
Oligonucleotide Therapeutics: From Discovery and Development to Patentability

Abstract
:1. Introduction
2. Oligonucleotides on the Market and in Clinical Development
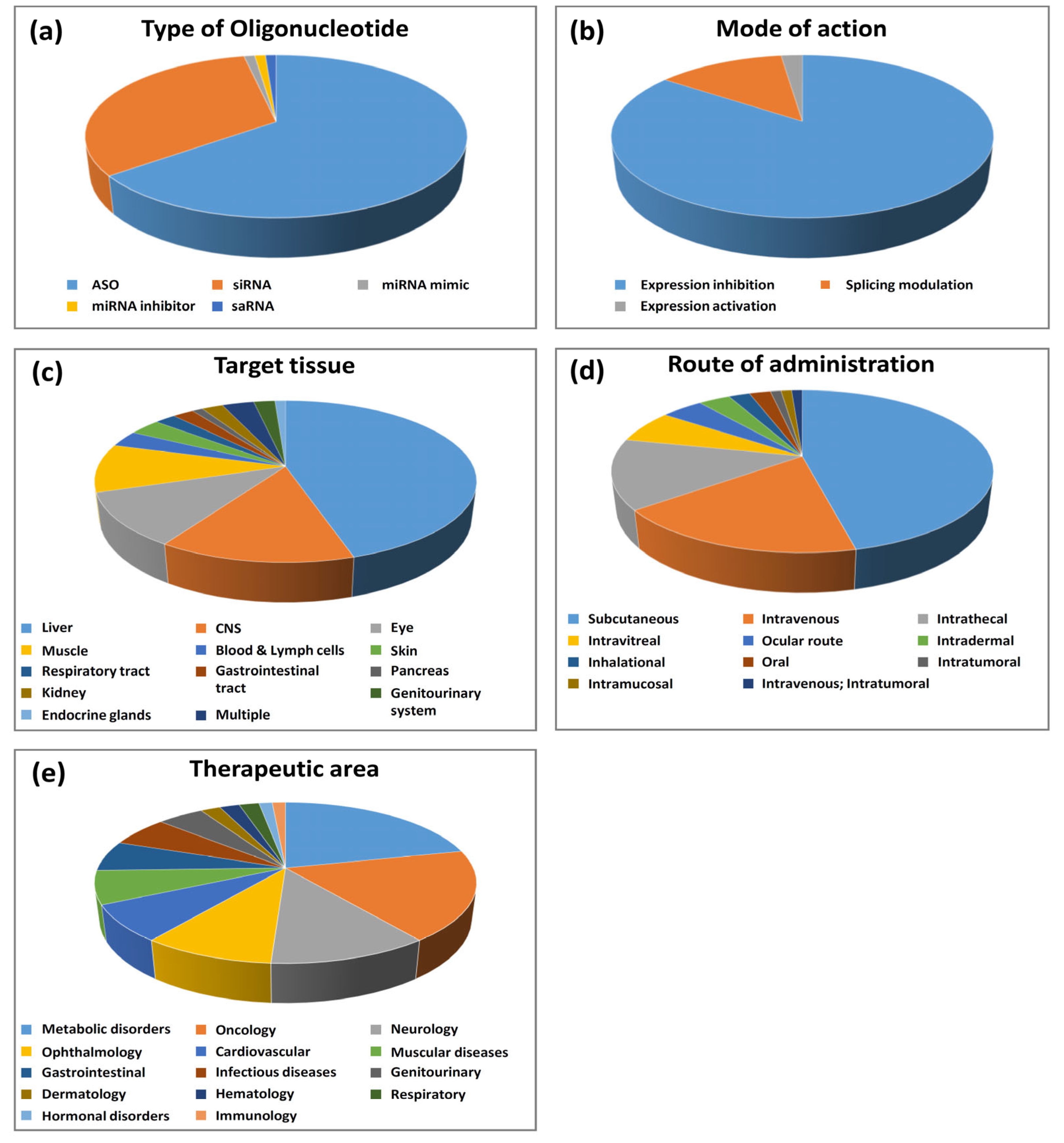
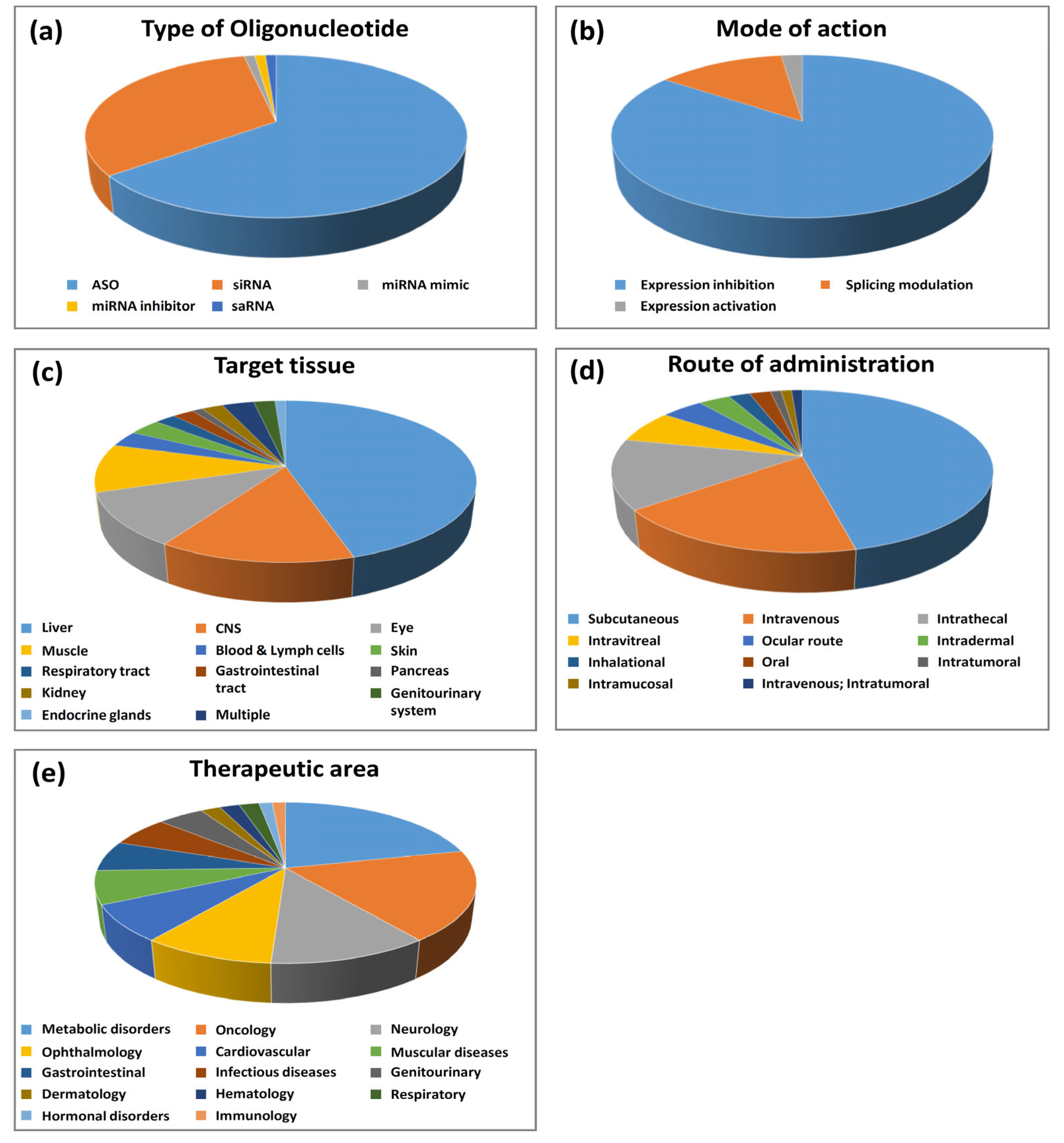
2.1. Type of Olignucleotides and Mode of Action
2.1.1. Antisense Oligonucleotides (ASOs)
- Gene expression-inhibiting ASOs
- Splicing–modifying ASOs
2.1.2. Small Interfering RNA (siRNA)
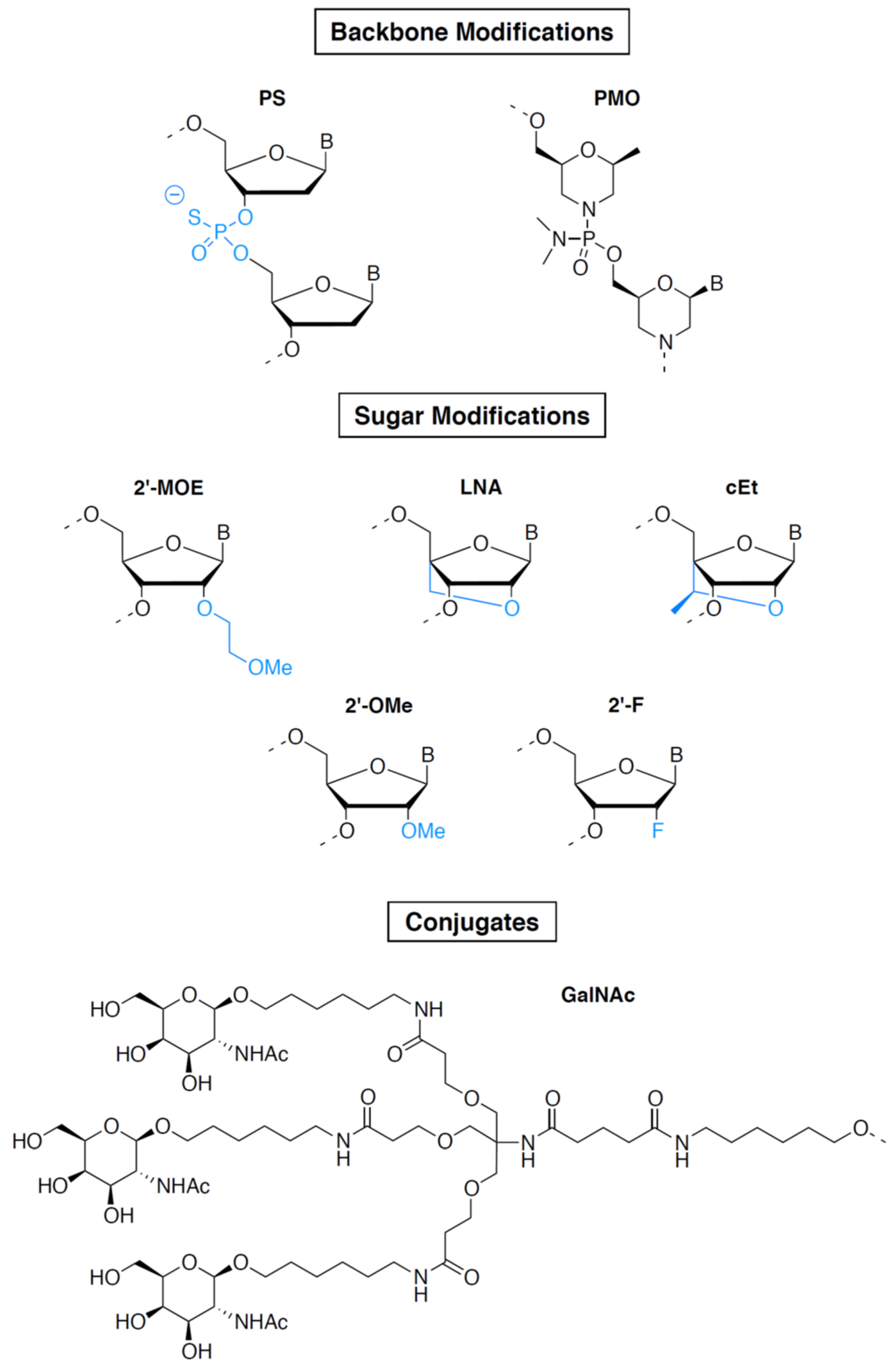
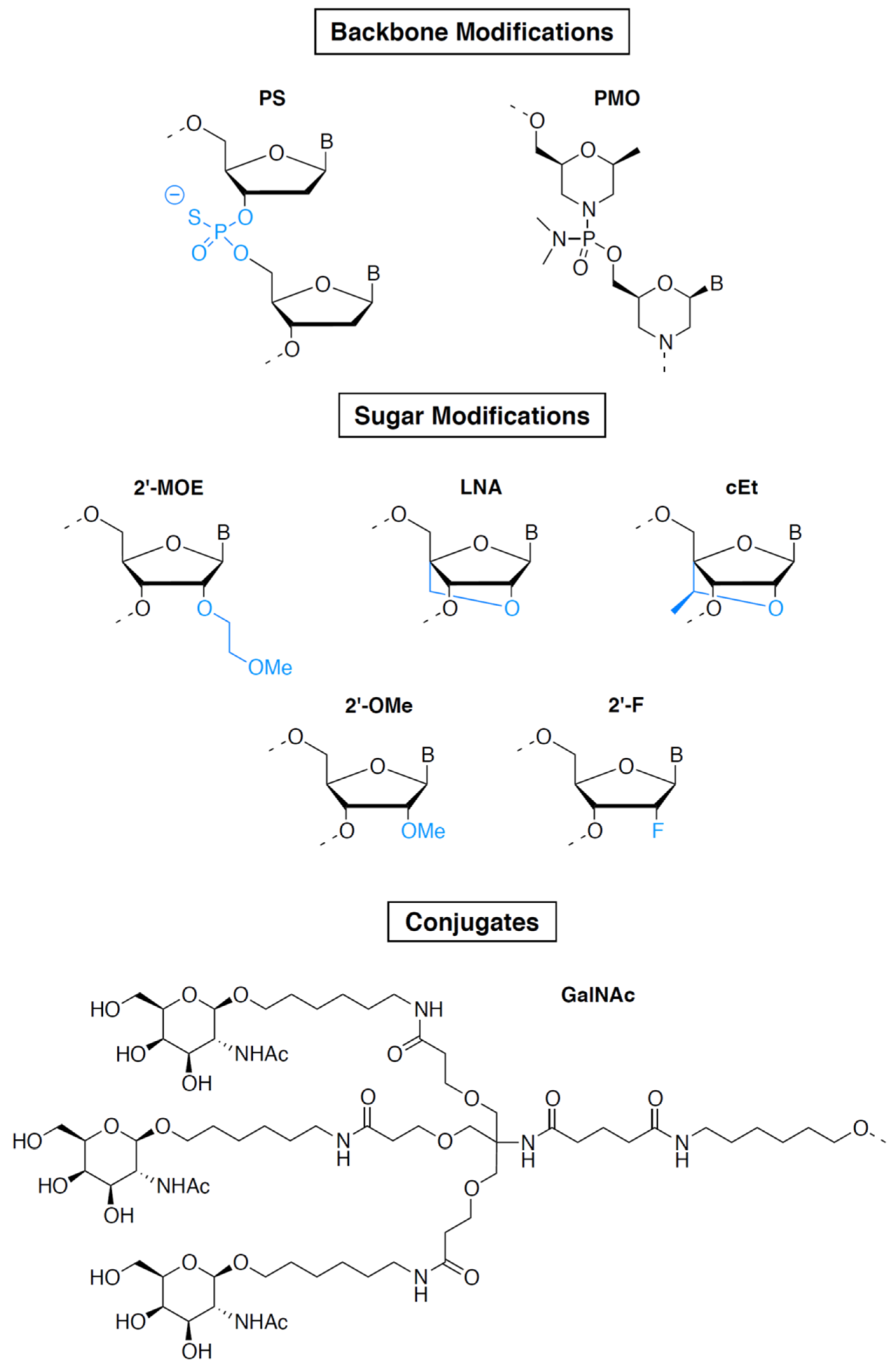
2.1.3. Oligonucleotides Other Than ASOs and siRNAs
2.2. Oligonucleotide Target Genes
2.3. Target Tissues and Routes of Administration
2.4. Therapeutic Areas and Indications
3. Patentability of Oligonucleotides
3.1. Oligonucleotide Therapeutics: How to Patent Them?
3.2. The Different Strategies of Oligonucleotide Therapeutics Protection
“An antisense oligonucleotide that reduces the expression and/or activity of [the target].”
“An antisense oligonucleotide that reduces the expression and/or activity of [the target] and targets the nucleic acids sequence X to XX of the nucleic acid sequence as set for SEQ ID NO: X.”
“An antisense oligonucleotide of gene X with a nucleic acid sequence as set forth in SEQ ID NO: 1.”
“Sequence listing example: SEQ ID NO: 1: acgcggtttattcggttaaa”
“An oligonucleotide with the following structure:atcgatc-(x)N-aattccgwherein (x) is any nucleic acid and N is comprised between 1 and 10,wherein the nucleic acids of the oligonucleotide are linked by covalent bonds, andwherein the oligonucleotide has no more than 60% guanosine nucleobases.”
3.3. Prosecution in Front of the EPO and USPTO
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hopkins, A.L.; Groom, C.R. The druggable genome. Nat. Rev. Drug Discov. 2002, 1, 727–730. [Google Scholar] [CrossRef] [PubMed]
- Crook, S.T.; Witztum, J.L.; Bennett, C.F.; Baker, B.F. RNA-Targeted Therapeutics. Cell Metab. 2018, 27, 714–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.C.; Okino, S.T.; Zhao, H.; Pookot, D.; Place, R.F.; Urakami, S.; Enokida, H.; Dahiya, R. Small dsRNAs induce transcriptional activation in human cells. Proc. Natl. Acad. Sci. USA 2006, 103, 17337–17342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winkle, M.; El-Daly, S.M.; Fabbri, M.; Calin, G.A. Noncoding RNA therapeutics—Challenges and potential solutions. Nat. Rev. Drug. Discov. 2021, 20, 629–651. [Google Scholar] [CrossRef]
- Stephenson, M.L.; Zamecnik, P.C. Inhibition of Rous sarcoma viral RNA translation by a specific oligodeoxyribonucleotide. Proc. Natl. Acad. Sci. USA 1978, 75, 285–288. [Google Scholar] [CrossRef] [Green Version]
- Roehr, B. Fomivirsen approved for CMV retinitis. J. Int. Assoc. Physicians AIDS Care 1998, 4, 14–16. [Google Scholar]
- Wadman, M. Antisense rescues babies from killer disease. Science 2016, 354, 1359–1360. [Google Scholar] [CrossRef]
- Hoy, S.M. Nusinersen: First Global Approval. Drugs 2017, 77, 473–479. [Google Scholar] [CrossRef]
- Bennett, C.F. Therapeutic Antisense Oligonucleotides Are Coming of Age. Annu. Rev. Med. 2019, 70, 307–321. [Google Scholar] [CrossRef]
- Eckstein, F. Phosphorothioate oligodeoxynucleotides: What is their origin and what is unique about them? Antisense Nucleic Acid Drug Dev. 2000, 10, 117–121. [Google Scholar] [CrossRef]
- Roberts, T.C.; Langer, R.; Wood, M.J.A. Advances in oligonucleotide drug delivery. Nat. Rev. Drug Discov. 2020, 19, 673–694. [Google Scholar] [CrossRef]
- Hair, P.; Cameron, F.; McKeage, K. Mipomersen sodium: First global approval. Drugs 2013, 73, 487–493. [Google Scholar] [CrossRef]
- Keam, S.J. Inotersen: First Global Approval. Drugs 2018, 78, 1371–1376. [Google Scholar] [CrossRef]
- Paik, J.; Duggan, S. Volanesorsen: First Global Approval. Drugs 2019, 79, 1349–1354. [Google Scholar] [CrossRef]
- Khorkova, O.; Wahlestedt, C. Oligonucleotide therapies for disorders of the nervous system. Nat. Biotechnol. 2017, 35, 249–263. [Google Scholar] [CrossRef]
- Dees, S.; Ganesan, R.; Singh, S.; Grewal, I.S. Regulatory T cell targeting in cancer: Emerging strategies in immunotherapy. Eur. J. Immunol. 2021, 51, 280–291. [Google Scholar] [CrossRef]
- Reilley, M.J.; McCoon, P.; Cook, C.; Lyne, P.; Kurzrock, R.; Kim, Y.; Woessner, R.; Younes, A.; Nemunaitis, J.; Fowler, N.; et al. STAT3 antisense oligonucleotide AZD9150 in a subset of patients with heavily pretreated lymphoma: Results of a phase 1b trial. J. Immunother. Cancer 2018, 6, 119. [Google Scholar] [CrossRef] [Green Version]
- De Velasco, M.A.; Kura, Y.; Sakai, K.; Hatanaka, Y.; Davies, B.R.; Campbell, H.; Klein, S.; Kim, Y.; MacLeod, A.R.; Sugimoto, K.; et al. Targeting castration-resistant prostate cancer with androgen receptor antisense oligonucleotide therapy. JCI Insight. 2019, 4, e122688. [Google Scholar] [CrossRef]
- Tsouka, A.N.; Tellis, C.C.; Tselepis, A.D. Pharmacology of PCSK9 Inhibitors: Current Status and Future Perspectives. Curr. Pharm. Des. 2018, 24, 3622–3633. [Google Scholar] [CrossRef]
- Pfeiffer, N.; Voykov, B.; Renieri, G.; Bell, K.; Richter, P.; Weigel, M.; Thieme, H.; Wilhelm, B.; Lorenz, K.; Feindor, M.; et al. First-in-human phase I study of ISTH0036, an antisense oligonucleotide selectively targeting transforming growth factor beta 2 (TGF-β2), in subjects with open-angle glaucoma undergoing glaucoma filtration surgery. PLoS ONE 2017, 12, e0188899. [Google Scholar] [CrossRef] [Green Version]
- Campbell, M.A.; Wengel, J. Locked vs. unlocked nucleic acids (LNA vs. UNA): Contrasting structures work towards common therapeutic goals. Chem. Soc. Rev. 2011, 40, 5680–5689. [Google Scholar] [CrossRef] [PubMed]
- Nair, J.K.; Willoughby, J.L.; Chan, A.; Charisse, K.; Alam, M.R.; Wang, Q.; Hoekstra, M.; Kandasamy, P.; Kel’in, A.V.; Milstein, S.; et al. Multivalent N-acetylgalactosamine-conjugated siRNA localizes in hepatocytes and elicits robust RNAi-mediated gene silencing. J. Am. Chem. Soc. 2014, 136, 16958–16961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Springer, A.D.; Dowdy, S.F. GalNAc-siRNA Conjugates: Leading the Way for Delivery of RNAi Therapeutics. Nucleic Acid Ther. 2018, 28, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Coelho, T.; Ando, Y.; Benson, M.D.; Berk, J.L.; Waddington-Cruz, M.; Dyck, P.J.; Gillmore, J.D.; Khella, S.L.; Litchy, W.J.; Obici, L.; et al. Design and Rationale of the Global Phase 3 NEURO-TTRansform Study of Antisense oligonucleotide AKCEA-TTR-L Rx (ION-682884-CS3) in Hereditary Transthyretin-Mediated Amyloid Polyneuropathy. Neurol. Ther. 2021, 10, 375–389. [Google Scholar] [CrossRef]
- Alexander, V.J.; Xia, S.; Hurh, E.; Hughes, S.G.; O’Dea, L.; Geary, R.S.; Witztum, J.L.; Tsimikas, S. N-acetyl galactosamine-conjugated antisense drug to APOC3 mRNA, triglycerides and atherogenic lipoprotein levels. Eur. Heart J. 2019, 40, 2785–2796. [Google Scholar] [CrossRef]
- Lefebvre, S.; Bürglen, L.; Reboullet, S.; Clermont, O.; Burlet, P.; Viollet, L.; Benichou, B.; Cruaud, C.; Millasseau, P.; Zeviani, M.; et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell 1995, 80, 155–165. [Google Scholar] [CrossRef] [Green Version]
- Monani, U.R.; Lorson, C.L.; Parsons, D.W.; Prior, T.W.; Androphy, E.J.; Burghes, A.H.; McPherson, J.D. A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum. Mol. Genet. 1999, 8, 1177–1183. [Google Scholar] [CrossRef]
- Hoy, S.M. Onasemnogene Abeparvovec: First Global Approval. Drugs 2019, 79, 1255–1262. [Google Scholar] [CrossRef]
- Dhillon, S. Risdiplam: First Approval. Drugs 2020, 80, 1853–1858. [Google Scholar] [CrossRef]
- Han, Z.; Chen, C.; Christiansen, A.; Ji, S.; Lin, Q.; Anumonwo, C.; Liu, C.; Leiser, S.C.; Meena Aznarez, I.; Liau, G.; et al. Antisense oligonucleotides increase Scn1a expression and reduce seizures and SUDEP incidence in a mouse model of Dravet syndrome. Sci. Transl. Med. 2020, 12, eaaz6100. [Google Scholar] [CrossRef]
- Matsuo, M. Antisense Oligonucleotide-Mediated Exon-skipping Therapies: Precision Medicine Spreading from Duchenne Muscular Dystrophy. JMA J. 2021, 4, 232–240. [Google Scholar]
- Duan, D.; Goemans, N.; Takeda, S.; Mercuri, E.; Aartsma-Rus, A. Duchenne muscular dystrophy. Nat. Rev. Dis. Prim. 2021, 7, 13. [Google Scholar] [CrossRef]
- Syed, Y.Y. Eteplirsen: First Global Approval. Drugs 2016, 76, 1699–1704. [Google Scholar] [CrossRef]
- Heo, Y.A. Golodirsen: First Approval. Drugs 2020, 80, 329–333. [Google Scholar] [CrossRef]
- Shirley, M. Casimersen: First Approval. Drugs 2021, 81, 875–879. [Google Scholar] [CrossRef]
- Dhillon, S. Viltolarsen: First Approval. Drugs 2020, 80, 1027–1031. [Google Scholar] [CrossRef]
- Tsoumpra, M.K.; Fukumoto, S.; Matsumoto, T.; Takeda, S.; Wood, M.J.A.; Aoki, Y. Peptide-conjugate antisense based splice-correction for Duchenne muscular dystrophy and other neuromuscular diseases. eBioMedicine 2019, 45, 630–645. [Google Scholar] [CrossRef] [Green Version]
- Cideciyan, A.V.; Jacobson, S.G.; Ho, A.C.; Garafalo, A.V.; Roman, A.J.; Sumaroka, A.; Krishnan, A.K.; Swider, M.; Schwartz, M.R.; Girach, A. Durable vision improvement after a single treatment with antisense oligonucleotide sepofarsen: A case report. Nat. Med. 2021, 27, 785–789. [Google Scholar] [CrossRef]
- Dulla, K.; Slijkerman, R.; van Diepen, H.C.; Albert, S.; Dona, M.; Beumer, W.; Turunen, J.J.; Chan, H.L.; Schulkens, I.A.; Vorthoren, L.; et al. Antisense oligonucleotide-based treatment of retinitis pigmentosa caused by USH2A exon 13 mutations. Mol. Ther. 2021, 29, 2441–2455. [Google Scholar] [CrossRef]
- Hu, B.; Zhong, L.; Weng, Y.; Peng, L.; Huang, Y.; Zhao, Y.; Liang, X.J. Therapeutic siRNA: State of the art. Signal Transduct. Target Ther. 2020, 5, 101. [Google Scholar] [CrossRef]
- Hoy, S.M. Patisiran: First Global Approval. Drugs 2018, 78, 1625–1631. [Google Scholar] [CrossRef]
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef]
- Elbashir, S.M.; Harborth, J.; Lendeckel, W.; Yalcin, A.; Weber, K.; Tuschl, T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 2001, 411, 494–498. [Google Scholar] [CrossRef]
- Blanco, E.; Shen, H.; Ferrari, M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat. Biotechnol. 2015, 33, 941–951. [Google Scholar] [CrossRef]
- Scott, L.J. Givosiran: First Approval. Drugs 2020, 80, 335–339. [Google Scholar] [CrossRef]
- Scott, L.J.; Keam, S.J. Lumasiran: First Approval. Drugs 2021, 81, 277–282. [Google Scholar] [CrossRef]
- Lamb, Y.N. Inclisiran: First Approval. Drugs 2021, 81, 389–395. [Google Scholar] [CrossRef]
- Cohen, J.C.; Boerwinkle, E.; Mosley THJr Hobbs, H.H. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N. Engl. J. Med. 2006, 354, 1264–1272. [Google Scholar] [CrossRef]
- Ray, K.K.; Wright, R.S.; Kallend, D.; Koenig, W.; Leiter, L.A.; Raal, F.J.; Bisch, J.A.; Richardson, T.; Jaros, M.; Wijngaard, P.L.J.; et al. Two Phase 3 Trials of Inclisiran in Patients with Elevated LDL Cholesterol. N. Engl. J. Med. 2020, 382, 1507–1519. [Google Scholar] [CrossRef]
- Gennemark, P.; Walter, K.; Clemmensen, N.; Rekić, D.; Nilsson, C.A.M.; Knöchel, J.; Hölttä, M.; Wernevik, L.; Rosengren, B.; Kakol-Palm, D.; et al. An oral antisense oligonucleotide for PCSK9 inhibition. Sci. Transl. Med. 2021, 13, eabe9117. [Google Scholar] [CrossRef]
- Lawitz, E.J.; Shevell, D.E.; Tirucherai, G.S.; Du, S.; Chen, W.; Kavita, U.; Coste, A.; Poordad, F.; Karsdal, M.; Nielsen, M.; et al. BMS-986263 in Patients with Advanced Hepatic Fibrosis: 36-Week Results from a Randomized, Placebo-Controlled Phase 2 Trial. Hepatology 2021. Online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. Metazoan MicroRNAs. Cell 2018, 173, 20–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallant-Behm, C.L.; Piper, J.; Lynch, J.M.; Seto, A.G.; Hong, S.J.; Mustoe, T.A.; Maari, C.; Pestano, L.A.; Dalby, C.M.; Jackson, A.L.; et al. A MicroRNA-29 Mimic (Remlarsen) Represses Extracellular Matrix Expression and Fibroplasia in the Skin. J. Investig. Dermatol. 2019, 139, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Daehn, I.S.; Duffield, J.S. The glomerular filtration barrier: A structural target for novel kidney therapies. Nat. Rev. Drug Discov. 2021, 20, 770–788. [Google Scholar] [CrossRef]
- Sarker, D.; Plummer, R.; Meyer, T.; Sodergren, M.H.; Basu, B.; Chee, C.E.; Huang, K.W.; Palmer, D.H.; Ma, Y.T.; Evans, T.R.J.; et al. MTL-CEBPA, a Small Activating RNA Therapeutic Upregulating C/EBP-α, in Patients with Advanced Liver Cancer: A First-in-Human, Multicenter, Open-Label, Phase I Trial. Clin. Cancer Res. 2020, 26, 3936–3946. [Google Scholar] [CrossRef]
- Gupta, A.; Kafetzis, K.N.; Tagalakis, A.D.; Yu-Wai-Man, C. RNA therapeutics in ophthalmology—translation to clinical trials. Exp. Eye Res. 2021, 205, 108482. [Google Scholar] [CrossRef]
- Tasfaout, H.; Buono, S.; Guo, S.; Kretz, C.; Messaddeq, N.; Booten, S.; Greenlee, S.; Monia, B.P.; Cowling, B.S.; Laporte, J. Antisense oligonucleotide-mediated Dnm2 knockdown prevents and reverts myotubular myopathy in mice. Nat. Commun. 2017, 8, 15661. [Google Scholar] [CrossRef] [Green Version]
- Soriano, V. Hepatitis B Gene Therapy Coming to Age. AIDS Rev. 2018, 20, 125–127. [Google Scholar]
- Yuen, M.F.; Heo, J.; Jang, J.W.; Yoon, J.H.; Kweon, Y.O.; Park, S.J.; Tami, Y.; You, S.; Yates, P.; Tao, Y.; et al. Safety, tolerability and antiviral activity of the antisense oligonucleotide bepirovirsen in patients with chronic hepatitis B: A phase 2 randomized controlled trial. Nat. Med. 2021, 27, 1725–1734. [Google Scholar] [CrossRef]
- Mak, L.Y.; Seto, W.K.; Yuen, M.F. Novel Antivirals in Clinical Development for Chronic Hepatitis B Infection. Viruses 2021, 13, 1169. [Google Scholar] [CrossRef]
- Amgen v. Sanofi, 872 F.3d 1367 (Fed. Cir. 2017). Available online: https://cafc.uscourts.gov/sites/default/files/opinions-orders/20-1074.OPINION.2-11-2021_1731739.pdf (accessed on 20 November 2021).
- Univ Western Australia. Antisense Oligonucleotides for Inducing Exon Skipping and Methods of Use Thereof. WO 2006000057, 28 June 2005. [Google Scholar]
- Alnylam Pharmaceuticals Inc. Compositions and Methods for Inhibiting Expression of Transthyretin. WO 2010048228, 20 October 2009. [Google Scholar]
- Isis Pharmaceuticals Inc. Compositions and Methods for Modulating hbv and ttr Expression. WO 2014179627, 1 May 2014. [Google Scholar]
- Alnylam Pharmaceuticals Inc. RNAi Agents, Compositions and Methods of Use Thereof for Treating Transthyretin (ttr) Associated Diseases. WO 2013075035, 16 November 2012. [Google Scholar]
- Ass’n v. Myriad, 569 U.S. 12–398 (U.S. Supreme Court 2013). Available online: https://www.justice.gov/sites/default/files/osg/briefs/2012/01/01/2012-0398.mer.ami.pdf (accessed on 20 November 2021).
- USPTO Guidelines, Nature-Based Products Examples, Example 3. Available online: https://www.uspto.gov/sites/default/files/documents/mdc_examples_nature-based_products.pdf (accessed on 20 November 2021).
- Walker, A.M. Silencing Innovation: The Patent Eligibility of siRNA Therapeutics. Minn. J. Law Sci. Technol. 2020, 21, 333–370. [Google Scholar]
- Tsimikas, S.; Viney, N.J.; Hughes, S.G.; Singleton, W.; Graham, M.J.; Baker, B.F.; Burkey, J.L.; Yang, Q.; Marcovina, S.M.; Geary, R.S.; et al. Antisense therapy targeting apolipoprotein(a): A randomised, double-blind, placebo-controlled phase 1 study. Lancet 2015, 386, 1472–1483. [Google Scholar] [CrossRef]
- Ray, K.K.; Landmesser, U.; Leiter, L.A.; Kallend, D.; Dufour, R.; Karakas, M.; Hall, T.; Troquay, R.P.; Turner, T.; Visseren, F.L.; et al. Inclisiran in patients at high cardiovascular risk with elevated LDL cholesterol. N. Engl. J. Med. 2017, 376, 1430–1440. [Google Scholar] [CrossRef] [Green Version]
- Ämmälä, C.; Drury, W.J., 3rd; Knerr, L.; Ahlstedt, I.; Stillemark-Billton, P.; Wennberg-Huldt, C.; Andersson, E.M.; Valeur, E.; Jansson-Löfmark, R.; Janzén, D.; et al. Targeted delivery of antisense oligonucleotides to pancreatic β-cells. Sci. Adv. 2018, 4, eaat3386. [Google Scholar] [CrossRef] [Green Version]
- Min, H.S.; Kim, H.J.; Naito, M.; Ogura, S.; Toh, K.; Hayashi, K.; Kim, B.S.; Fukushima, S.; Anraku, Y.; Miyata, K.; et al. Systemic Brain Delivery of Antisense Oligonucleotides across the Blood-Brain Barrier with a Glucose-Coated Polymeric Nanocarrier. Angew. Chem. Int. Ed. Engl. 2020, 59, 8173–8180. [Google Scholar] [CrossRef] [Green Version]
- Nagata, T.; Dwyer, C.A.; Yoshida-Tanaka, K.; Ihara, K.; Ohyagi, M.; Kaburagi, H.; Miyata, H.; Ebihara, S.; Yoshioka, K.; Ishii, T.; et al. Cholesterol-functionalized DNA/RNA heteroduplexes cross the blood-brain barrier and knock down genes in the rodent CNS. Nat. Biotechnol. 2021, 39, 1529–1536. [Google Scholar] [CrossRef]
- Maher, S.; Brayden, D.J.; Casettari, L.; Illum, L. Application of Permeation Enhancers in Oral Delivery of Macromolecules: An Update. Pharmaceutics 2019, 11, 41. [Google Scholar] [CrossRef] [Green Version]
- Lim, K.H.; Han, Z.; Jeon, H.Y.; Kach, J.; Jing, E.; Weyn-Vanhentenryck, S.; Downs, M.; Corrionero, A.; Oh, R.; Scharner, J.; et al. Antisense oligonucleotide modulation of non-productive alternative splicing upregulates gene expression. Nat. Commun. 2020, 11, 3501. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Drug Name | Target Gene | Mode of Action | Therapy Area | Latest Stage of Development | Company |
|---|---|---|---|---|---|
| Nusinersen | SMN2 | Splicing modulation | Neurology | Marketed | Biogen |
| Eteplirsen | DMD | Splicing modulation | Muscular disorders | Marketed | Sarepta Therapeutics |
| Inotersen | TTR | Expression inhibition | Metabolic disorders | Marketed | Akcea Therapeutics |
| Viltolarsen | DMD | Splicing modulation | Muscular disorders | Marketed | NS Pharma |
| Casimersen | DMD | Splicing modulation | Muscular disorders | Marketed | Sarepta Therapeutics |
| Golodirsen | DMD | Splicing modulation | Muscular disorders | Marketed | Sarepta Therapeutics |
| Mipomersen | APOB | Expression inhibition | Metabolic disorders | Marketed | Kastle Therapeutics |
| Volanesorsen | APOC3 | Expression inhibition | Metabolic disorders | Marketed | Akcea Therapeutics |
| Fomivirsen | CMV virus IE2 | Expression inhibition | Infectious disease | Marketed and withdrawn | Novartis |
| Aganirsen | IRS1 | Expression inhibition | Ophthalmology and metabolic disorders | Phase III | Gene Signal |
| Alicaforsen | ICAM1 | Expression inhibition | Gastrointestinal | Phase III | Atlantic Healthcare |
| Eplontersen | TTR | Expression inhibition | Metabolic disorders | Phase III | Akcea Therapeutics |
| ION-363 | FUS | Expression inhibition | Neurology | Phase III | Ionis Pharmaceuticals |
| Olezarsen | APOC3 | Expression inhibition | Cardiovascular and metabolic disorders | Phase III | Akcea Therapeutics |
| Pelacarsen | LPA | Expression inhibition | Cardiovascular and metabolic disorders | Phase III | Novartis |
| Sepofarsen | CEP290 | Splicing modulation | Ophthalmology | Phase III | ProQR Therapeutics |
| Tofersen | SOD1 | Expression inhibition | Neurology | Phase III | Biogen |
| Tominersen | HTT | Expression inhibition | Neurology | Phase III | Roche |
| Trabedersen | TGFB2 | Expression inhibition | Oncology | Phase III | Oncotelic |
| Zilganersen | GFAP | Expression inhibition | Neurology | Phase III | Ionis Pharmaceuticals |
| ASM-8 | CCR3 and CSF2RB | Expression inhibition | Respiratory | Phase II | Pharmaxis |
| Atesidorsen | GHR | Expression inhibition | Hormonal disorders | Phase II | Antisense Therapeutics |
| ATL-1102 | ITGA4 | Expression inhibition | Neurology and muscular disorders | Phase II | Antisense Therapeutics |
| AZD-8233 | PCSK9 | Expression inhibition | Metabolic disorders | Phase II | AstraZeneca |
| AZD-8701 | FOXP3 | Expression inhibition | Oncology | Phase II | AstraZeneca |
| Bepirovirsen | Viral HBV | Expression inhibition | Infectious disease | Phase II | Ionis Pharmaceuticals |
| BIIB-080 | MAPT | Expression inhibition | Neurology | Phase II | Biogen |
| Cepadacursen | PCSK9 | Expression inhibition | Metabolic disorders | Phase II | Civi Biopharma |
| Cimderlirsen | GHR | Expression inhibition | Hormonal disorders | Phase II | Ionis Pharmaceuticals |
| CODA-001 | GJA1 | Expression inhibition | Ophthalmology | Phase II | Eyevance Pharmaceuticals |
| Danvatirsen | STAT3 | Expression inhibition | Oncology | Phase II | AstraZeneca |
| Donidalorsen | KLKB1 | Expression inhibition | Immunology and infectious disease | Phase II | Ionis Pharmaceuticals |
| DYN-101 | DYN2 | Expression inhibition | Muscular disorders | Phase II | Dynacure |
| GTX-102 | UBE2A | Expression inhibition | Neurology | Phase II | GeneTx Biotherapeutics |
| ION-224 | DGAT2 | Expression inhibition | Gastrointestinal | Phase II | Ionis Pharmaceuticals |
| ION-253 | Undisclosed | Expression inhibition | Gastrointestinal | Phase II | Johnson & Johnson |
| ION-464 | SNCA | Expression inhibition | Neurology | Phase II | Ionis Pharmaceuticals |
| ION-541 | ATXN2 | Expression inhibition | Neurology | Phase II | Ionis Pharmaceuticals |
| ION-859 | LRRK2 | Expression inhibition | Neurology | Phase II | Ionis Pharmaceuticals |
| IONIS-AGTLRx | AGT | Expression inhibition | Cardiovascular | Phase II | Ionis Pharmaceuticals |
| IONIS-FB-LRx | CFB | Expression inhibition | Genitourinary system and ophthalmology | Phase II | Ionis Pharmaceuticals |
| IONIS-FXILRx | F11 | Expression inhibition | Cardiovascular, hematology, and genitourinary system | Phase II | Ionis Pharmaceuticals |
| IONIS-GCGRRx | GCGR | Expression inhibition | Metabolic disorders | Phase II | Ionis Pharmaceuticals |
| IONIS-HBVLRx | Viral HBV | Expression inhibition | Infectious disease | Phase II | Ionis Pharmaceuticals |
| IONIS-PKKRx | KLKB1 | Expression inhibition | Neurology | Phase II | Ionis Pharmaceuticals |
| IONISAR-2.5Rx | AR | Expression inhibition | Oncology | Phase II | Ionis Pharmaceuticals |
| IONISENAC-2.5Rx | SCNN1A | Expression inhibition | Respiratory | Phase II | Ionis Pharmaceuticals |
| IONISTMPRSS-6LRx | TMPRSS6 | Expression inhibition | Hematology | Phase II | Ionis Pharmaceuticals |
| ISTH-0036 | TGFB2 | Expression inhibition | Ophthalmology | Phase II | Isarna Therapeutics |
| NS-089 | DMD | Splicing modulation | Muscular disorders | Phase II | Nippon Shinyaku |
| Prexigebersen | GRB2 | Expression inhibition | Oncology | Phase II | Bio-Path Holdings |
| QR-1123 | RHO | Expression inhibition | Ophthalmology | Phase II | ProQR Therapeutics |
| QRX-421a | USH2A | Splicing modulation | Ophthalmology | Phase II | ProQR Therapeutics |
| Renadirsen | DMD | Splicing modulation | Muscular disorders | Phase II | Daiichi Sankyo |
| SRP-5051 | DMD | Splicing modulation | Muscular disorders | Phase II | Sarepta Therapeutics |
| STK-001 | SCN1A | Splicing modulation | Neurology | Phase II | Stoke Therapeutics |
| Vupanorsen | ANGPTL3 | Expression inhibition | Metabolic disorders | Phase II | Pfizer |
| WVE-003 | HTT | Expression inhibition | Neurology | Phase II | Wave Life Sciences |
| WVE-004 | C9orf72 | Expression inhibition | Neurology | Phase II | Wave Life Sciences |
| WVEN-531 | DMD | Splicing modulation | Muscular disorders | Phase II | Wave Life Sciences |
| Drug Name | Target Gene | Mode of Action | Therapy Area | Latest Stage of Development | Company |
|---|---|---|---|---|---|
| Patisiran | TTR | Expression inhibition | Metabolic disorders | Marketed | Alnylam Pharmaceuticals |
| Givosiran | ALAS1 | Expression inhibition | Metabolic disorders | Marketed | Alnylam Pharmaceuticals |
| Inclisiran | PCSK9 | Expression inhibition | Cardiovascular and metabolic disorders | Marketed | Novartis |
| Lumasiran | HAO1 | Expression inhibition | Genitourinary system | Marketed | Alnylam Pharmaceuticals |
| Vutrisiran | TTR | Expression inhibition | Cardiovascular and metabolic disorders | Pre-registration | Alnylam Pharmaceuticals |
| Fitusiran | SERPINC1 | Expression inhibition | Hematology | Phase III | Sanofi |
| Nedosiran | LDHA | Expression inhibition | Genitourinary system | Phase III | Dicerna Pharmaceuticals |
| QPI-1007 | CASP2 | Expression inhibition | Ophthalmology | Phase III | Quark Pharmaceuticals |
| Teprasiran | TP53 | Expression inhibition | Immunology | Phase III | Quark Pharmaceuticals |
| Tivanisiran | TRPV1 | Expression inhibition | Ophthalmology | Phase III | Sylentis |
| AB-729 | HBsAg | Expression inhibition | Infectious disease | Phase II | Arbutus Biopharma |
| ALNAAT-02 | SERPINA1 | Expression inhibition | Gastrointestinal and metabolic disorders | Phase II | Alnylam Pharmaceuticals |
| ARO-HSD | HSD17B13 | Expression inhibition | Gastrointestinal | Phase II | Arrowhead Pharmaceuticals |
| AROANG-3 | ANGPTL3 | Expression inhibition | Metabolic disorders | Phase II | Arrowhead Pharmaceuticals |
| AROAPOC-3 | APOC3 | Expression inhibition | Metabolic disorders | Phase II | Arrowhead Pharmaceuticals |
| Bamosiran | ADRB2 | Expression inhibition | Ophthalmology | Phase II | Sylentis |
| Belcesiran | SERPINA1 | Expression inhibition | Gastrointestinal | Phase II | Dicerna Pharmaceuticals |
| BMS-986263 | SERPINH1 | Expression inhibition | Gastrointestinal and respiratory | Phase II | Bristol-Myers Squibb |
| Cemdisiran | C5 | Expression inhibition | Genitourinary system | Phase II | Alnylam Pharmaceuticals |
| Fazirsiran | SERPINA1 | Expression inhibition | Metabolic disorders | Phase II | Arrowhead Pharmaceuticals |
| JNJ-3989 | viral HBV | Expression inhibition | Infectious disease | Phase II | Arrowhead Pharmaceuticals |
| MT-5745 | CHST15 | Expression inhibition | Gastrointestinal | Phase II | Mitsubishi Tanabe Pharma |
| Olpasiran | LPA | Expression inhibition | Cardiovascular | Phase II | Amgen |
| OLX-101 | CTGF | Expression inhibition | Dermatology | Phase II | Hugel/OliX Pharmaceuticals |
| RG-6346 | HBsAg | Expression inhibition | Infectious disease | Phase II | Dicerna Pharmaceuticals |
| siG-12D-LODER | KRAS | Expression inhibition | Oncology | Phase II | Silenseed |
| SR-063 | AR | Expression inhibition | Oncology | Phase II | Suzhou Ribo Life Sciences |
| STP-705 | PTGS2/TGFB1 | Expression inhibition | Oncology and dermatology | Phase II | Sirnaomics |
| VIR-2218 | HBsAg | Expression inhibition | Infectious disease | Phase II | Alnylam Pharmaceuticals |
| Zilebesiran | AGT | Expression inhibition | Cardiovascular | Phase II | Alnylam Pharmaceuticals |
| Drug Name | Target Gene | Mode of Action (Type of Compound) | Therapy Area | Latest Stage of Development | Company |
|---|---|---|---|---|---|
| Lademirsen | MIR21 | Expression inhibition (miRNA inhibitor) | Genitourinary system | Phase II | Sanofi |
| MTL-CEBPA | CEBPA | Expression activation (saRNA) | Oncology | Phase II | Mina Therapeutics |
| remlarsen | MIR29B1 | Expression activation (miRNA mimic) | Dermatology | Phase II | Miragen Therapeutics |
| Is the Target Known? | Is There at Least One Oligonucleotide against This Target? | Novelty? | Inventive Step? | ||
|---|---|---|---|---|---|
| No | No | Europe | Yes, because the target is new (functional definition possible; however, in the US the oligonucleotide must have a modification to be eligible). | Europe | Yes, because the target is new. |
| United States | United States | Yes, but the USPTO could refuse a patent on the target (comparison to the decision between Amgen v. Sanofi [61] relating to the protection of antibodies). | |||
| Yes | No | Europe | Yes, because it is the first oligonucleotide used in therapy (however, in the US, the oligonucleotide must have a modification to be eligible and a functional definition is not possible). | Europe | Yes, if the oligonucleotide has a particular functional characteristic. |
| United States | United States | ||||
| Yes | Yes | Europe | Yes, if the oligonucleotide has a different sequence/structure (e.g., with modifications) than oligonucleotides of the prior art (However, in the US, the oligonucleotide must have a modification to be eligible and a functional definition is not possible). | Europe | No, unless the oligonucleotide has an unexpected or new property compared to the oligonucleotides disclosed in the prior art (e.g., addition of a new modification, surprising effect) or if the oligonucleotide targets a particular region (e.g., a particular region of a gene). |
| United States | United States | ||||
| Type of Claim | Drug Name | Latest Stage of Development | Patent Application | Claims |
|---|---|---|---|---|
| Functional definition | Casimersen | Marketed | WO2006000057 [62] | 1. An antisense molecule capable of binding to a selected target site to induce exon skipping in the dystrophin gene, as set forth in SEQ ID NO: 1 to 202. |
| Patisiran | Marketed | WO2010048228 [63] | 1. A double-stranded ribonucleic acid (dsRNA) for inhibiting expression of transthyretin (TTR), wherein said dsRNA comprises a sense strand and an antisense strand, the antisense strand comprising a region complementary to a part of an mRNA encoding transthyretin (TTR), wherein said region of complementarity is less than 30 nucleotides in length and the antisense strand comprises 15 or more contiguous nucleotides of SEQ ID NO:170, SEQ ID NO:450, SEQ ID NO:730, or SEQ ID NO:1010. | |
| Structural definition | Inotersen | Marketed | WO2014179627 [64] | 1. A compound comprising a modified oligonucleotide and a conjugate group, wherein the modified oligonucleotide consists of eight to 80 linked nucleosides and has a nucleobase sequence at least 85%, 90%, 95%, or 100% complementary to SEQ ID NO: 2 encoding transthyretin (TTR). |
| Scaffold claim | Vutrisiran | Pre-registration | WO2013075035 [65] | 1. A double-stranded RNAi agent comprising a sense strand complementary to an antisense strand, wherein said antisense strand comprises a region complementary to part of an mRNA encoding transthyretin (TTR), wherein each strand has about 14 to about 30 nucleotides, wherein said double-stranded RNAi agent is represented by formula (III): sense: 5′ np -Na -(X X X);-Nb -Y Y Y -Nb -(Z Z Z)j -Na-nq 3′ antisense: 3′ np’-Na’-(X’X’X’)k-Nb’-Y’Y’Y’-Nb’-(Z’Z’Z’)i-Na’- nq’ 5′ (III) wherein i, j, k, and 1 are each independently 0 or 1; p, p’, q, and q’ are each independently 0–6; each Na and Na’ independently represents an oligonucleotide sequence comprising 0–25 nucleotides that are either modified or unmodified or combinations thereof, each sequence comprising at least two differently modified nucleotides; each Nb and Nb’ independently represents an oligonucleotide sequence comprising 0–10 nucleotides that are either modified or unmodified or combinations thereof; each np, np’, nq, and nq’ independently represents an overhang nucleotide; XXX, YYY, ZZZ, Χ’Χ’Χ’, ΥΎΎ’, and Z’Z’Z’ each independently represent one motif of three identical modifications on three consecutive nucleotides; modifications on Nb differ from the modification on Y and modifications on Nb’ differ from the modification on Y’; and wherein the sense strand is conjugated to at least one ligand. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moumné, L.; Marie, A.-C.; Crouvezier, N. Oligonucleotide Therapeutics: From Discovery and Development to Patentability. Pharmaceutics 2022, 14, 260. https://doi.org/10.3390/pharmaceutics14020260
Moumné L, Marie A-C, Crouvezier N. Oligonucleotide Therapeutics: From Discovery and Development to Patentability. Pharmaceutics. 2022; 14(2):260. https://doi.org/10.3390/pharmaceutics14020260
Chicago/Turabian StyleMoumné, Lara, Anne-Céline Marie, and Nicolas Crouvezier. 2022. "Oligonucleotide Therapeutics: From Discovery and Development to Patentability" Pharmaceutics 14, no. 2: 260. https://doi.org/10.3390/pharmaceutics14020260
APA StyleMoumné, L., Marie, A.-C., & Crouvezier, N. (2022). Oligonucleotide Therapeutics: From Discovery and Development to Patentability. Pharmaceutics, 14(2), 260. https://doi.org/10.3390/pharmaceutics14020260