ALDH3A1 Overexpression in Melanoma and Lung Tumors Drives Cancer Stem Cell Expansion, Impairing Immune Surveillance through Enhanced PD-L1 Output


 ,
,  , and
, and
Abstract
1. Introduction
2. Results
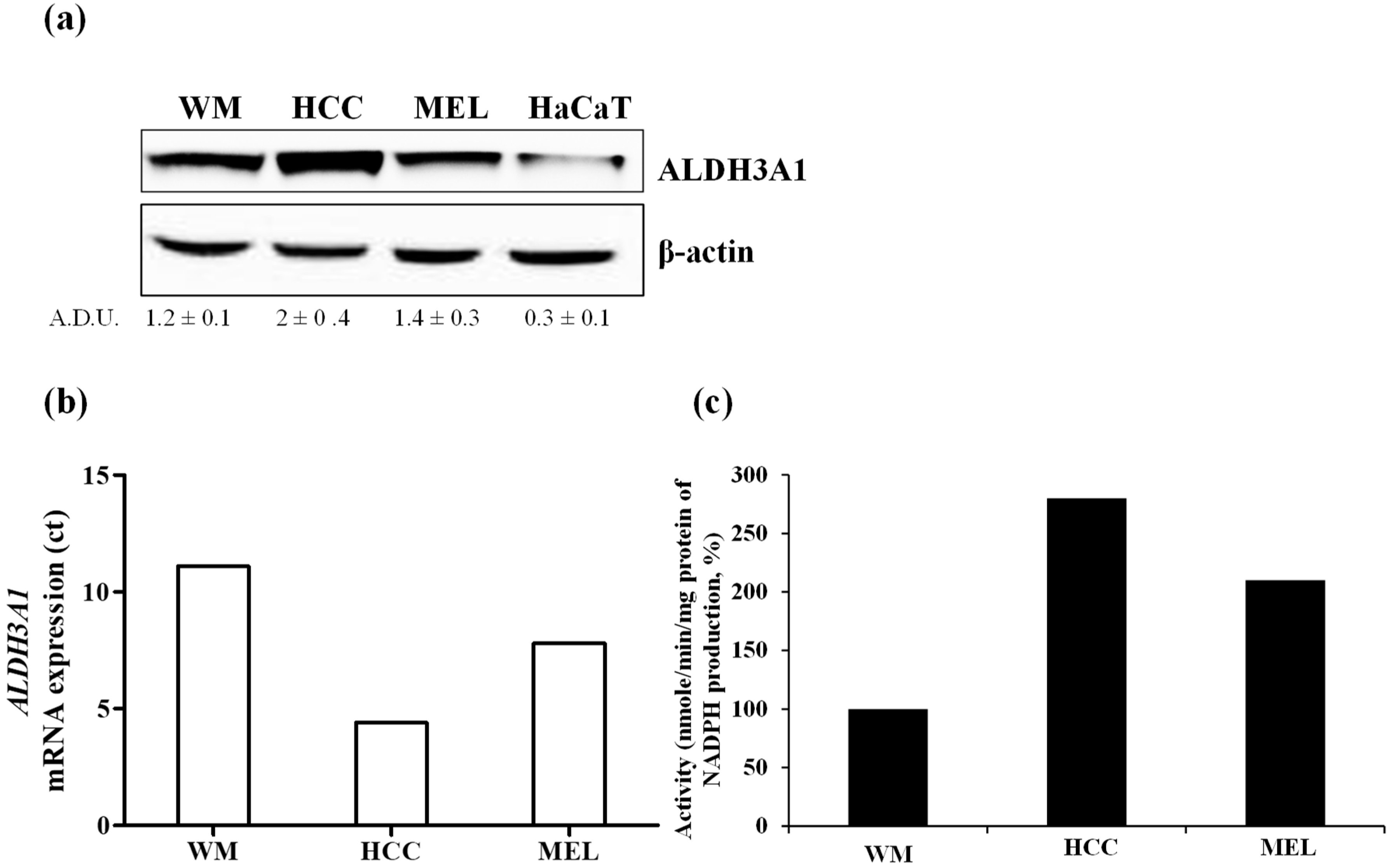
2.1. The Expression and Activity of ALDH3A1 in Melanoma and NSCLC Cells
2.2. ALDH3A1 Activity Elicits a Stem-Cell-Like Phenotype in WM266-4 and HCC4006 Cells
2.3. Epithelial Mesenchymal Transition (EMT) in Tumor Cells Is Associated with ALDH3A1 Expression
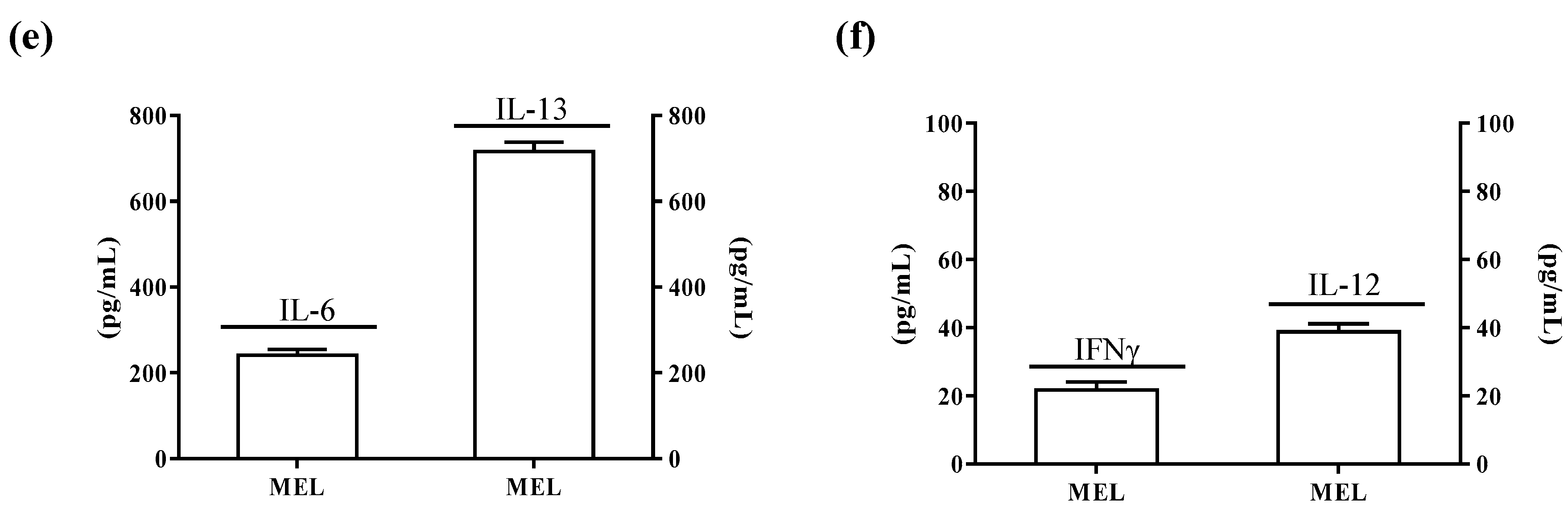
2.4. ALDH3A1 Affects Inflammatory Modulators of Immune Surveillance
2.5. ALDH3A1 Controls PD-L1 Expression in Tumor Cells
2.6. Tumor Intrinsic High ALDH3A1 Activity Reduces PBMC Proliferation
2.7. The Expression of ALDH3A1 Correlates with PD-L1 and COX-2 in Melanoma and NSCLC Tumor Samples
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Immunoblot Analysis
4.3. MTT Assay
4.4. Clonogenic Assay
4.5. Analysis of ALDH3A1-Catalyzed Dehydrogenase Activities from Cell Lysates
4.6. Prostaglandin E2 Express ELISA Assay
4.7. ELISA Immuno-Assay
4.8. Real-Time PCR
4.9. Immunofluorescence Analysis
4.10. Tumorsphere Formation In Vitro
4.11. Human Cytokine ELISA Plate Array
4.12. Conditioned Media from Tumor Cells
4.13. CFSE Cell Proliferation Assay
4.14. Tumor Sample Analysis
4.15. Invasion Assay
4.16. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Vasiliou, V.; Nerbert, D.W. Analysis and update of the human aldehyde dehydrogenase (ALDH) gene family. Hum. Genom. 2005, 2, 138–143. [Google Scholar]
- Vasiliou, V.; Thompson, D.C.; Smith, C.; Fujita, M.; Chen, Y. Aldehyde dehydrogenases: From eye crystallins to metabolic disease and cancer stem cells. Chem. Biol. Interact. 2013, 202, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Zavala, J.S.; Calleja, L.F.; Moreno-Sánchez, R.; Yoval-Sánchez, B. Role of Aldehyde Dehydrogenases in Physiopathological Processes. Chem. Res. Toxicol. 2019, 32, 405–420. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.P.; Sies, H. The Redox Code. Antioxid. Redox Signal. 2015, 23, 734–746. [Google Scholar] [CrossRef] [PubMed]
- Ma, I.; Allan, A.L. The role of human aldehyde dehydrogenase in normal and cancer stem cells. Stem Cell Rev. 2011, 7, 292–306. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Mou, Y.P.; Chen, K.; Cai, J.Q.; Zhou, Y.C.; Pan, Y.; Xu, X.W.; Zhou, W.; Gao, J.Q.; Chen, D.W.; et al. Aldehyde dehydrogenase 3A1 is robustly upregulated in gastric cancer stem-like cells and associated with tumorigenesis. Int J. Oncol. 2016, 49, 611–622. [Google Scholar] [CrossRef]
- Moreb, J.S.; Baker, H.V.; Chang, L.-J.; Amaya, M.; Lopez, M.C.; Ostmark, B.; Chou, W. ALDH isozymes downregulation affects cell growth, cell motility and gene expression in lung cancer cells. Mol. Cancer 2008, 7, 87. [Google Scholar] [CrossRef]
- Voulgaridou, G.P.; Kiziridou, M.; Mantso, T.; Chlichlia, K.; Galanis, A.; Koukourakis, M.I.; Franco, R.; Panayiotidis, M.I.; Pappa, A. Aldehyde dehydrogenase 3A1 promotes multi-modality resistance and alters gene expression profile in human breast adenocarcinoma MCF-7 cells. Int. J. Biochem. Cell Biol. 2016, 77, 120–128. [Google Scholar] [CrossRef]
- Boesch, J.S.; Lee, C.; Lindahl, R.G. Constitutive expression of class 3 aldehyde dehydrogenase in cultured rat corneal epithelium. J. Biol. Chem. 1996, 271, 5150–5157. [Google Scholar]
- Garaycoechea, J.I.; Crossan, G.P.; Langevin, F.; Mulderrig, L.; Louzada, S.; Yang, F.; Guilbaud, G.; Park, N.; Roerink, S.; Nik-Zainal, S.; et al. Alcohol and endogenous aldehydes damage chromosomes and mutate stem cells. Nature 2018, 553, 171–177. [Google Scholar] [CrossRef]
- Vassalli, G. Aldehyde Dehydrogenases: Not Just Markers, but Functional Regulators of Stem Cells. Stem Cells Int. 2019, 2019, 3904645. [Google Scholar] [CrossRef] [PubMed]
- Chute, J.P.; Muramoto, G.G.; Whitesides, J.; Colvin, M.; Safi, R.; Chao, N.J.; McDonnell, D.P. Inhibition of aldehyde dehydrogenase and retinoid signaling induces the expansion of human hematopoietic stem cells. Proc. Natl. Acad. Sci. USA 2006, 103, 11707–11712. [Google Scholar] [CrossRef] [PubMed]
- La Porta, C.A.M.; Zapperi, S. Complexity in cancer stem cells and tumor evolution: Toward precision medicine. Semin. Cancer Biol. 2017, 44, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Codony-Servat, J.; Rosell, R. Cancer stem cells and immunoresistance: Clinical implications and solutions. Transl. Lung Cancer Res. 2015, 4, 689–703. [Google Scholar] [PubMed]
- Bruttel, V.S.; Wischhusen, J. Cancer Stem Cell Immunology: Key to Understanding Tumorigenesis and Tumor Immune Escape? Front. Immunol. 2014, 5, 360. [Google Scholar] [CrossRef]
- Maccalli, C.; Volontè, A.; Cimminiello, C.; Parmiani, G. Immunology of cancer stem cells in solid tumours. A review. Eur. J. Cancer 2014, 50, 649–655. [Google Scholar] [CrossRef]
- Lourenço, A.R.; Coffer, P.J. SOX4: Joining the Master Regulators of Epithelial-to-Mesenchymal Transition? Trends Cancer 2017, 3, 571–582. [Google Scholar] [CrossRef]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef]
- Soundararajan, R.; Paranjape, A.N.; Maity, S.; Aparicio, A.; Mani, S.A. EMT, stemness and tumor plasticity in aggressive variant neuroendocrine prostate cancers. Biochim. Biophys. Acta 2018, 1870, 229–238. [Google Scholar] [CrossRef]
- Colotta, F.; Allavena, P.; Sica, A.; Garlanda, C.; Mantovani, A. Cancer-related inflammation, the seventh hallmark of cancer: Links to genetic instability. Carcinogenesis 2009, 30, 1073–1081. [Google Scholar] [CrossRef]
- Zelenay, S.; Van der Veen, A.G.; Böttcher, J.P.; Snelgrove, K.J.; Rogers, N.; Acton, S.E.; Chakravarty, P.; Girotti, M.R.; Marais, R.; Quezada, S.A.; et al. Cyclooxygenase-Dependent Tumor Growth through Evasion of Immunity. Cell 2015, 162, 1257–1270. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Roszik, J.; Cho, S.N.; Ogata, D.; Milton, D.R.; Peng, W.; Menter, D.G.; Ekmekcioglu, S.; Grimm, E.A. The COX2 Effector Microsomal PGE2 Synthase 1 is a Regulator of Immunosuppression in Cutaneous Melanoma. Clin. Cancer Res. 2019, 25, 1650–1663. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Fourcade, J.; Pagliano, O.; Chauvin, J.M.; Sander, C.; Kirkwood, J.M. IL10 and PD-1 cooperate to limit the activity of tumor-specific CD8+ T cells. Cancer Res. 2015, 75, 1635–1644. [Google Scholar] [CrossRef] [PubMed]
- Zou, W.; Chen, L. Inhibitory B7-family molecules in the tumor microenvironment. Nat. Rev. Immunol. 2008, 8, 467–477. [Google Scholar] [CrossRef]
- Chen, L.; Gibbons, D.L.; Goswami, S.; Cortez, M.A.; Ahn, Y.H.; Byers, L.A.; Zhang, X.; Yi, X.; Dwyer, D.; Lin, W.; et al. Metastasis is regulated via microRNA-200/ZEB1 axis control of tumor cell PD-L1 expression and intratumoral immunosuppression. Nat. Commun. 2014, 5, 5241. [Google Scholar] [CrossRef]
- Casey, S.C.; Tong, L.; Li, Y.; Do, R.; Walz, S.; Fitzgerald, K.N.; Gouw, A.M.; Baylot, V.; Gutgemann, I.; Eilers, M.; et al. MYC Regulates the Anti-Tumor Immune Response through CD47 and PD-L1. Science 2016, 352, 227–231. [Google Scholar] [CrossRef]
- Basu-Roy, U.; Seo, E.; Ramanathapuram, L.; Rapp, T.B.; Perry, J.A.; Orkin, S.H.; Mansukhani, A.; Basilico, C. Sox2 maintains self-renewal of tumor-initiating cells in osteosarcomas. Oncogene 2011, 31, 2270–2282. [Google Scholar] [CrossRef]
- Luo, Y.; Dallaglio, K.; Chen, Y.; Robinson, W.A.; Robinson, S.E.; McCarter, M.D.; Wang, J.; Gonzalez, R.; Thompson, D.C.; Norris, D.A.; et al. ALDH1A isozymes are markers of human melanoma stem cells and potential therapeutic targets. Stem Cells 2012, 30, 2100–2113. [Google Scholar] [CrossRef]
- Jiang, F.; Qiu, Q.; Khanna, A.; Todd, N.W.; Deepak, J.; Xing, L.; Wang, H.; Liu, Z.; Su, Y.; Stass, S.A.; et al. Aldehyde dehydrogenase 1 is a tumor stem cell-associated marker in lung cancer. Mol. Cancer Res. 2009, 7, 330–338. [Google Scholar] [CrossRef]
- Singh, S.; Brocker, C.; Koppaka, V.; Ying, C.; Jackson, B.; Matsumoto, A.; Thompson, D.C.; Vasiliou, V. Aldehyde Dehydrogenases in Cellular Responses to Oxidative/electrophilic Stress. Free Radic. Biol. Med. 2013, 56, 89–101. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer-immunesetpoint. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Terzuoli, E.; Finetti, F.; Costanza, F.; Giachetti, A.; Ziche, M.; Donnini, S. Linking of mPGES-1 and iNOS activates stem-like phenotype in EGFR-driven epithelial tumor cells. Nitric Oxide 2017, 66, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Finetti, F.; Terzuoli, E.; Giachetti, A.; Santi, R.; Villari, D.; Hanaka, H.; Radmark, O.; Ziche, M.; Donnini, S. mPGES-1 in prostate cancer controls stemness and amplifies epidermal growth factor receptor-driven oncogenicity. Endocr. Relat. Cancer 2015, 22, 665–678. [Google Scholar] [CrossRef] [PubMed]
- Terzuoli, E.; Donnini, S.; Finetti, F.; Nesi, G.; Villari, D.; Hanaka, H.; Radmark, O.; Giachetti, A.; Ziche, M. Linking microsomal prostaglandin E Synthase-1/PGE-2 pathway with miR-15a and -186 expression: Novel mechanism of VEGF modulation in prostate cancer. Oncotarget 2016, 7, 44350–44364. [Google Scholar] [CrossRef] [PubMed]
- Kurtova, A.V.; Xiao, J.; Mo, Q.; Pazhanisamy, S.; Krasnow, R.; Lerner, S.P.; Chen, F.; Roh, T.T.; Lay, E.; Ho, P.L. Blocking PGE2-induced tumour repopulation abrogates bladder cancer chemoresistance. Nature 2015, 517, 209–213. [Google Scholar] [CrossRef]
- Hsu, J.M.; Xia, W.; Hsu, Y.H.; Chan, L.C.; Yu, W.H.; Cha, J.H.; Chen, C.-T.; Liao, H.-W.; Kuo, C.W.; Khoo, K.H.; et al. STT3-dependent PD-L1 accumulation on cancer stem cells promotes immune evasion. Nat. Commun. 2018, 9, 1908. [Google Scholar] [CrossRef]
- Shao, C.; Sullivan, J.P.; Girard, L.; Augustyn, A.; Yenerall, P.; Rodriguez-Canales, J.; Liu, H.; Behrens, C.; Shay, J.W.; Wistuba, I.I.; et al. Essential role of aldehyde dehydrogenase 1A3 for the maintenance of non-small cell lung cancer stem cells is associated with the STAT3 pathway. Clin. Cancer Res. 2014, 20, 4154–4166. [Google Scholar] [CrossRef]
- Grimm, E.A. Immunology Comes Full Circle in Melanoma While Specific Immunity Is Unleashed to Eliminate Metastatic Disease, Inflammatory Products of Innate Immunity Promote Resistance. Crit. Rev. Oncog. 2016, 21, 57–63. [Google Scholar] [CrossRef]
- Berry, S.; Taube, J.M. Innate vs. Adaptive: PD-L1-mediated immune resistance by melanoma. Oncoimmunology 2015, 4, e1029704. [Google Scholar] [CrossRef]
- Donnini, S.; Finetti, F.; Terzuoli, E.; Giachetti, A.; Iñiguez, M.A.; Hanaka, H.; Fresno, M.; Rådmark, O.; Ziche, M. EGFR signaling upregulates expression of microsomal prostaglandin E synthase-1 in cancer cells leading to enhanced tumorigenicity. Oncogene 2012, 31, 3457–3466. [Google Scholar] [CrossRef]
- Kim, J.; Shin, J.H.; Chen, C.H.; Cruz, L.; Farnebo, L.; Yang, J.; Borges, P.; Kang, G.; Mochly-Rosen, D.; Sunwoo, J.B.; et al. Targeting aldehyde dehydrogenase activity in head and neck squamous cell carcinoma with a novel small molecule inhibitor. Oncotarget 2017, 8, 52345–52356. [Google Scholar] [CrossRef] [PubMed]
- Ciccone, V.; Terzuoli, E.; Donnini, S.; Giachetti, A.; Morbidelli, L.; Ziche, M. Stemness marker ALDH1A1 promotes tumor angiogenesis via retinoic acid/HIF-1α/VEGF signalling in MCF-7 breast cancer cells. J. Exp. Clin. Cancer Res. 2018, 37, 311. [Google Scholar] [CrossRef] [PubMed]
- Ciccone, V.; Monti, M.; Monzani, E.; Casella, L.; Morbidelli, L. The metal-nonoate Ni(SalPipNONO) inhibits in vitro tumor growth, invasiveness and angiogenesis. Oncotarget 2018, 9, 13353–13365. [Google Scholar] [CrossRef] [PubMed]
- Najjar, Y.G.; Menk, A.V.; Sander, C.; Rao, U.; Karunamurthy, A.; Bhatia, R.; Zhai, S.; Kirkwood, J.M.; Delgoffe, G.M. Tumor cell oxidative metabolism as a barrier to PD-1 blockade immunotherapy in melanoma. JCI Insight 2019, 4, e124989. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Liang, Y.; Anders, R.A.; Taube, J.M.; Qiu, X.; Mulgaonkar, A.; Liu, X.; Harrington, S.M.; Guo, J.; Xin, Y.; et al. PD-L1 on host cells is essential for PD-L1 blockade–mediated tumor regression. J. Clin. Invest. 2018, 128, 580–588. [Google Scholar] [CrossRef]
- Lin, H.; Wei, S.; Hurt, E.M.; Green, M.D.; Zhao, L.; Vatan, L.; Szeliga, W.; Herbst, R.; Harms, P.W.; Fecher, L.A.; et al. Host expression of PD-L1 determines efficacy of PD-L1 pathway blockade-mediated tumor regression. J. Clin. Invest. 2018, 128, 805–815. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cells | MFI | CFSE % LOW | CFSE % HIGH |
|---|---|---|---|
| PBMC | 929 | - | 100 |
| PBMC + L-2 + WM266-4 Ctr | 546 | 11.8 | 88.2 |
| PBMC + IL-2 + WM266-4 ALDH3A1high | 653 | 17.6 | 82.4 |
| PBMC + IL-2 + WM266-4 ALDH3A1low | 402 | 32.2 | 67.7 |
| PBMC + WM266-4 Ctr | 627 | 9.85 | 90.15 |
| PBMC + WM266-4 ALDH3A1high | 745 | 9.04 | 90.96 |
| PBMC + WM266-4 ALDH3A1low | 125 | 24.8 | 75.2 |
| PBMC + IL-2 + HCC4006 Ctr | 769 | 16.6 | 83.4 |
| PBMC + IL-2 + HCC4006 ALDH3A1low | 306 | 54.4 | 45.6 |
| PBMC + HCC4006 Ctr | 838 | 9.47 | 90.53 |
| PBMC + HCC4006 ALDH3A1low | 442 | 48.7 | 51.3 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Terzuoli, E.; Bellan, C.; Aversa, S.; Ciccone, V.; Morbidelli, L.; Giachetti, A.; Donnini, S.; Ziche, M. ALDH3A1 Overexpression in Melanoma and Lung Tumors Drives Cancer Stem Cell Expansion, Impairing Immune Surveillance through Enhanced PD-L1 Output. Cancers 2019, 11, 1963. https://doi.org/10.3390/cancers11121963
Terzuoli E, Bellan C, Aversa S, Ciccone V, Morbidelli L, Giachetti A, Donnini S, Ziche M. ALDH3A1 Overexpression in Melanoma and Lung Tumors Drives Cancer Stem Cell Expansion, Impairing Immune Surveillance through Enhanced PD-L1 Output. Cancers. 2019; 11(12):1963. https://doi.org/10.3390/cancers11121963
Chicago/Turabian StyleTerzuoli, Erika, Cristiana Bellan, Sara Aversa, Valerio Ciccone, Lucia Morbidelli, Antonio Giachetti, Sandra Donnini, and Marina Ziche. 2019. "ALDH3A1 Overexpression in Melanoma and Lung Tumors Drives Cancer Stem Cell Expansion, Impairing Immune Surveillance through Enhanced PD-L1 Output" Cancers 11, no. 12: 1963. https://doi.org/10.3390/cancers11121963
APA StyleTerzuoli, E., Bellan, C., Aversa, S., Ciccone, V., Morbidelli, L., Giachetti, A., Donnini, S., & Ziche, M. (2019). ALDH3A1 Overexpression in Melanoma and Lung Tumors Drives Cancer Stem Cell Expansion, Impairing Immune Surveillance through Enhanced PD-L1 Output. Cancers, 11(12), 1963. https://doi.org/10.3390/cancers11121963