol. Biol. 2014, 50, 1–6. [Google Scholar] [CrossRef]
- Jiang, W.G. E-cadherin and its associated protein catenins, cancer invasion and metastasis. Br. J. Surg. 1996, 83, 437–446. [Google Scholar] [CrossRef]
- Rimm, D.L.; Sinard, J.H.; Morrow, J.S. Reduced alpha-catenin and E-cadherin expression in breast cancer. Lab. Investig. 1995, 72, 506–512. [Google Scholar]
- Schackmann, R.C.; Tenhagen, M.; van de Ven, R.A.; Derksen, P.W. p120-catenin in cancer—Mechanisms, models and opportunities for intervention. J. Cell Sci. 2013, 126, 3515–3525. [Google Scholar] [CrossRef]
- Gao, C.; Wang, Y.; Broaddus, R.; Sun, L.; Xue, F.; Zhang, W. Exon 3 mutations of CTNNB1 drive tumorigenesis: A review. Oncotarget 2018, 9, 5492. [Google Scholar] [CrossRef]
- Kaler, P.; Augenlicht, L.; Klampfer, L. Activating mutations in β-catenin in colon cancer cells alter their interaction with macrophages; the role of snail. PLoS ONE 2012, 7, e45462. [Google Scholar] [CrossRef] [PubMed]
- Parry, M.L.; Blanck, G. Flat cells come full sphere: Are mutant cytoskeletal-related proteins oncoprotein-monsters or useful immunogens? Hum. Vaccines Immunother. 2016, 12, 120–123. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Schiewek, J.; Schumacher, U.; Lange, T.; Joosse, S.A.; Wikman, H.; Pantel, K.; Mikhaylova, M.; Kneussel, M.; Linder, S.; Schmalfeldt, B. Clinical relevance of cytoskeleton associated proteins for ovarian cancer. J. Cancer Res. Clin. Oncol. 2018, 144, 2195–2205. [Google Scholar] [CrossRef] [PubMed]
- Segarra, D.T.; Yavorski, J.M.; Blanck, G. Protected cytoskeletal-related proteins: Towards a resolution of contradictions regarding the role of the cytoskeleton in cancer. Biomed. Rep. 2017, 7, 163–168. [Google Scholar] [CrossRef]
- Xu, Y.; Bismar, T.A.; Su, J.; Xu, B.; Kristiansen, G.; Varga, Z.; Teng, L.; Ingber, D.E.; Mammoto, A.; Kumar, R.; et al. Filamin A regulates focal adhesion disassembly and suppresses breast cancer cell migration and invasion. J. Exp. Med. 2010, 207, 2421–2437. [Google Scholar] [CrossRef]
- Boland, M.L.; Chourasia, A.H.; Macleod, K.F. Mitochondrial dysfunction in cancer. Front. Oncol. 2013, 3, 292. [Google Scholar] [CrossRef]
- Jones, D.P. Radical-free biology of oxidative stress. Am. J. Physiol. Cell Physiol. 2008, 295, C849–C868. [Google Scholar] [CrossRef]
- Reuter, S.; Gupta, S.C.; Chaturvedi, M.M.; Aggarwal, B.B. Oxidative stress, inflammation, and cancer: How are they linked? Free Radic. Biol. Med. 2010, 49, 1603–1616. [Google Scholar] [CrossRef]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef]
- Sosa, V.; Moline, T.; Somoza, R.; Paciucci, R.; Kondoh, H.; ME, L.L. Oxidative stress and cancer: An overview. Ageing Res. Rev. 2013, 12, 376–390. [Google Scholar] [CrossRef]
- Valko, M.; Rhodes, C.J.; Moncol, J.; Izakovic, M.; Mazur, M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem. Biol. Interact. 2006, 160, 1–40. [Google Scholar] [CrossRef] [PubMed]
- Gill, J.G.; Piskounova, E.; Morrison, S.J. Cancer, Oxidative Stress, and Metastasis. Cold Spring Harb. Symp. Quant. Biol. 2016, 81, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Schroder, M.; Kaufman, R.J. ER stress and the unfolded protein response. Mutat. Res. 2005, 569, 29–63. [Google Scholar] [CrossRef] [PubMed]
- Yadav, R.K.; Chae, S.W.; Kim, H.R.; Chae, H.J. Endoplasmic reticulum stress and cancer. J. Cancer Prev. 2014, 19, 75–88. [Google Scholar] [CrossRef]
- Clarke, H.J.; Chambers, J.E.; Liniker, E.; Marciniak, S.J. Endoplasmic reticulum stress in malignancy. Cancer Cell 2014, 25, 563–573. [Google Scholar] [CrossRef]
- Calaf, G.M.; Urzua, U.; Termini, L.; Aguayo, F. Oxidative stress in female cancers. Oncotarget 2018, 9, 23824–23842. [Google Scholar] [CrossRef]
- Calvisi, D.F.; Ladu, S.; Hironaka, K.; Factor, V.M.; Thorgeirsson, S.S. Vitamin E down-modulates iNOS and NADPH oxidase in c-Myc/TGF-α transgenic mouse model of liver cancer. J. Hepatol. 2004, 41, 815–822. [Google Scholar] [CrossRef]
- Oliveira, C.P.; Kassab, P.; Lopasso, F.P.; Souza, H.P.; Janiszewski, M.; Laurindo, F.R.; Iriya, K.; Laudanna, A.A. Protective effect of ascorbic acid in experimental gastric cancer: Reduction of oxidative stress. World J. Gastroenterol. 2003, 9, 446. [Google Scholar] [CrossRef]
- Lee, J.; Kang, Y.; Khare, V.; Jin, Z.; Kang, M.; Yoon, Y.; Hyun, J.; Chung, M.; Cho, S.; Jun, J. The p53-inducible gene 3 (PIG3) contributes to early cellular response to DNA damage. Oncogene 2010, 29, 1431. [Google Scholar] [CrossRef]
- Herraiz, C.; Calvo, F.; Pandya, P.; Cantelli, G.; Rodriguez-Hernandez, I.; Orgaz, J.L.; Kang, N.; Chu, T.; Sahai, E.; Sanz-Moreno, V. Reactivation of p53 by a cytoskeletal sensor to control the balance between DNA damage and tumor dissemination. JNCI J. Natl. Cancer Inst. 2016, 108. [Google Scholar] [CrossRef]
- Huang, B.; Deng, S.; Loo, S.Y.; Datta, A.; Yap, Y.L.; Yan, B.; Ooi, C.H.; Dinh, T.D.; Zhuo, J.; Tochhawng, L. Gelsolin-mediated activation of PI3K/Akt pathway is crucial for hepatocyte growth factor-induced cell scattering in gastric carcinoma. Oncotarget 2016, 7, 25391. [Google Scholar] [CrossRef] [PubMed]
- Tochhawng, L.; Deng, S.; Pugalenthi, G.; Kumar, A.P.; Lim, K.H.; Tan, T.Z.; Yang, H.; Hooi, S.C.; Goh, Y.C.; Maciver, S.K. Gelsolin-Cu/ZnSOD interaction alters intracellular reactive oxygen species levels to promote cancer cell invasion. Oncotarget 2016, 7, 52832. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, J.; Tan, E.H.; Yan, B.; Tochhawng, L.; Jayapal, M.; Koh, S.; Tay, H.K.; Maciver, S.K.; Hooi, S.C.; Salto-Tellez, M. Gelsolin induces colorectal tumor cell invasion via modulation of the urokinase-type plasminogen activator cascade. PLoS ONE 2012, 7, e43594. [Google Scholar] [CrossRef] [PubMed]
- Girouard, M.P.; Pool, M.; Alchini, R.; Rambaldi, I.; Fournier, A.E. RhoA proteolysis regulates the actin cytoskeleton in response to oxidative stress. PLoS ONE 2016, 11, e0168641. [Google Scholar] [CrossRef]
- Munsie, L.N.; Desmond, C.R.; Truant, R. Cofilin nuclear-cytoplasmic shuttling affects cofilin-actin rod formation during stress. J. Cell Sci. 2012, 125, 3977–3988. [Google Scholar] [CrossRef]
- Farah, M.E.; Sirotkin, V.; Haarer, B.; Kakhniashvili, D.; Amberg, D.C. Diverse protective roles of the actin cytoskeleton during oxidative stress. Cytoskeleton 2011, 68, 340–354. [Google Scholar] [CrossRef]
- Tsubota, A.; Matsumoto, K.; Mogushi, K.; Nariai, K.; Namiki, Y.; Hoshina, S.; Hano, H.; Tanaka, H.; Saito, H.; Tada, N. IQGAP1 and vimentin are key regulator genes in naturally occurring hepatotumorigenesis induced by oxidative stress. Carcinogenesis 2009, 31, 504–511. [Google Scholar] [CrossRef]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef]
- Westermann, B. Mitochondrial fusion and fission in cell life and death. Nat. Rev. Mol. Cell Biol. 2010, 11, 872–884. [Google Scholar] [CrossRef]
- Friedman, J.R.; Lackner, L.L.; West, M.; DiBenedetto, J.R.; Nunnari, J.; Voeltz, G.K. ER tubules mark sites of mitochondrial division. Science 2011, 334, 358–362. [Google Scholar] [CrossRef]
- Korobova, F.; Ramabhadran, V.; Higgs, H.N. An actin-dependent step in mitochondrial fission mediated by the ER-associated formin INF2. Science 2013, 339, 464–467. [Google Scholar] [CrossRef] [PubMed]
- Manor, U.; Bartholomew, S.; Golani, G.; Christenson, E.; Kozlov, M.; Higgs, H.; Spudich, J.; Lippincott-Schwartz, J. A mitochondria-anchored isoform of the actin-nucleating spire protein regulates mitochondrial division. eLife 2015, 4, e08828. [Google Scholar] [CrossRef] [PubMed]
- Ji, W.K.; Hatch, A.L.; Merrill, R.A.; Strack, S.; Higgs, H.N. Actin filaments target the oligomeric maturation of the dynamin GTPase Drp1 to mitochondrial fission sites. eLife 2015, 4, e11553. [Google Scholar] [CrossRef]
- Nishimura, A.; Shimauchi, T.; Tanaka, T.; Shimoda, K.; Toyama, T.; Kitajima, N.; Ishikawa, T.; Shindo, N.; Numaga-Tomita, T.; Yasuda, S.; et al. Hypoxia-induced interaction of filamin with Drp1 causes mitochondrial hyperfission-associated myocardial senescence. Sci. Signal. 2018, 11. [Google Scholar] [CrossRef]
- Korobova, F.; Gauvin, T.J.; Higgs, H.N. A role for myosin II in mammalian mitochondrial fission. Curr. Biol. CB 2014, 24, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, R.; Ji, W.K.; Stan, R.V.; de Juan Sanz, J.; Ryan, T.A.; Higgs, H.N. INF2-mediated actin polymerization at the ER stimulates mitochondrial calcium uptake, inner membrane constriction, and division. J. Cell Biol. 2018, 217, 251–268. [Google Scholar] [CrossRef] [PubMed]
- Moore, A.S.; Wong, Y.C.; Simpson, C.L.; Holzbaur, E.L. Dynamic actin cycling through mitochondrial subpopulations locally regulates the fission-fusion balance within mitochondrial networks. Nat. Commun. 2016, 7, 12886. [Google Scholar] [CrossRef]
- Magri, A.; Reina, S.; De Pinto, V. VDAC1 as Pharmacological Target in Cancer and Neurodegeneration: Focus on Its Role in Apoptosis. Front. Chem. 2018, 6, 108. [Google Scholar] [CrossRef]
- Carre, M.; Andre, N.; Carles, G.; Borghi, H.; Brichese, L.; Briand, C.; Braguer, D. Tubulin is an inherent component of mitochondrial membranes that interacts with the voltage-dependent anion channel. J. Biol. Chem. 2002, 277, 33664–33669. [Google Scholar] [CrossRef]
- Rostovtseva, T.K.; Sheldon, K.L.; Hassanzadeh, E.; Monge, C.; Saks, V.; Bezrukov, S.M.; Sackett, D.L. Tubulin binding blocks mitochondrial voltage-dependent anion channel and regulates respiration. Proc. Natl. Acad. Sci. USA 2008, 105, 18746–18751. [Google Scholar] [CrossRef]
- Sheldon, K.L.; Gurnev, P.A.; Bezrukov, S.M.; Sackett, D.L. Tubulin tail sequences and post-translational modifications regulate closure of mitochondrial voltage-dependent anion channel (VDAC). J. Biol. Chem. 2015, 290, 26784–26789. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, E.N.; Lemasters, J.J. Warburg revisited: Regulation of mitochondrial metabolism by voltage-dependent anion channels in cancer cells. J. Pharmacol. Exp. Ther. 2012, 342, 637–641. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, E.N.; Patnaik, J.; Mullins, M.R.; Lemasters, J.J. Free tubulin modulates mitochondrial membrane potential in cancer cells. Cancer Res. 2010, 70, 10192–10201. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, E.N.; Sheldon, K.L.; DeHart, D.N.; Patnaik, J.; Manevich, Y.; Townsend, D.M.; Bezrukov, S.M.; Rostovtseva, T.K.; Lemasters, J.J. Voltage-dependent anion channels modulate mitochondrial metabolism in cancer cells: Regulation by free tubulin and erastin. J. Biol. Chem. 2013, 288, 11920–11929. [Google Scholar] [CrossRef]
- Roman, I.; Figys, J.; Steurs, G.; Zizi, M. Direct measurement of VDAC-actin interaction by surface plasmon resonance. Biochim. Biophys. Acta 2006, 1758, 479–486. [Google Scholar] [CrossRef][Green Version]
- Xu, X.; Forbes, J.G.; Colombini, M. Actin modulates the gating of Neurospora crassa VDAC. J. Membr. Biol. 2001, 180, 73–81. [Google Scholar] [CrossRef]
- Kusano, H.; Shimizu, S.; Koya, R.C.; Fujita, H.; Kamada, S.; Kuzumaki, N.; Tsujimoto, Y. Human gelsolin prevents apoptosis by inhibiting apoptotic mitochondrial changes via closing VDAC. Oncogene 2000, 19, 4807–4814. [Google Scholar] [CrossRef]
- Schroder, M. Endoplasmic reticulum stress responses. Cell. Mol. Life Sci. CMLS 2008, 65, 862–894. [Google Scholar] [CrossRef]
- Corazzari, M.; Gagliardi, M.; Fimia, G.M.; Piacentini, M. Endoplasmic Reticulum Stress, Unfolded Protein Response, and Cancer Cell Fate. Front. Oncol. 2017, 7. [Google Scholar] [CrossRef]
- Malhotra, J.D.; Kaufman, R.J. The endoplasmic reticulum and the unfolded protein response. Semin. Cell Dev. Biol. 2007, 18, 716–731. [Google Scholar] [CrossRef]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519. [Google Scholar] [CrossRef] [PubMed]
- Ishiwata-Kimata, Y.; Yamamoto, Y.H.; Takizawa, K.; Kohno, K.; Kimata, Y. F-actin and a type-II myosin are required for efficient clustering of the ER stress sensor Ire1. Cell Struct. Funct. 2013, 38, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Hao, H.X.; Xie, Y.; Zhang, Y.; Charlat, O.; Oster, E.; Avello, M.; Lei, H.; Mickanin, C.; Liu, D.; Ruffner, H. ZNRF3 promotes Wnt receptor turnover in an R-spondin-sensitive manner. Nature 2012, 485, 195. [Google Scholar] [CrossRef] [PubMed]
- van Vliet, A.R.; Giordano, F.; Gerlo, S.; Segura, I.; Van Eygen, S.; Molenberghs, G.; Rocha, S.; Houcine, A.; Derua, R.; Verfaillie, T. The ER stress sensor PERK coordinates ER-plasma membrane contact site formation through interaction with filamin-A and F-actin remodeling. Mol. Cell 2017, 65, 885–899.e6. [Google Scholar] [CrossRef] [PubMed]
- Goyal, U.; Blackstone, C. Untangling the web: Mechanisms underlying ER network formation. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2013, 1833, 2492–2498. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Chen, L.B. Dynamic behavior of endoplasmic reticulum in living cells. Cell 1988, 54, 37–46. [Google Scholar] [CrossRef]
- Ho, C.T.; Chang, Y.J.; Yang, L.X.; Wei, P.L.; Liu, T.Z.; Liu, J.J. A novel microtubule-disrupting agent induces endoplasmic reticular stress-mediated cell death in human hepatocellular carcinoma cells. PLoS ONE 2015, 10, e0136340. [Google Scholar] [CrossRef]
- Wang, J.; Yin, Y.; Hua, H.; Li, M.; Luo, T.; Xu, L.; Wang, R.; Liu, D.; Zhang, Y.; Jiang, Y. Blockade of GRP78 sensitizes breast cancer cells to microtubules-interfering agents that induce the unfolded protein response. J. Cell. Mol. Med. 2009, 13, 3888–3897. [Google Scholar] [CrossRef]
- Bambang, I.F.; Lu, D.; Li, H.; Chiu, L.L.; Lau, Q.C.; Koay, E.; Zhang, D. Cytokeratin 19 regulates endoplasmic reticulum stress and inhibits ERp29 expression via p38 MAPK/XBP-1 signaling in breast cancer cells. Exp. Cell Res. 2009, 315, 1964–1974. [Google Scholar] [CrossRef]
- Raab, M.S.; Breitkreutz, I.; Tonon, G.; Zhang, J.; Hayden, P.J.; Nguyen, T.; Fruehauf, J.H.; Lin, B.K.; Chauhan, D.; Hideshima, T. Targeting PKC: A novel role for beta-catenin in ER stress and apoptotic signaling. Blood 2009, 113, 1513–1521. [Google Scholar] [CrossRef]
- Cullinan, S.B.; Diehl, J.A. Coordination of ER and oxidative stress signaling: The PERK/Nrf2 signaling pathway. Int. J. Biochem. Cell Biol. 2006, 38, 317–332. [Google Scholar] [CrossRef] [PubMed]
- Ranganathan, A.C.; Zhang, L.; Adam, A.P.; Aguirre-Ghiso, J.A. Functional coupling of p38-induced up-regulation of BiP and activation of RNA-dependent protein kinase-like endoplasmic reticulum kinase to drug resistance of dormant carcinoma cells. Cancer Res. 2006, 66, 1702–1711. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.C.; Yang, F.; Thorne, R.F.; Zhu, B.K.; Hersey, P.; Zhang, X.D. Human melanoma cells under endoplasmic reticulum stress acquire resistance to microtubule-targeting drugs through XBP-1-mediated activation of Akt. Neoplasia 2009, 11, 436–447. [Google Scholar] [CrossRef]
- Brayford, S.; Schevzov, G.; Vos, J.; Gunning, P. The role of the actin cytoskeleton in cancer and its potential use as a therapeutic target. In The Cytoskeleton in Health and Disease; Springer: Berlin/Heidelberg, Germany, 2015; pp. 373–391. [Google Scholar]
- Fife, C.M.; McCarroll, J.A.; Kavallaris, M. Movers and shakers: Cell cytoskeleton in cancer metastasis. Br. J. Pharmacol. 2014, 171, 5507–5523. [Google Scholar] [CrossRef]
- Stanton, R.A.; Gernert, K.M.; Nettles, J.H.; Aneja, R. Drugs that target dynamic microtubules: A new molecular perspective. Med. Res. Rev. 2011, 31, 443–481. [Google Scholar] [CrossRef] [PubMed]
- Dumontet, C.; Jordan, M.A. Microtubule-binding agents: A dynamic field of cancer therapeutics. Nat. Rev. Drug Discov. 2010, 9, 790–803. [Google Scholar] [CrossRef] [PubMed]
- Trendowski, M. Exploiting the cytoskeletal filaments of neoplastic cells to potentiate a novel therapeutic approach. Biochim. Biophys. Acta (BBA) Rev. Cancer 2014, 1846, 599–616. [Google Scholar] [CrossRef] [PubMed]
- Jordan, M.A.; Wilson, L. Microtubules as a target for anticancer drugs. Nat. Rev. Cancer 2004, 4, 253–265. [Google Scholar] [CrossRef]
- Mukhtar, E.; Adhami, V.M.; Mukhtar, H. Targeting microtubules by natural agents for cancer therapy. Mol. Cancer Ther. 2014, 13, 275–284. [Google Scholar] [CrossRef]
- Pronzato, P. New therapeutic options for chemotherapy-resistant metastatic breast cancer: The epothilones. Drugs 2008, 68, 139–146. [Google Scholar] [CrossRef]
- Rohena, C.C.; Mooberry, S.L. Recent progress with microtubule stabilizers: New compounds, binding modes and cellular activities. Nat. Prod. Rep. 2014, 31, 335–355. [Google Scholar] [CrossRef] [PubMed]
- Hari, M.; Yang, H.; Zeng, C.; Canizales, M.; Cabral, F. Expression of class III β-tubulin reduces microtubule assembly and confers resistance to paclitaxel. Cell Motil. Cytoskelet. 2003, 56, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Kavallaris, M.; Tait, A.S.; Walsh, B.J.; He, L.; Horwitz, S.B.; Norris, M.D.; Haber, M. Multiple microtubule alterations are associated with Vinca alkaloid resistance in human leukemia cells. Cancer Res. 2001, 61, 5803–5809. [Google Scholar] [PubMed]
- Martello, L.A.; Verdier-Pinard, P.; Shen, H.J.; He, L.; Torres, K.; Orr, G.A.; Horwitz, S.B. Elevated levels of microtubule destabilizing factors in a Taxol-resistant/dependent A549 cell line with an α-tubulin mutation. Cancer Res. 2003, 63, 1207–1213. [Google Scholar]
- Wong, S.T.; Goodin, S. Overcoming drug resistance in patients with metastatic breast cancer. Pharmacother. J. Hum. Pharmacol. Drug Ther. 2009, 29, 954–965. [Google Scholar] [CrossRef] [PubMed]
- Altaha, R.; Fojo, T.; Reed, E.; Abraham, J. Epothilones: A novel class of non-taxane microtubule-stabilizing agents. Curr. Pharm. Des. 2002, 8, 1707–1712. [Google Scholar] [CrossRef]
- Gradishar, W. Management of advanced breast cancer with the epothilone B analog, ixabepilone. Drug Des. Dev. Ther. 2009, 3, 163–171. [Google Scholar] [CrossRef][Green Version]
- Zheng, Y.B.; Gong, J.H.; Liu, X.J.; Wu, S.Y.; Li, Y.; Xu, X.D.; Shang, B.Y.; Zhou, J.M.; Zhu, Z.L.; Si, S.Y. A novel nitrobenzoate microtubule inhibitor that overcomes multidrug resistance exhibits antitumor activity. Sci. Rep. 2016, 6, 31472. [Google Scholar] [CrossRef]
- Olson, M.F.; Sahai, E. The actin cytoskeleton in cancer cell motility. Clin. Exp. Metastasis 2009, 26, 273. [Google Scholar] [CrossRef]
- Bousquet, P.F.; Paulsen, L.A.; Fondy, C.; Lipski, K.M.; Loucy, K.J.; Fondy, T.P. Effects of cytochalasin B in culture and in vivo on murine Madison 109 lung carcinoma and on B16 melanoma. Cancer Res. 1990, 50, 1431–1439. [Google Scholar]
- Calaghan, S.C.; White, E.; Bedut, S.; Le Guennec, J.Y. Cytochalasin D reduces Ca2+ sensitivity and maximum tension via interactions with myofilaments in skinned rat cardiac myocytes. J. Physiol. 2000, 529 Pt 2, 405–411. [Google Scholar] [CrossRef]
- Chao, J.I.; Liu, H.F. The blockage of survivin and securin expression increases the cytochalasin B-induced cell death and growth inhibition in human cancer cells. Mol. Pharmacol. 2006, 69, 154–164. [Google Scholar] [CrossRef]
- Glinsukon, T.; Lekutai, S. Comparative toxicity in the rat of cytochalasins B and E. Toxicon 1979, 17, 137–144. [Google Scholar] [CrossRef]
- Sachs, H.G.; McDonald, T.F.; Springer, M. Cytochalasin B and embryonic heart muscle: Contractility, excitability and ultrastructure. J. Cell Sci. 1974, 14, 163–185. [Google Scholar] [PubMed]
- Van Goietsenoven, G.; Mathieu, V.; Andolfi, A.; Cimmino, A.; Lefranc, F.; Kiss, R.; Evidente, A. In vitro growth inhibitory effects of cytochalasins and derivatives in cancer cells. Planta Med. 2011, 77, 711–717. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, P.B.; Hanna, B.; Ohl, S.; Sellner, L.; Zenz, T.; Dohner, H.; Stilgenbauer, S.; Larsen, T.O.; Lichter, P.; Seiffert, M. Chaetoglobosin A preferentially induces apoptosis in chronic lymphocytic leukemia cells by targeting the cytoskeleton. Leukemia 2014, 28, 1289–1298. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Li, B.; Li, Z.; Cutler, S.J.; Rankin, G.O.; Chen, Y.C. Chaetoglobosin K inhibits tumor angiogenesis through downregulation of vascular epithelial growth factor-binding hypoxia-inducible factor 1α. Anti-Cancer Drugs 2013, 24, 715–724. [Google Scholar] [CrossRef] [PubMed]
- Ohtsubo, K.; Saito, M.; Sekita, S.; Yoshihira, K.; Natori, S. Acute toxic effects of chaetoglobosin A, a new cytochalasan compound produced by chetomium globosum, on mice and rats. Jpn. J. Exp. Med. 1978, 48, 105–110. [Google Scholar]
- Holzinger, A. Jasplakinolide: An actin-specific reagent that promotes actin polymerization. In Cytoskeleton Methods and Protocols; Springer: Berlin/Heidelberg, Germany, 2009; pp. 71–87. [Google Scholar]
- Odaka, C.; Sanders, M.L.; Crews, P. Jasplakinolide induces apoptosis in various transformed cell lines by a caspase-3-like protease-dependent pathway. Clin. Diagn. Lab. Immunol. 2000, 7, 947–952. [Google Scholar] [CrossRef]
- Schweikart, K.; Guo, L.; Shuler, Z.; Abrams, R.; Chiao, E.T.; Kolaja, K.L.; Davis, M. The effects of jaspamide on human cardiomyocyte function and cardiac ion channel activity. Toxicol. In Vitro Int. J. Publ. Assoc. BIBRA 2013, 27, 745–751. [Google Scholar] [CrossRef]
- Senderowicz, A.M.; Kaur, G.; Sainz, E.; Laing, C.; Inman, W.D.; Rodriguez, J.; Crews, P.; Malspeis, L.; Grever, M.R.; Sausville, E.A.; et al. Jasplakinolide’s inhibition of the growth of prostate carcinoma cells in vitro with disruption of the actin cytoskeleton. J. Natl. Cancer Inst. 1995, 87, 46–51. [Google Scholar] [CrossRef]
- Konishi, H.; Kikuchi, S.; Ochiai, T.; Ikoma, H.; Kubota, T.; Ichikawa, D.; Fujiwara, H.; Okamoto, K.; Sakakura, C.; Sonoyama, T.; et al. Latrunculin a has a strong anticancer effect in a peritoneal dissemination model of human gastric cancer in mice. Anticancer Res. 2009, 29, 2091–2097. [Google Scholar] [PubMed]
- Morton, W.M.; Ayscough, K.R.; McLaughlin, P.J. Latrunculin alters the actin-monomer subunit interface to prevent polymerization. Nat. Cell Biol. 2000, 2, 376. [Google Scholar] [CrossRef] [PubMed]
- Sayed, K.A.; Khanfar, M.A.; Shallal, H.M.; Muralidharan, A.; Awate, B.; Youssef, D.T.; Liu, Y.; Zhou, Y.D.; Nagle, D.G.; Shah, G. Latrunculin A and its C-17-O-carbamates inhibit prostate tumor cell invasion and HIF-1 activation in breast tumor cells. J. Nat. Prod. 2008, 71, 396–402. [Google Scholar] [CrossRef]
- Sierra-Paredes, G.; Oreiro-Garcia, T.; Nunez-Rodriguez, A.; Vazquez-Lopez, A.; Sierra-Marcuno, G. Seizures induced by in vivo latrunculin a and jasplakinolide microperfusion in the rat hippocampus. J. Mol. Neurosci. MN 2006, 28, 151–160. [Google Scholar] [CrossRef]
- Britten, C.D.; Rowinsky, E.K.; Baker, S.D.; Weiss, G.R.; Smith, L.; Stephenson, J.; Rothenberg, M.; Smetzer, L.; Cramer, J.; Collins, W.; et al. A Phase I and Pharmacokinetic Study of the Mitochondrial-specific Rhodacyanine Dye Analog MKT 077. Clin. Cancer Res. 2000, 6, 42–49. [Google Scholar] [PubMed]
- Koya, K.; Li, Y.; Wang, H.; Ukai, T.; Tatsuta, N.; Kawakami, M.; Shishido, T.; Chen, L.B. MKT-077, a novel rhodacyanine dye in clinical trials, exhibits anticarcinoma activity in preclinical studies based on selective mitochondrial accumulation. Cancer Res. 1996, 56, 538–543. [Google Scholar]
- Propper, D.J.; Braybrooke, J.P.; Taylor, D.J.; Lodi, R.; Styles, P.; Cramer, J.A.; Collins, W.C.; Levitt, N.C.; Talbot, D.C.; Ganesan, T.S.; et al. Phase I trial of the selective mitochondrial toxin MKT077 in chemo-resistant solid tumours. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 1999, 10, 923–927. [Google Scholar] [CrossRef]
- Lahat, G.; Zhu, Q.S.; Huang, K.L.; Wang, S.; Bolshakov, S.; Liu, J.; Torres, K.; Langley, R.R.; Lazar, A.J.; Hung, M.C.; et al. Vimentin is a novel anti-cancer therapeutic target; insights from in vitro and in vivo mice xenograft studies. PLoS ONE 2010, 5, e10105. [Google Scholar] [CrossRef]
- Mohan, R.; Hammers, H.J.; Bargagna-Mohan, P.; Zhan, X.H.; Herbstritt, C.J.; Ruiz, A.; Zhang, L.; Hanson, A.D.; Conner, B.P.; Rougas, J.; et al. Withaferin A is a potent inhibitor of angiogenesis. Angiogenesis 2004, 7, 115–122. [Google Scholar] [CrossRef]
- Hahm, E.R.; Singh, S.V. Withaferin A-induced apoptosis in human breast cancer cells is associated with suppression of inhibitor of apoptosis family protein expression. Cancer Lett. 2013, 334, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.J.; Zeng, J.; Zhu, G.D.; Zhang, L.L.; Zhang, D.; Li, L.; Fan, J.H.; Wang, X.Y.; He, D.L. Silibinin inhibits prostate cancer invasion, motility and migration by suppressing vimentin and MMP-2 expression. Acta Pharmacol. Sin. 2009, 30, 1162–1168. [Google Scholar] [CrossRef] [PubMed]
- Dong, T.T.; Zhou, H.M.; Wang, L.L.; Feng, B.; Lv, B.; Zheng, M.H. Salinomycin selectively targets ‘CD133+’ cell subpopulations and decreases malignant traits in colorectal cancer lines. Ann. Surg. Oncol. 2011, 18, 1797–1804. [Google Scholar] [CrossRef] [PubMed]
- Horst, D.; Kriegl, L.; Engel, J.; Kirchner, T.; Jung, A. CD133 expression is an independent prognostic marker for low survival in colorectal cancer. Br. J. Cancer 2008, 99, 1285–1289. [Google Scholar] [CrossRef]
- Raab-Westphal, S.; Marshall, J.F.; Goodman, S.L. Integrins as Therapeutic Targets: Successes and Cancers. Cancers 2017, 9, 110. [Google Scholar] [CrossRef]
- Agrez, M.V.; Bates, R.C.; Mitchell, D.; Wilson, N.; Ferguson, N.; Anseline, P.; Sheppard, D. Multiplicity of fibronectin-binding alpha V integrin receptors in colorectal cancer. Br. J. Cancer 1996, 73, 887–892. [Google Scholar] [CrossRef]
- Goodman, S.L.; Grote, H.J.; Wilm, C. Matched rabbit monoclonal antibodies against αv-series integrins reveal a novel αvβ3-LIBS epitope, and permit routine staining of archival paraffin samples of human tumors. Biol. Open 2012, 1, 329–340. [Google Scholar] [CrossRef]
- Wirth, M.; Heidenreich, A.; Gschwend, J.E.; Gil, T.; Zastrow, S.; Laniado, M.; Gerloff, J.; Zuhlsdorf, M.; Mordenti, G.; Uhl, W.; et al. A multicenter phase 1 study of EMD 525797 (DI17E6), a novel humanized monoclonal antibody targeting alphav integrins, in progressive castration-resistant prostate cancer with bone metastases after chemotherapy. Eur. Urol. 2014, 65, 897–904. [Google Scholar] [CrossRef]
- Jiang, Y.; Dai, J.; Yao, Z.; Shelley, G.; Keller, E.T. Abituzumab Targeting of αV-Class Integrins Inhibits Prostate Cancer Progression. Mol. Cancer Res. 2017, 15, 875–883. [Google Scholar] [CrossRef]
- Stehn, J.R.; Haass, N.K.; Bonello, T.; Desouza, M.; Kottyan, G.; Treutlein, H.; Zeng, J.; Nascimento, P.R.; Sequeira, V.B.; Butler, T.L.; et al. A novel class of anticancer compounds targets the actin cytoskeleton in tumor cells. Cancer Res. 2013, 73, 5169–5182. [Google Scholar] [CrossRef]
- Currier, M.A.; Stehn, J.R.; Swain, A.; Chen, D.; Hook, J.; Eiffe, E.; Heaton, A.; Brown, D.; Nartker, B.A.; Eaves, D.W.; et al. Identification of Cancer-Targeted Tropomyosin Inhibitors and Their Synergy with Microtubule Drugs. Mol. Cancer Ther. 2017, 16, 1555–1565. [Google Scholar] [CrossRef] [PubMed]
- Prunier, C.; Prudent, R.; Kapur, R.; Sadoul, K.; Lafanechère, L. LIM kinases: Cofilin and beyond. Oncotarget 2017, 8, 41749–41763. [Google Scholar] [CrossRef] [PubMed]
- Mardilovich, K.; Baugh, M.; Crighton, D.; Kowalczyk, D.; Gabrielsen, M.; Munro, J.; Croft, D.R.; Lourenco, F.; James, D.; Kalna, G.; et al. LIM kinase inhibitors disrupt mitotic microtubule organization and impair tumor cell proliferation. Oncotarget 2015, 6, 38469–38486. [Google Scholar] [CrossRef]
- Sun, B.; Fang, Y.; Li, Z.; Chen, Z.; Xiang, J. Role of cellular cytoskeleton in epithelial-mesenchymal transition process during cancer progression. Biomed. Rep. 2015, 3, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Kleeberger, W.; Bova, G.S.; Nielsen, M.E.; Herawi, M.; Chuang, A.Y.; Epstein, J.I.; Berman, D.M. Roles for the stem cell associated intermediate filament Nestin in prostate cancer migration and metastasis. Cancer Res. 2007, 67, 9199–9206. [Google Scholar] [CrossRef]
- Krupkova, O., Jr.; Loja, T.; Zambo, I.; Veselska, R. Nestin expression in human tumors and tumor cell lines. Neoplasma 2010, 57, 291–298. [Google Scholar] [CrossRef]
- Teranishi, N.; Naito, Z.; Ishiwata, T.; Tanaka, N.; Furukawa, K.; Seya, T.; Shinji, S.; Tajiri, T. Identification of neovasculature using nestin in colorectal cancer. Int. J. Oncol. 2007, 30, 593–603. [Google Scholar] [CrossRef][Green Version]
- Liu, W.; Zhang, Y.; Hao, J.; Liu, S.; Liu, Q.; Zhao, S.; Shi, Y.; Duan, H. Nestin protects mouse podocytes against high glucose-induced apoptosis by a Cdk5-dependent mechanism. J. Cell. Biochem. 2012, 113, 3186–3196. [Google Scholar] [CrossRef]
- Sahlgren, C.M.; Pallari, H.M.; He, T.; Chou, Y.H.; Goldman, R.D.; Eriksson, J.E. A nestin scaffold links Cdk5/p35 signaling to oxidant-induced cell death. EMBO J. 2006, 25, 4808–4819. [Google Scholar] [CrossRef]
- Franke, W.W.; Grund, C.; Kuhn, C.; Jackson, B.W.; Illmensee, K. Formation of cytoskeletal elements during mouse embryogenesis. III. Primary mesenchymal cells and the first appearance of vimentin filaments. Differ. Res. Biol. Divers. 1982, 23, 43–59. [Google Scholar] [CrossRef]
- Hamidi, H.; Pietilä, M.; Ivaska, J. The complexity of integrins in cancer and new scopes for therapeutic targeting. Br. J. Cancer 2016, 115, 1017–1023. [Google Scholar] [CrossRef] [PubMed]
- Adachi, M.; Taki, T.; Higashiyama, M.; Kohno, N.; Inufusa, H.; Miyake, M. Significance of Integrin α5 Gene Expression as a Prognostic Factor in Node-negative Non-Small Cell Lung Cancer. Clin. Cancer Res. 2000, 6, 96–101. [Google Scholar] [PubMed]
- Bates, R.C.; Bellovin, D.I.; Brown, C.; Maynard, E.; Wu, B.; Kawakatsu, H.; Sheppard, D.; Oettgen, P.; Mercurio, A.M. Transcriptional activation of integrin β6 during the epithelial-mesenchymal transition defines a novel prognostic indicator of aggressive colon carcinoma. J. Clin. Investig. 2005, 115, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Danen, E.; BERGE, P.T.; MUIJEN, G.V.; Van’t Hof-Grootenboer, A.; Bröcker, E.; Ruiter, D. Emergence of α5β1 fibronectin-and αvβ3 vitronectin-receptor expression in melanocytic tumour progression. Histopathology 1994, 24, 249–256. [Google Scholar] [CrossRef]
- Diaz, L.K.; Cristofanilli, M.; Zhou, X.; Welch, K.L.; Smith, T.L.; Yang, Y.; Sneige, N.; Sahin, A.A.; Gilcrease, M.Z. β4 integrin subunit gene expression correlates with tumor size and nuclear grade in early breast cancer. Mod. Pathol. 2005, 18, 1165. [Google Scholar] [CrossRef]
- Slack-Davis, J.K.; Atkins, K.A.; Harrer, C.; Hershey, E.D.; Conaway, M. Vascular Cell Adhesion Molecule-1 Is a Regulator of Ovarian Cancer Peritoneal Metastasis. Cancer Res. 2009, 69, 1469–1476. [Google Scholar] [CrossRef]
- Arun, A.S.; Tepper, C.G.; Lam, K.S. Identification of integrin drug targets for 17 solid tumor types. Oncotarget 2018, 9, 30146. [Google Scholar] [CrossRef]
- Koistinen, P.; Heino, J. Integrins in cancer cell invasion. In Madame Curie Bioscience Database [Internet]; Landes Bioscience: Austin, TX, USA, 2013. [Google Scholar]
- Desgrosellier, J.S.; Cheresh, D.A. Integrins in cancer: Biological implications and therapeutic opportunities. Nat. Rev. Cancer 2010, 10, 9–22. [Google Scholar] [CrossRef]
- Uhm, J.H.; Dooley, N.P.; Kyritsis, A.P.; Rao, J.S.; Gladson, C.L. Vitronectin, a glioma-derived extracellular matrix protein, protects tumor cells from apoptotic death. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 1999, 5, 1587–1594. [Google Scholar]
- Aoudjit, F.; Vuori, K. Integrin signaling inhibits paclitaxel-induced apoptosis in breast cancer cells. Oncogene 2001, 20, 4995–5004. [Google Scholar] [CrossRef]
- Scatena, M.; Almeida, M.; Chaisson, M.L.; Fausto, N.; Nicosia, R.F.; Giachelli, C.M. NF-kappaB mediates alphavbeta3 integrin-induced endothelial cell survival. J. Cell Biol. 1998, 141, 1083–1093. [Google Scholar] [CrossRef] [PubMed]
- Matter, M.L.; Ruoslahti, E. A Signaling Pathway from the α5β1 and αvβ3 Integrins That Elevatesbcl-2 Transcription. J. Biol. Chem. 2001, 276, 27757–27763. [Google Scholar] [CrossRef] [PubMed]
- Daemi, N.; Thomasset, N.; Lissitzky, J.C.; Dumortier, J.; Jacquier, M.F.; Pourreyron, C.; Rousselle, P.; Chayvialle, J.A.; Remy, L. Anti-β4 integrin antibodies enhance migratory and invasive abilities of human colon adenocarcinoma cells and their MMP-2 expression. Int. J. Cancer 2000, 85, 850–856. [Google Scholar] [CrossRef]
- Li, W.; Liu, Z.; Zhao, C.; Zhai, L. Binding of MMP-9-degraded fibronectin to beta6 integrin promotes invasion via the FAK-Src-related Erk1/2 and PI3K/Akt/Smad-1/5/8 pathways in breast cancer. Oncol. Rep. 2015, 34, 1345–1352. [Google Scholar] [CrossRef] [PubMed]
- Sil, H.; Sen, T.; Chatterjee, A. Fibronectin-integrin (alpha5beta1) modulates migration and invasion of murine melanoma cell line B16F10 by involving MMP-9. Oncol. Res. 2011, 19, 335–348. [Google Scholar] [CrossRef]
- Avraamides, C.J.; Garmy-Susini, B.; Varner, J.A. Integrins in angiogenesis and lymphangiogenesis. Nat. Rev. Cancer 2008, 8, 604–617. [Google Scholar] [CrossRef]
- Lu, X.; Lu, D.; Scully, M.; Kakkar, V. The role of integrins in cancer and the development of anti-integrin therapeutic agents for cancer therapy. Perspect. Med. Chem. 2008, 2, 57–73. [Google Scholar] [CrossRef]
- Posey, J.A.; Khazaeli, M.B.; DelGrosso, A.; Saleh, M.N.; Lin, C.Y.; Huse, W.; LoBuglio, A.F. A pilot trial of Vitaxin, a humanized anti-vitronectin receptor (anti alpha v beta 3) antibody in patients with metastatic cancer. Cancer Biother. Radiopharm. 2001, 16, 125–132. [Google Scholar] [CrossRef]
- Gramoun, A.; Shorey, S.; Bashutski, J.D.; Dixon, S.J.; Sims, S.M.; Heersche, J.N.M.; Manolson, M.F. Effects of Vitaxin®, a novel therapeutic in trial for metastatic bone tumors, on osteoclast functions in vitro. J. Cell. Biochem. 2007, 102, 341–352. [Google Scholar] [CrossRef]
- Coleman, K.R.; Braden, G.A.; Willingham, M.C.; Sane, D.C. Vitaxin, a humanized monoclonal antibody to the vitronectin receptor (alphavbeta3), reduces neointimal hyperplasia and total vessel area after balloon injury in hypercholesterolemic rabbits. Circ. Res. 1999, 84, 1268–1276. [Google Scholar] [CrossRef]
- Prunier, C.; Josserand, V.; Vollaire, J.; Beerling, E.; Petropoulos, C.; Destaing, O.; Montemagno, C.; Hurbin, A.; Prudent, R.; De Koning, L. LIM kinase inhibitor Pyr1 reduces the growth and metastatic load of breast cancers. Cancer Res. 2016, 76, 3541–3552. [Google Scholar] [CrossRef] [PubMed]
- Yoshioka, K.; Foletta, V.; Bernard, O.; Itoh, K. A role for LIM kinase in cancer invasion. Proc. Natl. Acad. Sci. USA 2003, 100, 7247–7252. [Google Scholar] [CrossRef] [PubMed]
- Yap, C.T.; Simpson, T.I.; Pratt, T.; Price, D.J.; Maciver, S.K. The motility of glioblastoma tumour cells is modulated by intracellular cofilin expression in a concentration-dependent manner. Cell Motil. Cytoskelet. 2005, 60, 153–165. [Google Scholar] [CrossRef] [PubMed]
- Popow-Woźniak, A.; Mazur, A.J.; Mannherz, H.G.; Malicka-Błaszkiewicz, M.; Nowak, D. Cofilin overexpression affects actin cytoskeleton organization and migration of human colon adenocarcinoma cells. Histochem. Cell Biol. 2012, 138, 725–736. [Google Scholar] [CrossRef] [PubMed]
- Bernard, O. Lim kinases, regulators of actin dynamics. Int. J. Biochem. Cell Biol. 2007, 39, 1071–1076. [Google Scholar] [CrossRef] [PubMed]
- Arber, S.; Barbayannis, F.A.; Hanser, H.; Schneider, C.; Stanyon, C.A.; Bernard, O.; Caroni, P. Regulation of actin dynamics through phosphorylation of cofilin by LIM-kinase. Nature 1998, 393, 805. [Google Scholar] [CrossRef]
- Gorovoy, M.; Niu, J.; Bernard, O.; Profirovic, J.; Minshall, R.; Neamu, R.; Voyno-Yasenetskaya, T. LIM kinase 1 coordinates microtubule stability and actin polymerization in human endothelial cells. J. Biol. Chem. 2005, 280, 26533–26542. [Google Scholar] [CrossRef]
- Oleinik, N.V.; Krupenko, N.I.; Krupenko, S.A. ALDH1L1 inhibits cell motility via dephosphorylation of cofilin by PP1 and PP2A. Oncogene 2010, 29, 6233. [Google Scholar] [CrossRef]
- Lu, H.; Chen, J.; Luo, Y.; Xu, H.; Xiong, L.; Fu, J. Curcolonol suppresses the motility of breast cancer cells by inhibiting LIM kinase 1 to downregulate cofilin 1 phosphorylation. Int. J. Oncol. 2018, 53, 2695–2704. [Google Scholar] [CrossRef]
- Drewry, D.H.; Willson, T.M.; Zuercher, W.J. Seeding collaborations to advance kinase science with the GSK Published Kinase Inhibitor Set (PKIS). Curr. Top. Med. Chem. 2014, 14, 340–342. [Google Scholar] [CrossRef]
- Janke, C.; Bulinski, J.C. Post-translational regulation of the microtubule cytoskeleton: Mechanisms and functions. Nat. Rev. Mol. Cell Biol. 2011, 12, 773. [Google Scholar] [CrossRef] [PubMed]
- Varland, S.; Vandekerckhove, J.; Drazic, A. Actin Post-translational Modifications: The Cinderella of Cytoskeletal Control. Trends Biochem. Sci. 2019, 44, 502–516. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, N.; Caron, C.; Matthias, G.; Hess, D.; Khochbin, S.; Matthias, P. HDAC-6 interacts with and deacetylates tubulin and microtubules in vivo. EMBO J. 2003, 22, 1168–1179. [Google Scholar] [CrossRef] [PubMed]
- Valenzuela-Fernandez, A.; Cabrero, J.R.; Serrador, J.M.; Sanchez-Madrid, F. HDAC6: A key regulator of cytoskeleton, cell migration and cell-cell interactions. Trends Cell Biol. 2008, 18, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Haggarty, S.J.; Koeller, K.M.; Wong, J.C.; Grozinger, C.M.; Schreiber, S.L. Domain-selective small-molecule inhibitor of histone deacetylase 6 (HDAC6)-mediated tubulin deacetylation. Proc. Natl. Acad. Sci. USA 2003, 100, 4389–4394. [Google Scholar] [CrossRef]
- Hideshima, T.; Bradner, J.E.; Wong, J.; Chauhan, D.; Richardson, P.; Schreiber, S.L.; Anderson, K.C. Small-molecule inhibition of proteasome and aggresome function induces synergistic antitumor activity in multiple myeloma. Proc. Natl. Acad. Sci. USA 2005, 102, 8567–8572. [Google Scholar] [CrossRef]
- Schemies, J.; Sippl, W.; Jung, M. Histone deacetylase inhibitors that target tubulin. Cancer Lett. 2009, 280, 222–232. [Google Scholar] [CrossRef]
- Itoh, Y.; Suzuki, T.; Kouketsu, A.; Suzuki, N.; Maeda, S.; Yoshida, M.; Nakagawa, H.; Miyata, N. Design, synthesis, structure—Selectivity relationship, and effect on human cancer cells of a novel series of histone deacetylase 6-selective inhibitors. J. Med. Chem. 2007, 50, 5425–5438. [Google Scholar] [CrossRef]
- Suzuki, K.; Bose, P.; Leong-Quong, R.Y.; Fujita, D.J.; Riabowol, K. REAP: A two minute cell fractionation method. BMC Res. Notes 2010, 3, 294. [Google Scholar] [CrossRef]
- Rao, D.M.; Shackleford, M.T.; Bordeaux, E.K.; Sottnik, J.L.; Ferguson, R.L.; Yamamoto, T.M.; Wellberg, E.A.; Bitler, B.G.; Sikora, M.J. Wnt family member 4 (WNT4) and WNT3A activate cell-autonomous Wnt signaling independent of porcupine O-acyltransferase or Wnt secretion. J. Biol. Chem. 2019, 294, 19950–19966. [Google Scholar] [CrossRef]
- Chen, E.Y.; DeRan, M.T.; Ignatius, M.S.; Grandinetti, K.B.; Clagg, R.; McCarthy, K.M.; Lobbardi, R.M.; Brockmann, J.; Keller, C.; Wu, X. Glycogen synthase kinase 3 inhibitors induce the canonical WNT/β-catenin pathway to suppress growth and self-renewal in embryonal rhabdomyosarcoma. Proc. Natl. Acad. Sci. USA 2014, 111, 5349–5354. [Google Scholar] [CrossRef] [PubMed]
- McGough, A.; Chiu, W.; Way, M. Determination of the gelsolin binding site on F-actin: Implications for severing and capping. Biophys. J. 1998, 74, 764–772. [Google Scholar] [CrossRef]
- Freireich, E.J. The history of leukemia therapy—A personal journey. Clin. Lymphoma Myeloma Leuk. 2012, 12, 386–392. [Google Scholar] [CrossRef] [PubMed]
- Foulkes, W.D.; Reis-Filho, J.S.; Narod, S.A. Tumor size and survival in breast cancer—A reappraisal. Nat. Rev. Clin. Oncol. 2010, 7, 348–353. [Google Scholar] [CrossRef]
- Zhang, J.; Gold, K.A.; Lin, H.Y.; Swisher, S.G.; Xing, Y.; Lee, J.J.; Kim, E.S.; William, W.N., Jr. Relationship between tumor size and survival in non-small-cell lung cancer (NSCLC): An analysis of the surveillance, epidemiology, and end results (SEER) registry. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2015, 10, 682–690. [Google Scholar] [CrossRef]
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef]
- Fisher, R.; Pusztai, L.; Swanton, C. Cancer heterogeneity: Implications for targeted therapeutics. Br. J. Cancer 2013, 108, 479–485. [Google Scholar] [CrossRef]
- Nguyen, L.V.; Vanner, R.; Dirks, P.; Eaves, C.J. Cancer stem cells: An evolving concept. Nat. Rev. Cancer 2012, 12, 133–143. [Google Scholar] [CrossRef]
- Plaks, V.; Kong, N.; Werb, Z. The cancer stem cell niche: How essential is the niche in regulating stemness of tumor cells? Cell Stem Cell 2015, 16, 225–238. [Google Scholar] [CrossRef]
- Gerharz, C.; Moll, R.; Meister, P.; Knuth, A.; Gabbert, H. Cytoskeletal heterogeneity of an epithelioid sarcoma with expression of vimentin, cytokeratins, and neurofilaments. Am. J. Surg. Pathol. 1990, 14, 274–283. [Google Scholar] [CrossRef]
- Raz, A.; Geiger, B. Altered organization of cell-substrate contacts and membrane-associated cytoskeleton in tumor cell variants exhibiting different metastatic capabilities. Cancer Res. 1982, 42, 5183–5190. [Google Scholar] [PubMed]



{kind=link}
{kind=link}
| Targeted Proteins | Drugs | Action and Cellular Effects | Ref. | |
|---|---|---|---|---|
| Microtubules | Taxanes (e.g., Docetaxel, Paclitaxel) |
| [5,91,97,98] | |
| Epothilones (e.g., Ixabepilone) | ||||
| Intermediate Filaments | Withaferin-A |
| [121,122,123] | |
| Silibinin |
| [124] | ||
| Salinomycin |
| [125,126] | ||
| Cytoskeletal-Associated Proteins | Integrins | Abituzumab |
| [127,128,129,130,131] |
| Tropomyosin | Anti-Tropomyosin Drug TR100 |
| [26,132,133] | |
| ATM-3507 |
| [133] | ||
| LIM kinase (LIMK) | 4-Pyridocarbazolone (LIMK1) |
| [134,135] | |
| 6-Damnacanthal (LIMK1 and LIMK2) |
| |||
| Drug | Mechanistic Action | Cellular Effects | Toxicity | Ref. |
|---|---|---|---|---|
| Cytochalasins |
|
|
| [101,102,103,104,105,106] |
| Chaetoglobosin |
|
|
| [107,108,109] |
| Jasplakinolide |
|
|
| [110,111,112,113] |
| Latrunculins |
|
|
| [114,115,116,117] |
| MKT-077 |
|
|
| [88,118,119,120] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ong, M.S.; Deng, S.; Halim, C.E.; Cai, W.; Tan, T.Z.; Huang, R.Y.-J.; Sethi, G.; Hooi, S.C.; Kumar, A.P.; Yap, C.T. Cytoskeletal Proteins in Cancer and Intracellular Stress: A Therapeutic Perspective. Cancers 2020, 12, 238. https://doi.org/10.3390/cancers12010238
Ong MS, Deng S, Halim CE, Cai W, Tan TZ, Huang RY-J, Sethi G, Hooi SC, Kumar AP, Yap CT. Cytoskeletal Proteins in Cancer and Intracellular Stress: A Therapeutic Perspective. Cancers. 2020; 12(1):238. https://doi.org/10.3390/cancers12010238
Chicago/Turabian StyleOng, Mei Shan, Shuo Deng, Clarissa Esmeralda Halim, Wanpei Cai, Tuan Zea Tan, Ruby Yun-Ju Huang, Gautam Sethi, Shing Chuan Hooi, Alan Prem Kumar, and Celestial T. Yap. 2020. "Cytoskeletal Proteins in Cancer and Intracellular Stress: A Therapeutic Perspective" Cancers 12, no. 1: 238. https://doi.org/10.3390/cancers12010238
APA StyleOng, M. S., Deng, S., Halim, C. E., Cai, W., Tan, T. Z., Huang, R. Y.-J., Sethi, G., Hooi, S. C., Kumar, A. P., & Yap, C. T. (2020). Cytoskeletal Proteins in Cancer and Intracellular Stress: A Therapeutic Perspective. Cancers, 12(1), 238. https://doi.org/10.3390/cancers12010238