
Differential Effects of Human Immunodeficiency Virus Nef Variants on Pulmonary Vascular Endothelial Cell Dysfunction

 ,
,  , , and
, , and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Transfections of Vascular Cells with HIV Nef Constructs
2.3. Gene Expression Analyses
2.4. Quantitation of Inflammatory Cytokines
2.5. Endothelial Nitric Oxide Synthase (eNOS) Measurement
2.6. Pulmonary Vascular Cell Co-Culture and Proliferation Monitoring
2.7. Western Blot Analysis
3. Results
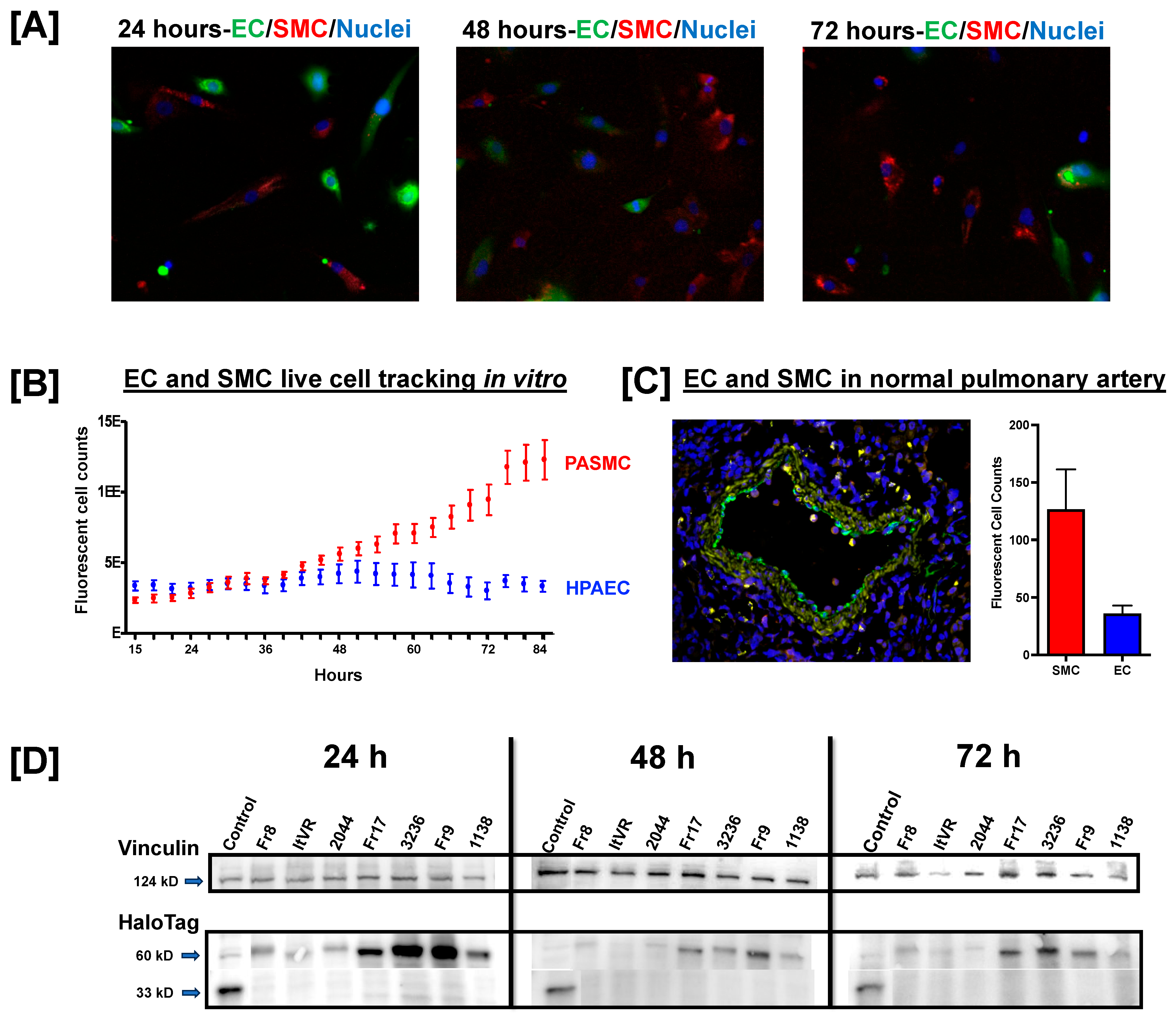
3.1. Pulmonary Vascular Direct Culture System
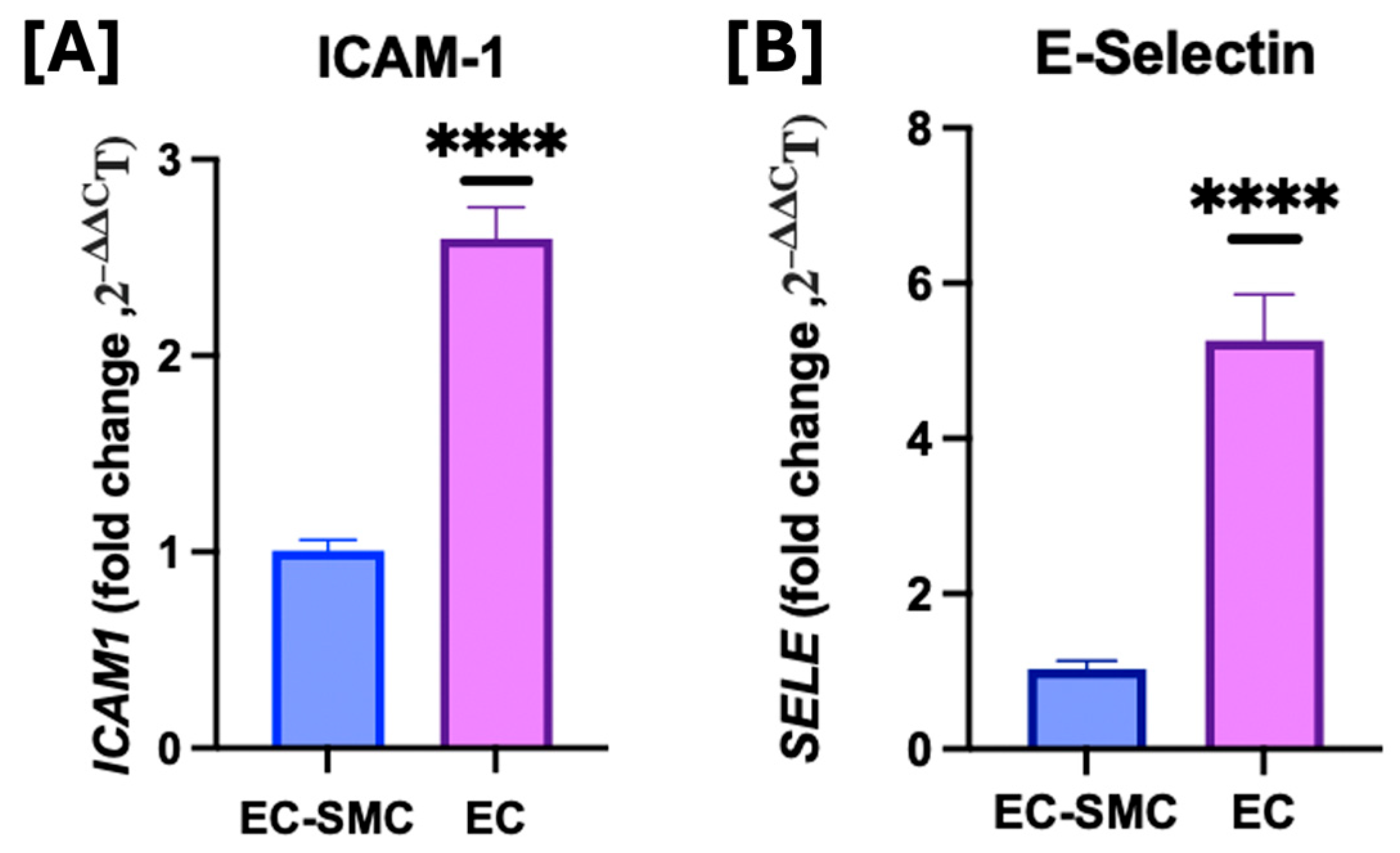
3.2. Effects of Co-Culture Model on Cell Adhesion Molecules
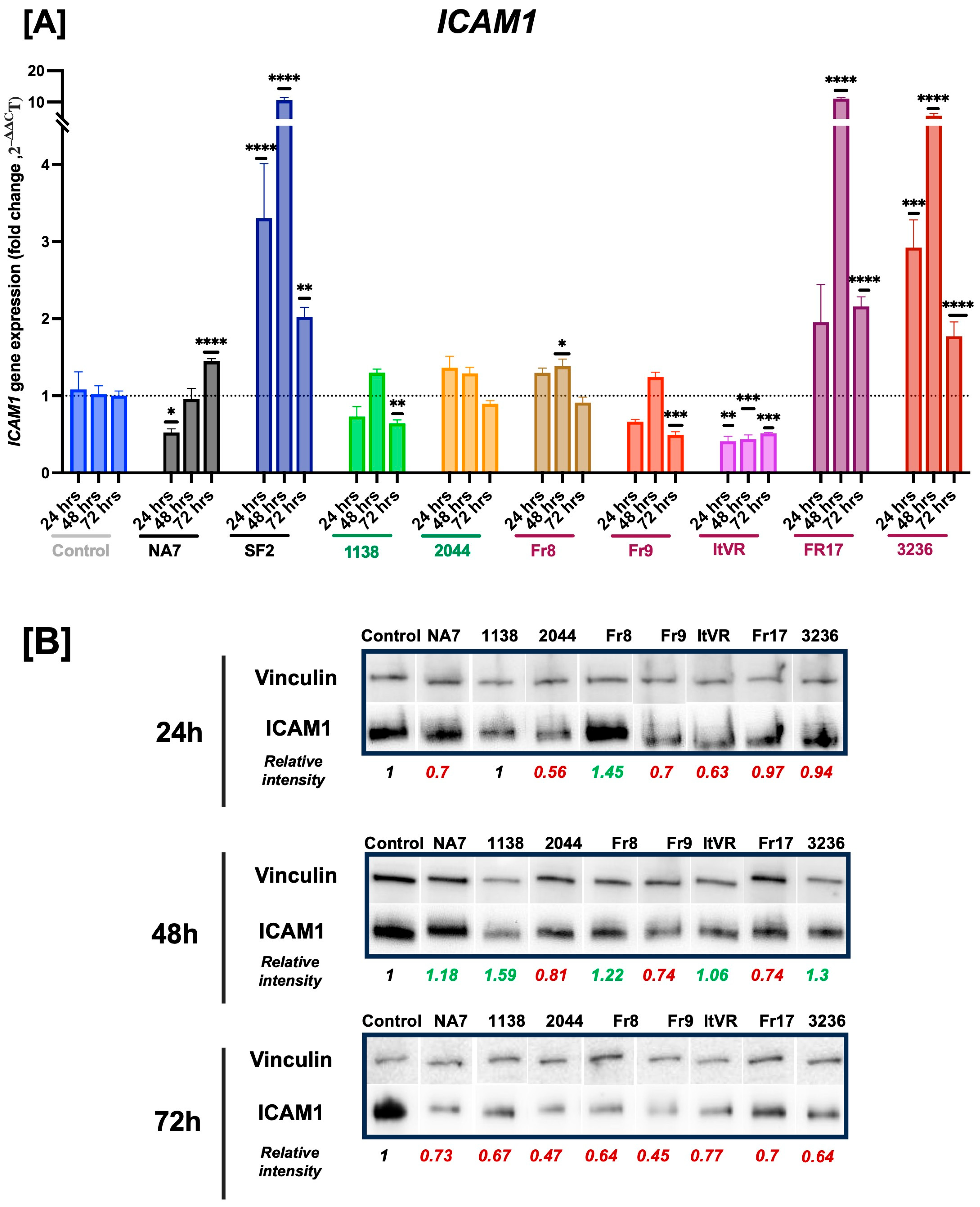
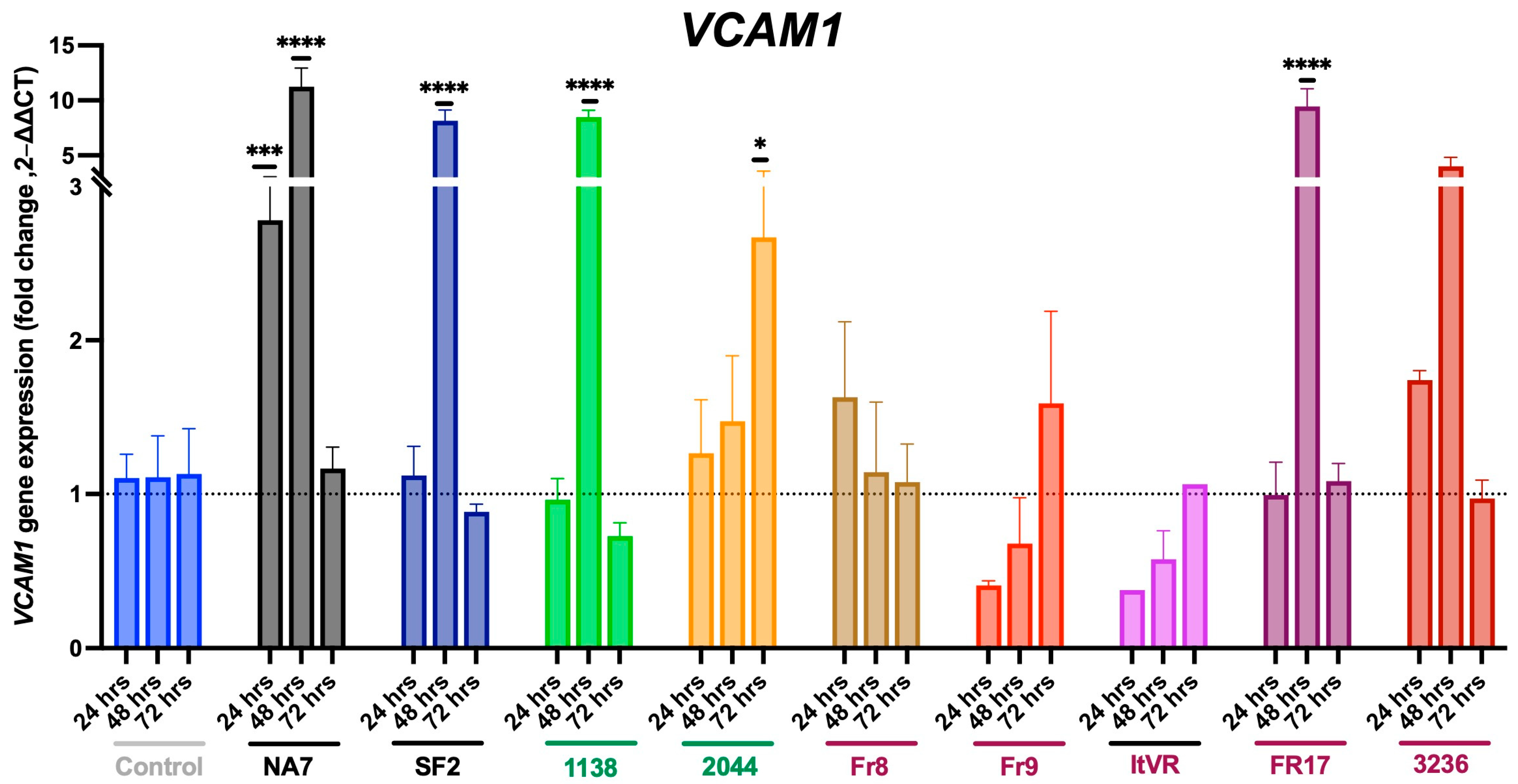
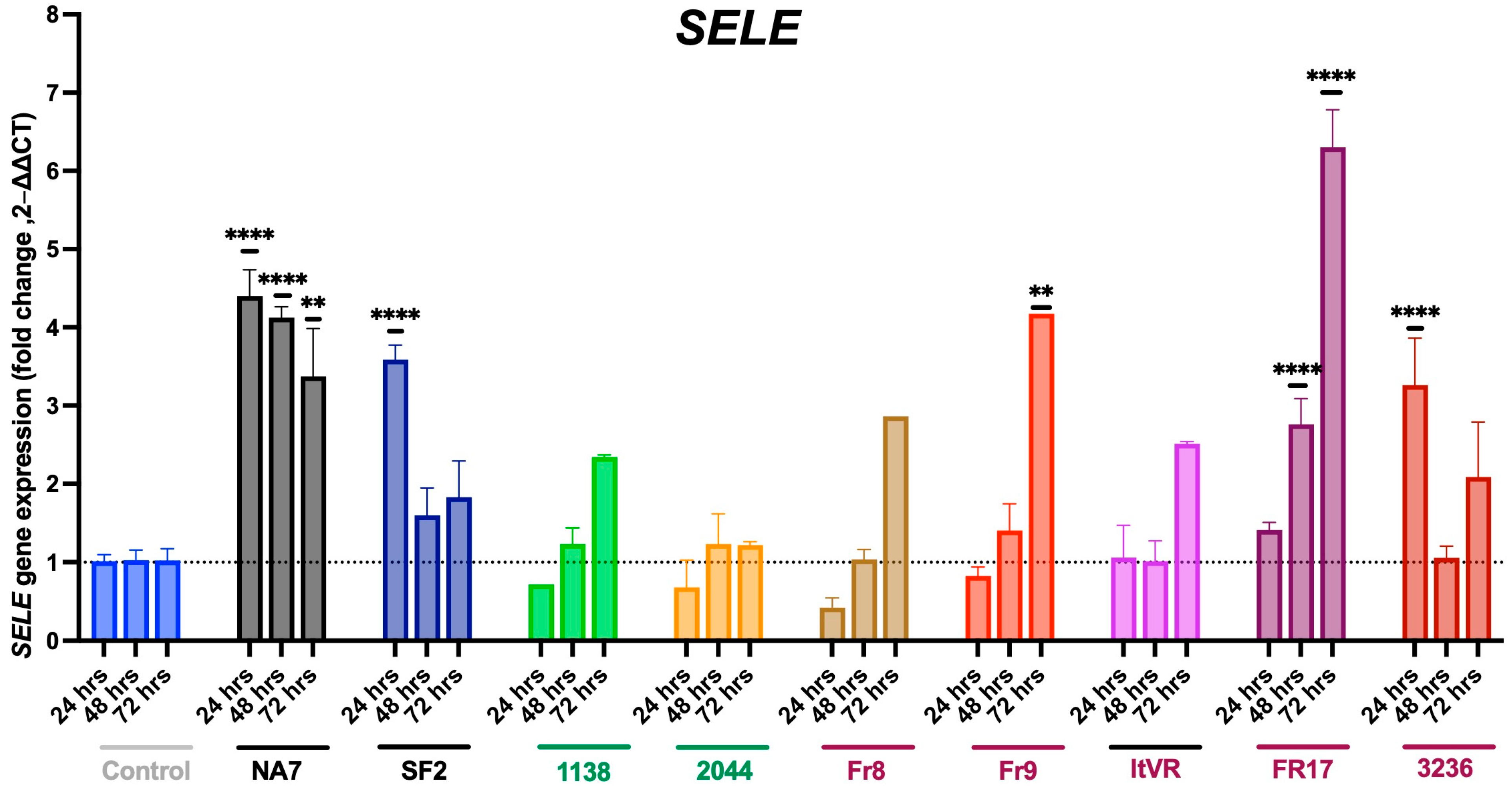
3.3. HIV Nef Impairs Pulmonary Vascular Cell Activation
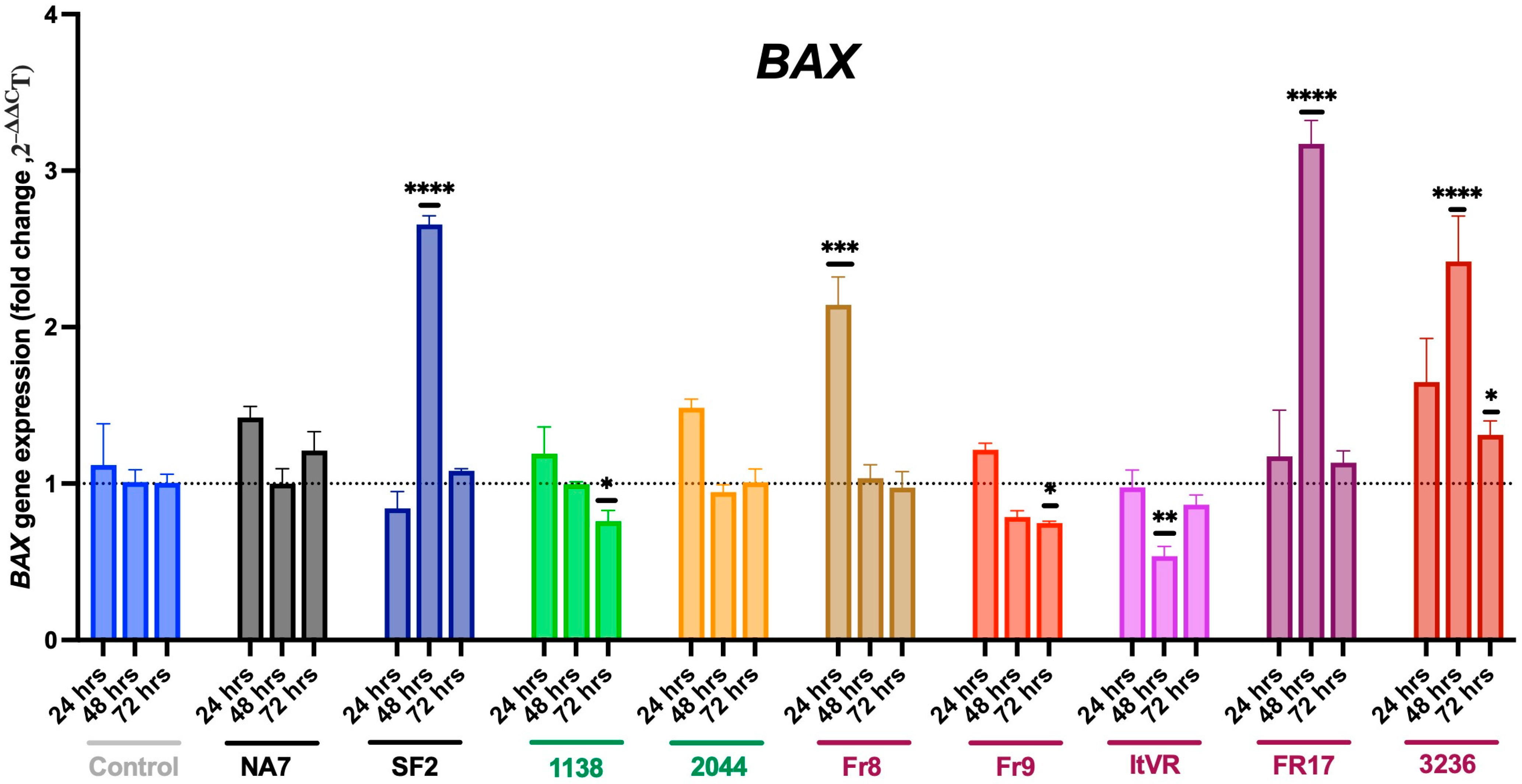
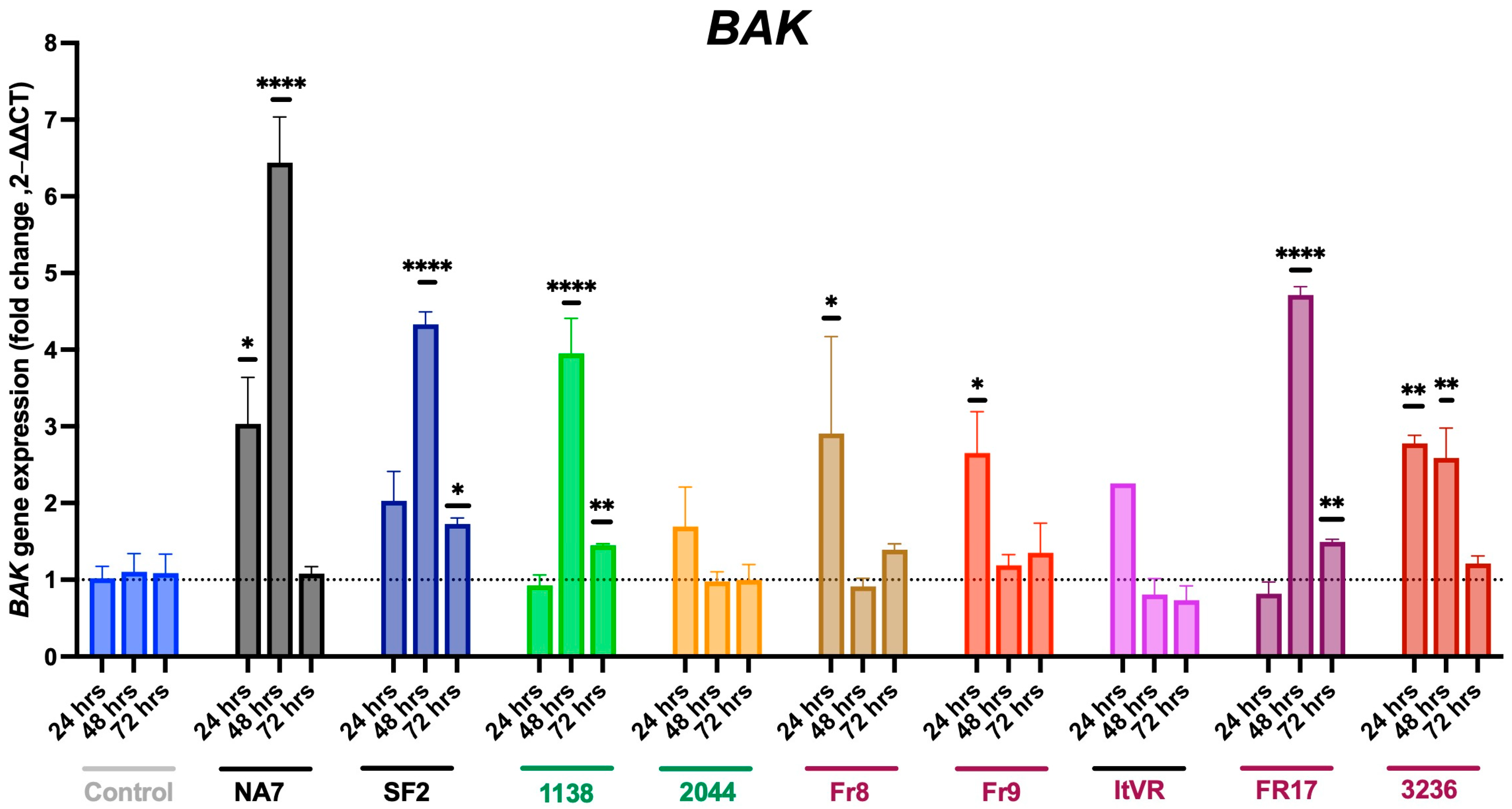
3.4. Expression of Pro-Apoptotic BAX and BAK Genes in Nef-Treated Cells
3.5. Expression of Vasoconstrictive Endothelin-1 Gene in Nef-Transfected Pulmonary Vascular Cells
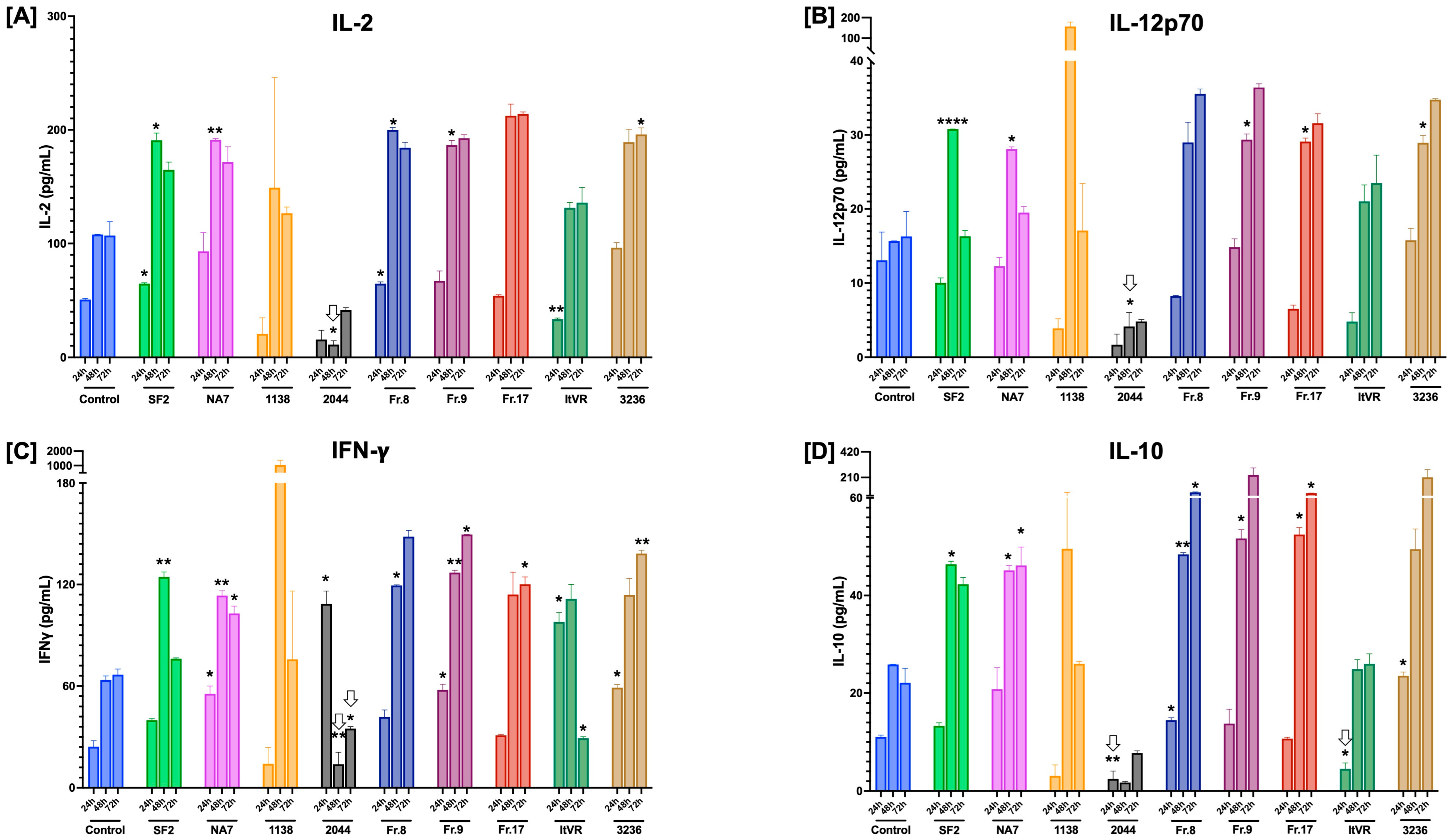
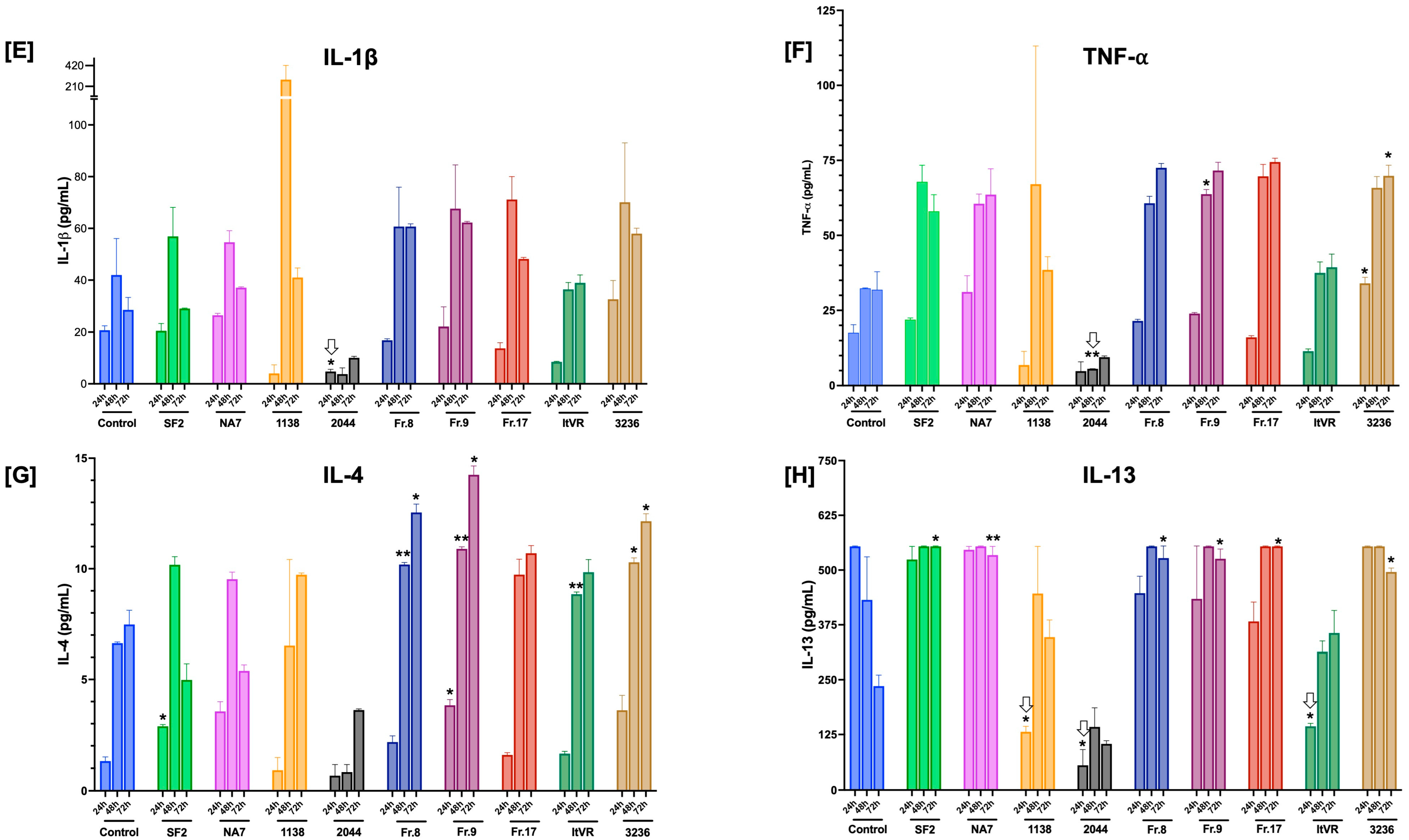
3.6. Characterization of Inflammatory Cytokine Release Patterns in Nef-Treated Pulmonary Vascular Cells
3.7. HIV Nef Induces a Transient Increase in Endothelial Nitric Oxide Synthase (eNOS) Production in Pulmonary Vascular Cells In Vitro
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| BAX | BCL2 associated X, apoptosis regulator |
| COPD | Chronic Obstructive Pulmonary Disease |
| DAF-2DA | 4,5-diaminofluorescein diacetate |
| EDN1 | Endothelin-1 |
| eNOS | endothelial Nitric Oxide Synthase |
| HIV | Human Immunodeficiency Virus |
| HIV-PH | Pulmonary Hypertension associated with HIV |
| ICAM1 | Intercellular Cell Adhesion Molecule-1 |
| MHC-1 | Major Histocompatibility Complex-1 |
| Nef | HIV NEgative regulatory Factor |
| PH | Pulmonary Hypertension |
| PLWH | People living with Human Immunodeficiency Virus |
| RPLP0 | Ribosomal protein lateral stalk subunit P0 |
| SELE | E-selectin |
| VCAM1 | Vascular Cell Adhesion Molecule-1 |
| VEGF | Vascular Endothelial Growth Factor |
References
- Trickey, A.; May, M.T.; Vehreschild, J.-J.; Obel, N.; Gill, M.J.; Crane, H.M.; Boesecke, C.; Patterson, S.; Grabar, S.; Cazanave, C.; et al. Survival of HIV-positive patients starting antiretroviral therapy between 1996 and 2013: A collaborative analysis of cohort studies. Lancet HIV 2017, 4, e349–e356. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, R.T.; Landovitz, R.J.; Sax, P.E.; Smith, D.M.; Springer, S.A.; Günthard, H.F.; Thompson, M.A.; Bedimo, R.J.; Benson, C.A.; Buchbinder, S.P.; et al. Antiretroviral Drugs for Treatment and Prevention of HIV in Adults: 2024 Recommendations of the International Antiviral Society–USA Panel. JAMA 2024, 333, 609–628. [Google Scholar] [CrossRef] [PubMed]
- Edwards, J.K.; Cole, S.R.; Breger, T.L.; Rudolph, J.E.; Filiatreau, L.M.; Buchacz, K.; Humes, E.; Rebeiro, P.F.; D’Souza, G.; Gill, M.J.; et al. Mortality Among Persons Entering HIV Care Compared with the General U.S. Population: An Observational Study. Ann. Intern. Med. 2021, 174, 1197–1206. [Google Scholar] [CrossRef] [PubMed]
- Jarrett, H.; Barnett, C. HIV-associated pulmonary hypertension. Curr. Opin. HIV AIDS 2017, 12, 566–571. [Google Scholar] [CrossRef]
- Thienemann, F.; Katoto, P.; Azibani, F.; Kodogo, V.; Mukasa, S.L.; Sani, M.U.; Karaye, K.M.; Mbanze, I.; Mocumbi, A.O.; Dzudie, A.; et al. Long-Term Follow-up of Human Immunodeficiency Virus-Associated Pulmonary Hypertension: Clinical Features and Survival Outcomes of the Pan Africa Pulmonary Hypertension Cohort (PAPUCO). Open Forum Infect. Dis. 2022, 9, ofac604. [Google Scholar] [CrossRef]
- Palakeel, J.J.; Ali, M.; Chaduvula, P.; Chhabra, S.; Lamsal Lamichhane, S.; Ramesh, V.; Opara, C.O.; Khan, F.Y.; Kabiraj, G.; Kauser, H.; et al. An Outlook on the Etiopathogenesis of Pulmonary Hypertension in HIV. Cureus 2022, 14, e27390. [Google Scholar] [CrossRef]
- Humbert, M.; Sitbon, O.; Chaouat, A.; Bertocchi, M.; Habib, G.; Gressin, V.; Yaici, A.; Weitzenblum, E.; Cordier, J.F.; Chabot, F.; et al. Pulmonary arterial hypertension in France: Results from a national registry. Am. J. Respir. Crit. Care Med. 2006, 173, 1023–1030. [Google Scholar] [CrossRef]
- Huluka, D.K.; Mekonnen, D.; Abebe, S.; Meshesha, A.; Mekonnen, D.; Deyessa, N.; Klinger, J.R.; Ventetuolo, C.E.; Schluger, N.W.; Sherman, C.B.; et al. Prevalence and risk factors of pulmonary hypertension among adult patients with HIV infection in Ethiopia. Pulm. Circ. 2020, 10, 2045894020971518. [Google Scholar] [CrossRef]
- Isasti, G.; Moreno, T.; Pérez, I.; Cabrera, F.; Palacios, R.; Santos, J. High prevalence of pulmonary arterial hypertension in a cohort of asymptomatic HIV-infected patients. AIDS Res. Hum. Retroviruses 2013, 29, 231–234. [Google Scholar] [CrossRef]
- Bigna, J.J.; Nansseu, J.R.; Noubiap, J.J. Pulmonary hypertension in the global population of adolescents and adults living with HIV: A systematic review and meta-analysis. Sci. Rep. 2019, 9, 7837. [Google Scholar] [CrossRef]
- Bigna, J.J.; Nansseu, J.R.; Um, L.N.; Noumegni, S.R.; Sime, P.S.; Aminde, L.N.; Koulla-Shiro, S.; Noubiap, J.J. Prevalence and incidence of pulmonary hypertension among HIV-infected people in Africa: A systematic review and meta-analysis. BMJ Open 2016, 6, e011921. [Google Scholar] [CrossRef] [PubMed]
- Jerjes-Sánchez, C.; Ramírez-Rivera, A.; Hernandez, N.Z.; Cueto Robledo, G.; García-Aguilar, H.; Gutiérrez-Fajardo, P.; Seoane García de León, M.; Moreno Hoyos-Abril, F.; Ernesto Beltrán Gámez, M.; Elizalde, J.; et al. Demographic, hemodynamic characteristics, and therapeutic trends of pulmonary hypertension patients: The Pulmonary Hypertension Mexican registry (REMEHIP). Pulm. Circ. 2024, 14, e12395. [Google Scholar] [CrossRef] [PubMed]
- Petrosillo, N.; Chinello, P.; Cicalini, S. Pulmonary hypertension in individuals with HIV infection. Aids 2006, 20, 2128–2129. [Google Scholar] [CrossRef] [PubMed]
- Youssef, M.; Boutros Salama, M.; Rehman, N.; Hanna, C.; Waniss, M.R.; Mbuagbaw, L. Pulmonary hypertension survival and hospitalisations in people living with HIV: A systematic review and meta-analysis. BMJ Open Respir. Res. 2024, 11, e002318. [Google Scholar] [CrossRef]
- Paternò Raddusa, M.S.; Marino, A.; Celesia, B.M.; Spampinato, S.; Giarratana, C.; Venanzi Rullo, E.; Cacopardo, B.; Nunnari, G. Atherosclerosis and Cardiovascular Complications in People Living with HIV: A Focused Review. Infect. Dis. Rep. 2024, 16, 846. [Google Scholar] [CrossRef]
- Kilroy, J.M.; Leal, A.A.; Henderson, A.J. Chronic HIV Transcription, Translation, and Persistent Inflammation. Viruses 2024, 16, 751. [Google Scholar] [CrossRef]
- Chelvanambi, S.; Bogatcheva, N.V.; Bednorz, M.; Agarwal, S.; Maier, B.; Alves, N.J.; Li, W.; Syed, F.; Saber, M.M.; Dahl, N.; et al. HIV-Nef Protein Persists in the Lungs of Aviremic Patients with HIV and Induces Endothelial Cell Death. Am. J. Respir. Cell Mol. Biol. 2019, 60, 357–366. [Google Scholar] [CrossRef]
- Wang, T.; Yi, R.; Green, L.A.; Chelvanambi, S.; Seimetz, M.; Clauss, M. Increased cardiovascular disease risk in the HIV-positive population on ART: Potential role of HIV-Nef and Tat. Cardiovasc. Pathol. 2015, 24, 279–282. [Google Scholar] [CrossRef]
- Lee, J.E.; Patel, K.; Almodovar, S.; Tuder, R.M.; Flores, S.C.; Sehgal, P.B. Dependence of Golgi apparatus integrity on nitric oxide in vascular cells: Implications in pulmonary arterial hypertension. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H1141–H1158. [Google Scholar] [CrossRef]
- Sehgal, P.B.; Mukhopadhyay, S.; Patel, K.; Xu, F.; Almodovar, S.; Tuder, R.M.; Flores, S.C. Golgi dysfunction is a common feature in idiopathic human pulmonary hypertension and vascular lesions in SHIV-nef-infected macaques. Am. J. Physiol. Lung Cell Mol. Physiol. 2009, 297, L729–L737. [Google Scholar] [CrossRef]
- Campbell, T.D.; Khan, M.; Huang, M.B.; Bond, V.C.; Powell, M.D. HIV-1 Nef protein is secreted into vesicles that can fuse with target cells and virions. Ethn. Dis. 2008, 18, S2-14-19. [Google Scholar]
- Clauss, M.; Chelvanambi, S.; Cook, C.; ElMergawy, R.; Dhillon, N. Viral Bad News Sent by EVAIL. Viruses 2021, 13, 1168. [Google Scholar] [CrossRef] [PubMed]
- Lenassi, M.; Cagney, G.; Liao, M.; Vaupotic, T.; Bartholomeeusen, K.; Cheng, Y.; Krogan, N.J.; Plemenitas, A.; Peterlin, B.M. HIV Nef is secreted in exosomes and triggers apoptosis in bystander CD4+ T cells. Traffic 2010, 11, 110–122. [Google Scholar] [CrossRef] [PubMed]
- Sami Saribas, A.; Cicalese, S.; Ahooyi, T.M.; Khalili, K.; Amini, S.; Sariyer, I.K. HIV-1 Nef is released in extracellular vesicles derived from astrocytes: Evidence for Nef-mediated neurotoxicity. Cell Death Dis. 2018, 8, e2542. [Google Scholar] [CrossRef]
- Wang, T.; Green, L.A.; Gupta, S.K.; Kim, C.; Wang, L.; Almodovar, S.; Flores, S.C.; Prudovsky, I.A.; Jolicoeur, P.; Liu, Z.; et al. Transfer of intracellular HIV Nef to endothelium causes endothelial dysfunction. PLoS ONE 2014, 9, e91063. [Google Scholar] [CrossRef]
- Marecki, J.C.; Cool, C.D.; Parr, J.E.; Beckey, V.E.; Luciw, P.A.; Tarantal, A.F.; Carville, A.; Shannon, R.P.; Cota-Gomez, A.; Tuder, R.M.; et al. HIV-1 Nef is associated with complex pulmonary vascular lesions in SHIV-nef-infected macaques. Am. J. Respir. Crit. Care Med. 2006, 174, 437–445. [Google Scholar] [CrossRef]
- Almodovar, S.; Knight, R.; Allshouse, A.A.; Roemer, S.; Lozupone, C.; McDonald, D.; Widmann, J.; Voelkel, N.F.; Shelton, R.J.; Suarez, E.B.; et al. Human Immunodeficiency Virus nef signature sequences are associated with pulmonary hypertension. AIDS Res. Hum. Retroviruses 2012, 28, 607–618. [Google Scholar] [CrossRef]
- Roeth, J.F.; Williams, M.; Kasper, M.R.; Filzen, T.M.; Collins, K.L. HIV-1 Nef disrupts MHC-I trafficking by recruiting AP-1 to the MHC-I cytoplasmic tail. J. Cell Biol. 2004, 167, 903–913. [Google Scholar] [CrossRef]
- Grzesiek, S.; Stahl, S.J.; Wingfield, P.T.; Bax, A. The CD4 determinant for downregulation by HIV-1 Nef directly binds to Nef. Mapping of the Nef binding surface by NMR. Biochemistry 1996, 35, 10256–10261. [Google Scholar] [CrossRef]
- Piguet, V.; Wan, L.; Borel, C.; Mangasarian, A.; Demaurex, N.; Thomas, G.; Trono, D. HIV-1 Nef protein binds to the cellular protein PACS-1 to downregulate class I major histocompatibility complexes. Nat. Cell Biol. 2000, 2, 163–167. [Google Scholar] [CrossRef]
- Shugars, D.C.; Smith, M.S.; Glueck, D.H.; Nantermet, P.V.; Seillier-Moiseiwitsch, F.; Swanstrom, R. Analysis of human immunodeficiency virus type 1 nef gene sequences present in vivo. J. Virol. 1993, 67, 4639–4650. [Google Scholar] [CrossRef] [PubMed]
- Saksela, K.; Cheng, G.; Baltimore, D. Proline-rich (PxxP) motifs in HIV-1 Nef bind to SH3 domains of a subset of Src kinases and are required for the enhanced growth of Nef+ viruses but not for down-regulation of CD4. EMBO J. 1995, 14, 484–491. [Google Scholar] [CrossRef] [PubMed]
- Wallace, C.S.; Truskey, G.A. Direct-contact co-culture between smooth muscle and endothelial cells inhibits TNF-alpha-mediated endothelial cell activation. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H338–H346. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Liu, C.; Qin, X.; Wang, Y.; Han, Y.; Zhou, Y. The role of ERK1/2 signaling pathway in Nef protein upregulation of the expression of the intercellular adhesion molecule 1 in endothelial cells. Angiology 2010, 61, 669–678. [Google Scholar] [CrossRef]
- Chelvanambi, S.; Gupta, S.K.; Chen, X.; Ellis, B.W.; Maier, B.F.; Colbert, T.M.; Kuriakose, J.; Zorlutuna, P.; Jolicoeur, P.; Obukhov, A.G.; et al. HIV-Nef Protein Transfer to Endothelial Cells Requires Rac1 Activation and Leads to Endothelial Dysfunction Implications for Statin Treatment in HIV Patients. Circ. Res. 2019, 125, 805–820. [Google Scholar] [CrossRef]
- Duffy, P.; Wang, X.; Lin, P.H.; Yao, Q.; Chen, C. HIV Nef protein causes endothelial dysfunction in porcine pulmonary arteries and human pulmonary artery endothelial cells. J. Surg. Res. 2009, 156, 257–264. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Gene ID | Protein Name | Primer Sequence | Amplicon Size |
|---|---|---|---|---|
| BAX | 581 | BCL2 associated X, apoptosis regulator | Fwd: 5′-GGACGAACTGGACAGTAACA-3′ Rev: 5′-ACCACCCTGGTCTTGGAT-3′ | 271 bp |
| BAK | 578 | BCL2 antagonist/killer 1 | Fwd: 5′-ACGCTATGACTCAGAGTTCC-3′ Rev: 5′-CTTCGTACCACAAACTGGCC-3′ | 360 bp |
| EDN1 | 1906 | Endothelin-1 | Fwd: 5′-AGAGTGTGTCTACTTCTGCC-3′ Rev: 5′-GTTGTGGGTCACATAACG-3′ | 442 bp |
| ICAM1 | 3383 | Intercellular cell adhesion molecule-1 | Fwd: 5′-AGCCAGTGGGCAAGAACCTT-3′ Rev: 5′-CGGCACGAGAAATTGGCTCC-3′ | 187 bp |
| SELE | 6401 | E-selectin | Fwd: 5′-GGCAGTGGACACAGCAAATC-3′ Rev: 5′-TGGACAGCATCGCATCTCA-3′ | 224 bp |
| VCAM1 | 7412 | Vascular cell adhesion molecule-1 | Fwd: 5′-TGGTCGTGATCCTTGGAGCC-3′ Rev: 5′-AGATGTGgTCCCCTCATTCGT-3′ | 221 bp |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garcia, A.K.; Lujea, N.C.; Baig, J.; Heath, E.; Nguyen, M.T.; Rodriguez, M.; Campbell, P.; Castro Piedras, I.; Suarez Martinez, E.; Almodovar, S. Differential Effects of Human Immunodeficiency Virus Nef Variants on Pulmonary Vascular Endothelial Cell Dysfunction. Infect. Dis. Rep. 2025, 17, 65. https://doi.org/10.3390/idr17030065
Garcia AK, Lujea NC, Baig J, Heath E, Nguyen MT, Rodriguez M, Campbell P, Castro Piedras I, Suarez Martinez E, Almodovar S. Differential Effects of Human Immunodeficiency Virus Nef Variants on Pulmonary Vascular Endothelial Cell Dysfunction. Infectious Disease Reports. 2025; 17(3):65. https://doi.org/10.3390/idr17030065
Chicago/Turabian StyleGarcia, Amanda K., Noelia C. Lujea, Javaria Baig, Eli Heath, Minh T. Nguyen, Mario Rodriguez, Preston Campbell, Isabel Castro Piedras, Edu Suarez Martinez, and Sharilyn Almodovar. 2025. "Differential Effects of Human Immunodeficiency Virus Nef Variants on Pulmonary Vascular Endothelial Cell Dysfunction" Infectious Disease Reports 17, no. 3: 65. https://doi.org/10.3390/idr17030065
APA StyleGarcia, A. K., Lujea, N. C., Baig, J., Heath, E., Nguyen, M. T., Rodriguez, M., Campbell, P., Castro Piedras, I., Suarez Martinez, E., & Almodovar, S. (2025). Differential Effects of Human Immunodeficiency Virus Nef Variants on Pulmonary Vascular Endothelial Cell Dysfunction. Infectious Disease Reports, 17(3), 65. https://doi.org/10.3390/idr17030065