
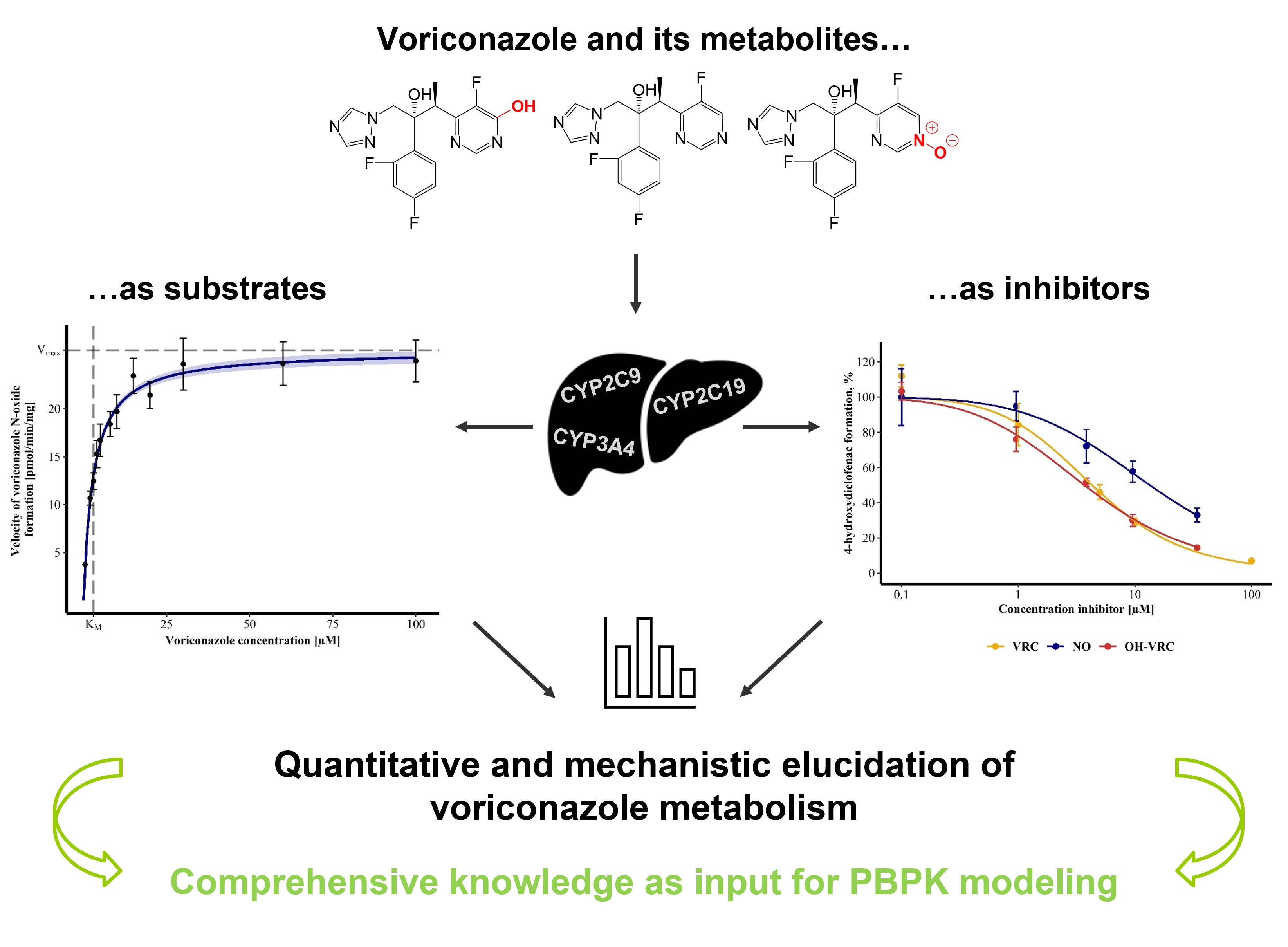
Towards the Elucidation of the Pharmacokinetics of Voriconazole: A Quantitative Characterization of Its Metabolism
 , ,
, , 
Abstract
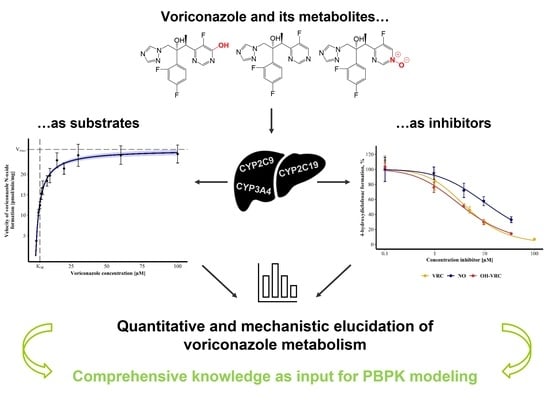
:
1. Introduction
2. Materials and Methods
2.1. Chemicals, Drugs and Enzyme Systems
2.2. Incubation Conditions and Assay Procedure
2.3. Bioanalysis
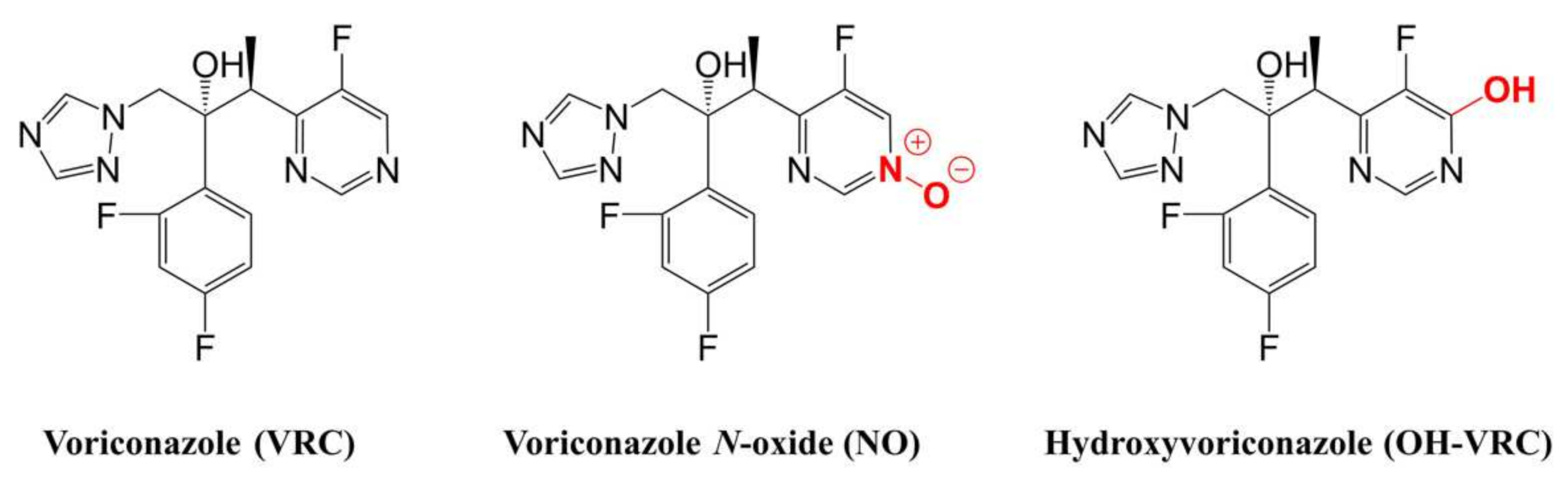
2.4. Voriconazole and Its Metabolites as Substrates
2.4.1. Michaelis–Menten Kinetics of Voriconazole N-oxidation
2.4.2. Contribution of Individual CYP Enzymes to Voriconazole N-oxidation
2.4.3. In Vitro In Vivo Extrapolation
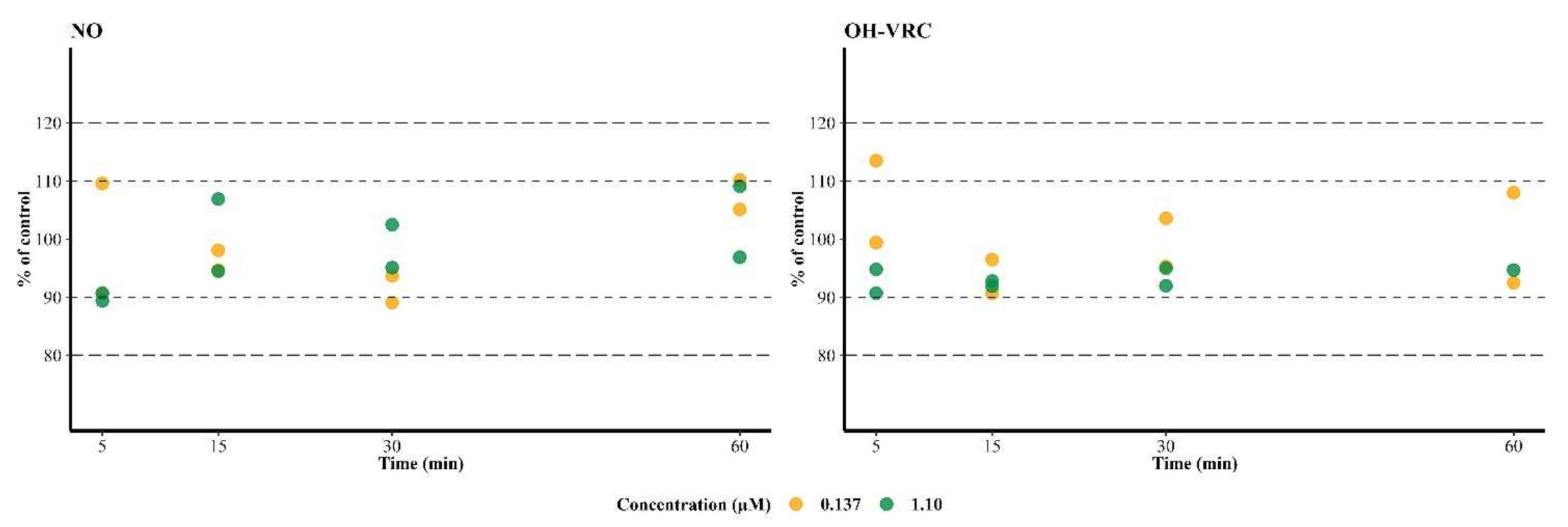
2.4.4. Metabolic Stability of Voriconazole N-oxide and Hydroxyvoriconazole
2.5. Voriconazole and Its Metabolites as Inhibitors
3. Results
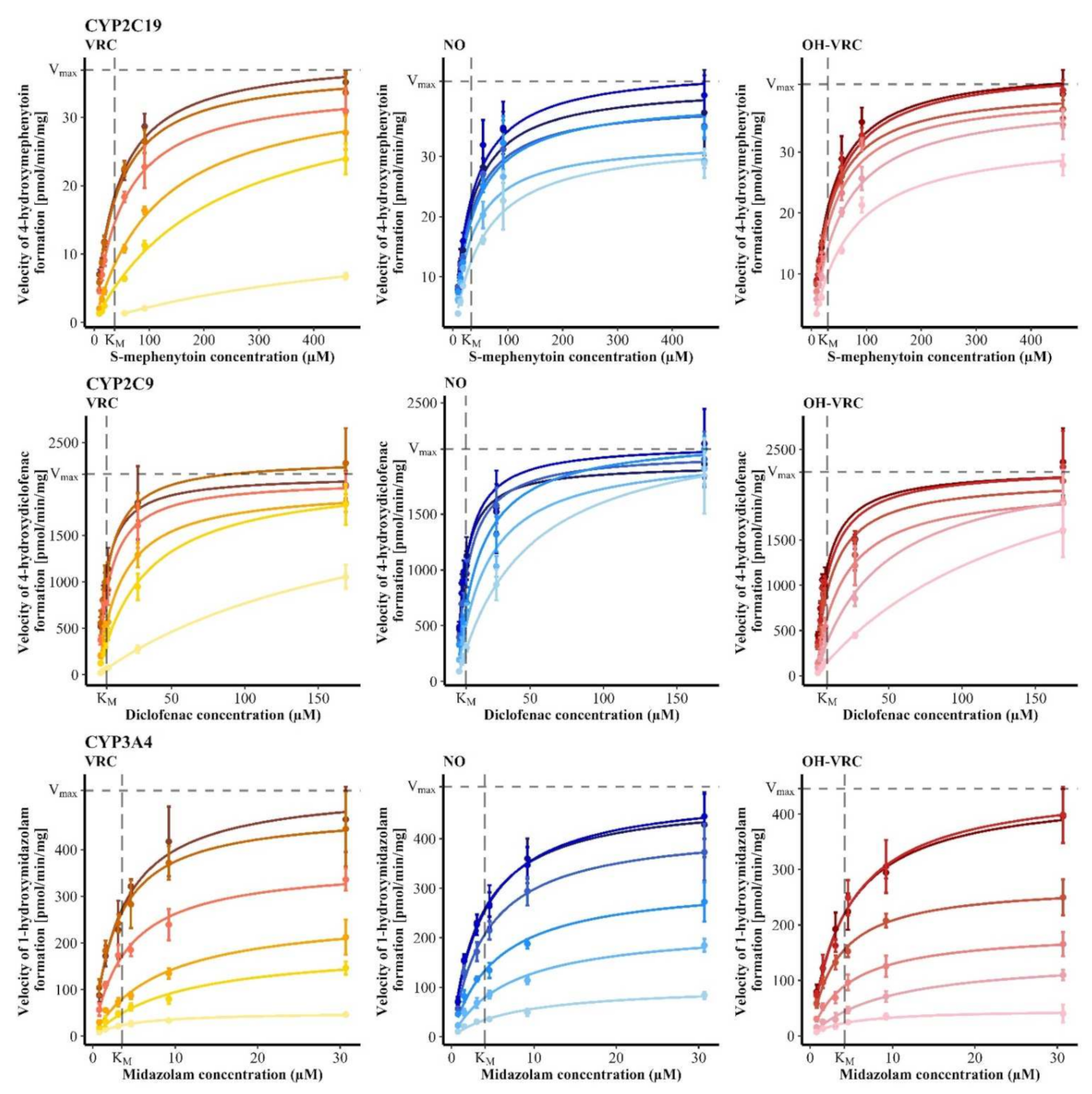
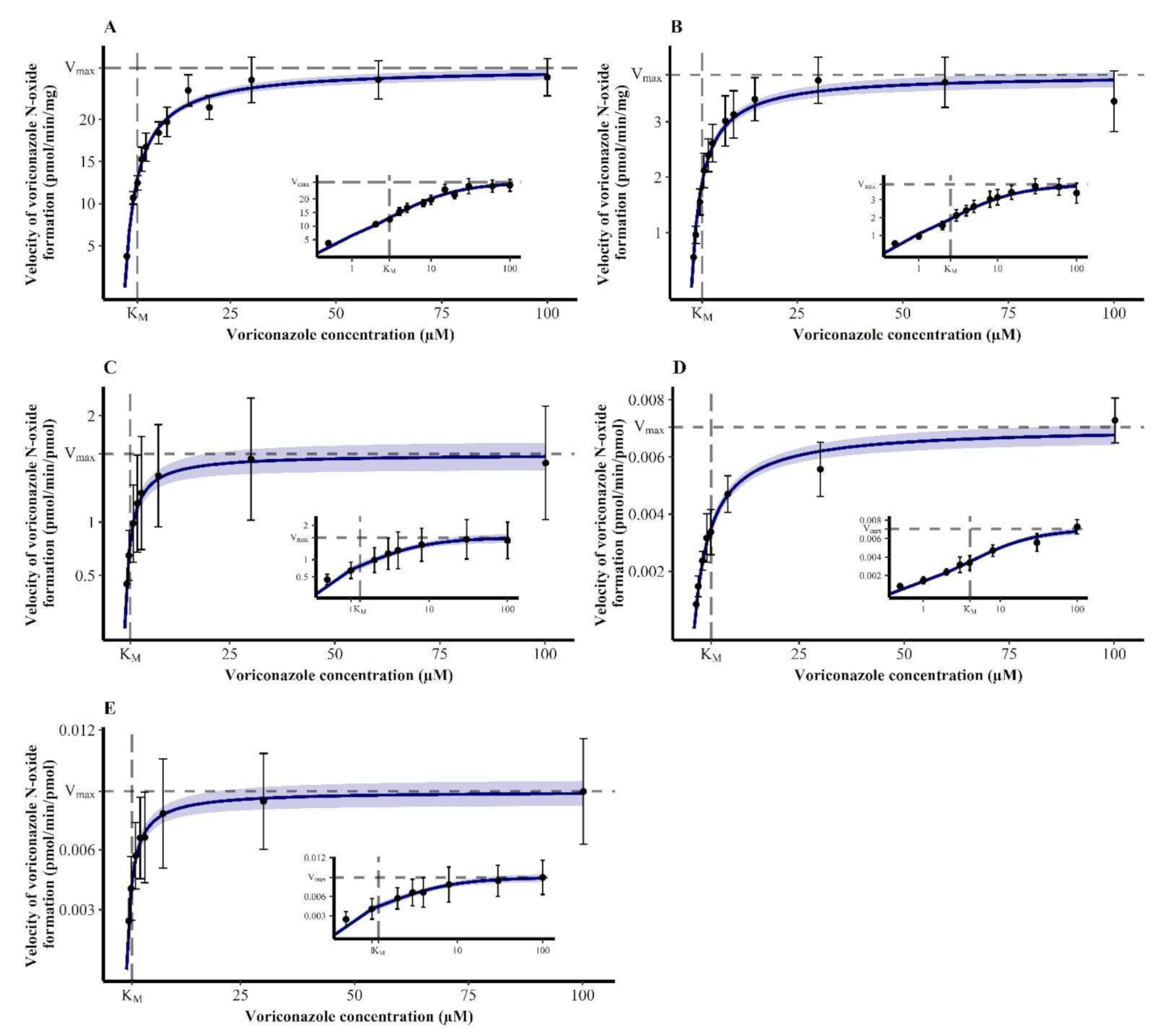
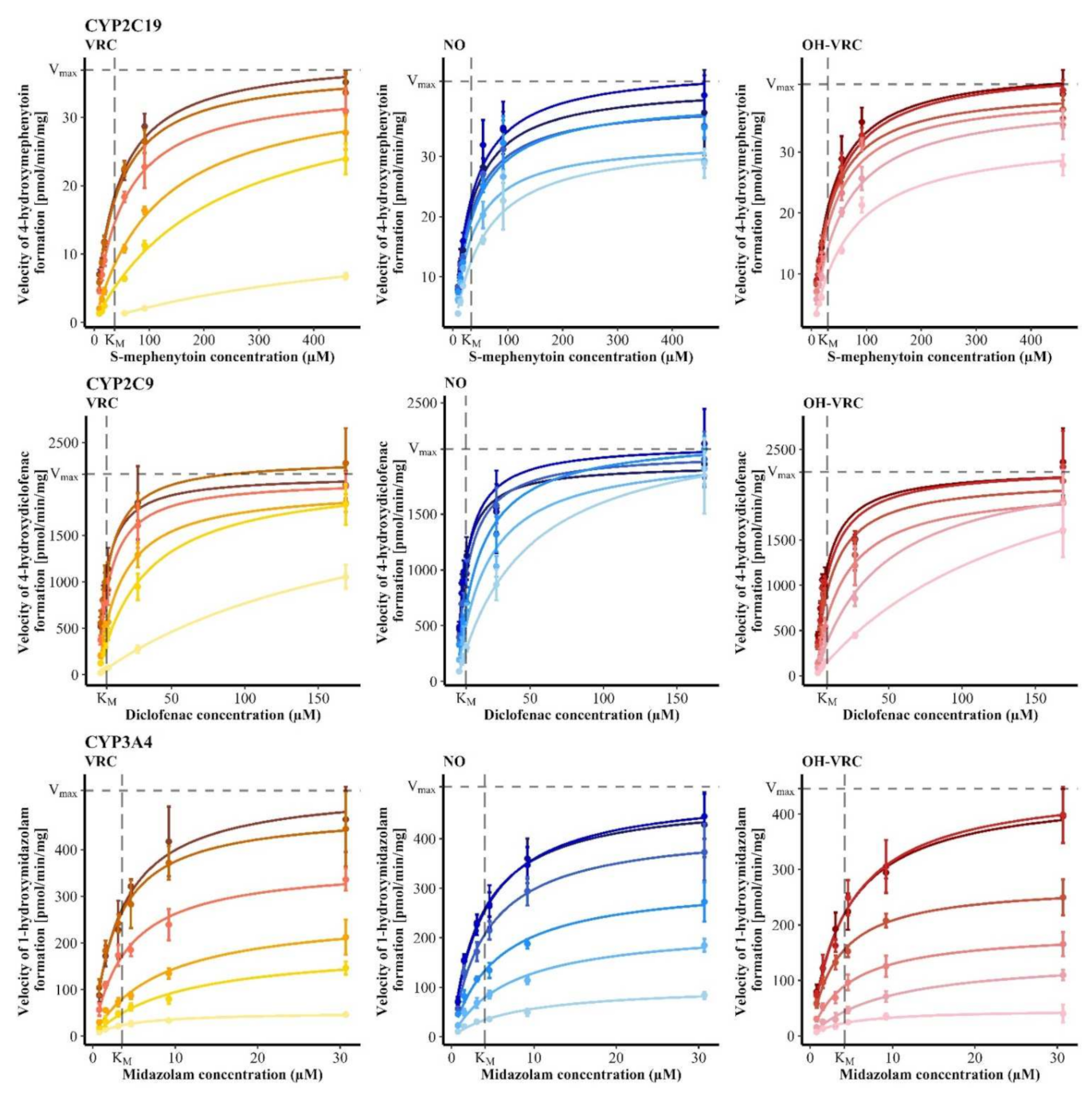
3.1. Michaelis-Menten Kinetics of Voriconazole N-oxidation
3.2. Contribution of Individual CYP Enzymes to Voriconazole N-oxidation
3.3. In Vitro In Vivo Extrapolation
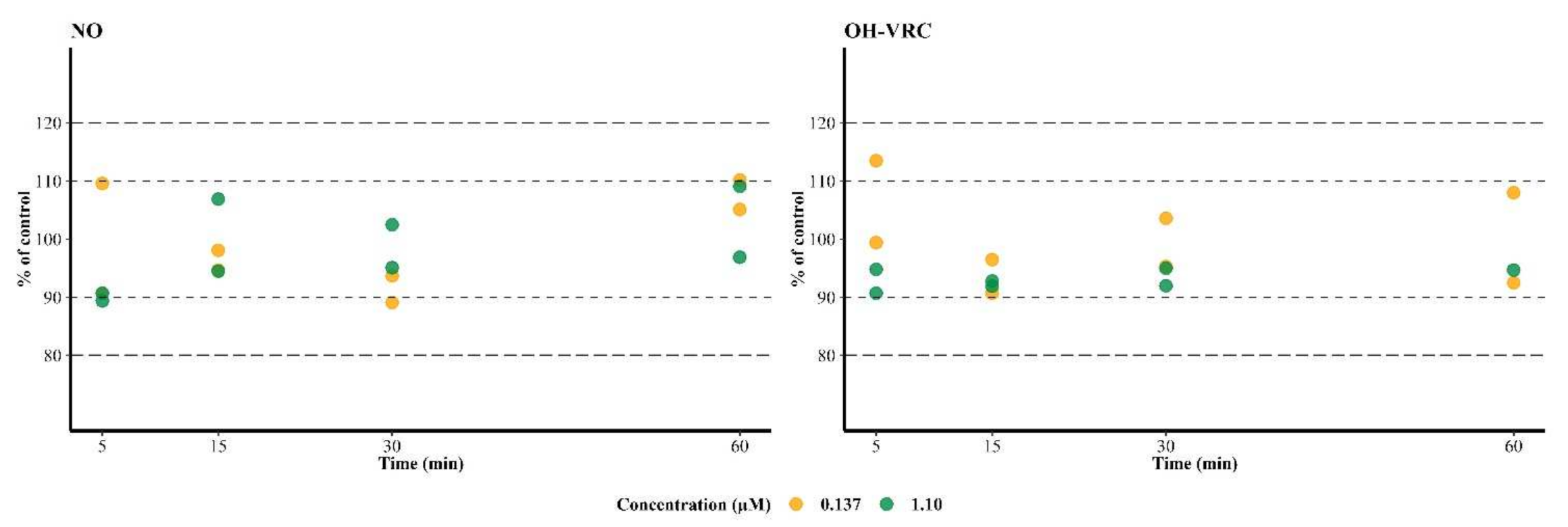
3.4. Metabolic Stability of Voriconazole N-oxide and Hydroxyvoriconazole
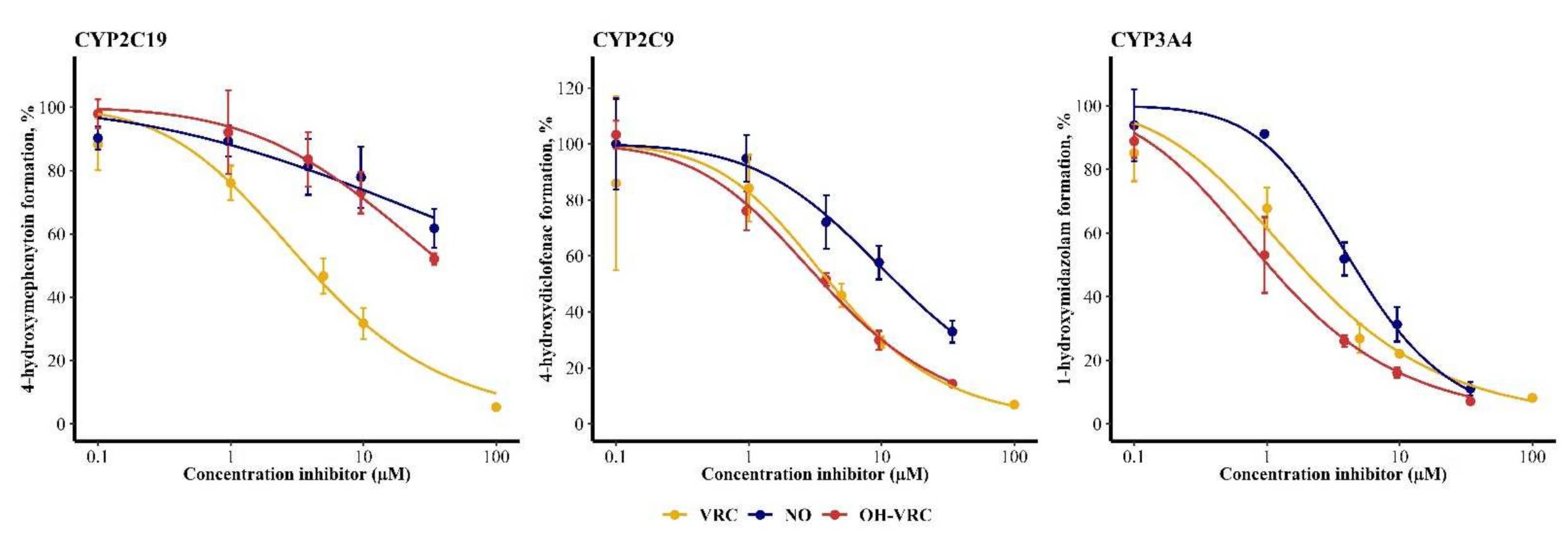
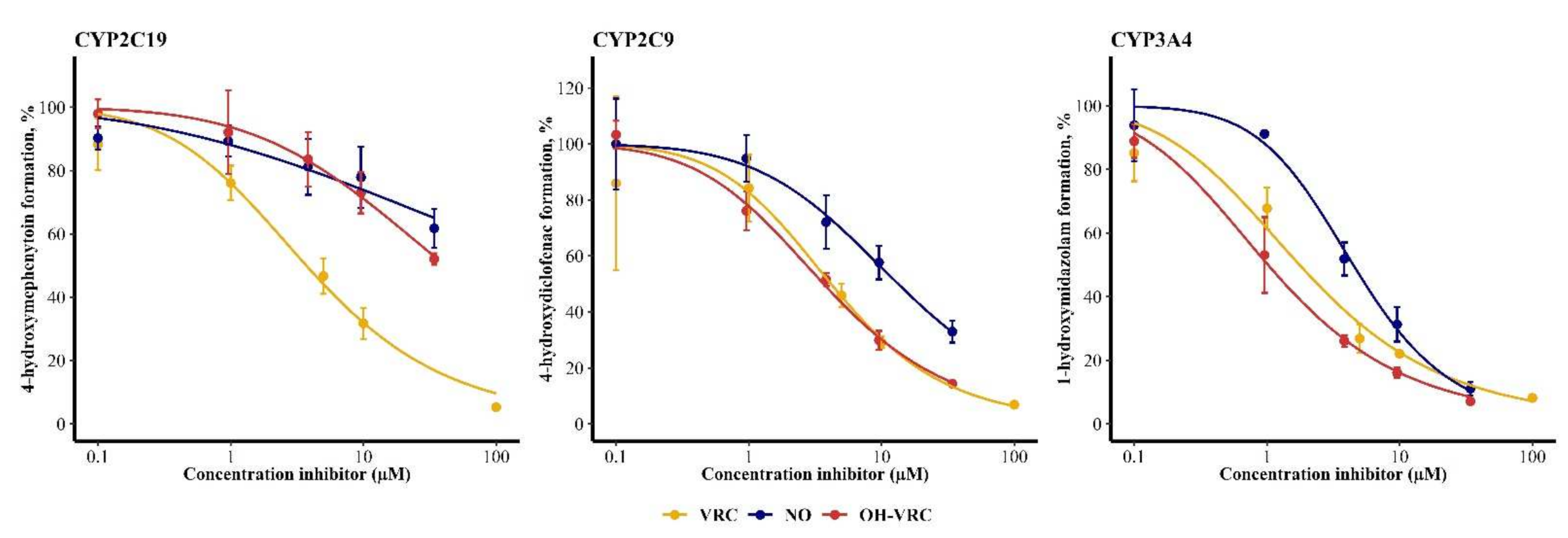
3.5. Voriconazole and Its Metabolites as Inhibitors
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Greer, N.D. Voriconazole: The Newest Triazole Antifungal Agent. Bayl. Univ. Med. Cent. Proc. 2003, 16, 241–248. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. WHO: Model List of Essential Medicines. Available online: https://apps.who.int/iris/rest/bitstreams/1374779/retrieve (accessed on 19 December 2021).
- World Health Organization. WHO: Model List of Essential Medicines for Children. Available online: https://apps.who.int/iris/rest/bitstreams/1145255/retrieve (accessed on 24 September 2021).
- Pfizer. VFEND: EPAR-Product Information. Available online: https://www.ema.europa.eu/documents/product-information/vfend-epar-product-information_en.pdf (accessed on 27 July 2021).
- Benedict, K.; Richardson, M.; Vallabhaneni, S.; Jackson, B.R.; Chiller, T. Emerging Issues, Challenges, and Changing Epidemiology of Fungal Disease Outbreaks. Lancet Infect. Dis. 2017, 17, e403–e411. [Google Scholar] [CrossRef]
- Jenks, J.D.; Cornely, O.A.; Chen, S.C.A.; Thompson, G.R.; Hoenigl, M. Breakthrough Invasive Fungal Infections: Who Is at Risk? Mycoses 2020, 63, 1021–1032. [Google Scholar] [CrossRef] [PubMed]
- Cole, D.C.; Govender, N.P.; Chakrabarti, A.; Sacarlal, J.; Denning, D.W. Improvement of Fungal Disease Identification and Management: Combined Health Systems and Public Health Approaches. Lancet Infect. Dis. 2017, 17, e412–e419. [Google Scholar] [CrossRef]
- Fisher, M.C.; Hawkins, N.J.; Sanglard, D.; Gurr, S.J. Worldwide Emergence of Resistance to Antifungal Drugs Challenges Human Health and Food Security. Science 2018, 360, 739–742. [Google Scholar] [CrossRef] [Green Version]
- Perlin, D.S.; Rautemaa-Richardson, R.; Alastruey-Izquierdo, A. The Global Problem of Antifungal Resistance: Prevalence, Mechanisms, and Management. Lancet Infect. Dis. 2017, 17, e383–e392. [Google Scholar] [CrossRef]
- Perfect, J.R.; Ghannoum, M. Emerging Issues in Antifungal Resistance. Infect. Dis. Clin. North Am. 2020, 34, 921–943. [Google Scholar] [CrossRef]
- Arastehfar, A.; Gabaldón, T.; Garcia-Rubio, R.; Jenks, J.D.; Hoenigl, M.; Salzer, H.J.F.; Ilkit, M.; Lass-Flörl, C.; Perlin, D.S. Drug-Resistant Fungi: An Emerging Challenge Threatening Our Limited Antifungal Armamentarium. Antibiotics 2020, 9, 877. [Google Scholar] [CrossRef]
- U. S. Food and Drug Adminstration. Vfend. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2003/021464s000_021466s000_VfendTOC.cfm (accessed on 27 July 2021).
- European Medicines Agency. Vfend. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/vfend#authorisation-details-section (accessed on 27 July 2021).
- Schulz, J.; Kluwe, F.; Mikus, G.; Michelet, R.; Kloft, C. Novel Insights into the Complex Pharmacokinetics of Voriconazole: A Review of Its Metabolism. Drug Metab. Rev. 2019, 51, 247–265. [Google Scholar] [CrossRef]
- Dote, S.; Sawai, M.; Nozaki, A.; Naruhashi, K.; Kobayashi, Y.; Nakanishi, H. A Retrospective Analysis of Patient-Specific Factors on Voriconazole Clearance. J. Pharm. Health Care Sci. 2016, 2, 10. [Google Scholar] [CrossRef] [Green Version]
- Gautier-Veyret, E.; Fonrose, X.; Tonini, J.; Thiebaut-Bertrand, A.; Bartoli, M.; Quesada, J.-L.; Bulabois, C.-E.; Cahn, J.-Y.; Stanke-Labesque, F. Variability of Voriconazole Plasma Concentrations after Allogeneic Hematopoietic Stem Cell Transplantation: Impact of Cytochrome P450 Polymorphisms and Comedications on Initial and Subsequent Trough Levels. Antimicrob. Agents Chemother. 2015, 59, 2305–2314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamadeh, I.S.; Klinker, K.P.; Borgert, S.J.; Richards, A.I.; Li, W.; Mangal, N.; Hiemenz, J.W.; Schmidt, S.; Langaee, T.Y.; Peloquin, C.A.; et al. Impact of the CYP2C19 Genotype on Voriconazole Exposure in Adults with Invasive Fungal Infections. Pharm. Genom. 2017, 27, 190–196. [Google Scholar] [CrossRef] [Green Version]
- Lamoureux, F.; Duflot, T.; Woillard, J.-B.; Metsu, D.; Pereira, T.; Compagnon, P.; Morisse-Pradier, H.; el Kholy, M.; Thiberville, L.; Stojanova, J.; et al. Impact of CYP2C19 Genetic Polymorphisms on Voriconazole Dosing and Exposure in Adult Patients with Invasive Fungal Infections. Int. J. Antimicrob. Agents 2016, 47, 124–131. [Google Scholar] [CrossRef]
- Wang, T.; Chen, S.; Sun, J.; Cai, J.; Cheng, X.; Dong, H.; Wang, X.; Xing, J.; Dong, W.; Yao, H.; et al. Identification of Factors Influencing the Pharmacokinetics of Voriconazole and the Optimization of Dosage Regimens Based on Monte Carlo Simulation in Patients with Invasive Fungal Infections. J. Antimicrob. Chemother. 2014, 69, 463–470. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Yu, C.; Wang, T.; Chen, K.; Zhai, S.; Tang, H. Effect of Cytochrome P450 2C19 Polymorphisms on the Clinical Outcomes of Voriconazole: A Systematic Review and Meta-Analysis. Eur. J. Clin. Pharmacol. 2016, 72, 1185–1193. [Google Scholar] [CrossRef] [PubMed]
- Shao, B.; Ma, Y.; Li, Q.; Wang, Y.; Zhu, Z.; Zhao, H.; Sun, J.; Dong, L.; Zhu, Y.; Zhao, N.; et al. Effects of Cytochrome P450 3A4 and Non-Genetic Factors on Initial Voriconazole Serum Trough Concentrations in Hematological Patients with Different Cytochrome P450 2C19 Genotypes. Xenobiotica 2017, 47, 1121–1129. [Google Scholar] [CrossRef]
- Mikus, G.; Scholz, I.M.; Weiss, J. Pharmacogenomics of the Triazole Antifungal Agent Voriconazole. Pharmacogenomics 2011, 12, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Scholz, I.; Oberwittler, H.; Riedel, K.-D.; Burhenne, J.; Weiss, J.; Haefeli, W.E.; Mikus, G. Pharmacokinetics, Metabolism and Bioavailability of the Triazole Antifungal Agent Voriconazole in Relation to CYP2C19 Genotype. Br. J. Clin. Pharmacol. 2009, 68, 906–915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geist, M.J.P.; Egerer, G.; Burhenne, J.; Riedel, K.-D.; Weiss, J.; Mikus, G. Steady-State Pharmacokinetics and Metabolism of Voriconazole in Patients. J. Antimicrob. Chemother. 2013, 68, 2592–2599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roffey, S.J.; Cole, S.; Comby, P.; Gibson, D.; Jezequel, S.G.; Nedderman, A.N.R.; Smith, D.A.; Walker, D.K.; Wood, N. The disposition of voriconazole in mouse, rat, rabbit, guinea pig, dog, and human. Drug Metab. Dispos. 2003, 31, 731–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theuretzbacher, U.; Ihle, F.; Derendorf, H. Pharmacokinetic/Pharmacodynamic Profile of Voriconazole. Clin. Pharmacokinet. 2006, 45, 649–663. [Google Scholar] [CrossRef] [PubMed]
- Epaulard, O.; Leccia, M.-T.; Blanche, S.; Chosidow, O.; Mamzer-Bruneel, M.-F.; Ravaud, P.; Thiebaut, A.; Villier, C.; Lortholary, O. Phototoxicity and Photocarcinogenesis Associated with Voriconazole. Médecine Et Mal. Infect. 2011, 41, 639–645. [Google Scholar] [CrossRef] [PubMed]
- Epaulard, O.; Villier, C.; Ravaud, P.; Chosidow, O.; Blanche, S.; Mamzer-Bruneel, M.-F.; Thiebaut, A.; Leccia, M.-T.; Lortholary, O. A Multistep Voriconazole-Related Phototoxic Pathway May Lead to Skin Carcinoma: Results From a French Nationwide Study. Clin. Infect. Dis. 2013, 57, e182–e188. [Google Scholar] [CrossRef]
- Ona, K.; Oh, D.H. Voriconazole N-Oxide and Its Ultraviolet B Photoproduct Sensitize Keratinocytes to Ultraviolet A. Br. J. Dermatol. 2015, 173, 751–759. [Google Scholar] [CrossRef] [PubMed]
- Zonios, D.; Yamazaki, H.; Murayama, N.; Natarajan, V.; Palmore, T.; Childs, R.; Skinner, J.; Bennett, J.E. Voriconazole Metabolism, Toxicity, and the Effect of Cytochrome P450 2C19 Genotype. J. Infect. Dis. 2014, 209, 1941–1948. [Google Scholar] [CrossRef] [Green Version]
- Hyland, R.; Jones, B.C.; Smith, D.A. Identification of the Cytochrome P450 Enzymes Involved in the N -Oxidation of Voriconazole. Drug Metab. Dispos. 2003, 31, 540–547. [Google Scholar] [CrossRef] [Green Version]
- Yanni, S.B.; Annaert, P.P.; Augustijns, P.; Bridges, A.; Gao, Y.; Benjamin, D.K.; Thakker, D.R. Role of Flavin-Containing Monooxygenase in Oxidative Metabolism of Voriconazole by Human Liver Microsomes. Drug Metab. Dispos. 2008, 36, 1119–1125. [Google Scholar] [CrossRef] [Green Version]
- Murayama, N.; Imai, N.; Nakane, T.; Shimizu, M.; Yamazaki, H. Roles of CYP3A4 and CYP2C19 in Methyl Hydroxylated and N-Oxidized Metabolite Formation from Voriconazole, a New Anti-Fungal Agent, in Human Liver Microsomes. Biochem. Pharmacol. 2007, 73, 2020–2026. [Google Scholar] [CrossRef]
- Geist, M.J.P.; Egerer, G.; Burhenne, J.; Mikus, G. Safety of Voriconazole in a Patient with CYP2C9*2/CYP2C9*2 Genotype. Antimicrob. Agents Chemother. 2006, 50, 3227–3228. [Google Scholar] [CrossRef] [Green Version]
- Dean, L. Voriconazole Therapy and CYP2C19 Genotype; Pratt, V., Scott, S., Primohamed, M., Eds.; Bethesda: Rockville, MD, USA, 2019. [Google Scholar]
- Niioka, T.; Fujishima, N.; Abumiya, M.; Yamashita, T.; Ubukawa, K.; Nara, M.; Fujishima, M.; Takahashi, N.; Miura, M. Relationship Between the CYP2C19 Phenotype Using the Voriconazole-to-Voriconazole N-Oxide Plasma Concentration Ratio and Demographic and Clinical Characteristics of Japanese Patients With Different CYP2C19 Genotypes. Ther. Drug Monit. 2017, 39, 514–521. [Google Scholar] [CrossRef]
- Chawla, P.K.; Nanday, S.R.; Dherai, A.J.; Soman, R.; Lokhande, R.V.; Naik, P.R.; Ashavaid, T.F. Correlation of CYP2C19 Genotype with Plasma Voriconazole Levels: A Preliminary Retrospective Study in Indians. Int. J. Clin. Pharm. 2015, 37, 925–930. [Google Scholar] [CrossRef] [PubMed]
- Weiss, J.; ten Hoevel, M.M.; Burhenne, J.; Walter-Sack, I.; Hoffmann, M.M.; Rengelshausen, J.; Haefeli, W.E.; Mikus, G. CYP2C19 Genotype Is a Major Factor Contributing to the Highly Variable Pharmacokinetics of Voriconazole. J. Clin. Pharmacol. 2009, 49, 196–204. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Lei, H.P.; Li, Z.; Tan, Z.R.; Guo, D.; Fan, L.; Chen, Y.; Hu, D.L.; Wang, D.; Zhou, H.H. The CYP2C19 Ultra-Rapid Metabolizer Genotype Influences the Pharmacokinetics of Voriconazole in Healthy Male Volunteers. Eur. J. Clin. Pharmacol. 2009, 65, 281–285. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, R.; Fujioka, T.; Suzuki, Y.; Iwao, M.; Itoh, H. A Prospective Study on the Usefulness of Initial Voriconazole Dose Adjustment Based on CYP2C19 Gene Polymorphism Analysis. Chemotherapy 2020, 65, 59–64. [Google Scholar] [CrossRef]
- Chung, H.; Lee, H.; Han, H.; An, H.; Lim, K.S.; Lee, Y.; Cho, J.Y.; Yoon, S.H.; Jang, I.J.; Yu, K.S. A Pharmacokinetic Comparison of Two Voriconazole Formulations and the Effect of CYP2C19 Polymorphism on Their Pharmacokinetic Profiles. Drug Des. Dev. Ther. 2015, 9, 2609–2616. [Google Scholar] [CrossRef] [Green Version]
- Hohmann, N.; Kreuter, R.; Blank, A.; Weiss, J.; Burhenne, J.; Haefeli, W.E.; Mikus, G. Autoinhibitory Properties of the Parent but Not of the N-Oxide Metabolite Contribute to Infusion Rate-Dependent Voriconazole Pharmacokinetics. Br. J. Clin. Pharmacol. 2017, 83, 1954–1965. [Google Scholar] [CrossRef] [Green Version]
- Jeong, S.; Nguyen, P.D.; Desta, Z. Comprehensive in vitro Analysis of Voriconazole Inhibition of Eight Cytochrome P450 (CYP) Enzymes: Major Effect on CYPs 2B6, 2C9, 2C19, and 3A. Antimicrob. Agents Chemother. 2009, 53, 541–551. [Google Scholar] [CrossRef] [Green Version]
- Niwa, T.; Shiraga, T.; Takagi, A. Effect of Antifungal Drugs on Cytochrome P450 (CYP) 2C9, CYP2C19, and CYP3A4 Activities in Human Liver Microsomes. Biol. Pharm. Bull. 2005, 28, 1805–1808. [Google Scholar] [CrossRef] [Green Version]
- Niwa, T.; Inoue-Yamamoto, S.; Shiraga, T.; Takagi, A. Effect of Antifungal Drugs on Cytochrome P450 (CYP) 1A2, CYP2D6, and CYP2E1 Activities in Human Liver Microsomes. Biol. Pharm. Bull. 2005, 28, 1813–1816. [Google Scholar] [CrossRef] [Green Version]
- Pasqualotto, A.C.; Xavier, M.O.; Andreolla, H.F.; Linden, R. Voriconazole Therapeutic Drug Monitoring: Focus on Safety. Expert Opin. Drug Saf. 2010, 9, 125–137. [Google Scholar] [CrossRef]
- Yamada, T.; Mino, Y.; Yagi, T.; Naito, T.; Kawakami, J. Saturated Metabolism of Voriconazole N-Oxidation Resulting in Nonlinearity of Pharmacokinetics of Voriconazole at Clinical Doses. Biol. Pharm. Bull. 2015, 38, 1496–1503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, H.; Chen, Y.; Gibson, C.; Heimbach, T.; Parrott, N.; Peters, S.; Snoeys, J.; Upreti, V.; Zheng, M.; Hall, S. Physiologically Based Pharmacokinetic Modeling in Drug Discovery and Development: A Pharmaceutical Industry Perspective. Clin. Pharmacol. Ther. 2015, 97, 247–262. [Google Scholar] [CrossRef] [PubMed]
- Michelet, R.; van Bocxlaer, J.; Allegaert, K.; Vermeulen, A. The Use of PBPK Modeling across the Pediatric Age Range Using Propofol as a Case. J. Pharmacokinet. Pharmacodyn. 2018, 45, 765–785. [Google Scholar] [CrossRef] [PubMed]
- Schulz, J.; Michelet, R.; Joseph, J.F.; Zeitlinger, M.; Schumacher, F.; Mikus, G.; Kloft, C. A Versatile High-Performance LC-MS/MS Assay for the Quantification of Voriconazole and Its N-Oxide Metabolite in Small Sample Volumes of Multiple Human Matrices for Biomedical Applications. J. Pharm. Biomed. Anal. 2022, 210, 114551. [Google Scholar] [CrossRef]
- European Medicines Agency. Guideline on Bioanalytical Method Validation. Available online: https://www.ema.europa.eu/documents/scientific-guideline/guideline-bioanalytical-method-validation_en.pdf (accessed on 27 July 2021).
- U.S. Food and Drug Administration. Drug Development and Drug Interactions|Table of Substrates, Inhibitors and Inducers. Available online: https://www.fda.gov/drugs/drug-interactions-labeling/drug-development-and-drug-interactions-table-substrates-inhibitors-and-inducers#table1-2 (accessed on 18 August 2021).
- European Medicines Agency. Guideline on the Investigation of Drug Interactions. Available online: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/07/WC500129606.pdf (accessed on 18 August 2021).
- Khojasteh, S.C.; Prabhu, S.; Kenny, J.R.; Halladay, J.S.; Lu, A.Y.H. Chemical Inhibitors of Cytochrome P450 Isoforms in Human Liver Microsomes: A Re-Evaluation of P450 Isoform Selectivity. Eur. J. Drug Metab. Pharmacokinet. 2011, 36, 1–16. [Google Scholar] [CrossRef]
- Foti, R.S.; Wahlstrom, J.L. CYP2C19 Inhibition: The Impact of Substrate Probe Selection on in vitro Inhibition Profiles. Drug Metab. Dispos. 2008, 36, 523–528. [Google Scholar] [CrossRef] [Green Version]
- Dekker, S.J.; Dohmen, F.; Vermeulen, N.P.E.; Commandeur, J.N.M. Characterization of Kinetics of Human Cytochrome P450s Involved in Bioactivation of Flucloxacillin: Inhibition of CYP3A-Catalysed Hydroxylation by Sulfaphenazole. Br. J. Pharmacol. 2019, 176, 466–477. [Google Scholar] [CrossRef] [Green Version]
- Kumar, V.; Wahlstrom, J.L.; Rock, D.A.; Warren, C.J.; Gorman, L.A.; Tracy, T.S. CYP2C9 Inhibition: Impact of Probe Selection and Pharmacogenetics on in vitro Inhibition Profiles. Drug Metab. Dispos. 2006, 34, 1966–1975. [Google Scholar] [CrossRef] [Green Version]
- Barecki, M.E.; Casciano, C.N.; Johnson, W.W.; Clement, R.P. In Vitro Characterization of the Inhibition Profile of Loratadine, Desloratadine, and 3-OH-Desloratadine for Five Human Cytochrome P-450 Enzymes. Drug Metab. Dispos. 2001, 29, 1173–1175. [Google Scholar]
- Chen, Y.; Liu, L.; Nguyen, K.; Fretland, A.J. Utility of Intersystem Extrapolation Factors in Early Reaction Phenotyping and the Quantitative Extrapolation of Human Liver Microsomal Intrinsic Clearance Using Recombinant Cytochromes P450. Drug Metab. Dispos. 2011, 39, 373–382. [Google Scholar] [CrossRef] [Green Version]
- Proctor, N.J.; Tucker, G.T.; Rostami-Hodjegan, A. Predicting Drug Clearance from Recombinantly Expressed CYPs: Intersystem Extrapolation Factors. Xenobiotica 2004, 34, 151–178. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Jamei, M.; Yeo, K.R.; Rostami-Hodjegan, A.; Tucker, G.T. Misuse of the Well-Stirred Model of Hepatic Drug Clearance. Drug Metab. Dispos. 2007, 35, 501–502. [Google Scholar] [CrossRef] [PubMed]
- Pang, K.S.; Han, Y.R.; Noh, K.; Lee, P.I.; Rowland, M. Hepatic Clearance Concepts and Misconceptions: Why the Well-Stirred Model Is Still Used Even Though It Is Not Physiologic Reality? Biochem. Pharmacol. 2019, 169, 113596. [Google Scholar] [CrossRef] [PubMed]
- Berry, L.; Zhao, Z. An Examination of IC50 and IC50-Shift Experiments in Assessing Time-Dependent Inhibition of CYP3A4, CYP2D6 and CYP2C9 in Human Liver Microsomes. Drug Metab. Lett. 2008, 2, 51–59. [Google Scholar] [CrossRef]
- Akaike, H. A New Look at the Statistical Model Identification. IEEE Trans. Autom. Control 1974, 19, 716–723. [Google Scholar] [CrossRef]
- Achour, B.; Barber, J.; Rostami-Hodjegan, A. Expression of Hepatic Drug-Metabolizing Cytochrome P450 Enzymes and Their Intercorrelations: A Meta-Analysis. Drug Metab. Dispos. 2014, 42, 1349–1356. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Gao, N.; Tian, X.; Liu, T.; Fang, Y.; Zhou, J.; Wen, Q.; Xu, B.; Qi, B.; Gao, J.; et al. Content and Activity of Human Liver Microsomal Protein and Prediction of Individual Hepatic Clearance in vivo. Sci. Rep. 2015, 5, 17671. [Google Scholar] [CrossRef] [Green Version]
- U.S. Food and Drug Administration. In Vitro Drug Interaction Studies: Cytochrome P450 Enzyme-and Transporter-Mediated Drug Interactions, Guidance for Industry. Available online: https://www.fda.gov/media/134582/download (accessed on 4 December 2021).
- Abdul-Aziz, M.H.; Alffenaar, J.-W.C.; Bassetti, M.; Bracht, H.; Dimopoulos, G.; Marriott, D.; Neely, M.N.; Paiva, J.-A.; Pea, F.; Sjovall, F.; et al. Antimicrobial Therapeutic Drug Monitoring in Critically Ill Adult Patients: A Position Paper. Intensive Care Med. 2020, 46, 1127–1153. [Google Scholar] [CrossRef]
- ter Avest, M.; Veringa, A.; van Hateren, K.; Koster, R.A.; Touw, D.J.; Alffenaar, J.-W. Method for Therapeutic Drug Monitoring of Voriconazole and Its Primary Metabolite Voriconazole-N-Oxide in Human Serum Using LC-MS/MS. J. Appl. Bioanal. 2018, 4, 114–123. [Google Scholar] [CrossRef]
- Nagar, S.; Argikar, U.A.; Tweedie, D.J. Enzyme Kinetics in Drug Metabolism: Fundamentals and Applications; Humana Press: Totowa, NJ, USA, 2014; ISBN 9781627037570. [Google Scholar]
- Chauret, N.; Gauthier, A.; Nicoll-Griffith, D.A. Effect of Common Organic Solvents on in vitro Cytochrome P450-Mediated Metabolic Activities in Human Liver Microsomes. Drug Metab. Dispos. 1998, 26, 1–4. [Google Scholar]
- McLure, J.A.; Miners, J.O.; Birkett, D.J. Nonspecific Binding of Drugs to Human Liver Microsomes. Br. J. Clin. Pharmacol. 2000, 49, 453–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinney, S.K.; Knopp, S.; Chang, C.; Hall, S.D.; Li, L. Integration of in vitro Binding Mechanism Into the Semiphysiologically Based Pharmacokinetic Interaction Model Between Ketoconazole and Midazolam. CPT Pharmacomet. Syst. Pharmacol. 2013, 2, e75. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Tang, X.; Yang, T.; Xu, J.; Zhang, J.; Liu, X.; Liu, L. Predicted Contributions of Cytochrome P450s to Drug Metabolism in Human Liver Microsomes Using Relative Activity Factor Were Dependent on Probes. Xenobiotica 2019, 49, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Siu, Y.A.; Lai, W.G. Impact of Probe Substrate Selection on Cytochrome P450 Reaction Phenotyping Using the Relative Activity Factor. Drug Metab. Dispos. 2017, 45, 183–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikus, G.; Schöwel, V.; Drzewinska, M.; Rengelshausen, J.; Ding, R.; Riedel, K.-D.; Burhenne, J.; Weiss, J.; Thomsen, T.; Haefeli, W.E. Potent Cytochrome P450 2C19 Genotype-Related Interaction between Voriconazole and the Cytochrome P450 3A4 Inhibitor Ritonavir. Clin. Pharmacol. Ther. 2006, 80, 126–135. [Google Scholar] [CrossRef]
- Kirbs, C.; Kluwe, F.; Drescher, F.; Lackner, E.; Matzneller, P.; Weiss, J.; Zeitlinger, M.; Kloft, C. High Voriconazole Target-Site Exposure after Approved Sequence Dosing Due to Nonlinear Pharmacokinetics Assessed by Long-Term Microdialysis. Eur. J. Pharm. Sci. 2019, 131, 218–229. [Google Scholar] [CrossRef] [PubMed]
- Paine, M.F.; Hart, H.L.; Ludington, S.S.; Haining, R.L.; Rettie, A.E.; Zeldin, D.C. The human intestinal cytochrome p450 “pie”. Drug Metab. Dispos. 2006, 34, 880–886. [Google Scholar] [CrossRef]
- Hohmann, N.; Kocheise, F.; Carls, A.; Burhenne, J.; Weiss, J.; Haefeli, W.E.; Mikus, G. Dose-Dependent Bioavailability and CYP3A Inhibition Contribute to Non-Linear Pharmacokinetics of Voriconazole. Clin. Pharmacokinet. 2016, 55, 1535–1545. [Google Scholar] [CrossRef]
- Niwa, T.; Imagawa, Y.; Yamazaki, H. Drug Interactions between Nine Antifungal Agents and Drugs Metabolized by Human Cytochromes P450. Curr. Drug Metab. 2015, 15, 651–679. [Google Scholar] [CrossRef]
- Luo, X.; Li, T.; Yu, Z.; Xue, X.; Zhao, H.; Li, N.; Ma, L.; Yang, C.; Huang, L.; Feng, W. The Impact of Azole Antifungal Drugs on Imatinib Metabolism in Human Liver Microsomes. Xenobiotica 2019, 49, 753–761. [Google Scholar] [CrossRef]
- Li, X.; Frechen, S.; Moj, D.; Lehr, T.; Taubert, M.; Hsin, C.H.; Mikus, G.; Neuvonen, P.J.; Olkkola, K.T.; Saari, T.I.; et al. A Physiologically Based Pharmacokinetic Model of Voriconazole Integrating Time-Dependent Inhibition of CYP3A4, Genetic Polymorphisms of CYP2C19 and Predictions of Drug–Drug Interactions. Clin. Pharmacokinet. 2020, 59, 781–808. [Google Scholar] [CrossRef] [PubMed]
- Parkinson, A.; Kazmi, F.; Buckley, D.B.; Yerino, P.; Paris, B.L.; Holsapple, J.; Toren, P.; Otradovec, S.M.; Ogilvie, B.W. An Evaluation of the Dilution Method for Identifying Metabolism-Dependent Inhibitors of Cytochrome P450 Enzymes. Drug Metab. Dispos. 2011, 39, 1370–1387. [Google Scholar] [CrossRef] [PubMed]
- Giri, P.; Naidu, S.; Patel, N.; Patel, H.; Srinivas, N.R. Evaluation of In Vitro Cytochrome P450 Inhibition and in vitro Fate of Structurally Diverse N-Oxide Metabolites: Case Studies with Clozapine, Levofloxacin, Roflumilast, Voriconazole and Zopiclone. Eur. J. Drug Metab. Pharmacokinet. 2017, 42, 677–688. [Google Scholar] [CrossRef] [PubMed]
- Peters, S.A.; Dolgos, H. Requirements to Establishing Confidence in Physiologically Based Pharmacokinetic (PBPK) Models and Overcoming Some of the Challenges to Meeting Them. Clin. Pharmacokinet. 2019, 58, 1355–1371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rostami-Hodjegan, A. Physiologically Based Pharmacokinetics Joined with in vitro–in vivo Extrapolation of ADME: A Marriage Under the Arch of Systems Pharmacology. Clin. Pharmacol. Ther. 2012, 92, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Jin, J.Y.; Mukadam, S.; Malhi, V.; Kenny, J.R. Application of IVIVE and PBPK Modeling in Prospective Prediction of Clinical Pharmacokinetics: Strategy and Approach during the Drug Discovery Phase with Four Case Studies. Biopharm. Drug Dispos. 2012, 33, 85–98. [Google Scholar] [CrossRef] [Green Version]
- Damle, B.; Varma, M.V.; Wood, N. Pharmacokinetics of Voriconazole Administered Concomitantly with Fluconazole and Population-Based Simulation for Sequential Use. Antimicrob. Agents Chemother. 2011, 55, 5172–5177. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Zhu, L.; Chen, L.; Li, N.; Qi, F. Assessment of Drug–Drug Interactions between Voriconazole and Glucocorticoids. J. Chemother. 2018, 30, 296–303. [Google Scholar] [CrossRef]
- Kluwe, F.; Michelet, R.; Mueller-Schoell, A.; Maier, C.; Klopp-Schulze, L.; Dyk, M.; Mikus, G.; Huisinga, W.; Kloft, C. Perspectives on Model-Informed Precision Dosing in the Digital Health Era: Challenges, Opportunities, and Recommendations. Clin. Pharmacol. Ther. 2021, 109, 29–36. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzymatic System | KM (95% CI) (µM) | Vmax,HLM/HIM (95% CI) (pmol/min·mg) or Vmax,rhCYP (95% CI) (pmol/min·pmol) |
|---|---|---|
| HLM | 2.98 (2.63–3.33) | 26.1 (25.4–26.8) |
| HIM | 2.53 (2.15–2.92) | 3.85 (3.70–4.00) |
| rhCYP2C19 | 1.31 (0.862–1.75) | 1.64 (1.50–1.77) |
| rhCYP2C9 | 4.06 (3.32–4.81) | 0.00705 (0.00665–0.00744) |
| rhCYP3A4 | 1.20 (0.830–1.58) | 0.00893 (0.00827–0.00958) |
| Voriconazole Concentration (µM) | Mean (SD) Reaction Velocity (pmol/min·mg) | % of Uninhibited Control | Mean (SD) % of Uninhibited Control | % Inhibition |
|---|---|---|---|---|
| Control incubation without inhibitor | ||||
| 0.5 | 2.92 (0.288) | 100 | 100 | 0 |
| 1 | 5.34 (0.574) | 100 | ||
| 2 | 7.79 (1.05) | 100 | ||
| 3 | 9.38 (1.11) | 100 | ||
| Incubation with CYP2C19 inhibitor loratadine | ||||
| 0.5 | 1.23 (0.118) | 42.0 | 38.2 (2.64) | 61.8 |
| 1 | 2.01 (0.267) | 37.7 | ||
| 2 | 2.86 (0.351) | 36.7 | ||
| 3 | 3.40 (0.311) | 36.2 | ||
| Incubation with CYP2C9 inhibitor sulfaphenazole | ||||
| 0.5 | 1.89 (0.124) | 64.8 | 63.8 (2.69) | 36.2 |
| 1 | 3.24 (0.431) | 60.7 | ||
| 2 | 4.88 (0.391) | 62.6 | ||
| 3 | 6.48 (0.839) | 66.9 | ||
| Incubation with CYP3A4 inhibitor ketoconazole | ||||
| 0.5 | 1.45 (0.140) | 49.8 | 52.4 (4.26) | 47.6 |
| 1 | 2.58 (0.248) | 48.4 | ||
| 2 | 4.17 (0.350) | 53.6 | ||
| 3 | 5.43 (0.393) | 57.9 | ||
| Incubation with a mixture of the CYP 2C19, 2C9 and 3A4 inhibitors loratadine, sulfaphenazole and ketoconazole | ||||
| 0.5 | 0.249 (0.0151) | 8.52 | 10.8 (2.02) | 89.2 |
| 1 | 0.525 (0.0442) | 9.83 | ||
| 2 | 0.910 (0.111) | 11.7 | ||
| 3 | 1.23 (0.149) | 13.1 | ||
| Enzyme | Inhibitor | IC50 (95% Confidence Interval) (µM) | IC50 NADPH (−) (95% Confidence Interval) (µM) | IC50 Shift NADPH (−) | IC50 NADPH (+) (95% Confidence Interval) (µM) | IC50 Shift NADPH (+) |
|---|---|---|---|---|---|---|
| CYP2C19 | VRC | 3.72 (2.85–4.78) | 5.59 (4.61–6.74) | 0.667 | 5.02 (4.12–6.08) | 0.741 |
| NO | 288 (65.0–31,623) | 450 (93.3–339,557) | 0.641 | 320 (52.0–364,870) | 0.900 | |
| OH–VRC | 41.7 (26.9–89.1) | 35.5 (26.8–53.6) | 1.17 | 33.6 (17.0–160) | 1.24 | |
| CYP2C9 | VRC | 4.17 (2.54–6.51) | 3.31 (2.70–4.01) | 1.29 | 3.16 (2.71–3.67) | 1.35 |
| NO | 13.4 (9.90–19.1) | 10.1 (8.28–12.4) | 1.34 | 14.9 (11.1–21.3) | 0.899 | |
| OH–VRC | 3.67 (3.16–4.26) | 3.68 (2.91–4.59) | 0.997 | 3.64 (3.01–4.39) | 1.01 | |
| CYP3A4 | VRC | 1.76 (1.26–2.36) | 2.90 (2.13–3.85) | 0.607 | 2.63 (1.97–3.40) | 0.669 |
| NO | 4.48 (3.78–5.29) | 6.96 (4.63–10.2) | 0.644 | 2.91 (1.36–6.19) | 1.54 | |
| OH–VRC | 1.02 (0.796–1.27) | 0.579 (0.323–0.966) | 1.76 | 1.57 (1.25–1.95) | 0.650 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schulz, J.; Thomas, A.; Saleh, A.; Mikus, G.; Kloft, C.; Michelet, R. Towards the Elucidation of the Pharmacokinetics of Voriconazole: A Quantitative Characterization of Its Metabolism. Pharmaceutics 2022, 14, 477. https://doi.org/10.3390/pharmaceutics14030477
Schulz J, Thomas A, Saleh A, Mikus G, Kloft C, Michelet R. Towards the Elucidation of the Pharmacokinetics of Voriconazole: A Quantitative Characterization of Its Metabolism. Pharmaceutics. 2022; 14(3):477. https://doi.org/10.3390/pharmaceutics14030477
Chicago/Turabian StyleSchulz, Josefine, Antonia Thomas, Ayatallah Saleh, Gerd Mikus, Charlotte Kloft, and Robin Michelet. 2022. "Towards the Elucidation of the Pharmacokinetics of Voriconazole: A Quantitative Characterization of Its Metabolism" Pharmaceutics 14, no. 3: 477. https://doi.org/10.3390/pharmaceutics14030477
APA StyleSchulz, J., Thomas, A., Saleh, A., Mikus, G., Kloft, C., & Michelet, R. (2022). Towards the Elucidation of the Pharmacokinetics of Voriconazole: A Quantitative Characterization of Its Metabolism. Pharmaceutics, 14(3), 477. https://doi.org/10.3390/pharmaceutics14030477