Abstract
Recently, the genetic background of pheochromocytomas/paragangliomas (PPGLs) has been rapidly revealed. These tumors have been referred to as the “ten percent tumor”; however, the frequency of genetic variants of PPGLs has turned out to be more common than expected. PPGLs are potentially hereditary tumors and appear clinically sporadic. Here, we report a case of bilateral pheochromocytoma (PCC) with a variant in the MYC-associated factor X (MAX) gene (c.295 + 1G > A). A male patient was diagnosed with adrenal pheochromocytoma (PCC) and underwent a left adrenalectomy at the age of 40. A new tumor in the right adrenal gland was detected at the age of 43. Urinary metanephrine and normetanephrine concentrations gradually increased. The size of the right adrenal PCC continued to increase one year after detection. Genetic testing of the peripheral blood revealed the presence of a pathogenic variant in MAX. The natural history of adrenal PCCs with the MAX variant has not yet been clarified, because the number of reported cases is not sufficient. Thus, clinicians should consider a MAX variant when they find bilateral or multiple PCCs.
1. Introduction
Collectively known as pheochromocytomas/paragangliomas (PPGLs), pheochromocytomas (PCCs) and paragangliomas (PGLs) are catecholamine-producing tumors that arise from chromaffin cells in the adrenal medulla or paraganglia, respectively [1]. Approximately 10% of PCCs were thought to be familial cases [2]. However, approximately 30–40% of PPGLs have been reported as hereditary tumors [3,4]. RET, VHL, SDHA, SDHB, SDHC, SDHAF2, NF1, and TMEM127 contribute to hereditary PPGLs [5]. In 2011, MAX variants were identified as one of the causes of hereditary PCCs [5]. We report a case of bilateral adrenal PCC with a germline variant in MYC-associated factor X (MAX) gene without a clear family history of PPGLs.
2. Case Presentation
A healthy 40-year-old man (height, 178 cm; weight, 68 kg) did not have any family history of PPGL, but his father had hypertension. He had a younger brother and two children with no relevant medical history (Figure 1). Two masses in the left adrenal gland, 16 mm (tumor 1 in Figure 2) and 13 mm (tumor 2 in Figure 2), respectively, were incidentally detected by computed tomography imaging during a health check-up program, which are commonly conducted at medical facilities in Japan and other Asian countries in order to identify risk factors and screen for diseases when people are still healthy. His body temperature was 36.5 °C, and pulse rate was 61 beats per minute. Although he had paroxysmal hypertension (up to approximately 200/100 mmHg), his blood pressure was 113/73 mmHg during his first visit to our hospital. Thyroid enlargement and café-au-lait spots on the skin were not observed. Blood and urine tests revealed the following: plasma adrenaline, 74 pg/mL; plasma noradrenaline, 2119 pg/mL; urinary metanephrine, 0.54 mg/day; and urinary normetanephrine, 1.32 mg/day. 123I-metaiodobenzylguanidine (MIBG) scintigraphy showed two PCCs in the left adrenal gland. After left adrenalectomy, urinary normetanephrine levels rapidly normalized. The histological pattern of both tumors showed a zellballen and high cellularity (>250 cells/ high power field (HPF)), respectively. Tumor 1 showed absence of vascular and capsular invasion, whereas tumor 2 indicated capsular invasion. The Ki-67 labeling indices of tumor 1 and 2 were 0.1% and 0.6%, respectively. The grading system for adrenal pheochromocytoma and paraganglioma (GAPP) scores of both tumors corresponded to moderately differentiated tumors (tumor 1, score 5; tumor 2, score 3). Three years later, a mass was found in the right adrenal gland. The tumor size increased from 17 to 25 mm in diameter and urinary normetanephrine levels increased from 0.26 mg/g Cr to 0.54 mg/g Cr in one year (Figure 3). 123I-MIBG scintigraphy indicated an accumulation in the right adrenal mass. He was diagnosed with right adrenal PCC. Although the family history was unclear, this case was highly suspected of being hereditary PCC because of the presence of bilateral and multiple masses. PCR-direct sequencing of the peripheral blood samples did not show pathological variants in RET and VHL. Subsequently, the PCR test revealed a heterozygous germline variant in MAX (NM_002382.5: c.295 + 1G > A). Immunohistochemistry of the left adrenal tumor tissue with a MAX C-terminus-specific antibody (ab101271, Abcam, Cambridge, United Kingdom) showed no staining of tumor cells in this case (Figure 2). Besides his parents, the patient had a younger brother (adult) and two young children. Despite our recommendation, his family members did not undergo the genetic tests. The patient was treated with an alpha-blocker. Written informed consent was obtained from the patient, and ethical approval for this study was obtained from the Institutional Review Board of Shizuoka General Hospital.
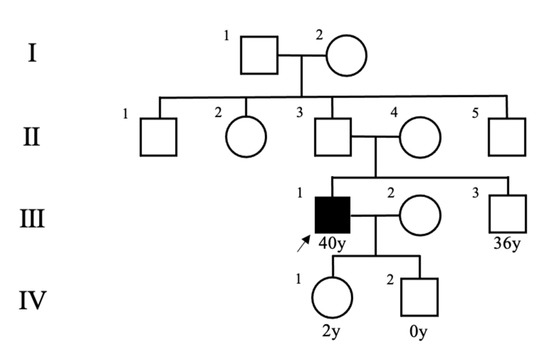
Figure 1.
Pedigree of the patient’s family. An arrow indicates the patient.
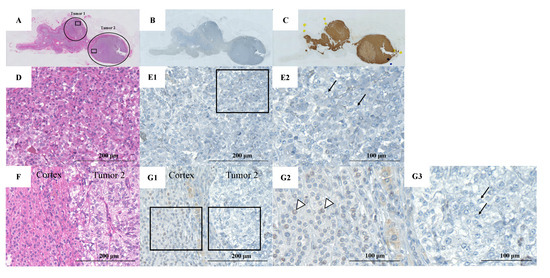
Figure 2.
Pathological findings of left adrenal tumors resected from the patient. (A) Hematoxylin-eosin (HE) staining of the left adrenal gland (circles show the location of the two tumors); (B) MAX staining of the left adrenal gland; (C) Chromogranin A staining of the left adrenal gland; (D) Hematoxylin and eosin staining of tumor 1 (rectangular area in the tumor 1); (E1) MAX staining in tumor 1 (rectangular area in the tumor 1); (E2) Rectangular area in panel E1. Pheochromocytoma (PCC) cells in the adrenal medulla show no MAX staining (arrows); (F) HE staining of tumor 2 (rectangular area in the tumor 2); (G1) MAX staining in tumor 2 (rectangular area in the tumor 2); (G2) Left rectangular area in panel G1. Adrenal cortex cells show MAX staining (arrowheads); (G3) Right rectangular area in panel G1. The PCC cells show no MAX staining (arrows).
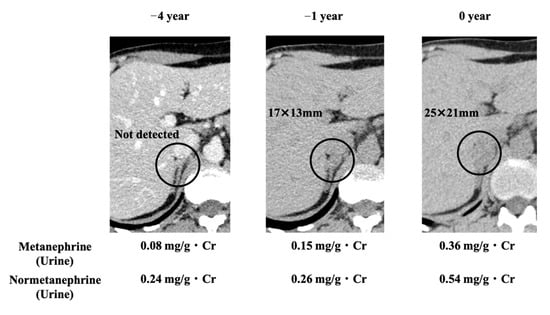
Figure 3.
CT images of the right adrenal gland and changes in biochemical test results. Circles indicate right adrenal tumor. Urinary metanephrine and normetanephrine outputs increased with increase in the tumor size.
3. Discussion
Among all PPGLs, the frequency of hereditary cases was reported to be 33.8% [3,4,6,7,8,9,10,11]. The Endocrine Society clinical practice guidelines state that genetic testing should be considered in all PPGL cases [12]. They also reported that 11–13% of clinically sporadic PCCs are hereditary tumors [13]. For patients, especially those with young, bilateral, multiple, extra-adrenal, or malignant PPGLs, identification of hereditary tumors might be beneficial, because clinical characteristics vary with each genetic background [14]. In this case, we found a germline variant in the MAX gene, which was seemingly sporadic. The characteristics of PPGLs with some genetic types, such as MAX gene, are difficult to describe because there have been few reports.
A large international study confirmed that MAX germline and somatic variants were responsible for PCCs in 1.12% and 1.65% of cases, respectively [15]. One small study, which included eight index patients and three relatives, showed a high penetration rate of 73% for up to 40 years of age, although the rate could be affected by a selection bias [16].
MAX is considered a tumor suppressor gene, forms the MYC-MAX-MXD1 network, and acts as a transcription factor that regulates cell proliferation, differentiation, and death [17]. Heterodimerization of MAX with MYC acts as a transcriptional activator, whereas the heterodimers of MAX with MXD1 repress MYC-dependent transcriptional activities by antagonizing the MYC-MAX function [18]. The MAX gene comprises five exons. The previously reported variants in MAX were distributed along the gene but were particularly frequent in exons 3 and 4, matching some of the crucial residues within the conserved basic helix-loop-helix leucine zipper (bHLH-Zip) domain of MAX [15].
The peripheral blood sample of our patient showed a heterozygous single nucleotide substitution in MAX (c.295 + 1G > A). The majority of MAX mutations result in truncated proteins [15]. A truncated protein was observed in the case of the same variant (c.295 + 1G > A), as reported previously [5]. In that case, the mutation site was in the intron but was located at the donor/acceptor splice site, leading to the skipping of exon 4. Therefore, we estimated that skipping exon 4 produced truncated proteins that had no ability to regulate cell proliferation, differentiation, and apoptosis and promoted the development of tumors in our case (Figure 4).
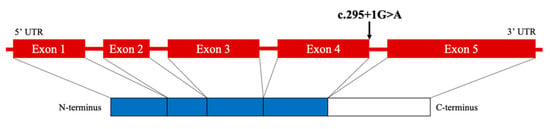
Figure 4.
Schematic diagram of MAX mutation in the patient. UTR, untranslated region.
A Pubmed search was performed using the key terms pheochromocytoma and variant. All searches were limited to reports published in the English language, dating from 1972 to April 2022. Among 499 studies, 213 articles reported variants related to PPGLs and 16 papers reported MAX variants. We summarized the characteristics of the cases with PPGLs in MAX variants [5,15,16,19,20,21,22,23,24,25,26,27,28,29,30,31] (Supplementary Table S1). Combining data from the 16 reports in MAX variants revealed that 42/71 cases (59.2%) had bilateral PCCs, 9/59 cases (8.5%) had PGLs, and 31/70 cases (44.3%) had an apparent family history of PPGLs. In this review, 11/65 patients (16.9%) had metastases. Unlike SDHB, MAX variants do not appear to be a high risk for malignancy, considering the frequency; however, the reported case of bilateral PCC with a variant in MAX (c.295 + 1G > A) had the same variant as our case, and was found to be malignant [5]. Apart from the aforementioned case [5], no other variants were exactly the same. However, another patient with a variant in MAX (c.295 + 1G > T) at the same site was presented with metastasis in another report [15]. Our search showed that there were 16 studies reporting MAX variants; only eight of them performed immunostaining for MAX. Thus, our report is valuable and contributes to the growing body of knowledge on this field.
In recent years, MAX variants have been reported to be associated with endocrine tumors, such as pituitary adenomas (prolactinoma and acromegaly) and parathyroid adenomas in addition to PPGLs [29]. MAX variants may also be associated with pancreatic neuroendocrine neoplasms [32]. In our case, hypercalcemia was not observed. There were no physical findings, clinical histories, or examinations suggestive of acromegaly or prolactinoma. Abdominal computed tomography did not exhibit tumors in the pancreas.
Burnichon et al. reported that urinary levels of normetanephrine were elevated in all patients with the MAX variant, with no difference between the group with the VHL and SDHB/D variant or the group with the RET/NF1 variant. In contrast, patients with the MAX variant had normal or moderately increased urinary outputs of metanephrine with an intermediate distribution. The metanephrine outputs were higher in the VHL/SDH group than in the RET/NF1 group [15]. In our case, the urinary metanephrine output was within the normal range but increased moderately compared to that in the VHL/SDH group. However, urinary normetanephrine output was increased. The urinary biochemical phenotype of our patient was consistent with that report.
Our case showed bilateral and multiple tumors without an apparent family history of PPGLs, and there were no findings suggestive of metastasis at this time. His tumors had typical features of PPGLs in MAX variants.
All patients with PPGL are recommended to be engaged in shared decision-making for genetic testing [12]. When a MAX pathogenic variant is found, Muth et al. recommended that all adult first-degree relatives be tested through targeted testing of the variant on DNA [33]. However, it is important to respect personal autonomy.
Four years after the first detection of the tumors, there were no findings suggesting metastasis in the case discussed; however, the possibility of malignancy or further tumor development cannot be ruled out. Variant classification according to the American College of Medical Genetics and Genomics Guidelines (ACMG) suggested that the variant was pathogenic, as our case satisfied PVS1, PM2, PP3, and PP4 [34]. Careful follow-up is required in this case.
Recent studies have indicated that the frequency of germline variants of PPGLs is very high among all human tumors. Besides MAX, multiple new PPGL-related genes such as CSDE1, H3F3A, MET, MERTK, UBTF-MAML3, SLC25A11, IRP1, DLST, and SUCLG2 have been discovered in recent years [35]. With the expansion of our knowledge of genetics, new biomarkers and artificial intelligence can also help assess the metastatic risk and overall prognosis of each individual. This disease is no longer the “ten percent tumor” in terms of genetics. Moreover, certain PPGL cases can promote metastasis. Whenever a case of PPGL is seen, we should consider the possibility of a familial or metastatic case. Finally, the possibility of hereditary PPGL including the MAX gene should be considered, particularly in cases of multiple or bilateral PPGLs, even without a clear family history such as in the case of this patient.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/clinpract12030035/s1, Table S1: Clinical features of PPGL with MAX variants.
Author Contributions
Conceptualization, S.H.; writing—original draft preparation, S.H.; writing—review and editing, S.H., M.A., H.T. and M.H.; funding acquisition, F.H., T.U. and E.K. interpretation of data, M.F. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and approved by Clinical Ethics Committee of Shizuoka General Hospital (protocol code, none.; date of approval, 22 August 2016).
Informed Consent Statement
Written informed consent was obtained from the patient.
Data Availability Statement
Not applicable.
Acknowledgments
We thank Yoshitaka Isojima for performing the immunohistochemical staining.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
Sex: F, female; M, male; PCC, adrenal pheochromocytoma; bPCC, bilateral adrenal pheochromocytoma; mPCC, multiple adrenal pheochromocytoma; PGL, paraganglioma. Other diseases: BrC, breast cancer; RO, renal oncocytoma; SCCT, squamous cell carcinoma of the tongue; CCH, C-cell hyperplasia; PA, pituitary adenoma; HPT, hyperparathyroidism; ReC, renal carcinoma; β-T, β-thalassemia; T1DM, type 1 diabetes; PRLoma, prolactinoma; Acro, acromegaly; TC, thyroid cancer; GNB, ganglio-neuroblastoma; NB, neuroblastoma; RiC, rib chondrosarcoma; LA, lung adenocarcinoma; IHC, immunohistochemistry. IHC: Pos, positive; Neg, negative.
References
- Pacak, K.; Taïeb, D. Pheochromocytoma (PHEO) and Paraganglioma (PGL). Cancers 2019, 11, 1391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maher, E.R.; Eng, C. The pressure rises: Update on the genetics of phaeochromocytoma. Hum. Mol. Genet. 2002, 11, 2347–2354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mannelli, M.; Castellano, M.; Schiavi, F.; Filetti, S.; Giacchè, M.; Mori, L.; Pignataro, V.; Bernini, G.; Giachè, V.; Bacca, A.; et al. Clinically guided genetic screening in a large cohort of Italian patients with pheochromocytomas and/or functional or nonfunctional paragangliomas. J. Clin. Endocrinol. Metab. 2009, 94, 1541–1547. [Google Scholar] [CrossRef] [Green Version]
- Cascón, A.; Pita, G.; Burnichon, N.; Landa, I.; López-Jiménez, E.; Monerto-Conde, C.; Leskelä, S.; Leandor-García, L.J.; Letón, R.; Rodriguez-Antona, C.; et al. Genetics of pheochromocytoma and paraganglioma in Spanish patients. J. Clin. Endocrinol. Metab. 2009, 94, 1701–1705. [Google Scholar] [CrossRef] [Green Version]
- Comino-Méndez, I.; Gracia-Aznárez, F.J.; Schiavi, F.; Landa, I.; Leandro-García, L.J.; Letón, R.; Honrado, E.; Ramos-Medina, R.; Caronia, D.; Pita, G.; et al. Exome sequencing identifies MAX mutations as a cause of hereditary pheochromocytoma. Nat. Genet. 2011, 43, 663–667. [Google Scholar] [CrossRef] [PubMed]
- Amar, L.; Bertherat, J.; Baudin, E.; Ajzenberg, C.; Bressac-de Paillerets, B.; Chabre, O.; Chamontin, B.; Delemer, B.; Giraud, S.; Murat, A.; et al. Genetic testing in pheochromocytoma or functional paraganglioma. J. Clin. Oncol. 2005, 23, 8812–8818. [Google Scholar] [CrossRef] [PubMed]
- Burnichon, N.; Rohmer, V.; Amar, L.; Herman, P.; Leboulleux, S.; Darrouzet, V.; Niccoli, P.; Gaillard, D.; Chabrier, G.; Chabolle, F.; et al. PGL.NET network. The succinate dehydrogenase genetic testing in a large prospective series of patients with paragangliomas. J. Clin. Endocrinol. Metab. 2009, 94, 2817–2827. [Google Scholar] [CrossRef] [PubMed]
- Jafri, M.; Whitworth, J.; Rattenberry, E.; Vialard, L.; Kilby, G.; Kumar, A.V.; Izatt, L.; Lalloo, F.; Brennan, P.; Cook, J.; et al. Evaluation of SDHB, SDHD and VHL gene susceptibility testing in the assessment of individuals with non-syndromic phaeochromocytoma, paraganglioma and head and neck paraganglioma. Clin. Endocrinol. 2013, 78, 898–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erlic, Z.; Rybicki, L.; Peczkowska, M.; Golcher, H.; Kann, P.H.; Brauckhoff, M.; Müssig, K.; Muresan, M.; Schäffler, A.; Reisch, N.; et al. European-American Pheochromocytoma Study Group. Clinical Predictors and Algorithm for the Genetic Diagnosis of Pheochromocytoma Patients. Clin. Cancer. Res. 2009, 15, 6378–6385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korpershoek, E.; Favier, J.; Gaal, J.; Burnichon, N.; van Gessel, B.; Oudijk, L.; Badoual, C.; Gadessaud, N.; Venisse, A.; Bayley, J.; et al. SDHA immunohistochemistry detects germline SDHA gene mutations in apparently sporadic paragangliomas and pheochromocytomas. J. Clin. Endocrinol. Metab. 2011, 96, E1472–E1476. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, S.; Borson-Chazot, F.; Boutry-Kryza, N.; Wion, N.; Schillo, F.; Peix, J.; Brunaud, L.; Finat, A.; Calender, A.; Giraud, S. Screening of mutations in genes that predispose to hereditary paragangliomas and pheochromocytomas. Horm. Metab. Res. 2012, 44, 334–338. [Google Scholar] [CrossRef] [PubMed]
- Lenders, J.W.; Duh, Q.Y.; Eisenhofer, G.; Gimenez-Roqueplo, A.P.; Grebe, S.K.; Murad, M.H.; Naruse, M.; Pacak, K.; Young, W.F. Endocrine Society. Pheochromocytoma and paraganglioma: An endocrine society clinical practice guideline. J. Clin. Endocrinol. Metab. 2014, 99, 1915–1942. [Google Scholar] [CrossRef]
- Brito, J.P.; Asi, N.; Bancos, I.; Gionfriddo, M.R.; Zeballos-Palacios, C.L.; Leppin, A.L.; Undavalli, C.; Wang, Z.; Domecq, J.P.; Prustsky, G.; et al. Testing for germline mutations in sporadic pheochromocytoma/paraganglioma: A systematic review. Clin. Endocrinol. 2015, 82, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Welander, J.; Söderkvist, P.; Gimm, O. Genetics and clinical characteristics of hereditary pheochromocytomas and paragangliomas. Endocr. Relat. Cancer. 2011, 18, R253–R276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burnichon, N.; Cascón, A.; Schiavi, F.; Morales, N.P.; Comino-Méndez, I.; Abermil, N.; Inglada-Pérez, L.; de Cubas, A.A.; Amar, L.; Barontini, M.; et al. MAX Mutations Cause Hereditary and Sporadic Pheochromocytoma and Paraganglioma. Clin. Cancer. Res. 2012, 18, 2828–2837. [Google Scholar] [CrossRef] [Green Version]
- Bausch, B.; Schiavi, F.; Ni, Y.; Welander, J.; Patocs, A.; Ngeow, J.; Wellner, U.; Malinoc, A.; Taschin, E.; Barbon, G.; et al. European-American-Asian Pheochromocytoma-Paraganglioma Registry Study Group. Clinical Characterization of the Pheochromocytoma and Paraganglioma Susceptibility Genes SDHA, TMEM127, MAX, and SDHAF2 for Gene-Informed Prevention. JAMA. Oncol. 2017, 3, 1204–1212. [Google Scholar] [CrossRef] [Green Version]
- Grandori, C.; Cowley, S.M.; James, L.P.; Eisenman, R.N. The Myc/Max/Mad network and the transcriptional control of cell behavior. Annu. Rev. Cell. Dev. Biol. 2000, 16, 653–699. [Google Scholar] [CrossRef]
- Diolaiti, D.; McFerrin, L.; Carroll, P.A.; Eisenman, R.N. Functional interactions among members of the MAX and MLX transcriptional network during oncogenesis. Biochim. Biophys. Acta. 2015, 1849, 484–500. [Google Scholar] [CrossRef] [Green Version]
- Shibata, M.; Inaishi, T.; Miyajima, N.; Adachi, Y.; Takano, Y.; Nakanishi, K.; Takeuchi, D.; Noda, S.; Aita, Y.; Takekoshi, K.; et al. Synchronous bilateral pheochromocytomas and paraganglioma with novel germline mutation in MAX: A case report. Surg. Case. Rep. 2017, 3, 131–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korpershoek, E.; Koffy, D.; Eussen, B.H.; Oudijk, L.; Papathomas, T.G.; van Nederveen, F.H.; Belt, E.J.; Franssen, G.J.; Restuccia, D.F.; Krol, N.M.; et al. Complex MAX Rearrangement in a Family with Malignant Pheochromocytoma, Renal Oncocytoma, and Erythrocytosis. J. Clin. Endocrinol. Metab. 2016, 101, 453–460. [Google Scholar] [CrossRef] [Green Version]
- Roszko, K.L.; Blouch, E.; Blake, M.; Powers, J.F.; Tischler, A.S.; Hodin, R.; Sadow, P.; Lawson, E.A. Case Report of a Prolactinoma in a Patient with a Novel MAX Mutation and Bilateral Pheochromocytomas. J. Endocr. Soc. 2017, 1, 1401–1407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romanet, P.; Guerin, C.; Pedini, P.; Essamet, W.; Castinetti, F.; Sebag, F.; Roche, P.; Cascon, A.; Tischler, A.S.; Pacak, K.; et al. Pathological and Genetic Characterization of Bilateral Adrenomedullary Hyperplasia in a Patient with Germline MAX Mutation. Endocr. Pathol. 2017, 28, 302–307. [Google Scholar] [CrossRef] [PubMed]
- Daly, A.F.; Castermans, E.; Oudijk, L.; Guitelman, M.A.; Beckers, P.; Potorac, I.; Neggers, S.J.; Sacre, N.; van der Lely, A.; Bours, V.; et al. Pheochromocytomas and pituitary adenomas in three patients with MAX exon deletions. Endocr. Relat. Cancer. 2018, 25, L37–L42. [Google Scholar] [CrossRef] [Green Version]
- Kobza, A.O.; Dizon, S.; Arnaout, A. Case Report of Bilateral Pheochromocytomas due to a Novel Max Mutation in a Patient Known to have a Pituitary Prolactinoma. AACE Clin. Case. Rep. 2018, 4, e453–e456. [Google Scholar] [CrossRef] [Green Version]
- Pozza, C.; Sesti, F.; Di Dato, C.; Sbardella, E.; Pofi, R.; Schiavi, F.; Bonifacio, V.; Isidori, A.M.; Faggiano, A.; Lenzi, A.; et al. A Novel MAX Gene Mutation Variant in a Patient with Multiple and "Composite" Neuroendocrine-Neuroblastic Tumors. Front. Endocrinol. 2020, 11, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Chang, X.; Li, Z.; Ma, X.; Cui, Y.; Chen, S.; Tong, A. A Novel Phenotype of Germline Pathogenic Variants in MAX: Concurrence of Pheochromocytoma and Ganglioneuroma in a Chinese Family and Literature Review. Front. Endocrinol. 2020, 11, 558–563. [Google Scholar] [CrossRef]
- Choi, H.; Kim, K.J.; Hong, N.; Shin, S.; Choi, J.R.; Kang, S.W.; Lee, S.T.; Rhee, Y. Genetic Analysis and Clinical Characteristics of Hereditary Pheochromocytoma and Paraganglioma Syndrome in Korean Population. Endocrinol. Metab. 2020, 35, 858–872. [Google Scholar] [CrossRef]
- Duarte, D.B.; Ferreira, L.; Santos, A.P.; Costa, C.; Lima, J.; Santos, C.; Afonso, M.; Teixeira, M.R.; Carvalho, R.; Cardoso, M.H. Case Report: Pheochromocytoma and Synchronous Neuroblastoma in a Family with Hereditary Pheochromocytoma Associated with a MAX Deleterious Variant. Front. Endocrinol. 2021, 12, 609263–609271. [Google Scholar] [CrossRef]
- Seabrook, A.J.; Harris, J.E.; Velosa, S.B.; Kim, E.; McInerney-Leo, A.M.; Dwight, T.; Hockings, J.I.; Hockings, N.G.; Kirk, J.; Leo, P.J.; et al. Multiple Endocrine Tumors Associated with Germline MAX Mutations: Multiple Endocrine Neoplasia Type 5? J. Clin. Endocrinol. Metab. 2021, 106, 1163–1182. [Google Scholar] [CrossRef] [PubMed]
- Mamedova, E.; Vasilyev, E.; Petrov, V.; Buryakina, S.; Tiulpakov, A.; Belaya, Z. Familial Acromegaly and Bilateral Asynchronous Pheochromocytomas in a Female Patient with a MAX Mutation: A Case Report. Front. Endocrinol. 2021, 12, 683492–683497. [Google Scholar] [CrossRef]
- Lam-Chung, C.E.; Rodríguez, L.L.; Vázquez, J.A.; Chávarri-Guerra, Y.; Arízaga-Ramírez, R.; Antonio, O.F.; González, J.D.; López-Hernández, M.A.; Weitzel, J.N.; Castillo, D.; et al. A Novel, Likely Pathogenic MAX Germline Variant in a Patient with Unilateral Pheochromocytoma. J. Endocr. Soc. 2021, 5, 1–6. [Google Scholar] [CrossRef]
- Petignot, S.; Daly, A.F.; Castermans, E.; Korpershoek, E.; Scagnol, I.; Beckers, P.; Dideberg, V.; Rohmer, V.; Bours, V.; Beckers, A. Pancreatic Neuroendocrine Neoplasm Associated with a Familial MAX Deletion. Horm. Metab. Res. 2020, 52, 784–787. [Google Scholar] [CrossRef]
- Muth, A.; Crona, J.; Gimm, O.; Elmgren, A.; Filipsson, K.; Askmalm, M.S.; Sandstedt, J.; Tengvar, M.; Tham, E. Genetic testing and surveillance guidelines in hereditary pheochromocytoma and paraganglioma. J. Intern. Med. 2019, 285, 187–204. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hedge, M.; Lyon, E.; Spector, E.; et al. ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [Green Version]
- Jhawar, S.; Arakawa, Y.; Kumar, S.; Varghese, D.; Kim, Y.S.; Roper, N.; Elloumi, F.; Pommier, Y.; Pacak, K.; Del Rivero, J. New Insights on the genetics of pheochromocytoma and paraganglioma and its clinical implications. Cancers 2022, 14, 594–608. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).