
DNA Tweezers with Replaceable Clamps for the Targeted Degradation of Cell Membrane Proteins

 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
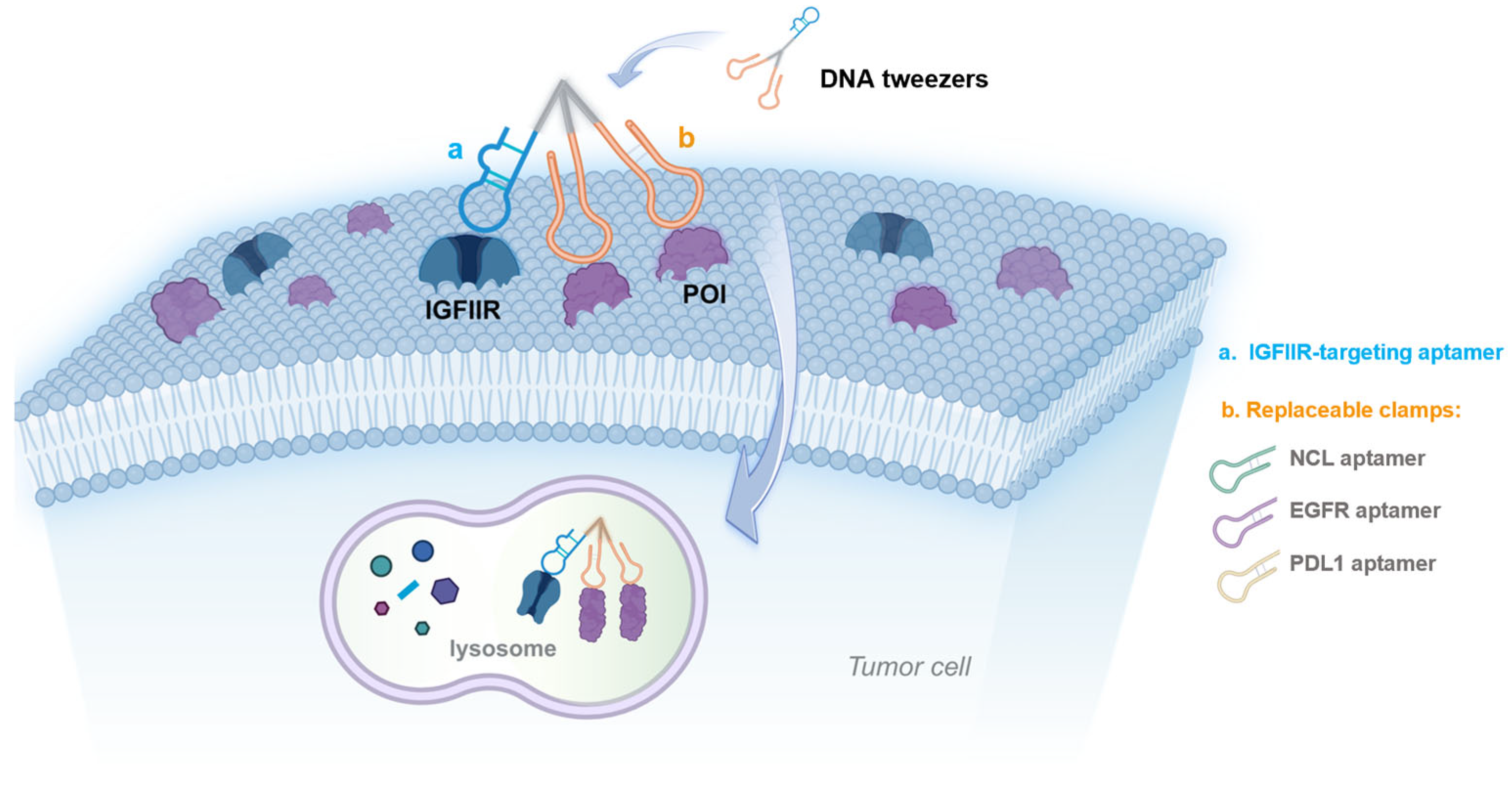
1. Introduction
2. Materials and Methods
2.1. Chemicals, Cell Lines, and Reagents
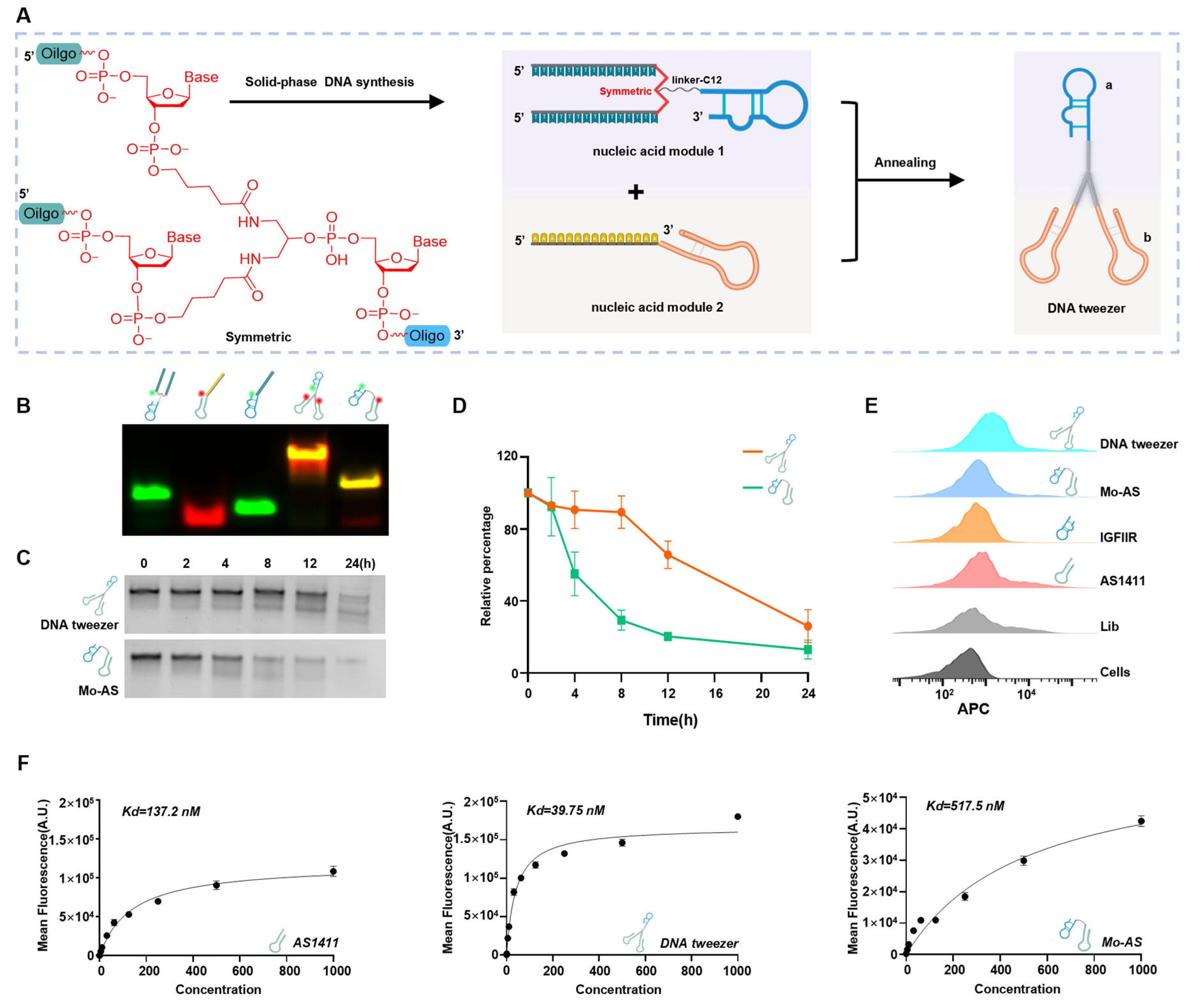
2.2. Construction of DNA Tweezers and Mo-AS in Buffer
2.3. Serum Stability Test
2.4. Flow Cytometry Analysis of Aptamer Binding to MCF-7 Cells
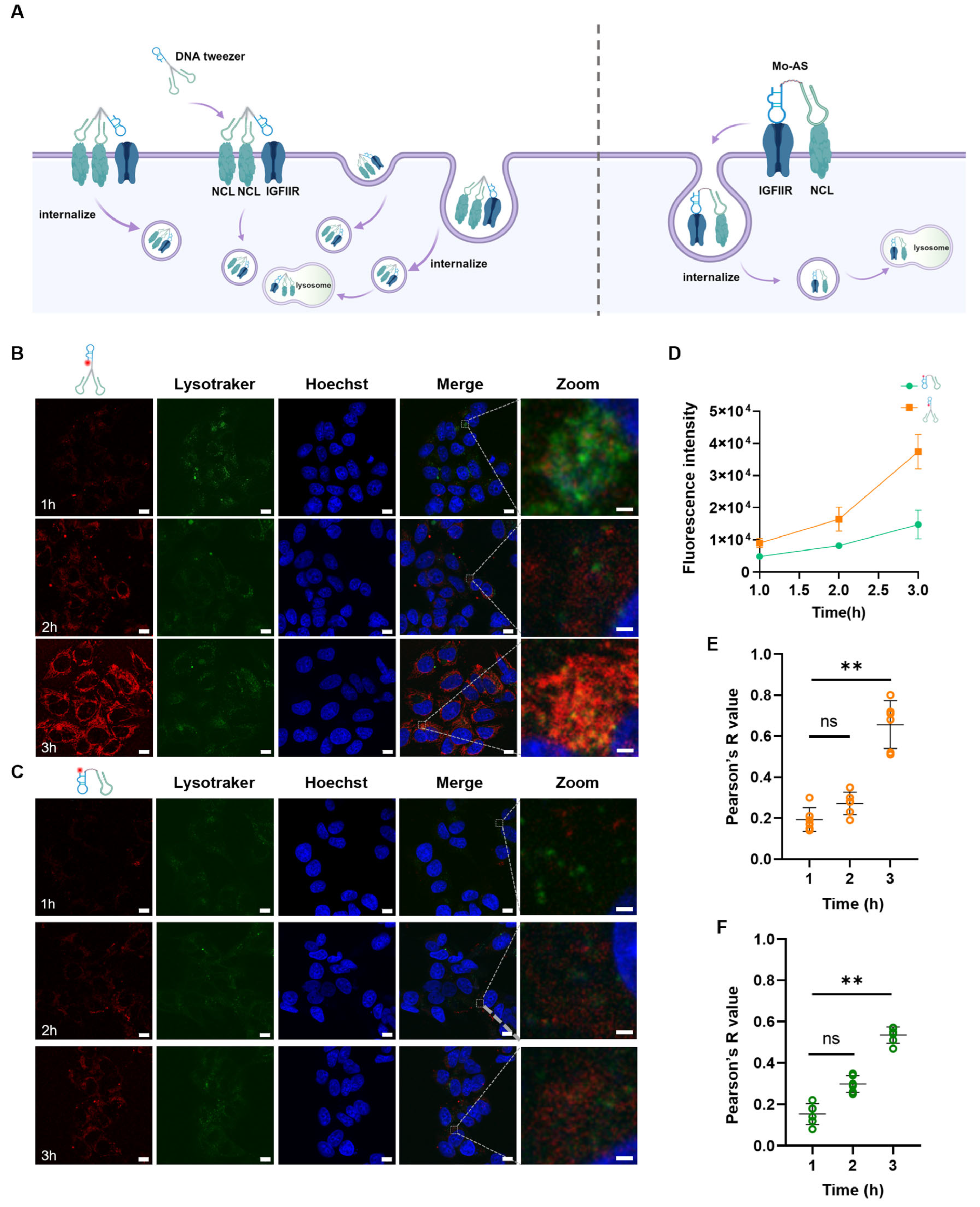
2.5. Lysosomal Colocalization Experiment Using Confocal Microscope
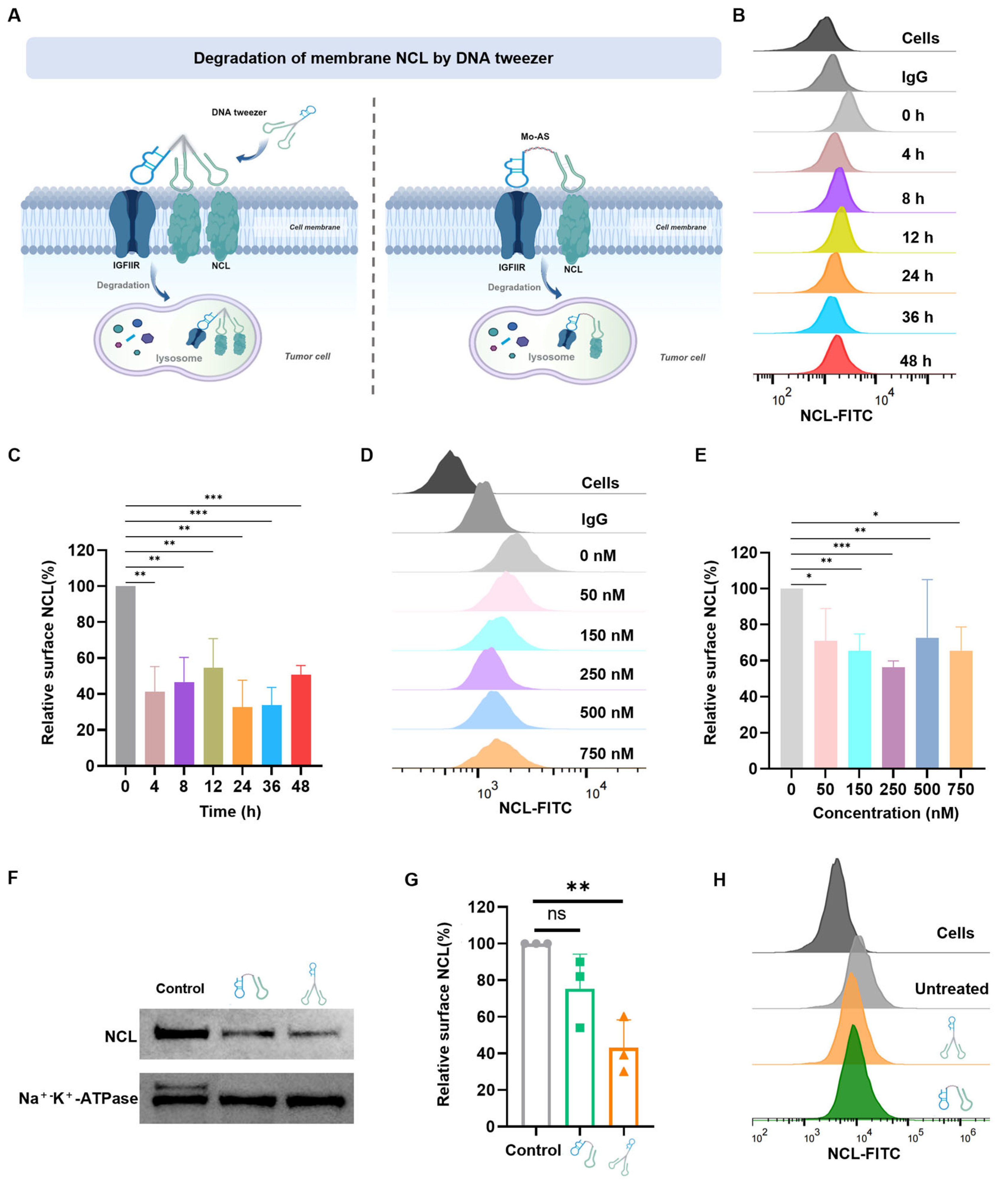
2.6. Detection of Cell-Surface Nucleolin by Flow Cytometry
2.7. Detection of Cell-Surface Nucleolin by Western Blot Analysis
2.8. Detection of Cell-Surface EGFR by Flow Cytometry
2.9. Western Blot Analysis
2.10. In Vivo Imaging
2.11. Immunohistochemistry
2.12. Animal Care and Husbandry
2.13. Xenograft Zebrafish Model
3. Results and Discussion
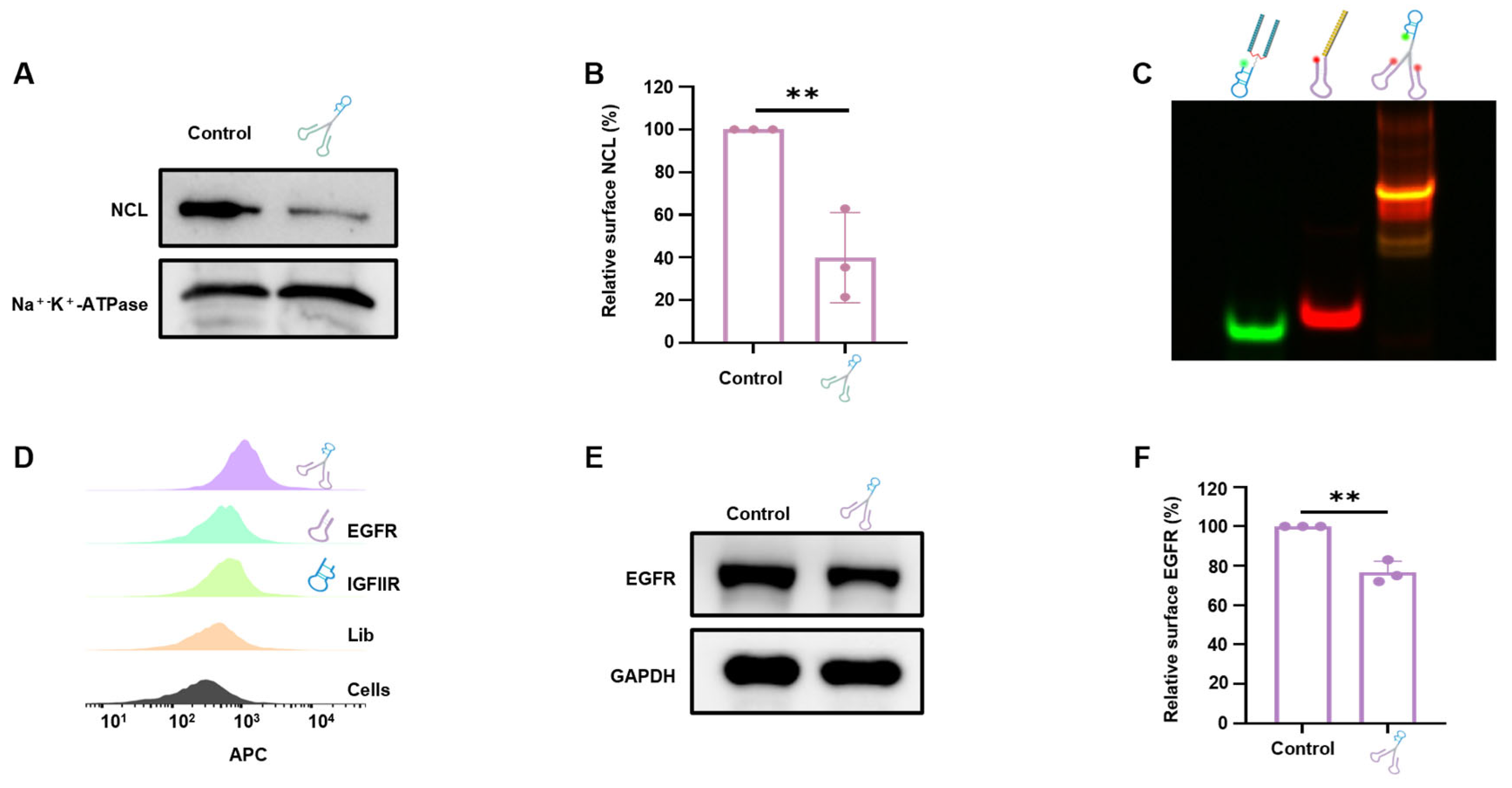
3.1. Synthesis and Characterization of the DNA Tweezers
3.2. Internalization and Lysosomal Colocalization of the DNA Tweezers
3.3. DNA Tweezer-Mediated Degradation of Membrane NCL
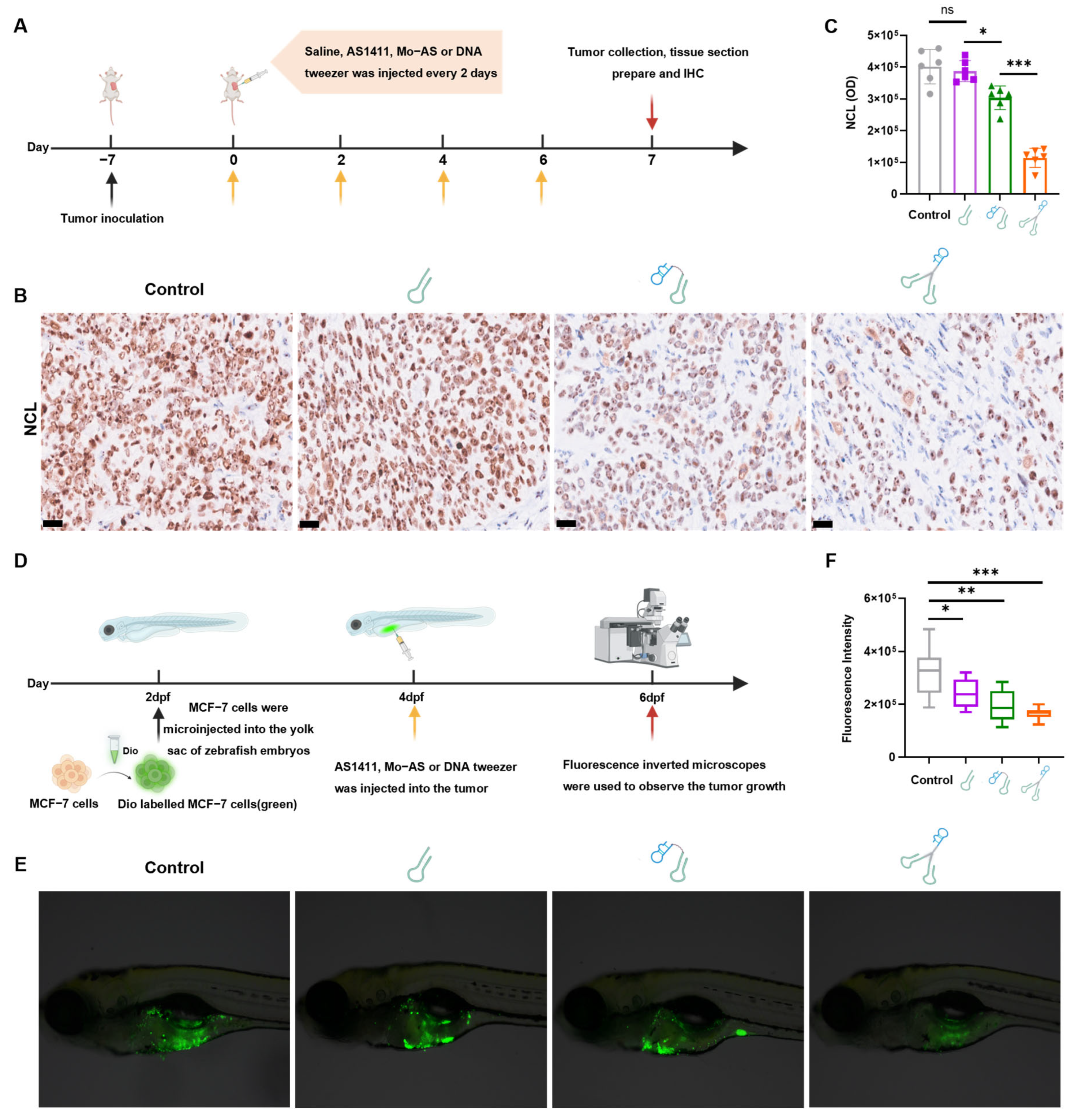
3.4. In Vivo Targeting and Biodistribution of the DNA Tweezers
3.5. In Vivo Interference Effect of DNA Tweezers on NCL and Its Antitumor Efficacy
3.6. Generality of Using DNA Tweezers for Protein Degradation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| LYTACs | Lysosome-targeting chimeras |
| IGFIIR | Insulin-like growth factor-2 receptor |
| NCL | Nucleolin |
| dpf | Days postfertilization |
| EGFR | Epidermal growth factor receptor |
| PDL1 | Programmed death ligand 1 |
| APS | Ammonium persulphate |
| DEPC | Diethyl pyrocarbonate |
| Mo-AS | Monovalent-AS1411 |
| RIPA | Radio immunoprecipitation assay |
| BCA | Bicinchoninic acid assay |
| TBST | Tris-borate-sodium Tween-20 |
| ECL | Electrochemiluminescence |
| HRP | Horseradish peroxidase |
| DAB | Diaminobenzidine |
| DiO | 3,3′-dioctadecyloxacarbocyanine perchlorate |
| FAS | Factor-related Apoptosis |
References
- Fagerberg, L.; Jonasson, K.; von Heijne, G.; Uhlén, M.; Berglund, L. Prediction of the human membrane proteome. Proteomics 2010, 10, 1141–1149. [Google Scholar] [CrossRef] [PubMed]
- Birch, J.; Cheruvara, H.; Gamage, N.; Harrison, P.J.; Lithgo, R.; Quigley, A. Changes in Membrane Protein Structural Biology. Biology 2020, 9, 401. [Google Scholar] [CrossRef] [PubMed]
- Rucevic, M.; Hixson, D.; Josic, D. Mammalian plasma membrane proteins as potential biomarkers and drug targets. Electrophoresis 2011, 32, 1549–1564. [Google Scholar] [CrossRef]
- Gomes, I.M.; Santos, C.R.; Maia, C.J. Expression of STEAP1 and STEAP1B in prostate cell lines, and the putative regulation of STEAP1 by post-transcriptional and post-translational mechanisms. Genes Cancer 2014, 5, 142–151. [Google Scholar] [CrossRef]
- Dale, B.; Cheng, M.; Park, K.S.; Kaniskan, H.; Xiong, Y.; Jin, J. Advancing targeted protein degradation for cancer therapy. Nat. Rev. Cancer 2021, 21, 638–654. [Google Scholar] [CrossRef]
- Neklesa, T.K.; Winkler, J.D.; Crews, C.M. Targeted protein degradation by PROTACs. Pharmacol. Ther. 2017, 174, 138–144. [Google Scholar] [CrossRef]
- Cromm, P.M.; Crews, C.M. Targeted Protein Degradation: From Chemical Biology to Drug Discovery. Cell Chem. Biol. 2017, 24, 1181–1190. [Google Scholar] [CrossRef]
- Li, X.; Song, Y. Proteolysis-targeting chimera (PROTAC) for targeted protein degradation and cancer therapy. J. Hematol. Oncol. 2020, 13, 50. [Google Scholar] [CrossRef] [PubMed]
- Banik, S.M.; Pedram, K.; Wisnovsky, S.; Ahn, G.; Riley, N.M.; Bertozzi, C.R. Lysosome-targeting chimaeras for degradation of extracellular proteins. Nature 2020, 584, 291–297. [Google Scholar] [CrossRef]
- Caianiello, D.F.; Zhang, M.; Ray, J.D.; Howell, R.A.; Swartzel, J.C.; Branham, E.M.J.; Chirkin, E.; Sabbasani, V.R.; Gong, A.Z.; McDonald, D.M.; et al. Bifunctional small molecules that mediate the degradation of extracellular proteins. Nat. Chem. Biol. 2021, 17, 947–953. [Google Scholar] [CrossRef]
- Zhou, Y.; Teng, P.; Montgomery, N.T.; Li, X.; Tang, W. Development of Triantennary N-Acetylgalactosamine Conjugates as Degraders for Extracellular Proteins. ACS Cent. Sci. 2021, 7, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.; Liu, B.; Liu, J.; Zhang, J.; Chen, K.; Liu, H. Cell-specific aptamers and their conjugation with nanomaterials for targeted drug delivery. Expert. Opin. Drug Deliv. 2015, 12, 493–506. [Google Scholar] [CrossRef] [PubMed]
- Kinghorn, A.B.; Fraser, L.A.; Lang, S.; Shiu, S.C.C.; Tanner, J.A. Aptamer Bioinformatics. Int. J. Mol. Sci. 2017, 18, 2516. [Google Scholar] [CrossRef]
- Miao, Y.; Gao, Q.; Mao, M.; Zhang, C.; Yang, L.; Yang, Y.; Han, D. Bispecific Aptamer Chimeras Enable Targeted Protein Degradation on Cell Membranes. Angew. Chem. Int. Ed. Engl. 2021, 60, 11267–11271. [Google Scholar] [CrossRef]
- Li, Y.; Liu, X.; Yu, L.; Huang, X.; Wang, X.; Han, D.; Yang, Y.; Liu, Z. Covalent LYTAC Enabled by DNA Aptamers for Immune Checkpoint Degradation Therapy. J. Am. Chem. Soc. 2023, 145, 24506–24521. [Google Scholar] [CrossRef]
- Fang, Y.; Zhang, Y.; Bi, S.; Peng, B.; Wang, C.; Ju, H.; Liu, Y. Securing LYTAC with Logic-Identification System for Cancer Cell-Selective Membrane Protein Degradation. Small 2024, 20, e2310039. [Google Scholar] [CrossRef]
- Sun, W.; Zhang, H.; Xie, W.; Ma, L.; Dang, Y.; Liu, Y.; Li, L.; Qu, F.; Tan, W. Development of Integrin-Facilitated Bispecific Aptamer Chimeras for Membrane Protein Degradation. J. Am. Chem. Soc. 2024, 146, 25490–25500. [Google Scholar] [CrossRef] [PubMed]
- Ahn, G.; Riley, N.M.; Kamber, R.A.; Wisnovsky, S.; Moncayo von Hase, S.; Bassik, M.C.; Banik, S.M.; Bertozzi, C.R. Elucidating the cellular determinants of targeted membrane protein degradation by lysosome-targeting chimeras. Science 2023, 382, eadf6249. [Google Scholar] [CrossRef]
- Sheng, W.; Chen, T.; Tan, W.; Fan, Z.H. Multivalent DNA nanospheres for enhanced capture of cancer cells in microfluidic devices. ACS Nano 2013, 7, 7067–7076. [Google Scholar] [CrossRef]
- Lin, Y.; Jiang, L.; Huang, Y.; Yang, Y.; He, Y.; Lu, C.; Yang, H. DNA-mediated reversible capture and release of circulating tumor cells with a multivalent dual-specific aptamer coating network. Chem. Commun. 2019, 55, 5387–5390. [Google Scholar] [CrossRef]
- Sun, Y.; Mo, L.; Hu, X.; Yu, D.; Xie, S.; Li, J.; Zhao, Z.; Fang, X.; Ye, M.; Qiu, L.; et al. Bispecific Aptamer-Based Recognition-then-Conjugation Strategy for PD1/PDL1 Axis Blockade and Enhanced Immunotherapy. ACS Nano 2022, 16, 21129–21138. [Google Scholar] [CrossRef] [PubMed]
- Cramer, P. Organization and regulation of gene transcription. Nature 2019, 573, 45–54. [Google Scholar] [CrossRef]
- Sun, Y.; Gao, F.; Yang, C.; Li, Y.; Jin, C.; Xie, S.; Lv, C.; Ding, D.; Han, D.; Li, J.; et al. Construction of Bispecific Aptamer-Drug Conjugate by a Hybrid Chemical and Biological Approach. Bioconjug Chem. 2020, 31, 1289–1294. [Google Scholar] [CrossRef]
- Li, S.; Zeng, T.; Wu, Z.; Huang, J.; Cao, X.; Liu, Y.; Bai, S.; Chen, Q.; Li, C.; Lu, C.; et al. DNA Tetrahedron-Driven Multivalent Proteolysis-Targeting Chimeras: Enhancing Protein Degradation Efficiency and Tumor Targeting. J. Am. Chem. Soc. 2025, 147, 2168–2181. [Google Scholar] [CrossRef]
- Chen, J.; Zuehlke, A.; Deng, B.; Peng, H.; Hou, X.; Zhang, H. A Target-Triggered DNAzyme Motor Enabling Homogeneous, Amplified Detection of Proteins. Anal. Chem. 2017, 89, 12888–12895. [Google Scholar] [CrossRef]
- Yue, L.; Wang, S.; Cecconello, A.; Lehn, J.M.; Willner, I. Orthogonal Operation of Constitutional Dynamic Networks Consisting of DNA-Tweezer Machines. ACS Nano 2017, 11, 12027–12036. [Google Scholar] [CrossRef]
- Thongchot, S.; Aksonnam, K.; Thuwajit, P.; Yenchitsomanus, P.T.; Thuwajit, C. Nucleolin-based targeting strategies in cancer treatment: Focus on cancer immunotherapy (Review). Int. J. Mol. Med. 2023, 52, 81. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Gao, W.; Wang, H.; Yang, J.; Zhang, Y.; Guo, S.; Bian, B.; Wang, H.; Wei, X.; Zhou, Y.; et al. The grading quality markers identification of Panax notoginseng under the guidance of traditional experience using untargeted metabolomics and anti-myocardial ischemia evaluation of zebrafish. Phytomedicine 2023, 111, 154674. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Miao, Y.; Guo, P.; Wang, J.; Han, D. Insulin-like Growth Factor 2-Tagged Aptamer Chimeras (ITACs) Modular Assembly for Targeted and Efficient Degradation of Two Membrane Proteins. Angew. Chem. Int. Ed. Engl. 2024, 63, e202316089. [Google Scholar] [CrossRef]
- Ahn, G.; Banik, S.M.; Bertozzi, C.R. Degradation from the outside in: Targeting extracellular and membrane proteins for degradation through the endolysosomal pathway. Cell Chem. Biol. 2021, 28, 1072–1080. [Google Scholar] [CrossRef]
- Pettersson, M.; Crews, C.M. PROteolysis TArgeting Chimeras (PROTACs)—Past, present and future. Drug Discov. Today Technol. 2019, 31, 15–27. [Google Scholar] [CrossRef] [PubMed]
- González-Cuesta, M.; Ortiz Mellet, C.; García Fernández, J.M. Carbohydrate supramolecular chemistry: Beyond the multivalent effect. Chem. Commun. 2020, 56, 5207–5222. [Google Scholar] [CrossRef]
- Wise, J.F.; Berkova, Z.; Mathur, R.; Zhu, H.; Braun, F.K.; Tao, R.H.; Sabichi, A.L.; Ao, X.; Maeng, H.; Samaniego, F. Nucleolin inhibits Fas ligand binding and suppresses Fas-mediated apoptosis in vivo via a surface nucleolin-Fas complex. Blood 2013, 121, 4729–4739. [Google Scholar] [CrossRef]
- Chen, X.; Liu, M.; Lou, H.; Lu, Y.; Zhou, M.T.; Ou, R.; Xu, Y.; Tang, K.F. Degradation of endogenous proteins and generation of a null-like phenotype in zebrafish using Trim-Away technology. Genome Biol. 2019, 20, 19. [Google Scholar] [CrossRef]
- Taha, M.S.; Nouri, K.; Milroy, L.G.; Moll, J.M.; Herrmann, C.; Brunsveld, L.; Piekorz, R.P.; Ahmadian, M.R. Subcellular fractionation and localization studies reveal a direct interaction of the fragile X mental retardation protein (FMRP) with nucleolin. PLoS ONE 2014, 9, e91465. [Google Scholar] [CrossRef] [PubMed]
- Ginisty, H.; Sicard, H.; Roger, B.; Bouvet, P. Structure and functions of nucleolin. J. Cell Sci. 1999, 112 Pt 6, 761–772. [Google Scholar] [CrossRef]
- Gauci, S.; Helbig, A.O.; Slijper, M.; Krijgsveld, J.; Heck, A.J.; Mohammed, S. Lys-N and trypsin cover complementary parts of the phosphoproteome in a refined SCX-based approach. Anal. Chem. 2009, 81, 4493–4501. [Google Scholar] [CrossRef] [PubMed]
- Vu, T.H.; Jirtle, R.L.; Hoffman, A.R. Cross-species clues of an epigenetic imprinting regulatory code for the IGF2R gene. Cytogenet. Genome Res. 2006, 113, 202–208. [Google Scholar] [CrossRef]
- Yaghoobi, E.; Zavvar, T.; Ramezani, M.; Alibolandi, M.; Rahimzadeh Oskuei, S.; Zahiri, M.; Alinezhad Nameghi, M.; Abnous, K.; Taghdisi, S.M. A multi-storey DNA nanostructure containing doxorubicin and AS1411 aptamer for targeting breast cancer cells. J. Drug Target. 2022, 30, 1106–1112. [Google Scholar] [CrossRef]
- Harjes, U. EGFR is going circular. Nat. Rev. Cancer 2021, 21, 280. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, Y.; Huang, Y.; Chen, D.; Hu, S.; Pan, T.; Liu, Y.; Wang, R.; Tan, W. DNA Tweezers with Replaceable Clamps for the Targeted Degradation of Cell Membrane Proteins. Pharmaceutics 2025, 17, 785. https://doi.org/10.3390/pharmaceutics17060785
Sun Y, Huang Y, Chen D, Hu S, Pan T, Liu Y, Wang R, Tan W. DNA Tweezers with Replaceable Clamps for the Targeted Degradation of Cell Membrane Proteins. Pharmaceutics. 2025; 17(6):785. https://doi.org/10.3390/pharmaceutics17060785
Chicago/Turabian StyleSun, Yang, Yichen Huang, Daiquan Chen, Shangjiu Hu, Tao Pan, Yuanding Liu, Ruowen Wang, and Weihong Tan. 2025. "DNA Tweezers with Replaceable Clamps for the Targeted Degradation of Cell Membrane Proteins" Pharmaceutics 17, no. 6: 785. https://doi.org/10.3390/pharmaceutics17060785
APA StyleSun, Y., Huang, Y., Chen, D., Hu, S., Pan, T., Liu, Y., Wang, R., & Tan, W. (2025). DNA Tweezers with Replaceable Clamps for the Targeted Degradation of Cell Membrane Proteins. Pharmaceutics, 17(6), 785. https://doi.org/10.3390/pharmaceutics17060785