
Cyclodextrin Complexation of Fenofibrate by Co-Grinding Method and Monitoring the Process Using Complementary Analytical Tools

 , , ,
, , ,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Preparation of Co-Ground and Kneaded Products
2.2.2. Differential Scanning Calorimetry
2.2.3. X-ray Powder Diffractometry
2.2.4. Infrared Spectroscopy
2.2.5. Correlation Analysis
2.2.6. Scanning Electron Microscopy
2.2.7. In Vitro Dissolution Rate Studies
2.2.8. In Vitro Diffusion Studies
3. Results and Discussion
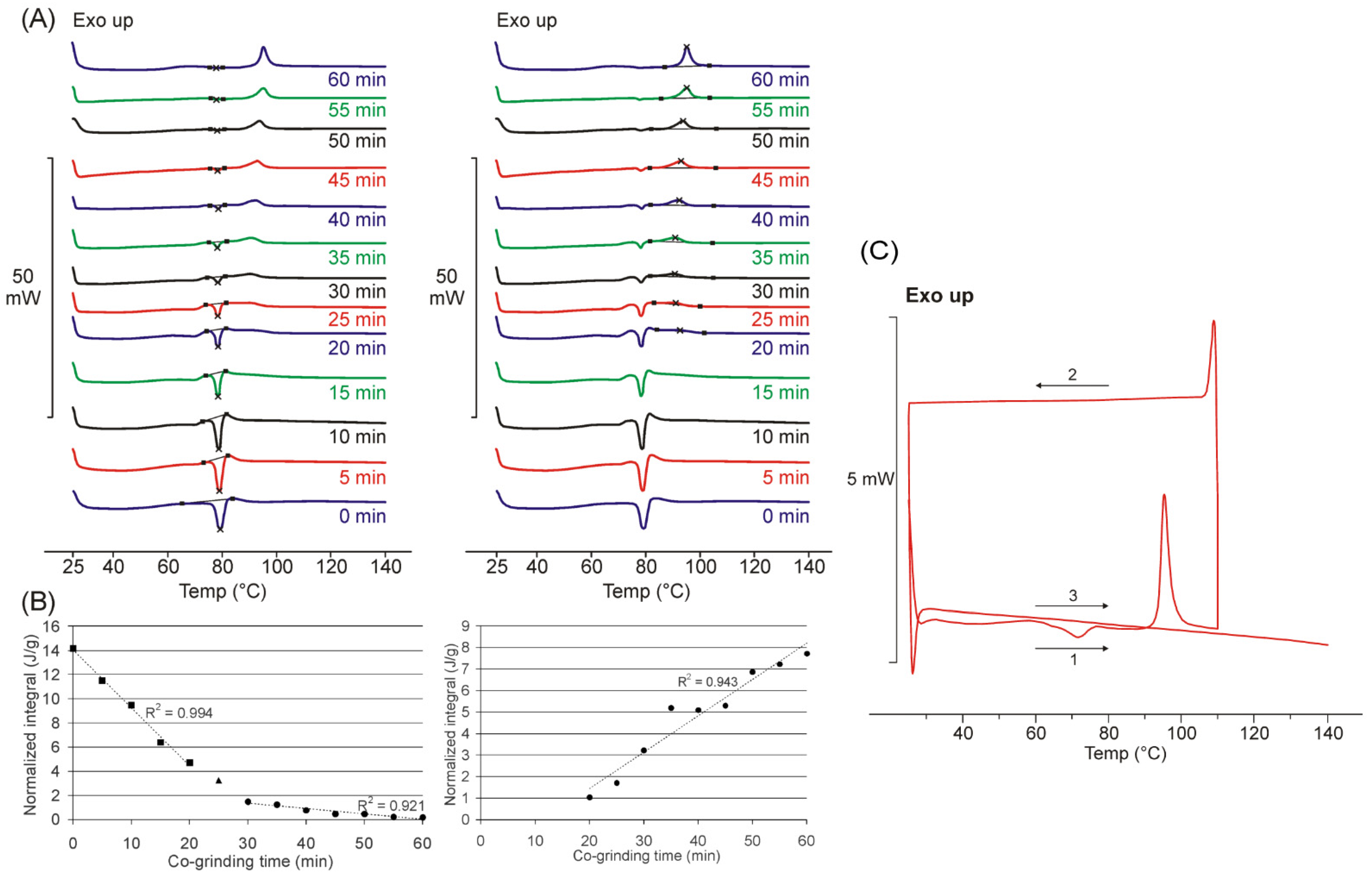
3.1. Differential Scanning Calorimetry
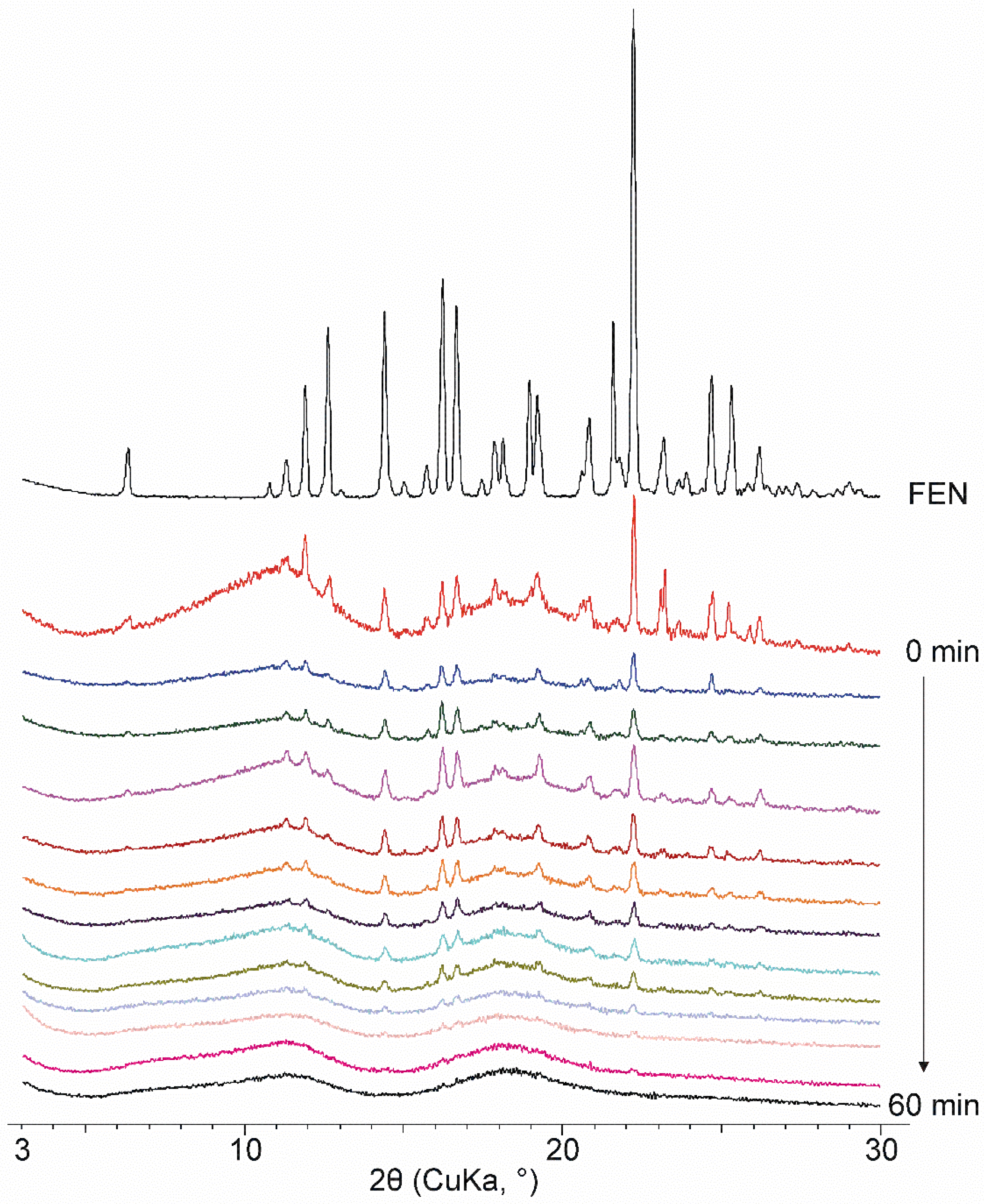
3.2. XRPD
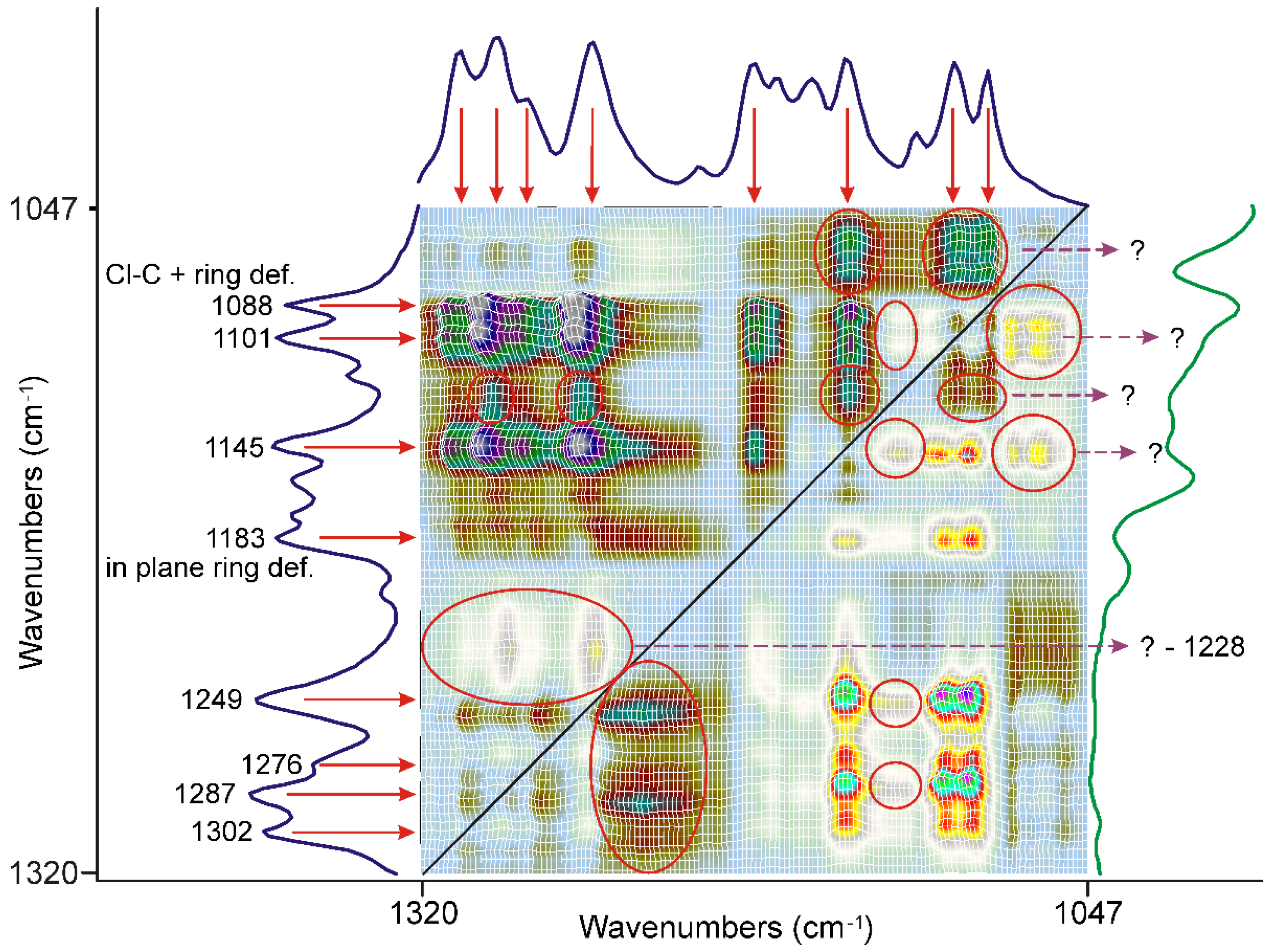
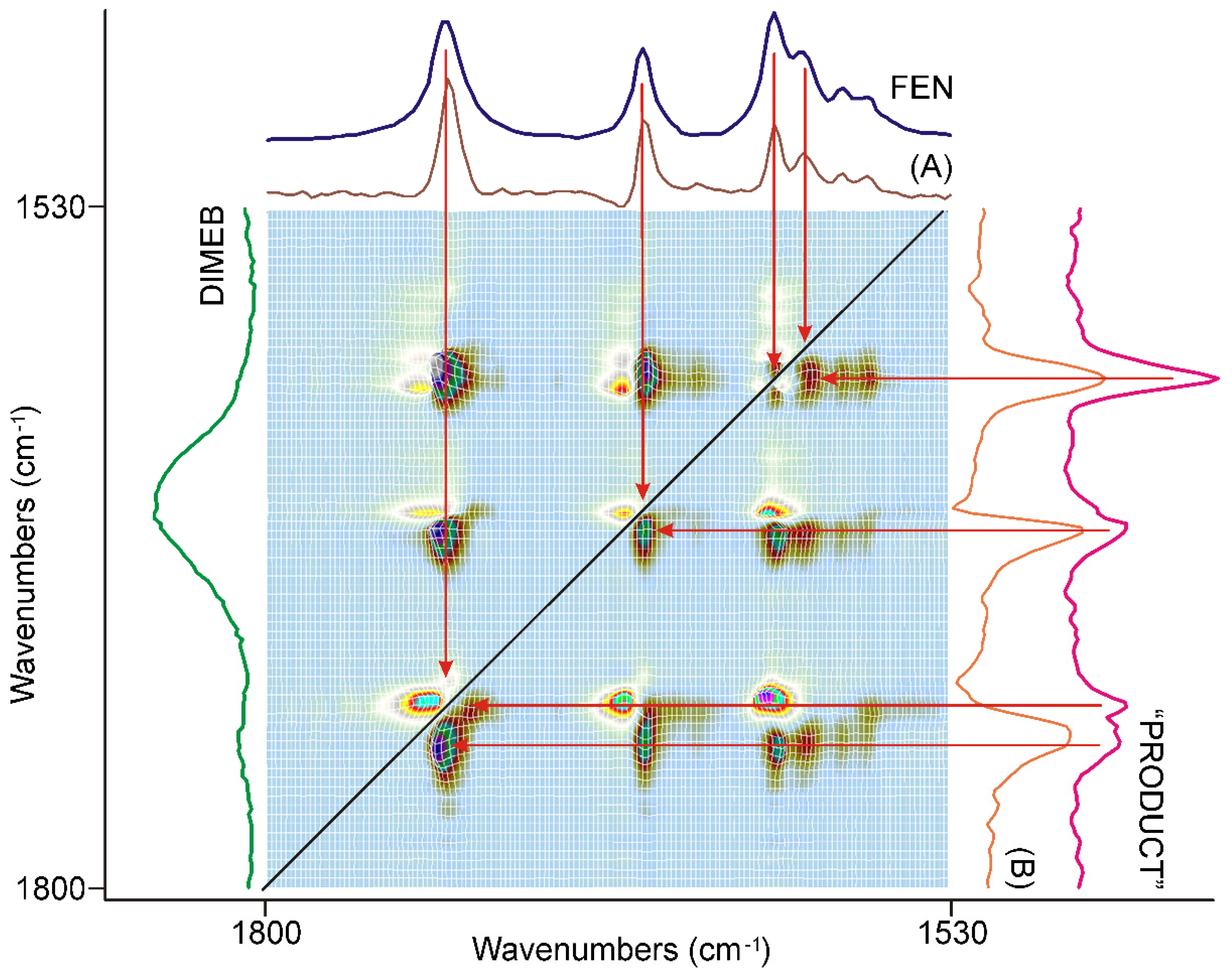
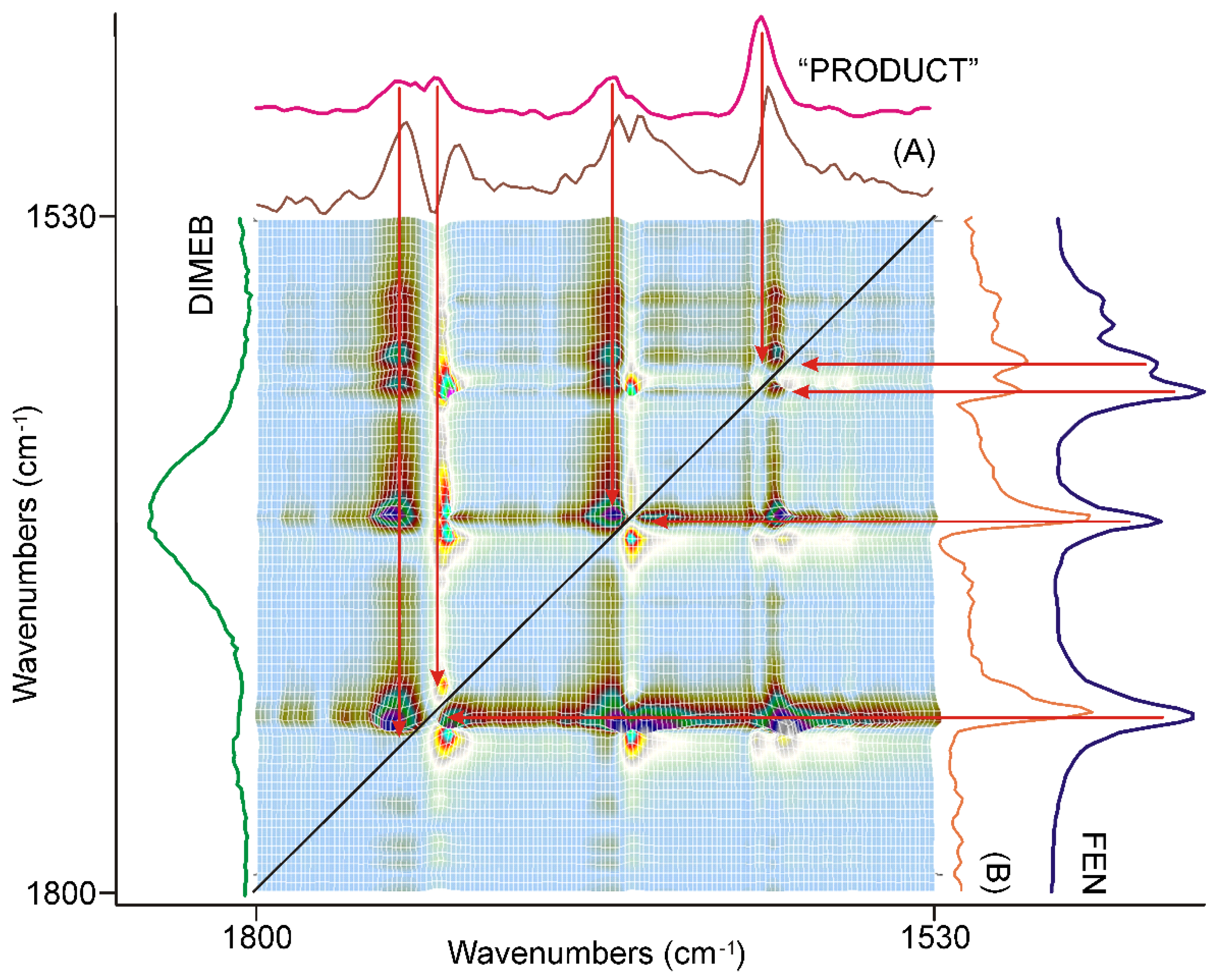
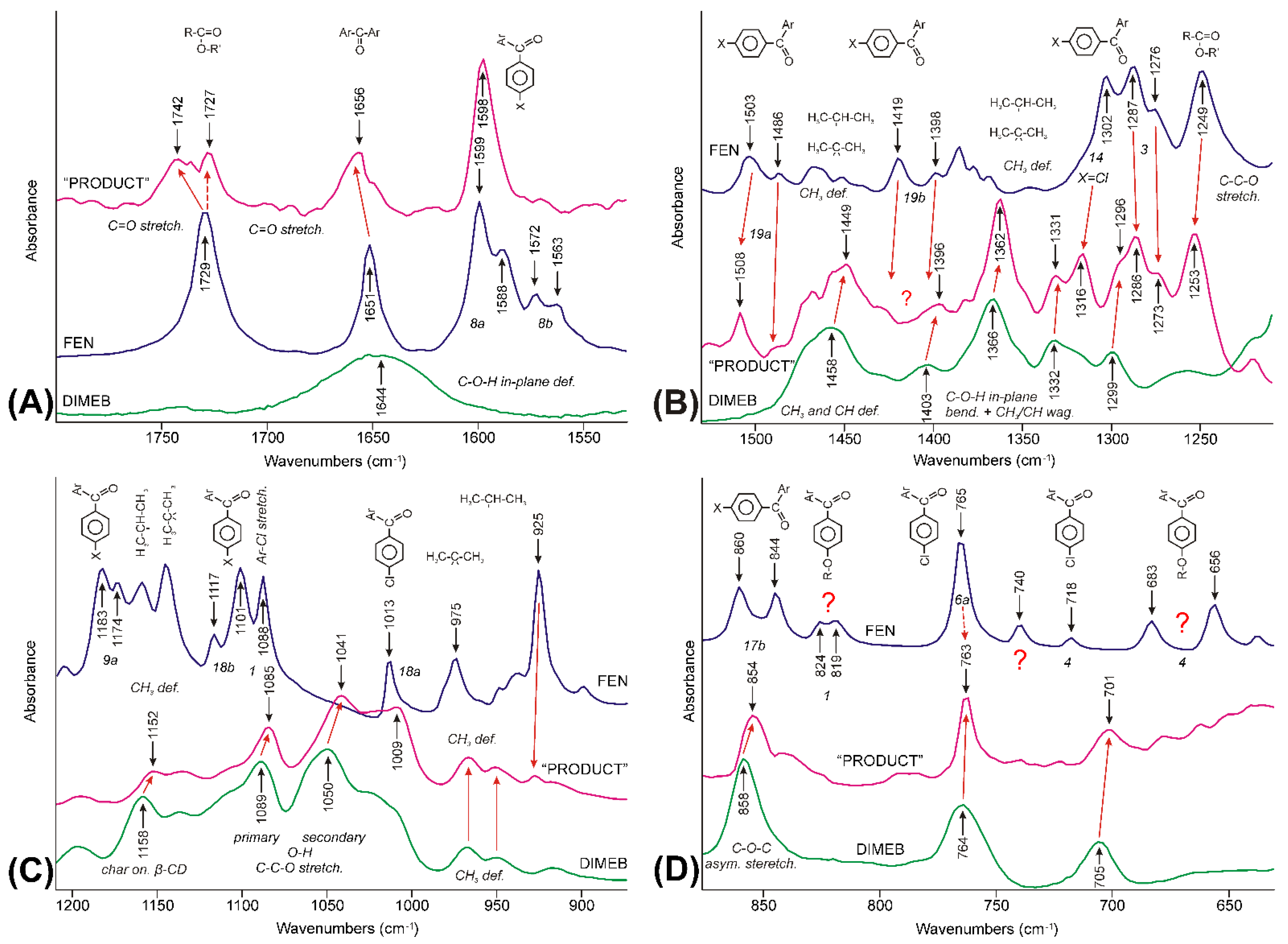
3.3. Evaluation of IR Spectroscopic Data
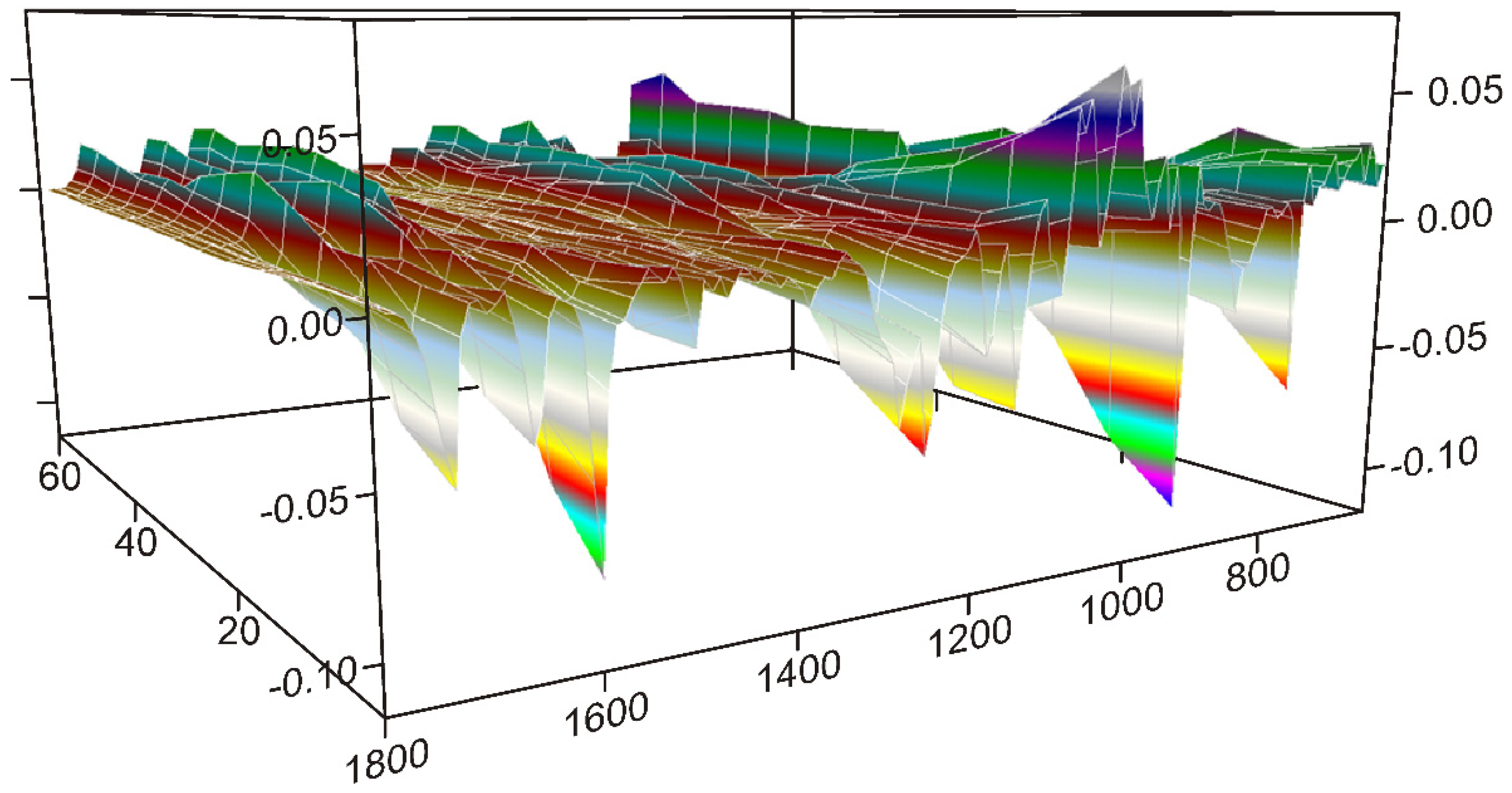
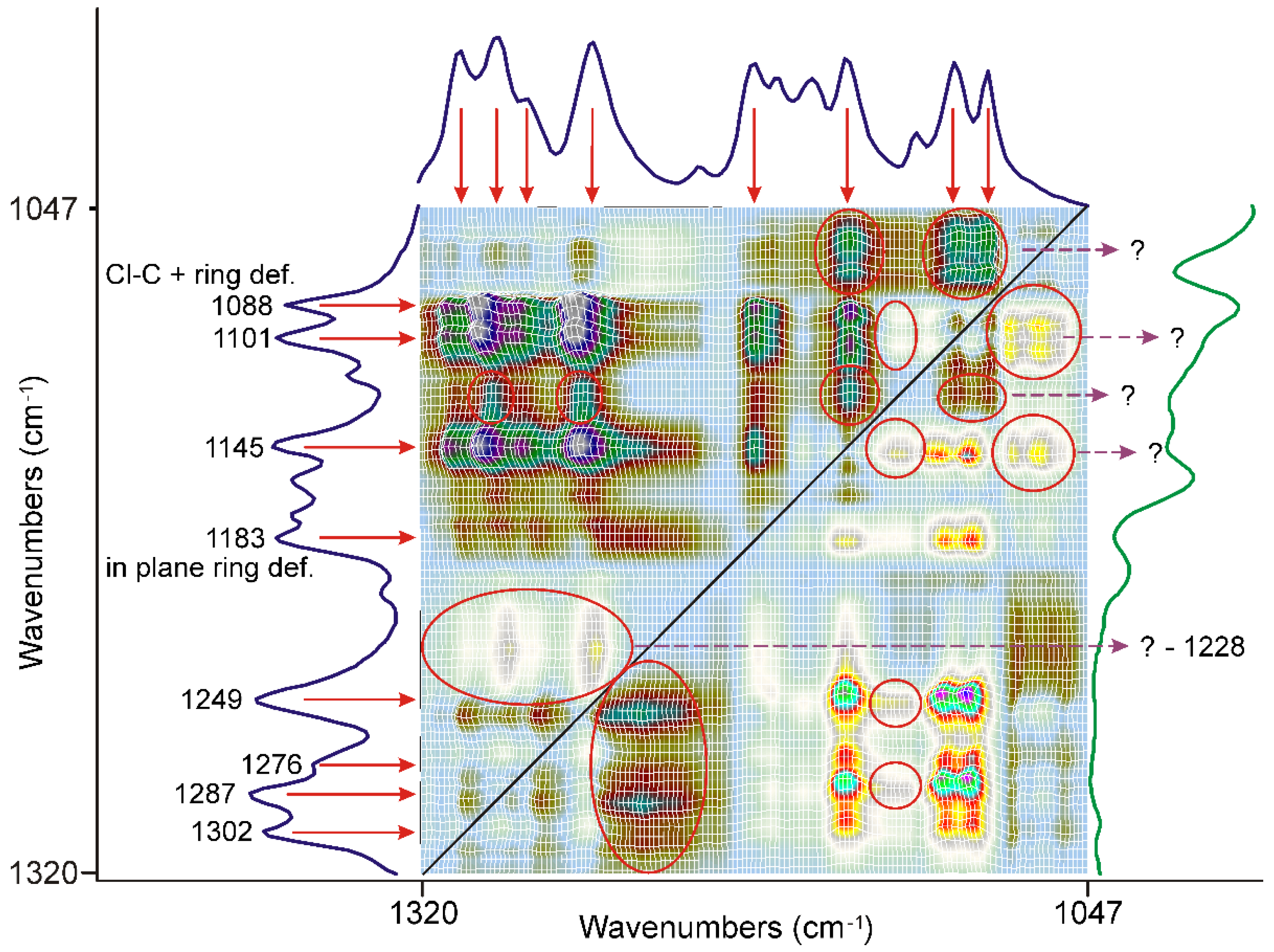
3.3.1. The First 20 min of Grinding
3.3.2. The Second 20 min of Grinding
3.3.3. The Last 20 min of Grinding
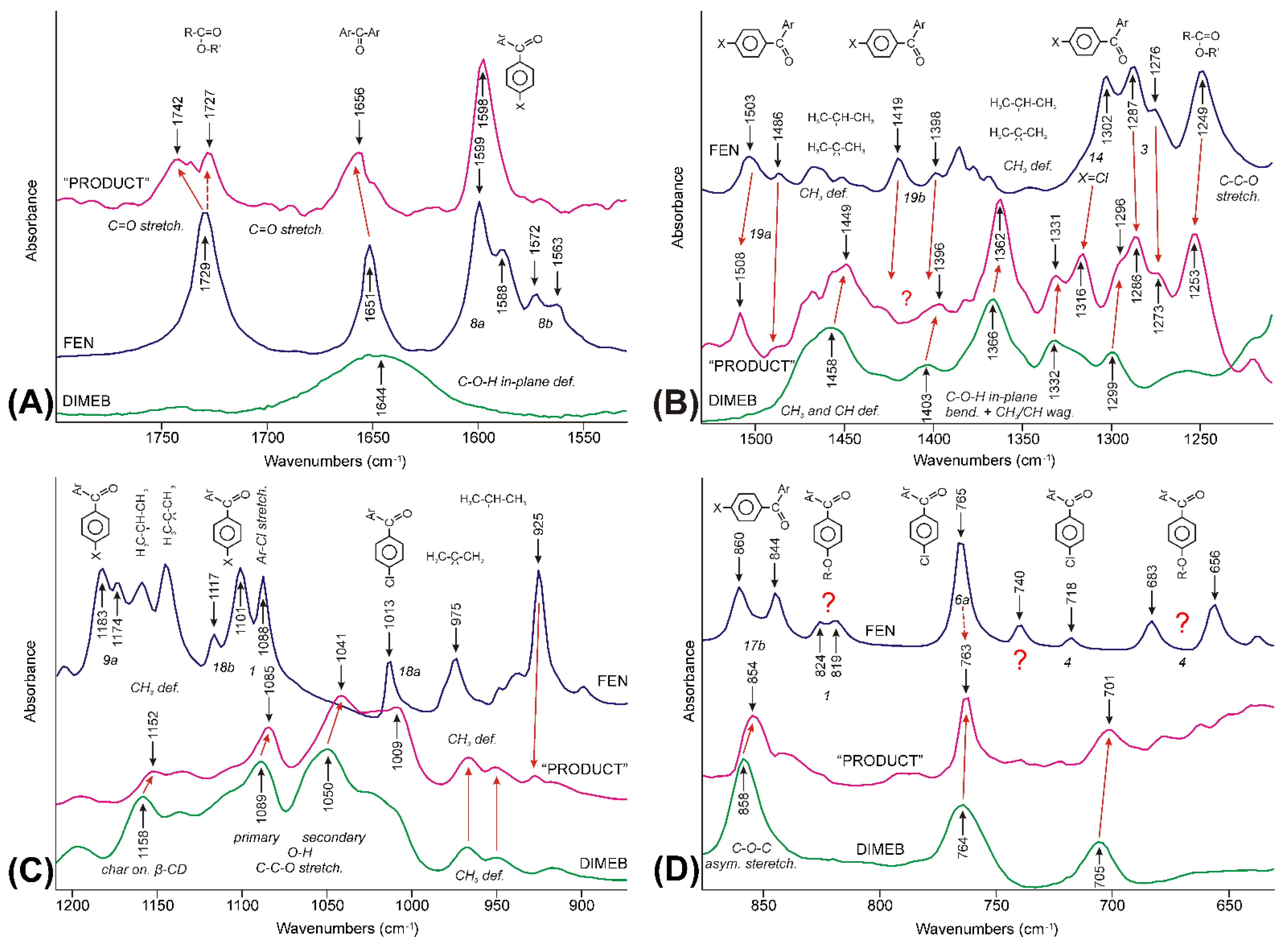
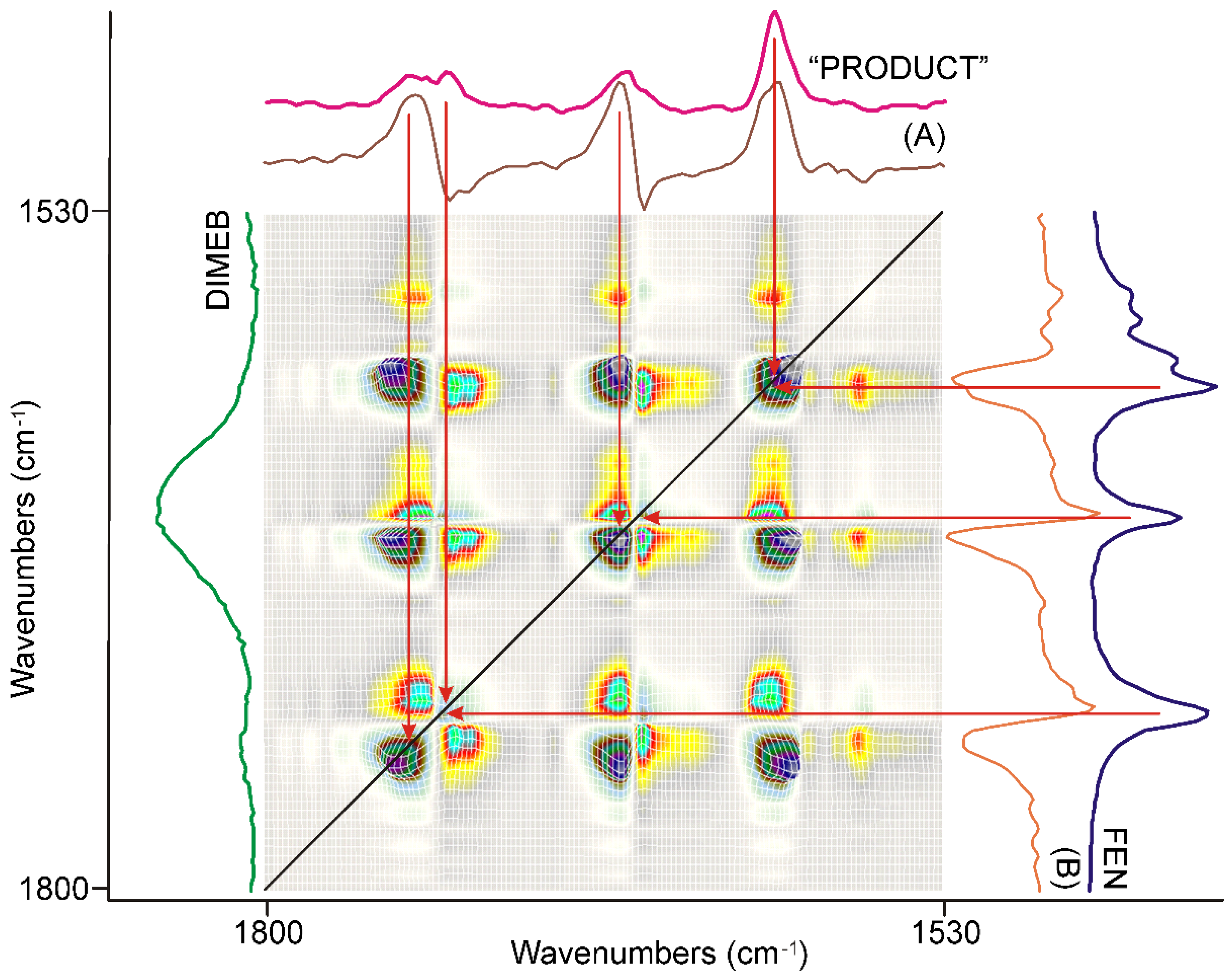
3.3.4. Molecular Considerations Based on the Spectra of FEN, DIMEB, and “PRODUCT”
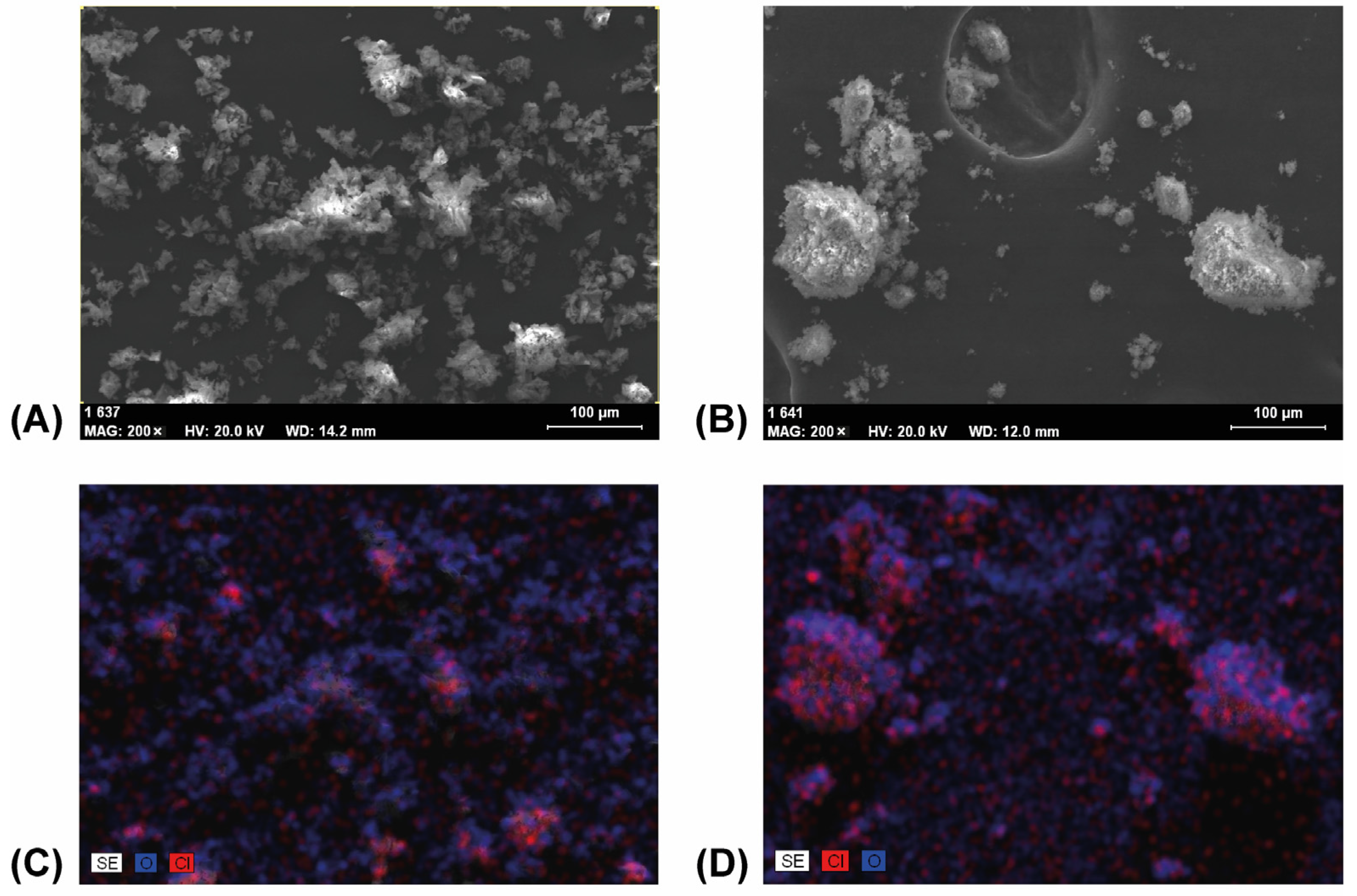
3.4. Scanning Electron Microscopy
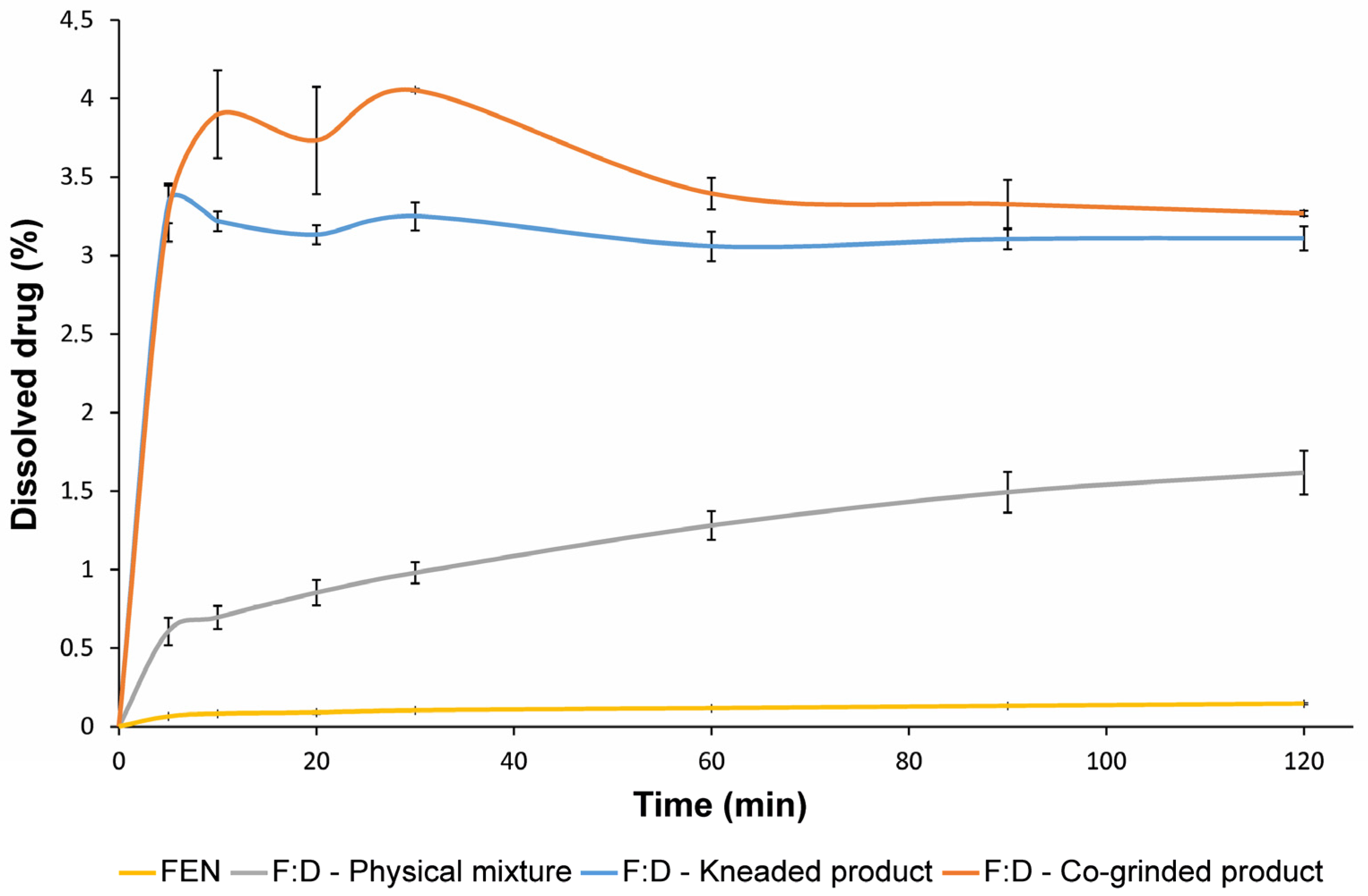
3.5. In Vitro Dissolution Studies
3.6. In Vitro Diffusion Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Staels, B.; Dallongeville, J.; Auwerx, J.; Schoonjans, K.; Leitersdorf, E.; Fruchart, J.-C. Mechanism of Action of Fibrates on Lipid and Lipoprotein Metabolism. Circulation 1998, 98, 2088–2093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogt, M.; Kunath, K.; Dressman, J.B. Dissolution Enhancement of Fenofibrate by Micronization, Cogrinding and Spray-Drying: Comparison with Commercial Preparations. Eur. J. Pharm. Biopharm. 2008, 68, 283–288. [Google Scholar] [CrossRef] [PubMed]
- Amidon, G.L. A Theoretical Basis for a Biopharmaceutic Drug Classification: The Correlation of in Vitro Drug Product Dissolution and in Vivo Bioavailability. Pharm. Res. 1995, 12, 413–420. [Google Scholar] [CrossRef] [Green Version]
- Lindenberg, M.; Kopp, S.; Dressman, J.B. Classification of Orally Administered Drugs on the World Health Organization Model List of Essential Medicines According to the Biopharmaceutics Classification System. Eur. J. Pharm. Biopharm. 2004, 58, 265–278. [Google Scholar] [CrossRef] [PubMed]
- Kawabata, Y.; Wada, K.; Nakatani, M.; Yamada, S.; Onoue, S. Formulation Design for Poorly Water-Soluble Drugs Based on Biopharmaceutics Classification System: Basic Approaches and Practical Applications. Int. J. Pharm. 2011, 420, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Tipduangta, P.; Takieddin, K.; Fábián, L.; Belton, P.; Qi, S. A New Low Melting-Point Polymorph of Fenofibrate Prepared via Talc Induced Heterogeneous Nucleation. Cryst. Growth Des. 2015, 15, 5011–5020. [Google Scholar] [CrossRef]
- Del Valle, E.M.M. Cyclodextrins and Their Uses: A Review. Process Biochem. 2004, 39, 1033–1046. [Google Scholar] [CrossRef]
- Hedges, A.R. Industrial Applications of Cyclodextrins. Chem. Rev. 1998, 98, 2035–2044. [Google Scholar] [CrossRef] [PubMed]
- Loftsson, T.; Jarho, P.; Másson, M.; Järvinen, T. Cyclodextrins in Drug Delivery. Expert Opin. Drug Deliv. 2005, 2, 335–351. [Google Scholar] [CrossRef]
- Jansook, P.; Ogawa, N.; Loftsson, T. Cyclodextrins: Structure, Physicochemical Properties and Pharmaceutical Applications. Int. J. Pharm. 2018, 535, 272–284. [Google Scholar] [CrossRef]
- Carrier, R.L.; Miller, L.A.; Ahmed, I. The Utility of Cyclodextrins for Enhancing Oral Bioavailability. J. Control. Release 2007, 123, 78–99. [Google Scholar] [CrossRef] [PubMed]
- Skrdla, P.J.; Floyd, P.D.; Dell’orco, P.C. Practical Estimation of Amorphous Solubility Enhancement Using Thermoanalytical Data: Determination of the Amorphous/Crystalline Solubility Ratio for Pure Indomethacin and Felodipine. J. Pharm. Sci. 2016, 105, 2625–2630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graeser, K.A.; Patterson, J.E.; Zeitler, J.A.; Gordon, K.C.; Rades, T. Correlating Thermodynamic and Kinetic Parameters with Amorphous Stability. Eur. J. Pharm. Biopharm. 2009, 37, 492–498. [Google Scholar] [CrossRef]
- Hancock, B.C.; Zografi, G. Characteristics and Significance of the Amorphous State in Pharmaceutical Systems. J. Pharm. Sci. 1997, 86, 1–12. [Google Scholar] [CrossRef]
- Jug, M.; Mura, P. Grinding as Solvent-Free Green Chemistry Approach for Cyclodextrin Inclusion Complex Preparation in the Solid State. Pharmaceutics 2018, 10, 189. [Google Scholar] [CrossRef] [Green Version]
- Cue, B.W.; Zhang, J. Green Process Chemistry in the Pharmaceutical Industry. Green Chem. Lett. Rev. 2009, 2, 193–211. [Google Scholar] [CrossRef]
- Ross, S.A.; Lamprou, D.A.; Douroumis, D. Engineering and Manufacturing of Pharmaceutical Co-Crystals: A Review of Solvent-Free Manufacturing Technologies. Chem. Commun. 2016, 52, 8772–8786. [Google Scholar] [CrossRef] [Green Version]
- Cugovčan, M.; Jablan, J.; Lovrić, J.; Cinčić, D.; Galić, N.; Jug, M. Biopharmaceutical Characterization of Praziquantel Cocrystals and Cyclodextrin Complexes Prepared by Grinding. J. Pharm. Biomed. Anal. 2017, 137, 42–53. [Google Scholar] [CrossRef]
- Mura, P. Analytical Techniques for Characterization of Cyclodextrin Complexes in Aqueous Solution: A Review. J. Pharm. Biomed. Anal. 2014, 101, 238–250. [Google Scholar] [CrossRef]
- Jablan, J.; Szalontai, G.; Jug, M. Comparative Analysis of Zaleplon Complexation with Cyclodextrins and Hydrophilic Polymers in Solution and in Solid State. J. Pharm. Biomed. Anal. 2012, 71, 35–44. [Google Scholar] [CrossRef]
- Mennini, N.; Bragagni, M.; Maestrelli, F.; Mura, P. Physico-Chemical Characterization in Solution and in the Solid State of Clonazepam Complexes with Native and Chemically-Modified Cyclodextrins. J. Pharm. Biomed. Anal. 2014, 89, 142–149. [Google Scholar] [CrossRef]
- Jug, M.; Kosalec, I.; Maestrelli, F.; Mura, P. Analysis of Triclosan Inclusion Complexes with β-Cyclodextrin and Its Water-Soluble Polymeric Derivative. J. Pharm. Biomed. Anal. 2011, 54, 1030–1039. [Google Scholar] [CrossRef]
- Jug, M.; Mennini, N.; Kövér, K.E.; Mura, P. Comparative Analysis of Binary and Ternary Cyclodextrin Complexes with Econazole Nitrate in Solution and in Solid State. J. Pharm. Biomed. Anal. 2014, 91, 81–91. [Google Scholar] [CrossRef]
- Aigner, Z.; Berkesi, O.; Farkas, G.; Szabó-Révész, P. DSC, X-Ray and FTIR Studies of a Gemfibrozil/Dimethyl-β-Cyclodextrin Inclusion Complex Produced by Co-Grinding. J. Pharm. Biomed. Anal. 2012, 57, 62–67. [Google Scholar] [CrossRef]
- Corti, G.; Capasso, G.; Maestrelli, F.; Cirri, M.; Mura, P. Physical–Chemical Characterization of Binary Systems of Metformin Hydrochloride with Triacetyl-β-Cyclodextrin. J. Pharm. Biomed. Anal. 2007, 45, 480–486. [Google Scholar] [CrossRef]
- Cirri, M.; Maestrelli, F.; Furlanetto, S.; Mura, P. Solid-State Characterization of Glyburide-Cyclodextrin Co-Ground Products. J. Therm. Anal. Calorim. 2004, 77, 413–422. [Google Scholar] [CrossRef]
- Jug, M.; Maestrelli, F.; Bragagni, M.; Mura, P. Preparation and Solid-State Characterization of Bupivacaine Hydrochloride Cyclodextrin Complexes Aimed for Buccal Delivery. J. Pharm. Biomed. Anal. 2010, 52, 9–18. [Google Scholar] [CrossRef]
- Aigner, Z.; Bencz, I.; Kata, M. Increasing the Solubility Characteristics of Fenofibrate with Cyclodextrin. J. Incl. Phenom. Macrocycl. Chem. 1995, 20, 241–252. [Google Scholar] [CrossRef]
- Mura, P. Analytical Techniques for Characterization of Cyclodextrin Complexes in the Solid State: A Review. J. Pharm. Biomed. Anal. 2015, 113, 226–238. [Google Scholar] [CrossRef]
- Noda, I. Generalized Two-Dimensional Correlation Method Applicable to Infrared, Raman, and Other Types of Spectroscopy. Appl. Spectrosc. 1993, 47, 1329–1336. [Google Scholar] [CrossRef]
- Nabet, A.; Pézolet, M. Two-Dimensional FT-IR Spectroscopy: A Powerful Method to Study the Secondary Structure of Proteins Using H-D Exchange. Appl. Spectrosc. 1997, 51, 466–469. [Google Scholar] [CrossRef]
- Szabo, Z.; Jost, K.; Soos, K.; Zarandi, M.; Kiss, J.T.; Penke, B. Solvent Effect on Aggregational Properties of b -Amyloid Polypeptides Studied by FT–IR Spectroscopy. J. Mol. Struct. 1999, 480, 481–487. [Google Scholar] [CrossRef]
- Yu, Z.W.; Liu, J.; Noda, I. Effect of Noise on the Evaluation of Correlation Coef Cients in Two- Dimensional Correlation Spectroscopy. Appl. Spectrosc. 2003, 57, 1605–1609. [Google Scholar] [CrossRef]
- Heinz, A.; Gordon, K.C.; McGoverin, C.M.; Rades, T.; Strachan, C.J. Understanding the Solid-State Forms of Fenofibrate—A Spectroscopic and Computational Study. Eur. J. Pharm. Biopharm. 2009, 71, 100–108. [Google Scholar] [CrossRef]
- Vazquez, I.N.; Rodriguez-Nunez, J.R.; Pena-Caballero, V.; Ruvalcaba, R.M.; Aceves-Hernandez, J.M. Theoretical and Experimental Study of Fenofibrate and Simvastatin. J. Mol. Struct. 2017, 1149, 683–693. [Google Scholar] [CrossRef]
- Krishnan, K. Different Accessories, Main Applications and Handling Techniques. In Fourier Transform Infrared Spectroscopy: Industrial Chemical and Biochemical Applications; Theophanides, T., Ed.; Springer: Dordrecht, The Netherlands, 1984; pp. 139–158. ISBN 978-94-009-6418-1. [Google Scholar]
- He, A.Q.; Zeng, Y.W.; Kang, X.Y.; Morita, S.; Xu, Y.Z.; Noda, I.; Ozaki, Y.; Wu, J.G. Novel Method of Constructing Two-Dimensional Correlation Spectroscopy without Subtracting a Reference Spectrum. J. Phys. Chem. A 2018, 122, 788–797. [Google Scholar] [CrossRef]
- Colthup, N.B.; Daly, L.H.; Wieberley, S.E. Introduction to Infrared and Raman Spectroscopy; Academic Press: New York, NY, USA, 1990; pp. 218, 236, 250, 261–283. [Google Scholar]
- Tipduangta, P.; Takieddin, K.; Fabian, L.; Belton, P.; Qi, S. Towards Controlling the Crystallisation Behaviour of Fenofibrate Melt: Triggers of Crystallisation and Polymorphic Transformation. Rsc. Adv. 2018, 8, 13513–13525. [Google Scholar] [CrossRef] [Green Version]
- Varsányi, G. Assignments for Vibrational Spectra of Seven Hundred Benzene Derivatives; Akadémiai Kiadó: Budapest, Hungary, 1973; pp. 12, 128, 163, 550–551. [Google Scholar]
- Wilson, E.B. The Normal Modes and Frequencies of Vibration of the Regular Plane Hexagon Model of the Benzene Molecule. Phys. Rev. 1934, 45, 706–714. [Google Scholar] [CrossRef]
- Egyed, O. Spectroscopic studies on β-cyclodextrin. Vib. Spectrosc. 1990, 1, 225–227. [Google Scholar] [CrossRef]
- Sabapathy, R.C.; Bhattacharyya, S.; Cleland, W.E.; Hussey, C.L. Host-Guest Complexation in Self-Assembled Monolayers: Inclusion of a Monolayer-Anchored Cationic Ferrocene-Based Guest by Cyclodextrin Hosts. Langmuir 1998, 14, 3797–3807. [Google Scholar] [CrossRef]
- Farcas, A.; Grigoras, N. Poly(Azomethine)s with Rotaxane Architecture Containing a Blocking Group in Every Structural Unit: Synthesis and Characterization. High Perform. Polym. 2001, 13, 149–158. [Google Scholar] [CrossRef]
- Billes, F.; Hernanz, A.; Mikosch, H.; Bratu, I. Structure and Vibrational Spectroscopy of the Fenbufen-Beta-Cyclodextrin Inclusion Complex. Vib. Spectrosc. 2013, 69, 30–39. [Google Scholar] [CrossRef]
- Henry, R.F.; Zhang, G.Z.; Gao, Y.; Buckner, I.S. Fenofibrate. Acta Crystallogr. E Struct. Rep. Online 2003, 59, o699–o700. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Ritter, H. Cyclodextrin Functionalized Polymers as Drug Delivery Systems. Polym. Chem. 2010, 1, 1552–1559. [Google Scholar] [CrossRef]
- Larkin, P.J. IR and Raman Spectroscopy, Principles and Spectral Interpretation; Elsevier Inc.: Oxford, UK, 2011. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DE 60 min (%) | DE 120 min (%) | |
|---|---|---|
| FEN | 0.09 ± 0.01 | 0.11 ± 0.01 |
| F:D—physical mixture | 0.94 ± 0.09 | 1.20 ± 0.12 |
| F:D—kneaded | 3.05 ± 0.10 | 3.07 ± 0.09 |
| F:D—co-ground | 3.59 ± 0.10 | 3.46 ± 0.11 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kondoros, B.A.; Berkesi, O.; Tóth, Z.; Aigner, Z.; Ambrus, R.; Csóka, I. Cyclodextrin Complexation of Fenofibrate by Co-Grinding Method and Monitoring the Process Using Complementary Analytical Tools. Pharmaceutics 2022, 14, 1329. https://doi.org/10.3390/pharmaceutics14071329
Kondoros BA, Berkesi O, Tóth Z, Aigner Z, Ambrus R, Csóka I. Cyclodextrin Complexation of Fenofibrate by Co-Grinding Method and Monitoring the Process Using Complementary Analytical Tools. Pharmaceutics. 2022; 14(7):1329. https://doi.org/10.3390/pharmaceutics14071329
Chicago/Turabian StyleKondoros, Balázs Attila, Ottó Berkesi, Zsolt Tóth, Zoltán Aigner, Rita Ambrus, and Ildikó Csóka. 2022. "Cyclodextrin Complexation of Fenofibrate by Co-Grinding Method and Monitoring the Process Using Complementary Analytical Tools" Pharmaceutics 14, no. 7: 1329. https://doi.org/10.3390/pharmaceutics14071329
APA StyleKondoros, B. A., Berkesi, O., Tóth, Z., Aigner, Z., Ambrus, R., & Csóka, I. (2022). Cyclodextrin Complexation of Fenofibrate by Co-Grinding Method and Monitoring the Process Using Complementary Analytical Tools. Pharmaceutics, 14(7), 1329. https://doi.org/10.3390/pharmaceutics14071329