The Role of Epithelial-to-Mesenchymal Plasticity in Ovarian Cancer Progression and Therapy Resistance

{kind=link}
{kind=link}
Abstract
1. Introduction
2. Epithelial-to-Mesenchymal Transition (EMT)
2.1. Definition of A Complex Process
2.2. EMT Transcription Factors
3. EMT and High-Grade Serous Ovarian Cancer Progression
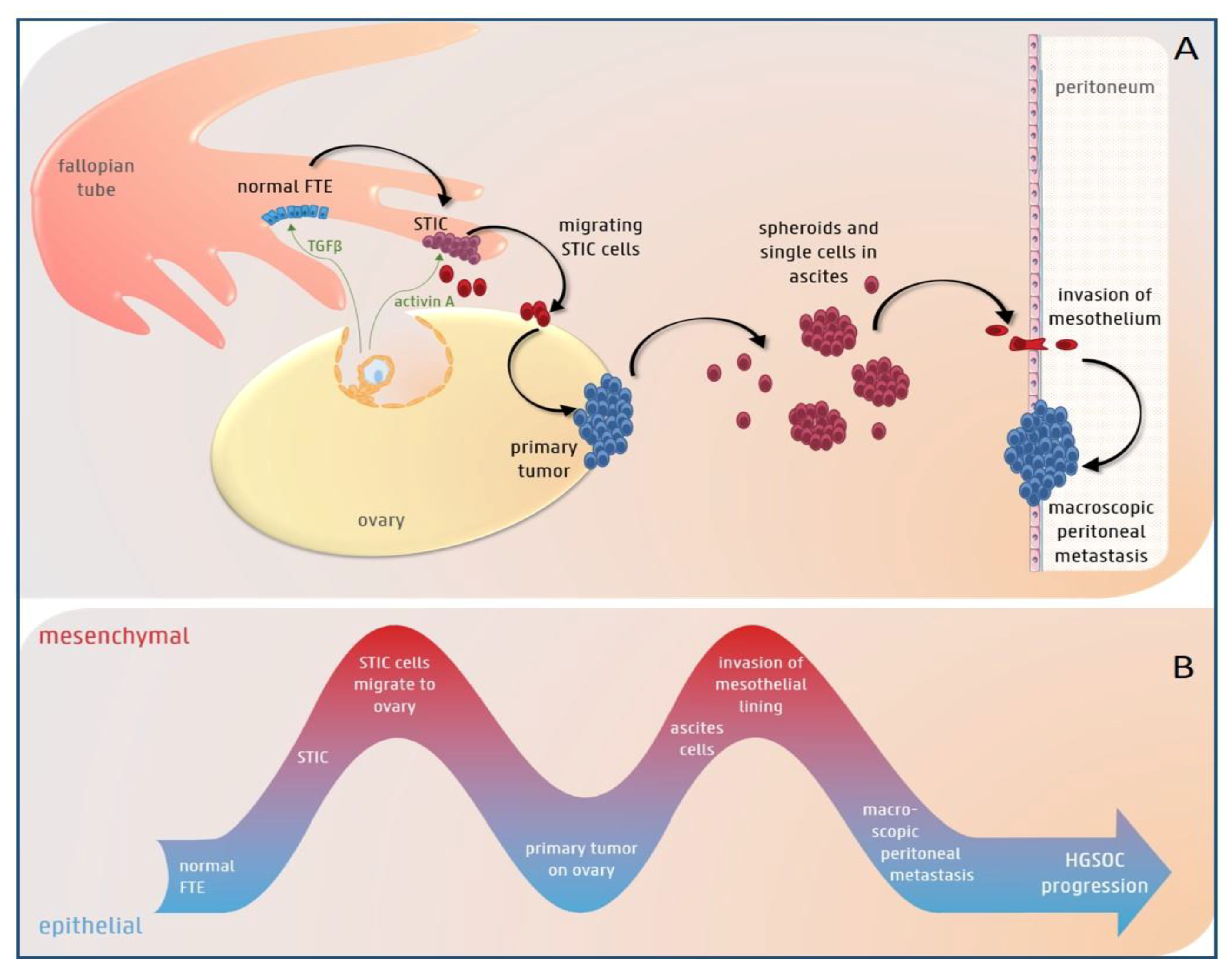
3.1. The Role of EMT in HGS Ovarian Cancer Initiation
3.2. EMT Plasticity during HGS Ovarian Cancer Progression
3.3. The Role of EMT in Low-grade Serous Ovarian Cancer Initiation and Progression
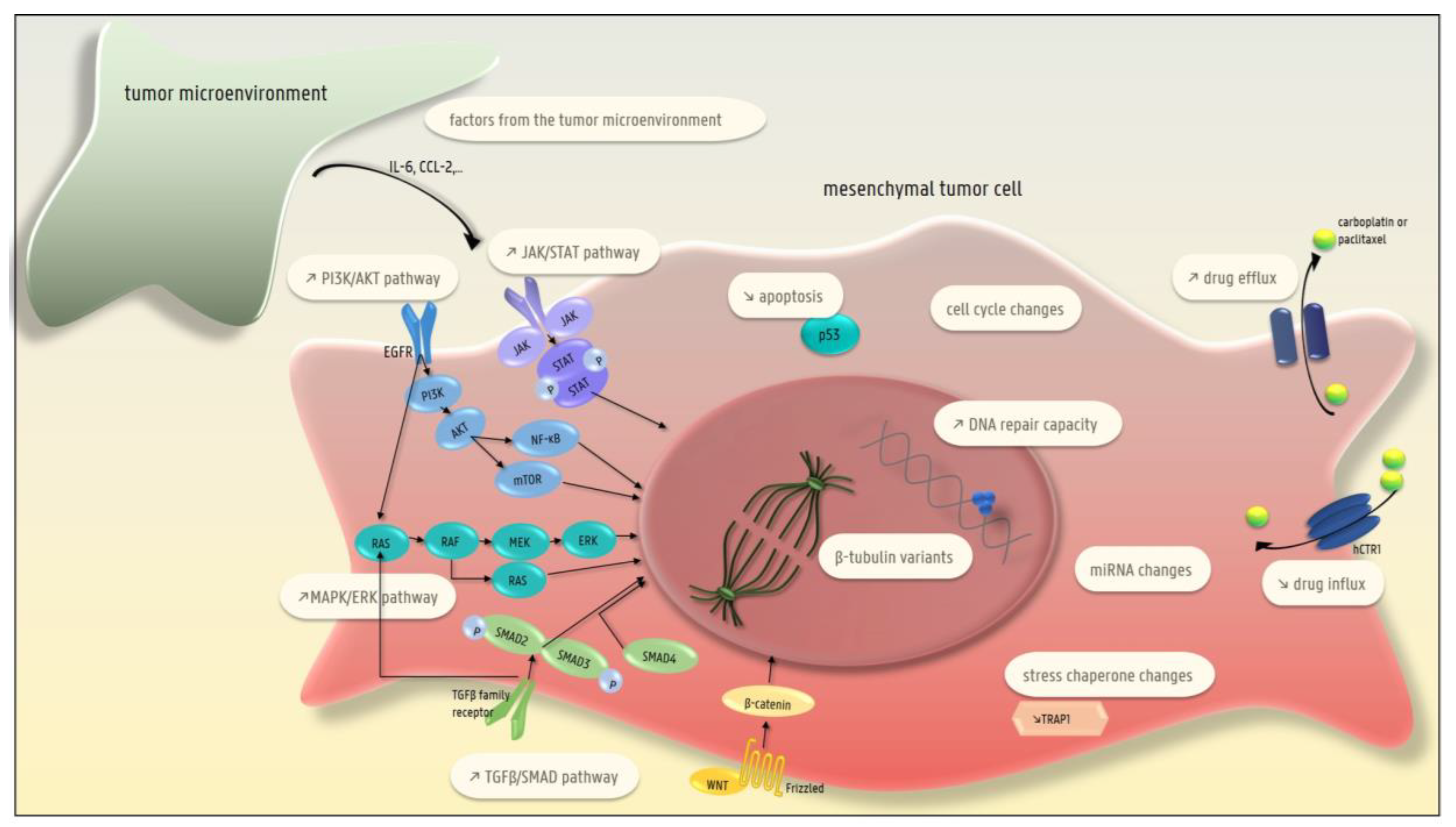
4. EMT and Chemotherapy Resistance
4.1. The Presence of β-Tubulin Variants (Taxane-Specific Resistance)
4.2. Lower Drug Uptake
4.3. Higher Efflux of the Drug
4.4. Higher DNA Repair Capacity
4.5. Decreased Apoptosis
4.6. Changes in the MAPK/ERK Pathway
4.7. Changes in EGFR Signaling
4.8. Changes in the TGFβ-SMAD Pathway
4.9. Changes in the PI3-K/AKT/NF-κB and JAK/STAT Pathways
4.10. Changes in the Cell Cycle
4.11. Changes in Micro-RNAs
4.12. Changes in Stress Chaperones
4.13. Factors Produced by the Tumor Microenvironment
5. EMT and Immunotherapy Resistance
6. EMT and Stemness
7. Targeting EMT in Ovarian Cancer
8. Controversy about the Role of EMT in Ovarian Cancer Progression and Therapy Resistance
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Pineros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 2019, 144, 1941–1953. [Google Scholar] [CrossRef] [PubMed]
- Allemani, C.; Weir, H.K.; Carreira, H.; Harewood, R.; Spika, D.; Wang, X.S.; Bannon, F.; Ahn, J.V.; Johnson, C.J.; Bonaventure, A.; et al. Global surveillance of cancer survival 1995–2009: Analysis of individual data for 25,676,887 patients from 279 population-based registries in 67 countries (CONCORD-2). Lancet 2015, 385, 977–1010. [Google Scholar] [CrossRef]
- Mor, G.; Alvero, A. The duplicitous origin of ovarian cancer. Rambam Maimonides Med. J. 2013, 4, e0006. [Google Scholar] [CrossRef] [PubMed]
- Oberaigner, W.; Minicozzi, P.; Bielska-Lasota, M.; Allemani, C.; de Angelis, R.; Mangone, L.; Sant, M.; Eurocare Working, G. Survival for ovarian cancer in Europe: The across-country variation did not shrink in the past decade. Acta Oncol. 2012, 51, 441–453. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, S.; Coward, J.I.; Bast, R.C., Jr.; Berchuck, A.; Berek, J.S.; Brenton, J.D.; Coukos, G.; Crum, C.C.; Drapkin, R.; Etemadmoghadam, D.; et al. Rethinking ovarian cancer: Recommendations for improving outcomes. Nat. Rev. Cancer 2011, 11, 719–725. [Google Scholar] [CrossRef] [PubMed]
- Kurman, R.J.; Carcangiu, M.L.; Herrington, C.S.; Young, R.H. WHO Classification of Tumours of Female Reproductive Organs, 4th ed.; WHO Press: Lyon, France, 2014. [Google Scholar]
- Meinhold-Heerlein, I.; Fotopoulou, C.; Harter, P.; Kurzeder, C.; Mustea, A.; Wimberger, P.; Hauptmann, S.; Sehouli, J. The new WHO classification of ovarian, fallopian tube, and primary peritoneal cancer and its clinical implications. Arch. Gynecol. Obstet. 2016, 293, 695–700. [Google Scholar] [CrossRef] [PubMed]
- Labidi-Galy, S.I.; Papp, E.; Hallberg, D.; Niknafs, N.; Adleff, V.; Noe, M.; Bhattacharya, R.; Novak, M.; Jones, S.; Phallen, J.; et al. High grade serous ovarian carcinomas originate in the fallopian tube. Nat. Commun. 2017, 8, 1093. [Google Scholar] [CrossRef]
- Walker, J.L.; Powell, C.B.; Chen, L.M.; Carter, J.; Bae Jump, V.L.; Parker, L.P.; Borowsky, M.E.; Gibb, R.K. Society of Gynecologic Oncology recommendations for the prevention of ovarian cancer. Cancer 2015, 121, 2108–2120. [Google Scholar] [CrossRef] [PubMed]
- Zeppernick, F.; Meinhold-Heerlein, I. The new FIGO staging system for ovarian, fallopian tube, and primary peritoneal cancer. Arch. Gynecol. Obstet. 2014, 290, 839–842. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research, N. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef]
- Kurman, R.J.; Shih Ie, M. Pathogenesis of ovarian cancer: Lessons from morphology and molecular biology and their clinical implications. Int. J. Gynecol. Pathol. Off. J. Int. Soc. Gynecol. Pathol. 2008, 27, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Rojas, V.; Hirshfield, K.M.; Ganesan, S.; Rodriguez-Rodriguez, L. Molecular Characterization of Epithelial Ovarian Cancer: Implications for Diagnosis and Treatment. Int. J. Mol. Sci. 2016, 17, 2113. [Google Scholar] [CrossRef] [PubMed]
- Berek, J.S.; Kehoe, S.T.; Kumar, L.; Friedlander, M. Cancer of the ovary, fallopian tube, and peritoneum. Int. J. Gynaecol. Obstet. Off. Organ Int. Fed. Gynaecol. Obstet. 2018, 143 (Suppl. 2), 59–78. [Google Scholar] [CrossRef]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef] [PubMed]
- Bierie, B.; Pierce, S.E.; Kroeger, C.; Stover, D.G.; Pattabiraman, D.R.; Thiru, P.; Liu Donaher, J.; Reinhardt, F.; Chaffer, C.L.; Keckesova, Z.; et al. Integrin-beta4 identifies cancer stem cell-enriched populations of partially mesenchymal carcinoma cells. Proc. Natl. Acad. Sci. USA 2017, 114, E2337–E2346. [Google Scholar] [CrossRef] [PubMed]
- Chaffer, C.L.; San Juan, B.P.; Lim, E.; Weinberg, R.A. EMT, cell plasticity and metastasis. Cancer Metastasis Rev. 2016, 35, 645–654. [Google Scholar] [CrossRef]
- Pastushenko, I.; Brisebarre, A.; Sifrim, A.; Fioramonti, M.; Revenco, T.; Boumahdi, S.; Van Keymeulen, A.; Brown, D.; Moers, V.; Lemaire, S.; et al. Identification of the tumour transition states occurring during EMT. Nature 2018, 556, 463–468. [Google Scholar] [CrossRef]
- Brozovic, A. The relationship between platinum drug resistance and epithelial-mesenchymal transition. Arch. Toxicol. 2017, 91, 605–619. [Google Scholar] [CrossRef]
- Guadamillas, M.C.; Cerezo, A.; Del Pozo, M.A. Overcoming anoikis—Pathways to anchorage-independent growth in cancer. J. Cell Sci. 2011, 124, 3189–3197. [Google Scholar] [CrossRef]
- De Craene, B.; Berx, G. Regulatory networks defining EMT during cancer initiation and progression. Nat. Rev. Cancer 2013, 13, 97–110. [Google Scholar] [CrossRef]
- Goossens, S.; Vandamme, N.; Van Vlierberghe, P.; Berx, G. EMT transcription factors in cancer development re-evaluated: Beyond EMT and MET. Biochim. Biophys. Acta Rev. Cancer 2017, 1868, 584–591. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A. Context-specific roles of EMT programmes in cancer cell dissemination. Nat. Cell Biol. 2017, 19, 416–418. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A.; Huang, R.Y.; Jackson, R.A.; Thiery, J.P. Emt: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [PubMed]
- Puisieux, A.; Brabletz, T.; Caramel, J. Oncogenic roles of EMT-inducing transcription factors. Nat. Cell Biol. 2014, 16, 488–494. [Google Scholar] [CrossRef] [PubMed]
- Stemmler, M.P.; Eccles, R.L.; Brabletz, S.; Brabletz, T. Non-redundant functions of EMT transcription factors. Nat. Cell Biol. 2019, 21, 102–112. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Tillo, E.; Lazaro, A.; Torrent, R.; Cuatrecasas, M.; Vaquero, E.C.; Castells, A.; Engel, P.; Postigo, A. ZEB1 represses E-cadherin and induces an EMT by recruiting the SWI/SNF chromatin-remodeling protein BRG1. Oncogene 2010, 29, 3490–3500. [Google Scholar] [CrossRef]
- Abubaker, K.; Latifi, A.; Luwor, R.; Nazaretian, S.; Zhu, H.; Quinn, M.A.; Thompson, E.W.; Findlay, J.K.; Ahmed, N. Short-term single treatment of chemotherapy results in the enrichment of ovarian cancer stem cell-like cells leading to an increased tumor burden. Mol. Cancer 2013, 12, 24. [Google Scholar] [CrossRef]
- Alwosaibai, K.; Abedini, A.; Al-Hujaily, E.M.; Tang, Y.; Garson, K.; Collins, O.; Vanderhyden, B.C. PAX2 maintains the differentiation of mouse oviductal epithelium and inhibits the transition to a stem cell-like state. Oncotarget 2017, 8, 76881–76897. [Google Scholar] [CrossRef]
- Bapat, S.A.; Mali, A.M.; Koppikar, C.B.; Kurrey, N.K. Stem and progenitor-like cells contribute to the aggressive behavior of human epithelial ovarian cancer. Cancer Res. 2005, 65, 3025–3029. [Google Scholar] [CrossRef]
- Hojo, N.; Huisken, A.L.; Wang, H.; Chirshev, E.; Kim, N.S.; Nguyen, S.M.; Campos, H.; Glackin, C.A.; Ioffe, Y.J.; Unternaehrer, J.J. Snail knockdown reverses stemness and inhibits tumour growth in ovarian cancer. Sci. Rep. 2018, 8, 8704. [Google Scholar] [CrossRef] [PubMed]
- Lupia, M.; Angiolini, F.; Bertalot, G.; Freddi, S.; Sachsenmeier, K.F.; Chisci, E.; Kutryb-Zajac, B.; Confalonieri, S.; Smolenski, R.T.; Giovannoni, R.; et al. CD73 Regulates Stemness and Epithelial-Mesenchymal Transition in Ovarian Cancer-Initiating Cells. Stem Cell Rep. 2018, 10, 1412–1425. [Google Scholar] [CrossRef] [PubMed]
- Park, G.B.; Kim, D. TLR5/7-mediated PI3K activation triggers epithelial-mesenchymal transition of ovarian cancer cells through WAVE3-dependent mesothelin or OCT4/SOX2 expression. Oncol. Rep. 2017, 38, 3167–3176. [Google Scholar] [CrossRef] [PubMed]
- Ricci, F.; Fratelli, M.; Guffanti, F.; Porcu, L.; Spriano, F.; Dell’Anna, T.; Fruscio, R.; Damia, G. Patient-derived ovarian cancer xenografts re-growing after a cisplatinum treatment are less responsive to a second drug re-challenge: A new experimental setting to study response to therapy. Oncotarget 2017, 8, 7441–7451. [Google Scholar] [CrossRef] [PubMed]
- Kenda Suster, N.; Smrkolj, S.; Virant-Klun, I. Putative stem cells and epithelial-mesenchymal transition revealed in sections of ovarian tumor in patients with serous ovarian carcinoma using immunohistochemistry for vimentin and pluripotency-related markers. J. Ovarian Res. 2017, 10, 11. [Google Scholar] [CrossRef] [PubMed]
- Pattabiraman, D.R.; Weinberg, R.A. Tackling the cancer stem cells—What challenges do they pose? Nat. Rev. Drug Discov. 2014, 13, 497–512. [Google Scholar] [CrossRef] [PubMed]
- Newsted, D.; Banerjee, S.; Watt, K.; Nersesian, S.; Truesdell, P.; Blazer, L.L.; Cardarelli, L.; Adams, J.J.; Sidhu, S.S.; Craig, A.W. Blockade of TGF-beta signaling with novel synthetic antibodies limits immune exclusion and improves chemotherapy response in metastatic ovarian cancer models. Oncoimmunology 2019, 8, e1539613. [Google Scholar] [CrossRef] [PubMed]
- Antoniou, A.; Pharoah, P.D.; Narod, S.; Risch, H.A.; Eyfjord, J.E.; Hopper, J.L.; Loman, N.; Olsson, H.; Johannsson, O.; Borg, A.; et al. Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case Series unselected for family history: A combined analysis of 22 studies. Am. J. Hum. Genet. 2003, 72, 1117–1130. [Google Scholar] [CrossRef] [PubMed]
- King, M.C.; Marks, J.H.; Mandell, J.B.; The New York Breast Cancer Study Group. Breast and ovarian cancer risks due to inherited mutations in BRCA1 and BRCA2. Science 2003, 302, 643–646. [Google Scholar] [CrossRef]
- Sengodan, S.K.; Sreelatha, K.H.; Nadhan, R.; Srinivas, P. Regulation of epithelial to mesenchymal transition by BRCA1 in breast cancer. Crit. Rev. Oncol./Hematol. 2018, 123, 74–82. [Google Scholar] [CrossRef]
- Lakhani, S.R.; Manek, S.; Penault-Llorca, F.; Flanagan, A.; Arnout, L.; Merrett, S.; McGuffog, L.; Steele, D.; Devilee, P.; Klijn, J.G.; et al. Pathology of ovarian cancers in BRCA1 and BRCA2 carriers. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2004, 10, 2473–2481. [Google Scholar] [CrossRef]
- Wang, H.; Bierie, B.; Li, A.G.; Pathania, S.; Toomire, K.; Dimitrov, S.D.; Liu, B.; Gelman, R.; Giobbie-Hurder, A.; Feunteun, J.; et al. BRCA1/FANCD2/BRG1-Driven DNA Repair Stabilizes the Differentiation State of Human Mammary Epithelial Cells. Mol. Cell 2016, 63, 277–292. [Google Scholar] [CrossRef] [PubMed]
- Huen, M.S.; Sy, S.M.; Chen, J. BRCA1 and its toolbox for the maintenance of genome integrity. Nat. Rev. Mol. Cell Biol. 2010, 11, 138–148. [Google Scholar] [CrossRef] [PubMed]
- Sinha, A.; Paul, B.T.; Sullivan, L.M.; Sims, H.; El Bastawisy, A.; Yousef, H.F.; Zekri, A.N.; Bahnassy, A.A.; ElShamy, W.M. BRCA1-IRIS overexpression promotes and maintains the tumor initiating phenotype: Implications for triple negative breast cancer early lesions. Oncotarget 2017, 8, 10114–10135. [Google Scholar] [CrossRef] [PubMed]
- Figueras, A.; Alsina-Sanchis, E.; Lahiguera, A.; Abreu, M.; Muinelo-Romay, L.; Moreno-Bueno, G.; Casanovas, O.; Graupera, M.; Matias-Guiu, X.; Vidal, A.; et al. A Role for CXCR4 in Peritoneal and Hematogenous Ovarian Cancer Dissemination. Mol. Cancer Ther. 2018, 17, 532–543. [Google Scholar] [CrossRef] [PubMed]
- Klymenko, Y.; Kim, O.; Stack, M.S. Complex Determinants of Epithelial: Mesenchymal Phenotypic Plasticity in Ovarian Cancer. Cancers 2017, 9, 104. [Google Scholar] [CrossRef] [PubMed]
- Sawada, K.; Mitra, A.K.; Radjabi, A.R.; Bhaskar, V.; Kistner, E.O.; Tretiakova, M.; Jagadeeswaran, S.; Montag, A.; Becker, A.; Kenny, H.A.; et al. Loss of E-cadherin promotes ovarian cancer metastasis via alpha 5-integrin, which is a therapeutic target. Cancer Res. 2008, 68, 2329–2339. [Google Scholar] [CrossRef]
- Tone, A.A. Taking the Tube: From Normal Fallopian Tube Epithelium to Ovarian High-grade Serous Carcinoma. Clin. Obstet. Gynecol. 2017, 60, 697–710. [Google Scholar] [CrossRef]
- Perets, R.; Wyant, G.A.; Muto, K.W.; Bijron, J.G.; Poole, B.B.; Chin, K.T.; Chen, J.Y.; Ohman, A.W.; Stepule, C.D.; Kwak, S.; et al. Transformation of the fallopian tube secretory epithelium leads to high-grade serous ovarian cancer in Brca;Tp53;Pten models. Cancer Cell 2013, 24, 751–765. [Google Scholar] [CrossRef]
- Dean, M.; Davis, D.A.; Burdette, J.E. Activin A stimulates migration of the fallopian tube epithelium, an origin of high-grade serous ovarian cancer, through non-canonical signaling. Cancer Lett. 2017, 391, 114–124. [Google Scholar] [CrossRef]
- Tan, D.S.; Agarwal, R.; Kaye, S.B. Mechanisms of transcoelomic metastasis in ovarian cancer. Lancet Oncol. 2006, 7, 925–934. [Google Scholar] [CrossRef]
- Al Habyan, S.; Kalos, C.; Szymborski, J.; McCaffrey, L. Multicellular detachment generates metastatic spheroids during intra-abdominal dissemination in epithelial ovarian cancer. Oncogene 2018, 37, 5127–5135. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, I.; Gurkan, U.A.; Tasoglu, S.; Alagic, N.; Celli, J.P.; Mensah, L.B.; Mai, Z.; Demirci, U.; Hasan, T. Flow induces epithelial-mesenchymal transition, cellular heterogeneity and biomarker modulation in 3D ovarian cancer nodules. Proc. Natl. Acad. Sci. USA 2013, 110, E1974–E1983. [Google Scholar] [CrossRef] [PubMed]
- Frisch, S.M.; Schaller, M.; Cieply, B. Mechanisms that link the oncogenic epithelial-mesenchymal transition to suppression of anoikis. J. Cell Sci. 2013, 126, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Yang, F. Akt2/ZEB2 may be a biomarker for exfoliant cells in ascitic fluid in advanced grades of serous ovarian carcinoma. Tumour Biol. 2015, 36, 7213–7219. [Google Scholar] [CrossRef] [PubMed]
- Veatch, A.L.; Carson, L.F.; Ramakrishnan, S. Differential expression of the cell-cell adhesion molecule E-cadherin in ascites and solid human ovarian tumor cells. Int. J. Cancer 1994, 58, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Burleson, K.M.; Hansen, L.K.; Skubitz, A.P. Ovarian carcinoma spheroids disaggregate on type I collagen and invade live human mesothelial cell monolayers. Clin. Exp. Metastasis 2004, 21, 685–697. [Google Scholar] [CrossRef] [PubMed]
- Rosso, M.; Majem, B.; Devis, L.; Lapyckyj, L.; Besso, M.J.; Llaurado, M.; Abascal, M.F.; Matos, M.L.; Lanau, L.; Castellvi, J.; et al. E-cadherin: A determinant molecule associated with ovarian cancer progression, dissemination and aggressiveness. PLoS ONE 2017, 12. [Google Scholar] [CrossRef]
- Ocana, O.H.; Corcoles, R.; Fabra, A.; Moreno-Bueno, G.; Acloque, H.; Vega, S.; Barrallo-Gimeno, A.; Cano, A.; Nieto, M.A. Metastatic colonization requires the repression of the epithelial-mesenchymal transition inducer Prrx1. Cancer Cell 2012, 22, 709–724. [Google Scholar] [CrossRef]
- Rafehi, S.; Ramos, Y.V.; Bertrand, M.; McGee, J.; Prefontaine, M.; Sugimoto, A.; DiMattia, G.E.; Shepherd, T.G. TGFβ signaling regulates epithelial-mesenchymal plasticity in ovarian cancer ascites-derived spheroids. Endocr-Relat Cancer 2016, 23, 147–159. [Google Scholar] [CrossRef]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef] [PubMed]
- Brabletz, T. To differentiate or not—Routes towards metastasis. Nat. Rev. Cancer 2012, 12, 425–436. [Google Scholar] [CrossRef] [PubMed]
- Koensgen, D.; Freitag, C.; Klaman, I.; Dahl, E.; Mustea, A.; Chekerov, R.; Braicu, I.; Lichtenegger, W.; Sehouli, J. Expression and localization of e-cadherin in epithelial ovarian cancer. AntiCancer Res. 2010, 30, 2525–2530. [Google Scholar] [PubMed]
- Pradeep, S.; Kim, S.W.; Wu, S.Y.; Nishimura, M.; Chaluvally-Raghavan, P.; Miyake, T.; Pecot, C.V.; Kim, S.J.; Choi, H.J.; Bischoff, F.Z.; et al. Hematogenous metastasis of ovarian cancer: Rethinking mode of spread. Cancer Cell 2014, 26, 77–91. [Google Scholar] [CrossRef] [PubMed]
- Strauss, R.; Li, Z.Y.; Liu, Y.; Beyer, I.; Persson, J.; Sova, P.; Moller, T.; Pesonen, S.; Hemminki, A.; Hamerlik, P.; et al. Analysis of epithelial and mesenchymal markers in ovarian cancer reveals phenotypic heterogeneity and plasticity. PLoS ONE 2011, 6, e16186. [Google Scholar] [CrossRef]
- Klymenko, Y.; Johnson, J.; Bos, B.; Lombard, R.; Campbell, L.; Loughran, E.; Stack, M.S. Heterogeneous Cadherin Expression and Multicellular Aggregate Dynamics in Ovarian Cancer Dissemination. Neoplasia 2017, 19, 549–563. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.Y.; Wong, M.K.; Tan, T.Z.; Kuay, K.T.; Ng, A.H.; Chung, V.Y.; Chu, Y.S.; Matsumura, N.; Lai, H.C.; Lee, Y.F.; et al. An EMT spectrum defines an anoikis-resistant and spheroidogenic intermediate mesenchymal state that is sensitive to e-cadherin restoration by a src-kinase inhibitor, saracatinib (AZD0530). Cell Death Dis. 2013, 4, e915. [Google Scholar] [CrossRef] [PubMed]
- Hudson, L.G.; Zeineldin, R.; Stack, M.S. Phenotypic plasticity of neoplastic ovarian epithelium: Unique cadherin profiles in tumor progression. Clin. Exp. Metastasis 2008, 25, 643–655. [Google Scholar] [CrossRef] [PubMed]
- Vang, R.; Shih Ie, M.; Kurman, R.J. Ovarian low-grade and high-grade serous carcinoma: Pathogenesis, clinicopathologic and molecular biologic features, and diagnostic problems. Adv. Anat. Pathol. 2009, 16, 267–282. [Google Scholar] [CrossRef]
- Sadlecki, P.; Grabiec, M.; Grzanka, D.; Jozwicki, J.; Antosik, P.; Walentowicz-Sadlecka, M. Expression of zinc finger transcription factors (ZNF143 and ZNF281) in serous borderline ovarian tumors and low-grade ovarian cancers. J. Ovarian Res. 2019, 12, 23. [Google Scholar] [CrossRef]
- Sadlecki, P.; Jozwicki, J.; Antosik, P.; Grabiec, M. Expression of selected epithelial-mesenchymal transition transcription factors in serous borderline ovarian tumors and type I ovarian cancers. Tumour Biol. 2018, 40, 1010428318784807. [Google Scholar] [CrossRef] [PubMed]
- Salamanca, C.M.; Maines-Bandiera, S.L.; Leung, P.C.; Hu, Y.L.; Auersperg, N. Effects of epidermal growth factor/hydrocortisone on the growth and differentiation of human ovarian surface epithelium. J. Soc. Gynecol. Investig. 2004, 11, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.C.; Auersperg, N.; Leung, P.C. Inhibition of p53 induces invasion of serous borderline ovarian tumor cells by accentuating PI3K/Akt-mediated suppression of E-cadherin. Oncogene 2011, 30, 1020–1031. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Thomas, J.P.; Lautermann, J.; Liedert, B.; Seiler, F.; Thomale, J. High accumulation of platinum-DNA adducts in strial marginal cells of the cochlea is an early event in cisplatin but not carboplatin ototoxicity. Mol. Pharmacol. 2006, 70, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Judson, P.L.; Watson, J.M.; Gehrig, P.A.; Fowler, W.C., Jr.; Haskill, J.S. Cisplatin inhibits paclitaxel-induced apoptosis in cisplatin-resistant ovarian cancer cell lines: Possible explanation for failure of combination therapy. Cancer Res. 1999, 59, 2425–2432. [Google Scholar] [PubMed]
- Siddik, Z.H. Cisplatin: Mode of cytotoxic action and molecular basis of resistance. Oncogene 2003, 22, 7265–7279. [Google Scholar] [CrossRef] [PubMed]
- Rowinsky, E.K.; Donehower, R.C. Paclitaxel (taxol). N. Engl. J. Med. 1995, 332, 1004–1014. [Google Scholar] [CrossRef] [PubMed]
- Baribeau, S.; Chaudhry, P.; Parent, S.; Asselin, E. Resveratrol inhibits cisplatin-induced epithelial-to-mesenchymal transition in ovarian cancer cell lines. PLoS ONE 2014, 9, e86987. [Google Scholar] [CrossRef]
- Brozovic, A.; Duran, G.E.; Wang, Y.C.; Francisco, E.B.; Sikic, B.I. The miR-200 family differentially regulates sensitivity to paclitaxel and carboplatin in human ovarian carcinoma OVCAR-3 and MES-OV cells. Mol. Oncol. 2015, 9, 1678–1693. [Google Scholar] [CrossRef]
- Chowanadisai, W.; Messerli, S.M.; Miller, D.H.; Medina, J.E.; Hamilton, J.W.; Messerli, M.A.; Brodsky, A.S. Cisplatin Resistant Spheroids Model Clinically Relevant Survival Mechanisms in Ovarian Tumors. PLoS ONE 2016, 11, e0151089. [Google Scholar] [CrossRef]
- Haslehurst, A.M.; Koti, M.; Dharsee, M.; Nuin, P.; Evans, K.; Geraci, J.; Childs, T.; Chen, J.; Li, J.; Weberpals, J.; et al. EMT transcription factors snail and slug directly contribute to cisplatin resistance in ovarian cancer. BMC Cancer 2012, 12, 91. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Shen, H.; Yin, X.; Long, L.; Xie, C.; Liu, Y.; Hui, L.; Lin, X.; Fang, Y.; Cao, Y.; et al. miR-186 regulation of Twist1 and ovarian cancer sensitivity to cisplatin. Oncogene 2016, 35, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Izutsu, N.; Maesawa, C.; Shibazaki, M.; Oikawa, H.; Shoji, T.; Sugiyama, T.; Masuda, T. Epigenetic modification is involved in aberrant expression of class III beta-tubulin, TUBB3, in ovarian cancer cells. Int. J. Oncol. 2008, 32, 1227–1235. [Google Scholar] [PubMed]
- Cochrane, D.R.; Howe, E.N.; Spoelstra, N.S.; Richer, J.K. Loss of miR-200c: A Marker of Aggressiveness and Chemoresistance in Female Reproductive Cancers. J. Oncol. 2010, 2010, 821717. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Michels, J.; Brenner, C.; Szabadkai, G.; Harel-Bellan, A.; Castedo, M.; Kroemer, G. Systems biology of cisplatin resistance: Past, present and future. Cell Death Dis. 2014, 5, e1257. [Google Scholar] [CrossRef] [PubMed]
- Sonego, M.; Pellizzari, I.; Dall’Acqua, A.; Pivetta, E.; Lorenzon, I.; Benevol, S.; Bomben, R.; Spessotto, P.; Sorio, R.; Gattei, V.; et al. Common biological phenotypes characterize the acquisition of platinum-resistance in epithelial ovarian cancer cells. Sci. Rep. 2017, 7, 7104. [Google Scholar] [CrossRef] [PubMed]
- Chiu, W.T.; Huang, Y.F.; Tsai, H.Y.; Chen, C.C.; Chang, C.H.; Huang, S.C.; Hsu, K.F.; Chou, C.Y. FOXM1 confers to epithelial-mesenchymal transition, stemness and chemoresistance in epithelial ovarian carcinoma cells. Oncotarget 2015, 6, 2349–2365. [Google Scholar] [CrossRef]
- Saxena, M.; Stephens, M.A.; Pathak, H.; Rangarajan, A. Transcription factors that mediate epithelial-mesenchymal transition lead to multidrug resistance by upregulating ABC transporters. Cell Death Dis. 2011, 2, e179. [Google Scholar] [CrossRef]
- Feng, T.; Wang, Y.; Lang, Y.; Zhang, Y. KDM5A promotes proliferation and EMT in ovarian cancer and closely correlates with PTX resistance. Mol. Med. Rep. 2017, 16, 3573–3580. [Google Scholar] [CrossRef]
- Zhang, Y.; Huang, S.; Guo, Y.; Li, L. MiR-1294 confers cisplatin resistance in ovarian Cancer cells by targeting IGF1R. Biomed. Pharmacother. Biomed. Pharmacother. 2018, 106, 1357–1363. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhu, Y.; Fan, X.; Zhang, C.; Wang, Y.; Zhang, L.; Zhang, H.; Wen, T.; Zhang, K.; Huo, X.; et al. NID1, a new regulator of EMT required for metastasis and chemoresistance of ovarian cancer cells. Oncotarget 2017, 8, 33110–33121. [Google Scholar] [CrossRef] [PubMed]
- Koti, M.; Gooding, R.J.; Nuin, P.; Haslehurst, A.; Crane, C.; Weberpals, J.; Childs, T.; Bryson, P.; Dharsee, M.; Evans, K.; et al. Identification of the IGF1/PI3K/NF kappaB/ERK gene signalling networks associated with chemotherapy resistance and treatment response in high-grade serous epithelial ovarian cancer. BMC Cancer 2013, 13, 549. [Google Scholar] [CrossRef] [PubMed]
- Dmitriev, O.Y. Mechanism of tumor resistance to cisplatin mediated by the copper transporter ATP7B. Biochem. Cell Biol. Biochim. Biol. Cell. 2011, 89, 138–147. [Google Scholar] [CrossRef] [PubMed]
- Dwivedi, S.K.; Mustafi, S.B.; Mangala, L.S.; Jiang, D.; Pradeep, S.; Rodriguez-Aguayo, C.; Ling, H.; Ivan, C.; Mukherjee, P.; Calin, G.A.; et al. Therapeutic evaluation of microRNA-15a and microRNA-16 in ovarian cancer. Oncotarget 2016, 7, 15093–15104. [Google Scholar] [CrossRef] [PubMed]
- Clarke, M.F.; Dick, J.E.; Dirks, P.B.; Eaves, C.J.; Jamieson, C.H.; Jones, D.L.; Visvader, J.; Weissman, I.L.; Wahl, G.M. Cancer stem cells—Perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res. 2006, 66, 9339–9344. [Google Scholar] [CrossRef]
- Samardzija, C.; Greening, D.W.; Escalona, R.; Chen, M.; Bilandzic, M.; Luwor, R.; Kannourakis, G.; Findlay, J.K.; Ahmed, N. Knockdown of stem cell regulator Oct4A in ovarian cancer reveals cellular reprogramming associated with key regulators of cytoskeleton-extracellular matrix remodelling. Sci. Rep. 2017, 7, 46312. [Google Scholar] [CrossRef] [PubMed]
- Bae, J.S.; Noh, S.J.; Kim, K.M.; Park, S.H.; Hussein, U.K.; Park, H.S.; Park, B.H.; Ha, S.H.; Lee, H.; Chung, M.J.; et al. SIRT6 Is Involved in the Progression of Ovarian Carcinomas via beta-Catenin-Mediated Epithelial to Mesenchymal Transition. Front. Oncol. 2018, 8, 538. [Google Scholar] [CrossRef]
- Liu, G.; Yang, D.; Rupaimoole, R.; Pecot, C.V.; Sun, Y.; Mangala, L.S.; Li, X.; Ji, P.; Cogdell, D.; Hu, L.; et al. Augmentation of response to chemotherapy by microRNA-506 through regulation of RAD51 in serous ovarian cancers. J. Natl. Cancer Inst. 2015, 107. [Google Scholar] [CrossRef]
- Kurrey, N.K.; Jalgaonkar, S.P.; Joglekar, A.V.; Ghanate, A.D.; Chaskar, P.D.; Doiphode, R.Y.; Bapat, S.A. Snail and slug mediate radioresistance and chemoresistance by antagonizing p53-mediated apoptosis and acquiring a stem-like phenotype in ovarian cancer cells. Stem Cells 2009, 27, 2059–2068. [Google Scholar] [CrossRef]
- Fraser, M.; Bai, T.; Tsang, B.K. Akt promotes cisplatin resistance in human ovarian cancer cells through inhibition of p53 phosphorylation and nuclear function. Int. J. Cancer 2008, 122, 534–546. [Google Scholar] [CrossRef]
- Vega, S.; Morales, A.V.; Ocana, O.H.; Valdes, F.; Fabregat, I.; Nieto, M.A. Snail blocks the cell cycle and confers resistance to cell death. Genes Dev. 2004, 18, 1131–1143. [Google Scholar] [CrossRef] [PubMed]
- Rosano, L.; Cianfrocca, R.; Spinella, F.; Di Castro, V.; Nicotra, M.R.; Lucidi, A.; Ferrandina, G.; Natali, P.G.; Bagnato, A. Acquisition of chemoresistance and EMT phenotype is linked with activation of the endothelin A receptor pathway in ovarian carcinoma cells. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2011, 17, 2350–2360. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Ding, H.; Tian, J.; Wu, L.; Wang, Y.; Xing, Y.; Chen, M. Forkhead Box Protein C2 (FOXC2) Promotes the Resistance of Human Ovarian Cancer Cells to Cisplatin In Vitro and In Vivo. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2016, 39, 242–252. [Google Scholar] [CrossRef] [PubMed]
- Latifi, A.; Abubaker, K.; Castrechini, N.; Ward, A.C.; Liongue, C.; Dobill, F.; Kumar, J.; Thompson, E.W.; Quinn, M.A.; Findlay, J.K.; et al. Cisplatin treatment of primary and metastatic epithelial ovarian carcinomas generates residual cells with mesenchymal stem cell-like profile. J. Cell. Biochem. 2011, 112, 2850–2864. [Google Scholar] [CrossRef] [PubMed]
- Hou, L.; Hou, X.; Wang, L.; Li, Z.; Xin, B.; Chen, J.; Gao, X.; Mu, H. PD98059 impairs the cisplatin-resistance of ovarian cancer cells by suppressing ERK pathway and epithelial mesenchymal transition process. Cancer Biomark. Sect. A Dis. Markers 2017, 21, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Granados, M.L.; Hudson, L.G.; Samudio-Ruiz, S.L. Contributions of the Epidermal Growth Factor Receptor to Acquisition of Platinum Resistance in Ovarian Cancer Cells. PLoS ONE 2015, 10, e0136893. [Google Scholar] [CrossRef] [PubMed]
- Tung, S.L.; Huang, W.C.; Hsu, F.C.; Yang, Z.P.; Jang, T.H.; Chang, J.W.; Chuang, C.M.; Lai, C.R.; Wang, L.H. miRNA-34c-5p inhibits amphiregulin-induced ovarian cancer stemness and drug resistance via downregulation of the AREG-EGFR-ERK pathway. Oncogenesis 2017, 6, e326. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Zhao, J.; Ruan, Y.; Sun, L.; Xu, C.; Jiang, H. Sialyltransferase ST3GAL1 promotes cell migration, invasion, and TGF-beta1-induced EMT and confers paclitaxel resistance in ovarian cancer. Cell Death Dis. 2018, 9, 1102. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Cong, Q.; Zhang, X.Y.; Zhang, M.X.; Lu, Y.Y.; Xu, C.J. Pyruvate dehydrogenase kinase 1 contributes to cisplatin resistance of ovarian cancer through EGFR activation. J. Cell. Physiol. 2019, 234, 6361–6370. [Google Scholar] [CrossRef]
- Morrison, J.; Thoma, C.; Goodall, R.J.; Lyons, T.J.; Gaitskell, K.; Wiggans, A.J.; Bryant, A. Epidermal growth factor receptor blockers for the treatment of ovarian cancer. Cochrane Database Syst. Rev. 2018, 10, CD007927. [Google Scholar] [CrossRef]
- Xu, J.; Lamouille, S.; Derynck, R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 2009, 19, 156–172. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Zhang, J.; Liu, M.; Huang, Y.; Yin, L. SMAD3 inducing the transcription of STYK1 to promote the EMT process and improve the tolerance of ovarian carcinoma cells to paclitaxel. J. Cell. Biochem. 2019. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yang, B.; Zhao, J.; Yu, X.; Liu, X.; Zhang, L.; Zhang, Y.; Li, X.; Zhai, Z. Epithelialmesenchymal transition induced by bone morphogenetic protein 9 hinders cisplatin efficacy in ovarian cancer cells. Mol. Med. Rep. 2019, 19, 1501–1508. [Google Scholar] [CrossRef] [PubMed]
- Sakata, J.; Utsumi, F.; Suzuki, S.; Niimi, K.; Yamamoto, E.; Shibata, K.; Senga, T.; Kikkawa, F.; Kajiyama, H. Inhibition of ZEB1 leads to inversion of metastatic characteristics and restoration of paclitaxel sensitivity of chronic chemoresistant ovarian carcinoma cells. Oncotarget 2017, 8, 99482–99494. [Google Scholar] [CrossRef] [PubMed]
- Altomare, D.A.; Wang, H.Q.; Skele, K.L.; De Rienzo, A.; Klein-Szanto, A.J.; Godwin, A.K.; Testa, J.R. AKT and mTOR phosphorylation is frequently detected in ovarian cancer and can be targeted to disrupt ovarian tumor cell growth. Oncogene 2004, 23, 5853–5857. [Google Scholar] [CrossRef] [PubMed]
- Shayesteh, L.; Lu, Y.; Kuo, W.L.; Baldocchi, R.; Godfrey, T.; Collins, C.; Pinkel, D.; Powell, B.; Mills, G.B.; Gray, J.W. PIK3CA is implicated as an oncogene in ovarian cancer. Nat. Genet. 1999, 21, 99–102. [Google Scholar] [CrossRef] [PubMed]
- De Donato, M.; Petrillo, M.; Martinelli, E.; Filippetti, F.; Zannoni, G.F.; Scambia, G.; Gallo, D. Uncovering the role of nuclear Lysyl oxidase (LOX) in advanced high grade serous ovarian cancer. Gynecol. Oncol. 2017, 146, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Bugide, S.; Gonugunta, V.K.; Penugurti, V.; Malisetty, V.L.; Vadlamudi, R.K.; Manavathi, B. HPIP promotes epithelial-mesenchymal transition and cisplatin resistance in ovarian cancer cells through PI3K/AKT pathway activation. Cell. Oncol. 2017, 40, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Qiu, L.; Zhang, G.F.; Yu, L.; Wang, H.Y.; Jia, X.J.; Wang, T.J. Novel oncogenic and chemoresistance-inducing functions of resistin in ovarian cancer cells require miRNAs-mediated induction of epithelial-to-mesenchymal transition. Sci. Rep. 2018, 8, 12522. [Google Scholar] [CrossRef] [PubMed]
- Sheng, W.J.; Jiang, H.; Wu, D.L.; Zheng, J.H. Early responses of the STAT3 pathway to platinum drugs are associated with cisplatin resistance in epithelial ovarian cancer. Braz. J. Med. Biol. Res. Rev. Bras. Pesqui. Med. Biol. 2013, 46, 650–658. [Google Scholar] [CrossRef]
- Jagadish, N.; Fatima, R.; Sharma, A.; Devi, S.; Suri, V.; Kumar, V.; Suri, A. Sperm associated antigen 9 (SPAG9) a promising therapeutic target of ovarian carcinoma. Tumour Biol. 2018, 40, 1010428318773652. [Google Scholar] [CrossRef] [PubMed]
- Duran, G.E.; Wang, Y.C.; Moisan, F.; Francisco, E.B.; Sikic, B.I. Decreased levels of baseline and drug-induced tubulin polymerisation are hallmarks of resistance to taxanes in ovarian cancer cells and are associated with epithelial-to-mesenchymal transition. Br. J. Cancer 2017, 116, 1318–1328. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Zhang, Q.; Miao, C.; Dongol, S.; Li, Y.; Jin, C.; Dong, R.; Li, Y.; Yang, X.; Kong, B. CCNG1 (Cyclin G1) regulation by mutant-P53 via induction of Notch3 expression promotes high-grade serous ovarian cancer (HGSOC) tumorigenesis and progression. Cancer Med. 2019, 8, 351–362. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.N.; Chang, R.; Lin, L.T.; Chern, C.U.; Tsai, H.W.; Wen, Z.H.; Li, Y.H.; Li, C.J.; Tsui, K.H. MicroRNA in Ovarian Cancer: Biology, Pathogenesis, and Therapeutic Opportunities. Int. J. Environ. Res. Public Health 2019, 16, 1510. [Google Scholar] [CrossRef] [PubMed]
- Deb, B.; Uddin, A.; Chakraborty, S. miRNAs and ovarian cancer: An overview. J. Cell. Physiol. 2018, 233, 3846–3854. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Zheng, H.; Chan, M.T.; Wu, W.K. Modulation of chemoresponsiveness to platinum-based agents by microRNAs in cancer. Am. J. Cancer Res. 2017, 7, 1769–1778. [Google Scholar]
- Boac, B.M.; Xiong, Y.; Marchion, D.C.; Abbasi, F.; Bush, S.H.; Ramirez, I.J.; Khulpateea, B.R.; Clair McClung, E.; Berry, A.L.; Bou Zgheib, N.; et al. Micro-RNAs associated with the evolution of ovarian cancer cisplatin resistance. Gynecol. Oncol. 2016, 140, 259–263. [Google Scholar] [CrossRef]
- Jurmeister, S.; Baumann, M.; Balwierz, A.; Keklikoglou, I.; Ward, A.; Uhlmann, S.; Zhang, J.D.; Wiemann, S.; Sahin, O. MicroRNA-200c represses migration and invasion of breast cancer cells by targeting actin-regulatory proteins FHOD1 and PPM1F. Mol. Cell. Biol. 2012, 32, 633–651. [Google Scholar] [CrossRef]
- Mezencev, R.; Wartell, R.M. Cisplatin binds to pre-miR-200b and impairs its processing to mature microRNA. Neoplasma 2018, 65, 222–227. [Google Scholar] [CrossRef]
- Cao, L.; Wan, Q.; Li, F.; Tang, C.E. MiR-363 inhibits cisplatin chemoresistance of epithelial ovarian cancer by regulating snail-induced epithelial-mesenchymal transition. BMB Rep. 2018, 51, 456–461. [Google Scholar] [CrossRef]
- Liu, Y.; Han, S.; Li, Y.; Liu, Y.; Zhang, D.; Li, Y.; Zhang, J. MicroRNA-20a contributes to cisplatin-resistance and migration of OVCAR3 ovarian cancer cell line. Oncol. Lett. 2017, 14, 1780–1786. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Xu, Q.H.; Dong, Y.H.; Li, G.X.; Yang, L.; Wang, L.W.; Li, H.Y. MiR-181a upregulation is associated with epithelial-to-mesenchymal transition (EMT) and multidrug resistance (MDR) of ovarian cancer cells. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 2004–2010. [Google Scholar] [PubMed]
- Xia, H.; Ooi, L.L.; Hui, K.M. MicroRNA-216a/217-induced epithelial-mesenchymal transition targets PTEN and SMAD7 to promote drug resistance and recurrence of liver cancer. Hepatology 2013, 58, 629–641. [Google Scholar] [CrossRef] [PubMed]
- Jin, P.; Liu, Y.; Wang, R. STAT3 regulated miR-216a promotes ovarian cancer proliferation and cisplatin resistance. Biosci. Rep. 2018, 38. [Google Scholar] [CrossRef] [PubMed]
- Han, M.; Liu, M.; Wang, Y.; Chen, X.; Xu, J.; Sun, Y.; Zhao, L.; Qu, H.; Fan, Y.; Wu, C. Antagonism of miR-21 reverses epithelial-mesenchymal transition and cancer stem cell phenotype through AKT/ERK1/2 inactivation by targeting PTEN. PLoS ONE 2012, 7, e39520. [Google Scholar] [CrossRef] [PubMed]
- Au Yeung, C.L.; Co, N.N.; Tsuruga, T.; Yeung, T.L.; Kwan, S.Y.; Leung, C.S.; Li, Y.; Lu, E.S.; Kwan, K.; Wong, K.K.; et al. Exosomal transfer of stroma-derived miR21 confers paclitaxel resistance in ovarian cancer cells through targeting APAF1. Nat. Commun. 2016, 7, 11150. [Google Scholar] [CrossRef] [PubMed]
- Voth, W.; Jakob, U. Stress-Activated Chaperones: A First Line of Defense. Trends Biochem. Sci. 2017, 42, 899–913. [Google Scholar] [CrossRef]
- Amoroso, M.R.; Matassa, D.S.; Agliarulo, I.; Avolio, R.; Lu, H.; Sisinni, L.; Lettini, G.; Gabra, H.; Landriscina, M.; Esposito, F. TRAP1 downregulation in human ovarian cancer enhances invasion and epithelial-mesenchymal transition. Cell Death Dis. 2016, 7, e2522. [Google Scholar] [CrossRef]
- Amoroso, M.R.; Matassa, D.S.; Agliarulo, I.; Avolio, R.; Maddalena, F.; Condelli, V.; Landriscina, M.; Esposito, F. Stress-Adaptive Response in Ovarian Cancer Drug Resistance: Role of TRAP1 in Oxidative Metabolism-Driven Inflammation. Adv. Protein Chem. Struct. Biol. 2017, 108, 163–198. [Google Scholar] [CrossRef]
- Aust, S.; Bachmayr-Heyda, A.; Pateisky, P.; Tong, D.; Darb-Esfahani, S.; Denkert, C.; Chekerov, R.; Sehouli, J.; Mahner, S.; Van Gorp, T.; et al. Role of TRAP1 and estrogen receptor alpha in patients with ovarian cancer -a study of the OVCAD consortium. Mol. Cancer 2012, 11, 69. [Google Scholar] [CrossRef]
- Na, Y.; Kaul, S.C.; Ryu, J.; Lee, J.S.; Ahn, H.M.; Kaul, Z.; Kalra, R.S.; Li, L.; Widodo, N.; Yun, C.O.; et al. Stress chaperone mortalin contributes to epithelial-mesenchymal transition and cancer metastasis. Cancer Res. 2016, 76, 2754–2765. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Li, H.; Jiang, Y.; Zuo, J.; Liu, W. Inhibition of mortalin expression reverses cisplatin resistance and attenuates growth of ovarian cancer cells. Cancer Lett. 2013, 336, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Borovski, T.; De Sousa, E.M.F.; Vermeulen, L.; Medema, J.P. Cancer stem cell niche: The place to be. Cancer Res. 2011, 71, 634–639. [Google Scholar] [CrossRef] [PubMed]
- Ghoneum, A.; Afify, H.; Salih, Z.; Kelly, M.; Said, N. Role of tumor microenvironment in ovarian cancer pathobiology. Oncotarget 2018, 9, 22832–22849. [Google Scholar] [CrossRef] [PubMed]
- Yeung, T.L.; Leung, C.S.; Li, F.; Wong, S.S.; Mok, S.C. Targeting Stromal-Cancer Cell Crosstalk Networks in Ovarian Cancer Treatment. Biomolecules 2016, 6, 3. [Google Scholar] [CrossRef]
- Hansen, J.M.; Coleman, R.L.; Sood, A.K. Targeting the tumour microenvironment in ovarian cancer. Eur. J. Cancer 2016, 56, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Chen, F.; Xu, Q.; Han, L.; Xu, J.; Gao, L.; Sun, X.; Li, Y.; Li, Y.; Qian, M.; et al. Revisiting ovarian cancer microenvironment: A friend or a foe? Protein Cell 2018, 9, 674–692. [Google Scholar] [CrossRef]
- Sullivan, N.J.; Sasser, A.K.; Axel, A.E.; Vesuna, F.; Raman, V.; Ramirez, N.; Oberyszyn, T.M.; Hall, B.M. Interleukin-6 induces an epithelial-mesenchymal transition phenotype in human breast cancer cells. Oncogene 2009, 28, 2940–2947. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, F.; Cui, J.Y.; Chen, L.; Chen, Y.T.; Liu, B.W. CAFs enhance paclitaxel resistance by inducing EMT through the IL6/JAK2/STAT3 pathway. Oncol. Rep. 2018, 39, 2081–2090. [Google Scholar] [CrossRef]
- Browning, L.; Patel, M.R.; Horvath, E.B.; Tawara, K.; Jorcyk, C.L. IL-6 and ovarian cancer: Inflammatory cytokines in promotion of metastasis. Cancer Manag. Res. 2018, 10, 6685–6693. [Google Scholar] [CrossRef]
- Coffman, L.G.; Choi, Y.J.; McLean, K.; Allen, B.L.; di Magliano, M.P.; Buckanovich, R.J. Human carcinoma-associated mesenchymal stem cells promote ovarian cancer chemotherapy resistance via a BMP4/HH signaling loop. Oncotarget 2016, 7, 6916–6932. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Guo, Y.; Huang, Z.; Cai, J.; Wang, Z. Adiposederived mesenchymal stem cells attenuate cisplatininduced apoptosis in epithelial ovarian cancer cells. Mol. Med. Rep. 2017, 16, 9587–9592. [Google Scholar] [CrossRef] [PubMed]
- Moisan, F.; Francisco, E.B.; Brozovic, A.; Duran, G.E.; Wang, Y.C.; Chaturvedi, S.; Seetharam, S.; Snyder, L.A.; Doshi, P.; Sikic, B.I. Enhancement of paclitaxel and carboplatin therapies by CCL2 blockade in ovarian cancers. Mol. Oncol. 2014, 8, 1231–1239. [Google Scholar] [CrossRef] [PubMed]
- Sato, E.; Olson, S.H.; Ahn, J.; Bundy, B.; Nishikawa, H.; Qian, F.; Jungbluth, A.A.; Frosina, D.; Gnjatic, S.; Ambrosone, C.; et al. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc. Natl. Acad. Sci. USA 2005, 102, 18538–18543. [Google Scholar] [CrossRef] [PubMed]
- Odunsi, K. Immunotherapy in ovarian cancer. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2017, 28, viii1–viii7. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Liu, C.; Bai, H.; Cao, G.; Cui, R.; Zhang, Z. Combinatorial therapy of immune checkpoint and cancer pathways provides a novel perspective on ovarian cancer treatment. Oncol. Lett. 2019, 17, 2583–2591. [Google Scholar] [CrossRef] [PubMed]
- Akalay, I.; Janji, B.; Hasmim, M.; Noman, M.Z.; Andre, F.; De Cremoux, P.; Bertheau, P.; Badoual, C.; Vielh, P.; Larsen, A.K.; et al. Epithelial-to-mesenchymal transition and autophagy induction in breast carcinoma promote escape from T-cell-mediated lysis. Cancer Res. 2013, 73, 2418–2427. [Google Scholar] [CrossRef]
- Chae, Y.K.; Chang, S.; Ko, T.; Anker, J.; Agte, S.; Iams, W.; Choi, W.M.; Lee, K.; Cruz, M. Epithelial-mesenchymal transition (EMT) signature is inversely associated with T-cell infiltration in non-small cell lung cancer (NSCLC). Sci. Rep. 2018, 8, 2918. [Google Scholar] [CrossRef]
- Chen, L.; Gibbons, D.L.; Goswami, S.; Cortez, M.A.; Ahn, Y.H.; Byers, L.A.; Zhang, X.; Yi, X.; Dwyer, D.; Lin, W.; et al. Metastasis is regulated via microRNA-200/ZEB1 axis control of tumour cell PD-L1 expression and intratumoral immunosuppression. Nat. Commun. 2014, 5, 5241. [Google Scholar] [CrossRef]
- Lou, Y.; Diao, L.; Cuentas, E.R.; Denning, W.L.; Chen, L.; Fan, Y.H.; Byers, L.A.; Wang, J.; Papadimitrakopoulou, V.A.; Behrens, C.; et al. Epithelial-Mesenchymal Transition Is Associated with a Distinct Tumor Microenvironment Including Elevation of Inflammatory Signals and Multiple Immune Checkpoints in Lung Adenocarcinoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 3630–3642. [Google Scholar] [CrossRef]
- Alsuliman, A.; Colak, D.; Al-Harazi, O.; Fitwi, H.; Tulbah, A.; Al-Tweigeri, T.; Al-Alwan, M.; Ghebeh, H. Bidirectional crosstalk between PD-L1 expression and epithelial to mesenchymal transition: Significance in claudin-low breast cancer cells. Mol. Cancer 2015, 14, 149. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Wang, Q.Q.; Chen, B.; Zhao, Y.Y.; Shen, B.; Wang, H.; Xu, J.; Zhu, M.L.; Zhao, X.D.; Xu, C.G.; et al. Gastric cancer mesenchymal stem cells derived IL-8 induces PD-L1 expression in gastric cancer cells via STAT3/mTOR-c-Myc signal axis. Cell Death Dis. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Connolly, E.; Smyth, J.W.; Akhurst, R.J.; Derynck, R. TGF-beta-induced activation of mTOR complex 2 drives epithelial-mesenchymal transition and cell invasion. J. Cell Sci. 2012, 125, 1259–1273. [Google Scholar] [CrossRef] [PubMed]
- Wendt, M.K.; Balanis, N.; Carlin, C.R.; Schiemann, W.P. STAT3 and epithelial-mesenchymal transitions in carcinomas. JAK-STAT 2014, 3, e28975. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Chen, Y.; Li, F.; Bao, L.; Liu, W. Atezolizumab and Bevacizumab Attenuate Cisplatin Resistant Ovarian Cancer Cells Progression Synergistically via Suppressing Epithelial-Mesenchymal Transition. Front. Immunol. 2019, 10, 867. [Google Scholar] [CrossRef]
- Dean, M. Cancer stem cells: Redefining the paradigm of cancer treatment strategies. Mol. Interv. 2006, 6, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Morrison, R.; Schleicher, S.M.; Sun, Y.; Niermann, K.J.; Kim, S.; Spratt, D.E.; Chung, C.H.; Lu, B. Targeting the mechanisms of resistance to chemotherapy and radiotherapy with the cancer stem cell hypothesis. J. Oncol. 2011, 2011, 941876. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, R.; Mitra, T.; Ray Chaudhuri, S.; Roy, S.S. Mesenchymal splice isoform of CD44 (CD44s) promotes EMT/invasion and imparts stem-like properties to ovarian cancer cells. J. Cell. Biochem. 2018, 119, 3373–3383. [Google Scholar] [CrossRef] [PubMed]
- Ip, C.K.; Li, S.S.; Tang, M.Y.; Sy, S.K.; Ren, Y.; Shum, H.C.; Wong, A.S. Stemness and chemoresistance in epithelial ovarian carcinoma cells under shear stress. Sci. Rep. 2016, 6, 26788. [Google Scholar] [CrossRef] [PubMed]
- Shahin, S.A.; Wang, R.; Simargi, S.I.; Contreras, A.; Parra Echavarria, L.; Qu, L.; Wen, W.; Dellinger, T.; Unternaehrer, J.; Tamanoi, F.; et al. Hyaluronic acid conjugated nanoparticle delivery of siRNA against TWIST reduces tumor burden and enhances sensitivity to cisplatin in ovarian cancer. Nanomed. Nanotechnol. Biol. Med. 2018, 14, 1381–1394. [Google Scholar] [CrossRef] [PubMed]
- Ozpolat, B.; Sood, A.K.; Lopez-Berestein, G. Liposomal siRNA nanocarriers for cancer therapy. Adv. Drug Deliv. Rev. 2014, 66, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Deng, S.; Wong, C.K.C.; Lai, H.C.; Wong, A.S.T. Ginsenoside-Rb1 targets chemotherapy-resistant ovarian cancer stem cells via simultaneous inhibition of Wnt/beta-catenin signaling and epithelial-to-mesenchymal transition. Oncotarget 2017, 8, 25897–25914. [Google Scholar] [CrossRef] [PubMed]
- Chau, W.K.; Ip, C.K.; Mak, A.S.; Lai, H.C.; Wong, A.S. c-Kit mediates chemoresistance and tumor-initiating capacity of ovarian cancer cells through activation of Wnt/beta-catenin-ATP-binding cassette G2 signaling. Oncogene 2013, 32, 2767–2781. [Google Scholar] [CrossRef] [PubMed]
- Dia, V.P.; Pangloli, P. Epithelial-to-Mesenchymal Transition in Paclitaxel-Resistant Ovarian Cancer Cells Is Downregulated by Luteolin. J. Cell. Physiol. 2017, 232, 391–401. [Google Scholar] [CrossRef] [PubMed]
- Tan, T.Z.; Miow, Q.H.; Huang, R.Y.; Wong, M.K.; Ye, J.; Lau, J.A.; Wu, M.C.; Bin Abdul Hadi, L.H.; Soong, R.; Choolani, M.; et al. Functional genomics identifies five distinct molecular subtypes with clinical relevance and pathways for growth control in epithelial ovarian cancer. EMBO Mol. Med. 2013, 5, 1051–1066. [Google Scholar] [CrossRef] [PubMed]
- Miow, Q.H.; Tan, T.Z.; Ye, J.; Lau, J.A.; Yokomizo, T.; Thiery, J.P.; Mori, S. Epithelial-mesenchymal status renders differential responses to cisplatin in ovarian cancer. Oncogene 2015, 34, 1899–1907. [Google Scholar] [CrossRef] [PubMed]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Loret, N.; Denys, H.; Tummers, P.; Berx, G. The Role of Epithelial-to-Mesenchymal Plasticity in Ovarian Cancer Progression and Therapy Resistance. Cancers 2019, 11, 838. https://doi.org/10.3390/cancers11060838
Loret N, Denys H, Tummers P, Berx G. The Role of Epithelial-to-Mesenchymal Plasticity in Ovarian Cancer Progression and Therapy Resistance. Cancers. 2019; 11(6):838. https://doi.org/10.3390/cancers11060838
Chicago/Turabian StyleLoret, Nele, Hannelore Denys, Philippe Tummers, and Geert Berx. 2019. "The Role of Epithelial-to-Mesenchymal Plasticity in Ovarian Cancer Progression and Therapy Resistance" Cancers 11, no. 6: 838. https://doi.org/10.3390/cancers11060838
APA StyleLoret, N., Denys, H., Tummers, P., & Berx, G. (2019). The Role of Epithelial-to-Mesenchymal Plasticity in Ovarian Cancer Progression and Therapy Resistance. Cancers, 11(6), 838. https://doi.org/10.3390/cancers11060838