
Punicalagin in Cancer Prevention—Via Signaling Pathways Targeting


Abstract

1. Pomegranate against Pathologies Development
2. Anti-Cancer Activities of Punicalagin (Pug)
2.1. Punicalagin against Cervical Cancer
2.2. Punicalagin against Ovarian Cancer
2.3. Punicalagin against Breast Cancer
2.4. Punicalagin against Colorectal Cancer
2.5. Punicalagin against Thyroid Cancer
2.6. Punicalagin against Lung Cancer
2.7. Punicalagin against Osteosarcoma
2.8. Punicalagin against Glioma
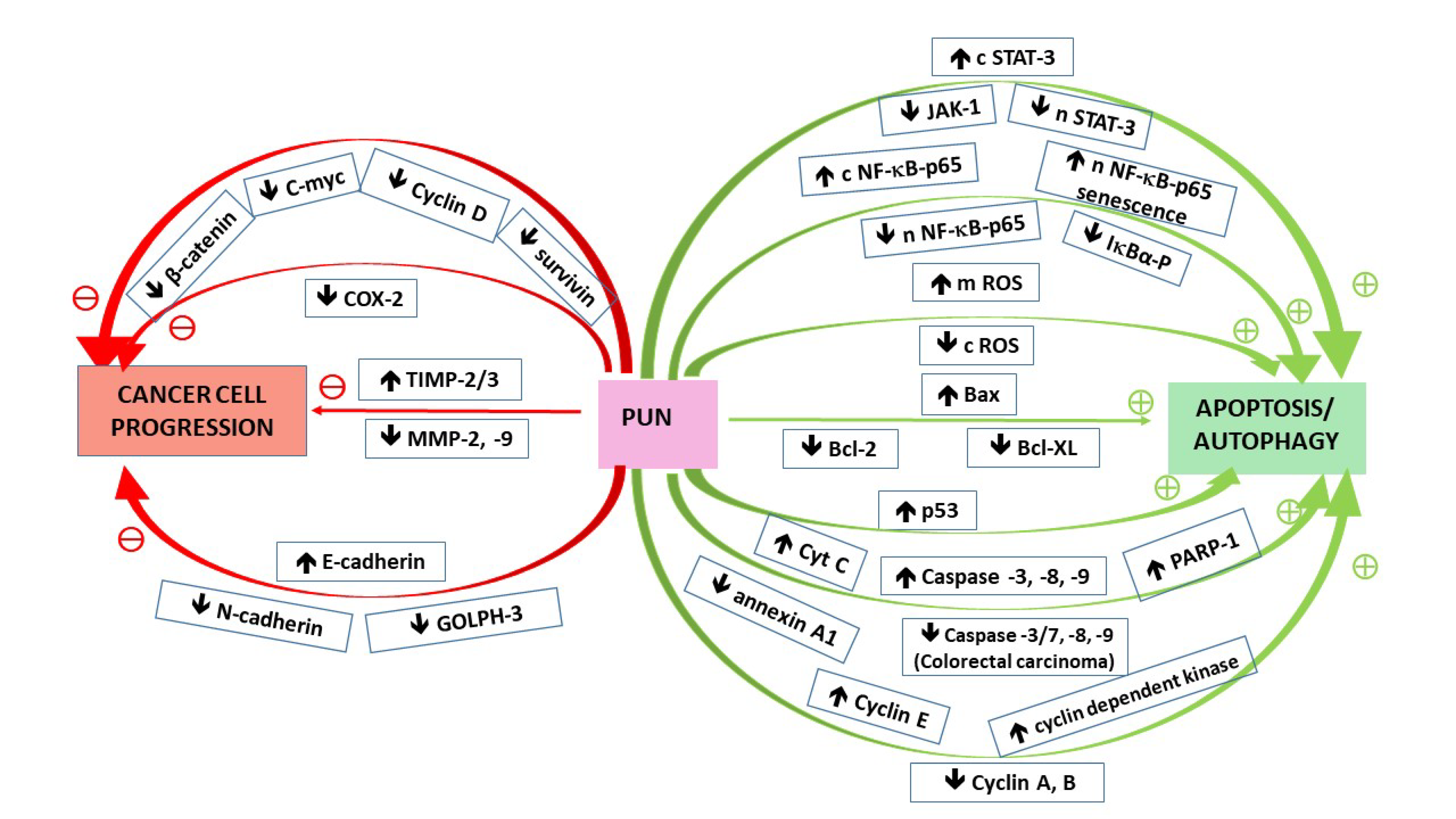
3. Punicalagin-Attenuated Signaling Pathways with Reference to Hallmarks of Cancer
3.1. Hallmarks of Cancer
3.1.1. Resisting Cell Death/Evading Growth Suppressors
3.1.2. Enabling Replicative Immortality/Dedifferentiation
3.1.3. Activating Invasion and Metastasis/Transdifferentiation
4. Punicalagin Application in Anti-Cancer Therapy?
4.1. Which Substance Actually Works?
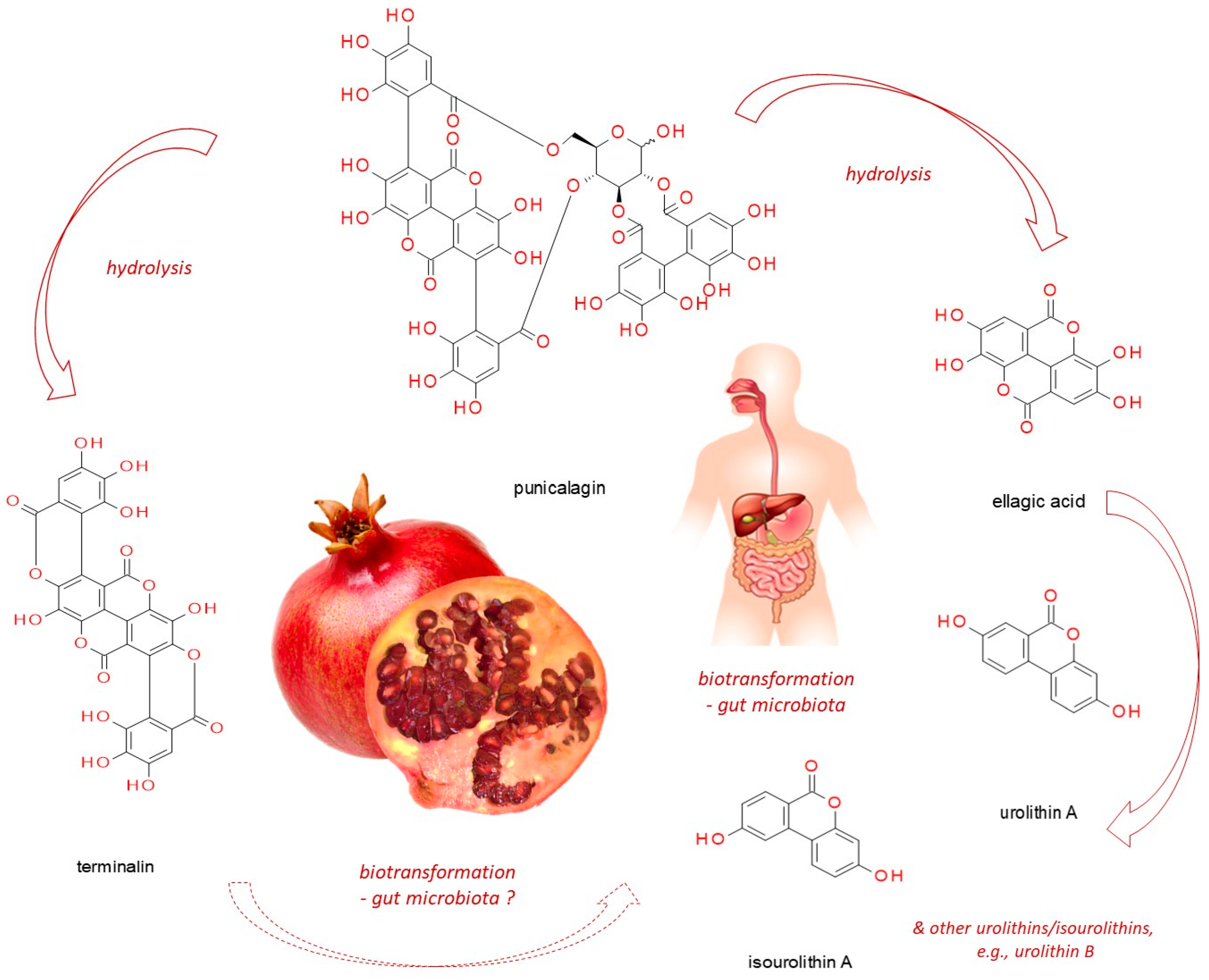
4.2. What Happens after Punicalagin Ingestion?
4.2.1. Ellagitannins Metabolism by Microbiota
4.2.2. Urolithins Phase II Biotransformation
4.2.3. Punicalagin Biotransformation
5. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Pham, N.M.; Do, V.V.; Lee, A.H. Polyphenol-rich foods and risk of gestational diabetes: A systematic review and meta-analysis. Eur. J. Clin. Nutr. 2019, 73, 647–656. [Google Scholar] [CrossRef] [PubMed]
- Palma-Duran, S.A.; Vlassopoulos, A.; Lean, M.; Govan, L.; Combet, E. Nutritional intervention and impact of polyphenol on glycohemoglobin (HbA1c) in non-diabetic and type 2 diabetic subjects: Systematic review and meta-analysis. Crit. Rev. Food Sci. Nutr. 2017, 57, 975–986. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Tresserra-Rimbau, A.; Estruch, R.; Martínez-González, M.A.; Medina-Remón, A.; Fitó, M.; Corella, D.; Salas-Salvadó, J.; Portillo, M.P.; Moreno, J.J.; et al. Polyphenol Levels Are Inversely Correlated with Body Weight and Obesity in an Elderly Population after 5 Years of Follow Up (The Randomised PREDIMED Study). Nutrients 2017, 9, 452. [Google Scholar] [CrossRef]
- Yamagata, K. Polyphenols Regulate Endothelial Functions and Reduce the Risk of Cardiovascular Disease. Curr. Pharm. Des. 2019, 25, 2443–2458. [Google Scholar] [CrossRef] [PubMed]
- Gu, H.F.; Mao, X.Y.; Du, M. Prevention of breast cancer by dietary polyphenols-role of cancer stem cells. Crit. Rev. Food Sci. Nutr. 2020, 60, 810–825. [Google Scholar] [CrossRef]
- Miyata, Y.; Shida, Y.; Hakariya, T.; Sakai, H. Anti-Cancer Effects of Green Tea Polyphenols against Prostate Cancer. Molecules 2019, 24, 193. [Google Scholar] [CrossRef] [PubMed]
- Ge, S.; Duo, L.; Wang, J.; Yang, J.; Li, Z.; Tu, Y. A unique understanding of traditional medicine of pomegranate, Punica granatum L. and its current research status. J. Ethnopharmacol. 2021, 271, 113877. [Google Scholar] [CrossRef]
- Pirzadeh, M.; Caporaso, N.; Rauf, A.; Shariati, M.A.; Yessimbekov, Z.; Khan, M.U.; Imran, M.; Mubarak, M.S. Pomegranate as a source of bioactive constituents: A review on their characterization, properties and applications. Crit. Rev. Food Sci. Nutr. 2021, 61, 982–999. [Google Scholar] [CrossRef]
- Satomi, H.; Umemura, K.; Ueno, A.; Hatano, T.; Okuda, T.; Noro, T. Carbonic anhydrase inhibitors from the pericarps of Punica granatum L. Biol. Pharm. Bull. 1993, 16, 787–790. [Google Scholar] [CrossRef]
- Viuda-Martos, M.; Fernández-Lóaez, J.; Pérez-álvarez, J.A. Pomegranate and its Many Functional Components as Related to Human Health: A Review. Compr. Rev. Food Sci. Food Saf. 2010, 9, 635–654. [Google Scholar] [CrossRef]
- Sharma, P.; McClees, S.F.; Afaq, F. Pomegranate for prevention and treatment of cancer: An update. Molecules 2017, 22, 177. [Google Scholar] [CrossRef]
- Danesi, F.; Ferguson, L.R. Could pomegranate juice help in the control of inflammatory diseases? Nutrients 2017, 9, 958. [Google Scholar] [CrossRef]
- Khwairakpam, A.D.; Bordoloi, D.; Thakur, K.K.; Monisha, J.; Arfuso, F.; Sethi, G.; Mishra, S.; Kumar, A.P.; Kunnumakkara, A.B. Possible use of Punica granatum (Pomegranate) in cancer therapy. Pharmacol. Res. 2018, 133, 53–64. [Google Scholar] [CrossRef]
- Yoshida, T.; Amakura, Y.; Yoshimura, M. Structural features and biological properties of ellagitannins in some plant families of the order Myrtales. Int. J. Mol. Sci. 2010, 11, 79–106. [Google Scholar] [CrossRef]
- Fecka, I.; Włodarczyk, M.; Starzec, A. Isolation and structure elucidation of cistusin: A new ellagitannin from Cistus × incanus L. leaves. Ind. Crops Prod. 2020. [Google Scholar] [CrossRef]
- Oelrichs, P.B.; Pearce, C.M.; Zhu, J.; Filippich, L.J. Isolation and structure determination of terminalin A toxic condensed tannin from Terminalia oblongata. Nat. Toxins 1994, 2, 144–150. [Google Scholar] [CrossRef]
- Zahin, M.; Ahmad, I.; Gupta, R.C.; Aqil, F. Punicalagin and ellagic acid demonstrate anti-mutagenic activity and inhibition of benzo[a]pyrene induced DNA adducts. Biomed. Res. Int. 2014, 2014, 467465. [Google Scholar] [CrossRef] [PubMed]
- Seeram, N.P.; Adams, L.S.; Henning, S.M.; Niu, Y.; Zhang, Y.; Nair, M.G.; Heber, D. In vitro antiproliferative, apoptotic and antioxidant activities of punicalagin, ellagic acid and a total pomegranate tannin extract are enhanced in combination with other polyphenols as found in pomegranate juice. J. Nutr. Biochem. 2005, 16, 360–367. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Chinnathambi, A.; Alharbi, S.A.; Veeraraghavan, V.P.; Mohan, S.K.; Zhang, G. Punicalagin promotes the apoptosis in human cervical cancer (ME-180) cells through mitochondrial pathway and by inhibiting the NF-kB signaling pathway. Saudi J. Biol. Sci. 2020, 27, 1100–1106. [Google Scholar] [CrossRef]
- Tang, J.; Li, B.; Hong, S.; Liu, C.; Min, J.; Hu, M.; Li, Y.; Liu, Y.; Hong, L. Punicalagin suppresses the proliferation and invasion of cervical cancer cells through inhibition of the β-catenin pathway. Mol. Med. Rep. 2017, 16, 1439–1444. [Google Scholar] [CrossRef] [PubMed]
- Tilborghs, S.; Corthouts, J.; Verhoeven, Y.; Arias, D.; Rolfo, C.; Trinh, X.B.; van Dam, P.A. The role of Nuclear Factor-kappa B signaling in human cervical cancer. Crit. Rev. Oncol. Hematol. 2017, 120, 141–150. [Google Scholar] [CrossRef]
- Lalle, G.; Twardowski, J.; Grinberg-Bleyer, Y. NF-κB in Cancer Immunity: Friend or Foe? Cells 2021, 10, 355. [Google Scholar] [CrossRef] [PubMed]
- Puar, Y.R.; Shanmugam, M.K.; Fan, L.; Arfuso, F.; Sethi, G.; Tergaonkar, V. Evidence for the Involvement of the Master Transcription Factor NF-κB in Cancer Initiation and Progression. Biomedicines 2018, 6, 82. [Google Scholar] [CrossRef] [PubMed]
- Tegowski, M.; Baldwin, A. Noncanonical NF-κB in Cancer. Biomedicines 2018, 6, 66. [Google Scholar] [CrossRef] [PubMed]
- Khan, H.; Ullah, H.; Castilho, P.C.M.F.; Gomila, A.S.; D’Onofrio, G.; Filosa, R.; Wang, F.; Nabavi, S.M.; Daglia, M.; Silva, A.S.; et al. Targeting NF-κB signaling pathway in cancer by dietary polyphenols. Crit. Rev. Food Sci. Nutr. 2020, 60, 2790–2800. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.; Duan, Y.; Ma, F.; Lou, L. Punicalagin inhibits the viability, migration, invasion, and EMT by regulating GOLPH3 in breast cancer cells. J. Recept. Signal. Transduct. 2020, 40, 173–180. [Google Scholar] [CrossRef]
- Tang, J.M.; Min, J.; Li, B.S.; Hong, S.S.; Liu, C.; Hu, M.; Li, Y.; Yang, J.; Hong, L. Therapeutic Effects of Punicalagin Against Ovarian Carcinoma Cells in Association with β-Catenin Signaling Inhibition. Int. J. Gynecol. Cancer 2016, 26, 1557–1563. [Google Scholar] [CrossRef]
- Fang, L.; Wang, H.; Zhang, J.; Fang, X. Punicalagin induces ROS-mediated apoptotic cell death through inhibiting STAT3 translocation in lung cancer A549 cells. J. Biochem. Mol. Toxicol. 2021, 35, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Berköz, M.; Krośniak, M. Punicalagin induces apoptosis in a549 cell line through mitochondria-mediated pathway. Gen. Physiol. Biophys. 2020, 39, 557–567. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.; Zhang, X.; Wang, H. Punicalagin inhibited proliferation, invasion and angiogenesis of osteosarcoma through suppression of NF-κB signaling. Mol. Med. Rep. 2020, 22, 2386–2394. [Google Scholar] [CrossRef]
- Ganesan, T.; Sinniah, A.; Chik, Z.; Alshawsh, M.A. Punicalagin regulates apoptosis-autophagy switch via modulation of annexin a1 in colorectal cancer. Nutrients 2020, 12, 2430. [Google Scholar] [CrossRef]
- Adams, L.S.; Seeram, N.P.; Aggarwal, B.B.; Takada, Y.; Sand, D.; Heber, D. Pomegranate juice, total pomegranate ellagitannins, and punicalagin suppress inflammatory cell signaling in colon cancer cells. J. Agric. Food Chem. 2006, 54, 980–985. [Google Scholar] [CrossRef] [PubMed]
- Larrosa, M.; Tomás-Barberán, F.A.; Espín, J.C. The dietary hydrolysable tannin punicalagin releases ellagic acid that induces apoptosis in human colon adenocarcinoma Caco-2 cells by using the mitochondrial pathway. J. Nutr. Biochem. 2006, 17, 611–625. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Gao, Y.; Yao, X.; Yu, H.; Bao, J.; Guan, H.; Sun, Y.; Zhang, L. Punicalagin induces apoptosis-independent autophagic cell death in human papillary thyroid carcinoma BCPAP cells. RSC Adv. 2016, 6, 68485–68493. [Google Scholar] [CrossRef]
- Yao, X.; Cheng, X.; Zhang, L.; Yu, H.; Bao, J.; Guan, H.; Lu, R. Punicalagin from pomegranate promotes human papillary thyroid carcinoma BCPAP cell death by triggering ATM-mediated DNA damage response. Nutr. Res. 2017, 47, 63–71. [Google Scholar] [CrossRef]
- Cheng, X.; Yao, X.; Xu, S.; Pan, J.; Yu, H.; Bao, J.; Guan, H.; Lu, R.; Zhang, L. Punicalagin induces senescent growth arrest in human papillary thyroid carcinoma BCPAP cells via NF-κB signaling pathway. Biomed. Pharmacother. 2018, 103, 490–498. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.G.; Huang, M.H.; Li, J.H.; Lai, F.I.; Lee, H.M.; Hsu, Y.N. Punicalagin induces apoptotic and autophagic cell death in human U87MG glioma cells. Acta Pharmacol. Sin. 2013, 34, 1411–1419. [Google Scholar] [CrossRef]
- Sechi, S.; Frappaolo, A.; Karimpour-Ghahnavieh, A.; Piergentili, R.; Giansanti, M.G. Onco-genic roles of GOLPH3 in the physiopathology of cancer. Int. J. Mol. Sci. 2020, 21, 933. [Google Scholar] [CrossRef]
- Zeng, Z.; Lin, H.; Zhao, X.; Liu, G.; Wang, X.; Xu, R.; Chen, K.; Li, J.; Song, L. Overexpression of GOLPH3 promotes proliferation and tumorigenicity in breast cancer via suppression of the FOXO1 transcription factor. Clin. Cancer Res. 2012, 18, 4059–4069. [Google Scholar] [CrossRef]
- Tokuda, E.; Itoh, T.; Hasegawa, J.; Ijuin, T.; Takeuchi, Y.; Irino, Y.; Fukumoto, M.; Takenawa, T. Phosphatidylinositol 4-phosphate in the Golgi apparatus regulates cell-cell adhesion and invasive cell migration in human breast cancer. Cancer Res. 2014, 74, 3054–3066. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Pan, H.; Wei, W.; Yang, H.; Liu, J.; Yang, R. GOLPH3: A novel biomarker that correlates with poor survival and resistance to chemotherapy in breast cancer. Oncotarget 2017, 8, 105155–105169. [Google Scholar] [CrossRef]
- Fu, Z.; Zhang, S.; Wang, B.; Huang, W.; Zheng, L.; Cheng, A. Annexin A1: A double-edged sword as novel cancer biomarker. Clin. Chim. Acta 2020, 504, 36–42. [Google Scholar] [CrossRef]
- Patton, K.T.; Chen, H.M.; Joseph, L.; Yang, X.J. Decreased annexin I expression in prostatic adenocarcinoma and in high-grade prostatic intraepithelial neoplasia. Histopathology 2005, 47, 597–601. [Google Scholar] [CrossRef]
- Moghanibashi, M.; Jazii, F.R.; Soheili, Z.S.; Zare, M.; Karkhane, A.; Parivar, K.; Mohamadynejad, P. Proteomics of a new esophageal cancer cell line established from Persian patient. Gene 2012, 500, 124–133. [Google Scholar] [CrossRef]
- Suo, A.; Zhang, M.; Yao, Y.; Zhang, L.; Huang, C.; Nan, K.; Zhang, W. Proteome analysis of the effects of sorafenib on human hepatocellular carcinoma cell line HepG2. Med. Oncol. 2012, 29, 1827–1836. [Google Scholar] [CrossRef] [PubMed]
- Roth, U.; Razawi, H.; Hommer, J.; Engelmann, K.; Schwientek, T.; Uller, S.M.; Baldus, S.E.; Patsos, G.; Corfield, A.P.; Paraskeva, C.; et al. Differential expression proteomics of human colorectal cancer based on a syngeneic cellular model for the progression of adenoma to carcinoma. Proteomics 2010, 10, 194–202. [Google Scholar] [CrossRef]
- Lecona, E.; Barrasa, J.I.; Olmo, N.; Llorente, B.; Turnay, J.; Lizarbe, M.A. Upregulation of Annexin A1 Expression by Butyrate in Human Colon Adenocarcinoma Cells: Role of p53, NF-Y, and p38 Mitogen-Activated Protein Kinase. Mol. Cell. Biol. 2008, 28, 4665–4674. [Google Scholar] [CrossRef][Green Version]
- Nielsen, M.; Kæstel, C.G.; Eriksen, K.W.; Woetmann, A.; Stokkedal, T.; Kaltoft, K.; Geisler, C.; Röpke, C.; ØDum, N. Inhibition of constitutively activated Stat3 correlates with altered Bcl-2/Bax expression and induction of apoptosis in mycosis fungoides tumor cells. Leukemia 1999, 13, 735–738. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Senga, S.S.; Grose, R.P. Hallmarks of cancer-the new testament. Open Biol. 2021, 11, 200358. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Buzun, K.; Gornowicz, A.; Lesyk, R.; Bielawski, K.; Bielawska, A. Autophagy Modulators in Cancer Therapy. Int. J. Mol. Sci. 2021, 22, 5804. [Google Scholar] [CrossRef]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef]
- Chen, C.; Gao, H.; Su, X. Autophagy-related signaling pathways are involved in cancer (Review). Exp. Ther. Med. 2021, 22, 710. [Google Scholar] [CrossRef]
- Zada, S.; Hwang, J.S.; Ahmed, M.; Lai, T.H.; Pham, T.M.; Elashkar, O.; Kim, D.R. Cross talk between autophagy and oncogenic signaling pathways and implications for cancer therapy. Biochim. Biophys. Acta Rev. Cancer 2021, 1876, 188565. [Google Scholar] [CrossRef]
- Qu, X.; Yu, J.; Bhagat, G.; Furuya, N.; Hibshoosh, H.; Troxel, A.; Rosen, J.; Eskelinen, E.L.; Mizushima, N.; Ohsumi, Y.; et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J. Clin. Invest. 2003, 112, 1809–1820. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.H.; Jackson, S.; Seaman, M.; Brown, K.; Kempkes, B.; Hibshoosh, H.; Levine, B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999, 402, 672–676. [Google Scholar] [CrossRef]
- Shen, Y.; Li, D.D.; Wang, L.L.; Deng, R.; Zhu, X.F. Decreased expression of autophagy-related proteins in malignant epithelial ovarian cancer. Autophagy 2008, 4, 1067–1068. [Google Scholar] [CrossRef]
- Shen, M.X.; Ding, J.B. Expression levels and roles of EMC-6, Beclin1, and Rab5a in the cervical cancer. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 3038–3046. [Google Scholar] [PubMed]
- Yu, S.; Cheng, C.; Wang, J.; Wang, J.; Qu, Z.; Ren, H.; Li, Y.; Ning, Q.; Chen, M.; Hu, T. Loss of Beclin1 Expression and Nrf2 Overexpression are Associated with Poor Survival of Patients with Non-Small Cell Lung Cancer. Anticancer Agents Med. Chem. 2018, 18, 1680–1687. [Google Scholar] [CrossRef] [PubMed]
- Qiu, D.M.; Wang, G.L.; Chen, L.; Xu, Y.Y.; He, S.; Cao, X.L.; Qin, J.; Zhou, J.M.; Zhang, Y.X.; E, Q. The expression of beclin-1, an autophagic gene, in hepatocellular carcinoma associated with clinical pathological and prognostic significance. BMC Cancer 2014, 14, 327. [Google Scholar] [CrossRef]
- Ding, Z.B.; Shi, Y.H.; Zhou, J.; Qiu, S.J.; Xu, Y.; Dai, Z.; Shi, G.M.; Wang, X.Y.; Ke, A.W.; Wu, B.; et al. Association of autophagy defect with a malignant phenotype and poor prognosis of hepatocellular carcinoma. Cancer Res. 2008, 68, 9167–9175. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Shao, Z.; Xiong, L.; Che, B.; Deng, C.; Xu, W. Expression of Beclin1 in osteosarcoma and the effects of down-regulation of autophagy on the chemotherapeutic sensitivity. J. Huazhong Univ. Sci. Technol. Med. Sci. 2009, 29, 737–740. [Google Scholar] [CrossRef]
- Huang, X.; Bai, H.M.; Chen, L.; Li, B.; Lu, Y.C. Reduced expression of LC3B-II and Beclin 1 in glioblastoma multiforme indicates a down-regulated autophagic capacity that relates to the progression of astrocytic tumors. J. Clin. Neurosci. 2010, 17, 1515–1519. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Sun, C.; Tian, D.; Li, Y.; Gao, X.; He, S.; Li, T. Expression and clinical significances of Beclin1, LC3 and mTOR in colorectal cancer. Int. J. Clin. Exp. Pathol. 2015, 8, 3882–3891. [Google Scholar] [PubMed]
- Cai, M.; Hu, Z.; Liu, J.; Gao, J.; Liu, C.; Liu, D.; Tan, M.; Zhang, D.; Lin, B. Beclin 1 expression in ovarian tissues and its effects on ovarian cancer prognosis. Int. J. Mol. Sci. 2014, 15, 5292–5303. [Google Scholar] [CrossRef]
- Yun, C.W.; Jeon, J.; Go, G.; Lee, J.H.; Lee, S.H. The Dual Role of Autophagy in Cancer Development and a Therapeutic Strategy for Cancer by Targeting Autophagy. Int. J. Mol. Sci. 2020, 22, 179. [Google Scholar] [CrossRef]
- Rahman, M.A.; Hannan, M.A.; Dash, R.; Rahman, M.H.; Islam, R.; Uddin, M.J.; Sohag, A.A.M.; Rahman, M.H.; Rhim, H. Phytochemicals as a Complement to Cancer Chemotherapy: Pharmacological Modulation of the Autophagy-Apoptosis Pathway. Front. Pharmacol. 2021, 12, 639628. [Google Scholar] [CrossRef]
- Li, X.; He, S.; Ma, B. Autophagy and autophagy-related proteins in cancer. Mol. Cancer 2020, 19, 12. [Google Scholar] [CrossRef]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 1997, 88, 593–602. [Google Scholar] [CrossRef]
- Bian, Y.; Wei, J.; Zhao, C.; Li, G. Natural Polyphenols Targeting Senescence: A Novel Prevention and Therapy Strategy for Cancer. Int. J. Mol. Sci. 2020, 21, 684. [Google Scholar] [CrossRef]
- Kim, Y.Y.; Um, J.H.; Shin, D.J.; Jeong, D.J.; Hong, Y.B.; Yun, J. p53-mediated regulation of mitochondrial dynamics plays a pivotal role in the senescence of various normal cells as well as cancer cells. FASEB J. 2021, 35, e21319. [Google Scholar] [CrossRef] [PubMed]
- McMellen, A.; Woodruff, E.R.; Corr, B.R.; Bitler, B.G.; Moroney, M.R. Wnt signaling in gynecologic malignancies. Int. J. Mol. Sci. 2020, 21, 4272. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Wang, M.; Li, X.; Xie, Y.; Xia, X.; Tian, J.; Zhang, K.; Tang, A. Wnt signaling in cervical cancer? J. Cancer 2018, 9, 1277–1286. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, I.; Thavathiru, E.; Ramalingam, S.; Natarajan, G.; Mills, W.K.; Benbrook, D.M.; Zuna, R.; Lightfoot, S.; Reis, A.; Anant, S.; et al. Wnt inhibitory factor 1 induces apoptosis and inhibits cervical cancer growth, invasion and angiogenesis in vivo. Oncogene 2012, 31, 2725–2737. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.Y.; Shen, J.X.; Wang, Y.; Liu, Y.; Shen, D.Y.; Quan, S. Tankyrase Promotes Aerobic Glycolysis and Proliferation of Ovarian Cancer through Activation of Wnt/β-Catenin Signaling. Biomed. Res. Int. 2019, 2019, 2686340. [Google Scholar] [CrossRef] [PubMed]
- Arend, R.C.; Londoño-Joshi, A.I.; Straughn, J.M.; Buchsbaum, D.J. The Wnt/β-catenin path-way in ovarian cancer: A review. Gynecol. Oncol. 2013, 131, 772–779. [Google Scholar] [CrossRef] [PubMed]
- Teeuwssen, M.; Fodde, R. Wnt Signaling in Ovarian Cancer Stemness, EMT, and Therapy Resistance. J. Clin. Med. 2019, 8, 1658. [Google Scholar] [CrossRef]
- Nguyen, V.H.L.; Hough, R.; Bernaudo, S.; Peng, C. Wnt/β-catenin signalling in ovarian can-cer: Insights into its hyperactivation and function in tumorigenesis. J. Ovarian Res. 2019, 12, 122. [Google Scholar] [CrossRef] [PubMed]
- Lou, Y.; Jiang, H.; Cui, Z.; Wang, L.; Wang, X.; Tian, T. Linc-ROR induces epithelial-to-mesenchymal transition in ovarian cancer by increasing Wnt/β-catenin signaling. Oncotarget 2017, 8, 69983–69994. [Google Scholar] [CrossRef]
- Chung, M.T.; Lai, H.C.; Sytwu, H.K.; Yan, M.D.; Shih, Y.L.; Chang, C.C.; Yu, M.H.; Liu, H.S.; Chu, D.W.; Lin, Y.W. SFRP1 and SFRP2 suppress the transformation and invasion abilities of cervical cancer cells through Wnt signal pathway. Gynecol. Oncol. 2009, 112, 646–653. [Google Scholar] [CrossRef]
- Li, J.; Zhou, B.P. Activation of β-catenin and Akt pathways by Twist are critical for the maintenance of EMT associated cancer stem cell-like characters. BMC Cancer 2011, 11, 49. [Google Scholar] [CrossRef]
- Huang, J.; Li, H.; Ren, G. Epithelial-mesenchymal transition and drug resistance in breast cancer (Review). Int. J. Oncol. 2015, 47, 840–848. [Google Scholar] [CrossRef]
- Roxanis, I. Occurrence and significance of epithelial-mesenchymal transition in breast cancer. J. Clin. Pathol. 2013, 66, 517–521. [Google Scholar] [CrossRef]
- Chen, Y.M.; Liu, Y.; Wei, H.Y.; Lv, K.Z.; Fu, P. Linc-ROR induces epithelial-mesenchymal transition and contributes to drug resistance and invasion of breast cancer cells. Tumour Biol. 2016, 37, 10861–10870. [Google Scholar] [CrossRef]
- Nieto, M.A.; Huang, R.Y.Y.J.; Jackson, R.A.A.; Thiery, J.P.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef]
- Babaei, G.; Aziz, S.G.; Jaghi, N.Z.Z. EMT, cancer stem cells and autophagy; The three main axes of metastasis. Biomed. Pharmacother. 2021, 133, 110909. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, V.; Brabletz, T.; Ceppi, P. Targeting EMT in Cancer with Repurposed Metabolic Inhibitors. Trends Cancer 2020, 6, 942–950. [Google Scholar] [CrossRef] [PubMed]
- Voon, D.C.; Huang, R.Y.; Jackson, R.A.; Thiery, J.P. The EMT spectrum and therapeutic opportunities. Mol. Oncol. 2017, 11, 878–891. [Google Scholar] [CrossRef] [PubMed]
- García-Villalba, R.; Selma, M.V.; Espín, J.C.; Tomás-Barberán, F.A. Identification of Novel Urolithin Metabolites in Human Feces and Urine after the Intake of a Pomegranate Extract. J. Agric. Food Chem. 2019, 67, 11099–11107. [Google Scholar] [CrossRef]
- Tomás-Barberán, F.A.; González-Sarrías, A.; García-Villalba, R.; Núñez-Sánchez, M.A.; Selma, M.V.; García-Conesa, M.T.; Espín, J.C. Urolithins, the rescue of “old” metabolites to understand a “new” concept: Metabotypes as a nexus among phenolic metabolism, microbiota dysbiosis, and host health status. Mol. Nutr. Food Res. 2017, 61, 1–74. [Google Scholar] [CrossRef]
- Selma, M.V.; González-Sarrías, A.; Salas-Salvadó, J.; Andrés-Lacueva, C.; Alasalvar, C.; Örem, A.; Tomás-Barberán, F.A.; Espín, J.C. The gut microbiota metabolism of pomegranate or walnut ellagitannins yields two urolithin-metabotypes that correlate with cardiometabolic risk biomarkers: Comparison between normoweight, overweight-obesity and metabolic syndrome. Clin. Nutr. 2018, 37, 897–905. [Google Scholar] [CrossRef]
- Romo-Vaquero, M.; Cortés-Martín, A.; Loria-Kohen, V.; Ramírez-de-Molina, A.; García-Mantrana, I.; Collado, M.C.; Espín, J.C.; Selma, M.V. Deciphering the Human Gut Microbiome of Urolithin Metabotypes: Association with Enterotypes and Potential Cardiometabolic Health Implications. Mol. Nutr. Food Res. 2019, 63, e1800958. [Google Scholar] [CrossRef]
- Beltrán, D.; Romo-Vaquero, M.; Espín, J.C.; Tomás-Barberán, F.A.; Selma, M.V. Ellagibacter isourolithinifaciens gen. nov., sp. nov., a new member of the family Eggerthellaceae, isolated from human gut. Int. J. Syst. Evol. Microbiol. 2018, 68, 1707–1712. [Google Scholar] [CrossRef]
- Selma, M.V.; Beltrán, D.; Luna, M.C.; Romo-Vaquero, M.; García-Villalba, R.; Mira, A.; Espín, J.C.; Tomás-Barberán, F.A. Isolation of Human Intestinal Bacteria Capable of Producing the Bioactive Metabolite Isourolithin A from Ellagic Acid. Front. Microbiol. 2017, 8, 1521. [Google Scholar] [CrossRef] [PubMed]
- Nuñez-Sánchez, M.A.; García-Villalba, R.; Monedero-Saiz, T.; García-Talavera, N.V.; Gómez-Sánchez, M.B.; Sánchez-Álvarez, C.; García-Albert, A.M.; Rodríguez-Gil, F.J.; Ruiz-Marín, M.; Pastor-Quirante, F.A.; et al. Targeted metabolic profiling of pomegranate polyphenols and urolithins in plasma, urine and colon tissues from colorectal cancer patients. Mol. Nutr. Food Res. 2014, 58, 1199–1211. [Google Scholar] [CrossRef]
- Piwowarski, J.P.; Stanisławska, I.; Granica, S.; Stefańska, J.; Kiss, A.K. Phase II Conjugates of Urolithins Isolated from Human Urine and Potential Role of β-Glucuronidases in Their Disposition. Drug. Metab. Dispos. 2017, 45, 657–665. [Google Scholar] [CrossRef] [PubMed]
- Seeram, N.P.; Henning, S.M.; Zhang, Y.; Suchard, M.; Li, Z.; Heber, D. Pomegranate juice ellagitannin metabolites are present in human plasma and some persist in urine for up to 48 hours. J. Nutr. 2006, 136, 2481–2485. [Google Scholar] [CrossRef]
- Bialonska, D.; Kasimsetty, S.G.; Schrader, K.K.; Ferreira, D. The effect of pomegranate (Punica granatum L.) byproducts and ellagitannins on the growth of human gut bacteria. J. Agric. Food Chem. 2009, 57, 8344–8349. [Google Scholar] [CrossRef]
- Al-Harbi, S.A.; Abdulrahman, A.O.; Zamzami, M.A.; Khan, M.I. Urolithins: The Gut Based Polyphenol Metabolites of Ellagitannins in Cancer Prevention, a Review. Front. Nutr. 2021, 8, 647582. [Google Scholar] [CrossRef] [PubMed]
- Norden, E.; Heiss, E.H. Urolithin A gains in antiproliferative capacity by reducing the glycolytic potential via the p53/TIGAR axis in colon cancer cells. Carcinogenesis 2019, 40, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Shi, F.; Guo, Z.; Zhao, J.; Song, X.; Yang, H. Metabolite of ellagitannins, urolithin A induces autophagy and inhibits metastasis in human sw620 colorectal cancer cells. Mol. Carcinog. 2018, 57, 193–200. [Google Scholar] [CrossRef]
- Giménez-Bastida, J.A.; Ávila-Gálvez, M.Á.; Espín, J.C.; González-Sarrías, A. The gut microbiota metabolite urolithin A, but not other relevant urolithins, induces p53-dependent cellular senescence in human colon cancer, cells. Food Chem. Toxicol. 2020, 139, 111260. [Google Scholar] [CrossRef]
- El-Wetidy, M.S.; Ahmad, R.; Rady, I.; Helal, H.; Rady, M.I.; Vaali-Mohammed, M.A.; Al-Khayal, K.; Traiki, T.B.; Abdulla, M.H. Urolithin A induces cell cycle arrest and apoptosis by inhibiting Bcl-2, increasing p53-p21 proteins and reactive oxygen species production in colorectal cancer cells. Cell Stress Chaperones 2021, 26, 473–493. [Google Scholar] [CrossRef]
- González-Sarrías, A.; Tomé-Carneiro, J.; Bellesia, A.; Tomás-Barberán, F.A.; Espín, J.C. The ellagic acid-derived gut microbiota metabolite, urolithin A, potentiates the anticancer effects of 5-fluorouracil chemotherapy on human colon cancer cells. Food Funct. 2015, 6, 1460–1469. [Google Scholar] [CrossRef]
- González-Sarrías, A.; Giménez-Bastida, J.A.; Núñez-Sánchez, M.Á.; Larrosa, M.; García-Conesa, M.T.; Tomás-Barberán, F.A.; Espín, J.C. Phase-II metabolism limits the antiproliferative activity of urolithins in human colon cancer cells. Eur. J. Nutr. 2014, 53, 853–864. [Google Scholar] [CrossRef]
- Alzahrani, A.M.; Shait Mohammed, M.R.; Alghamdi, R.A.; Ahmad, A.; Zamzami, M.A.; Choudhry, H.; Khan, M.I. Urolithin A and B Alter Cellular Metabolism and Induce Metabolites Associated with Apoptosis in Leukemic Cells. Int. J. Mol. Sci. 2021, 22, 5465. [Google Scholar] [CrossRef]
- Cheng, F.; Dou, J.; Zhang, Y.; Wang, X.; Wei, H.; Zhang, Z.; Cao, Y.; Wu, Z. Urolithin A Inhibits Epithelial-Mesenchymal Transition in Lung Cancer Cells via P53-Mdm2-Snail Pathway. OncoTargets Ther. 2021, 14, 3199–3208. [Google Scholar] [CrossRef]
- Totiger, T.M.; Srinivasan, S.; Jala, V.R.; Lamichhane, P.; Dosch, A.R.; Gaidarski, A.A.; Joshi, C.; Rangappa, S.; Castellanos, J.; Vemula, P.K.; et al. Urolithin A, a Novel Natural Compound to Target PI3K/AKT/mTOR Pathway in Pancreatic Cancer. Mol. Cancer Ther. 2019, 18, 301–311. [Google Scholar] [CrossRef]
- Khan, H.; Reale, M.; Ullah, H.; Sureda, A.; Tejada, S.; Wang, Y.; Zhang, Z.J.; Xiao, J. Anti-cancer effects of polyphenols via targeting p53 signaling pathway: Updates and future directions. Biotechnol. Adv. 2020, 38, 107385. [Google Scholar] [CrossRef] [PubMed]
- Shirode, A.B.; Bharali, D.J.; Nallanthighal, S.; Coon, J.K.; Mousa, S.A.; Reliene, R. Nanoencapsulation of pomegranate bioactive compounds for breast cancer chemoprevention. Int. J. Nanomedicine 2015, 10, 475–484. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Cancer Type | Experimental Model | Up-Regulation ↑ | Down-Regulation ↓ | Final Effect | Ref |
|---|---|---|---|---|---|
| Cervical carcinoma | ME-180 cervical cancer cell line | Bax; Casp-3 and 9; p53; cytosolic NF-κB-p65 | Bcl-2; nuclear NF-κB-p65 | Inhibition of cell proliferation and induction of apoptosis via suppressing NF-κB signaling | [19] |
| Cervix epitheloid carcinoma | HeLa cervical cancer cell line | Bax; TIMP2/3 | Bcl-2; β-catenin, C-myc, cyclin D1; MMP2/9 | Inhibition of cell proliferation and migration Cell cycle arrest at G1 phase Suppression of β-catenin pathway Induction of apoptosis | [20] |
| Breast adenocarcinoma | MCF-7 and MDA-MB-231 cell lines | E-cadherin | GOLPH3; MMP2/9; N-cadherin | Suppression of cell viability, EMT and migration via the regulation of GOLPH3 | [26] |
| Ovarian cancer | A2780 ovarian cancer cells | Bax; TIMP2/3 | Bcl-2; β-catenin, cyclin D1, survivin; MMP2/9 | Inhibition of cell viability and migration Cell cycle arrest at G1 phase Suppression of β-catenin pathway Induction of apoptosis | [27] |
| Lung carcinoma | Lung cancer A549 cell line | Bax; Casp-3 and 9; cytochrome C; ROS; cytosolic STAT-3 | Bcl-2; Jak-1; nuclear STAT-3 | Inhibition of cell proliferation and induction of apoptosis via suppressing STAT-3 activation | [28] |
| Lung carcinoma | Lung cancer A549 cell line | Casp-3, 8, and 9; PARP-1; mitochondrial ROS | Cytosolic ROS | Induction of apoptosis; cell cycle arrest at G1/S | [29] |
| Osteosarcoma | Cell lines U2OS, SaOS2 | - | Phosphorylated IκBα; nuclear NF-κB-p65; IL-6; IL-8 | Inhibition of cell proliferation and induction of apoptosis possibly via suppressing NF-κB signaling Reduction of invasion potential | [30] |
| Colorectal carcinoma | Cell line HCT116 | Cytochrome C | Annexin A1; caspases 3/7, 8 and 9 | Induction of cell death via apoptosis and autophagy | [31] |
| Colon adenocarcinoma | Cell line HT-29 | - | COX-2 | Suppression of inflammatory cell signaling | [32] |
| Colon adenocarcinoma | Cell line Caco-2 | Casp-3 and 9; Cytochrome C; Cyclin E | Bcl-XL; Cyclin A and B1 | Cell cycle arrest at S phase; induction of apoptosis via the intrinsic-mitochondrial pathway stimulation | [33] |
| Papillary thyroid carcinoma | Cell line BCPAP | LC3-II conversion, beclin-1; phosphorylated ERK 1/2 and p38 | p62; phosphorylated p70, S6, and 4E-BP1 | Induction of apoptosis-independent cell death via autophagy through the activation of MAPK and inhibition of mTOR signaling | [34] |
| Papillary thyroid carcinoma | Cell line BCPAP | p-H2A.X; p-ATM | - | Induction of cells death via the ATM-mediated DNA damage response | [35] |
| Papillary thyroid carcinoma | Cell line BCPAP | SA-beta-Gal; cyclin-dependent kinase inhibitor p21; IκBα; nuclear NF-κB-p65; IL-6; IL-1β | - | Induction of senescent growth arrest and senescence-associated secretory phenotype (SASP) through the activation of NF-κB | [36] |
| Glioblastoma astrocytoma | Cell line U87MG | Casp-3 and 9; PARP; Cyclin E; LC3-II cleavage, AMPK-P, p27-P | Bcl-2; Cyclin A and B | Cell cycle arrest at G2/M phase; induction of cell death via apoptosis and autophagy | [37] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Berdowska, I.; Matusiewicz, M.; Fecka, I. Punicalagin in Cancer Prevention—Via Signaling Pathways Targeting. Nutrients 2021, 13, 2733. https://doi.org/10.3390/nu13082733
Berdowska I, Matusiewicz M, Fecka I. Punicalagin in Cancer Prevention—Via Signaling Pathways Targeting. Nutrients. 2021; 13(8):2733. https://doi.org/10.3390/nu13082733
Chicago/Turabian StyleBerdowska, Izabela, Małgorzata Matusiewicz, and Izabela Fecka. 2021. "Punicalagin in Cancer Prevention—Via Signaling Pathways Targeting" Nutrients 13, no. 8: 2733. https://doi.org/10.3390/nu13082733
APA StyleBerdowska, I., Matusiewicz, M., & Fecka, I. (2021). Punicalagin in Cancer Prevention—Via Signaling Pathways Targeting. Nutrients, 13(8), 2733. https://doi.org/10.3390/nu13082733