Discovery of Novel Thiosemicarbazides Containing 1,3,5-Triazines Derivatives as Potential Synergists against Fluconazole-Resistant Candida albicans

 ,
,
Abstract
1. Introduction
2. Experimental Section
2.1. Chemistry
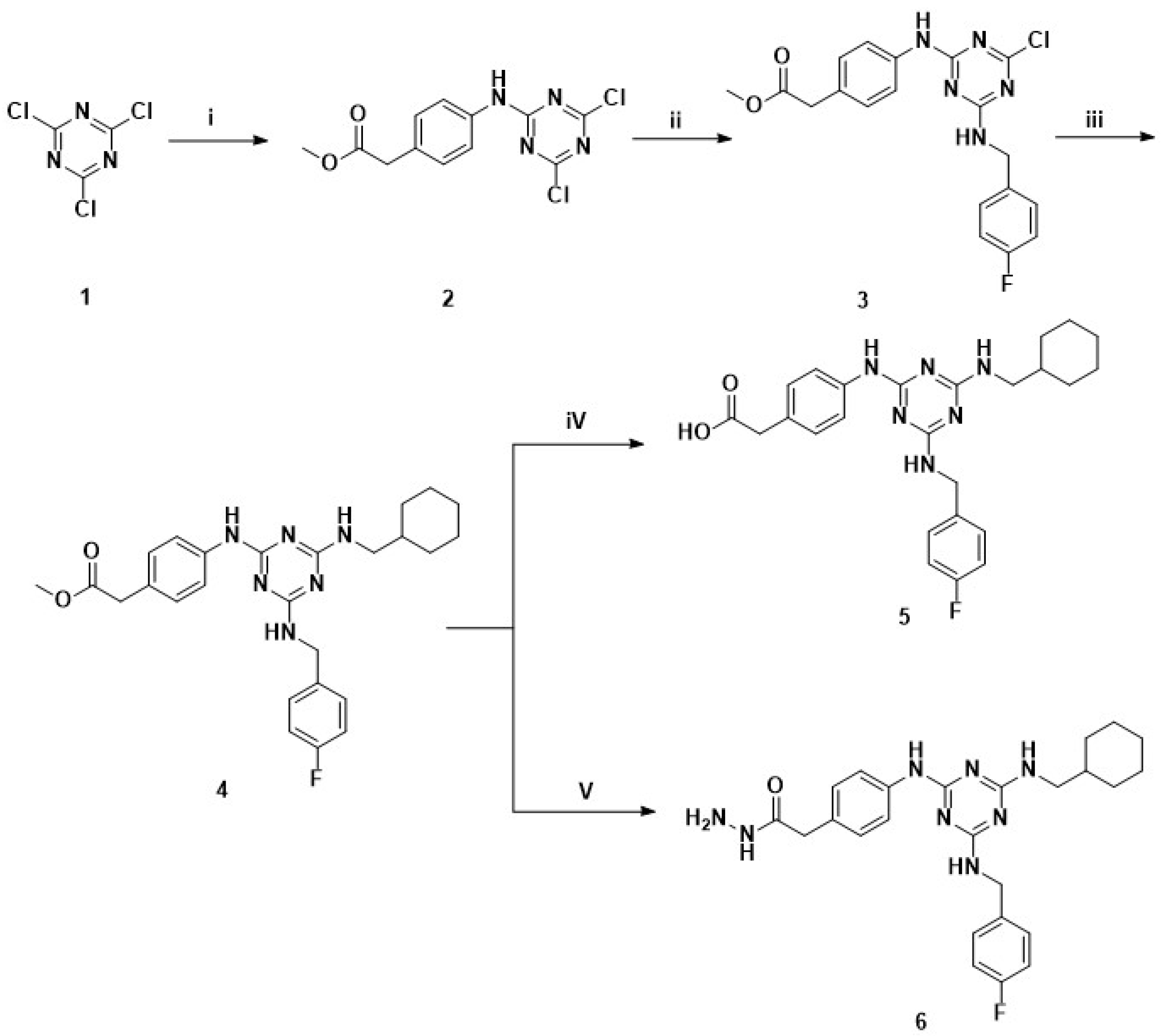
2.1.1. General Procedure for Synthesis of Methyl 2-(4-((4,6-Dichloro-1,3,5-triazin-2-yl)amino)phenyl)acetate (2)
2.1.2. General Procedure for Synthesis of Methyl 2-(4-((4-Chloro-6-((4-fluorobenzyl)amino)-1,3,5-triazin-2-yl)amino)phenyl)acetate (3)
2.1.3. General Procedure for Synthesis of Methyl 2-(4-((4-((Cyclohexylmethyl)amino)-6-((4-fluorobenzyl)amino)-1,3,5-triazin-2-yl)amino)phenyl)acetate (4)
2.1.4. General Procedure for Synthesis of 2-(4-((4-((Cyclohexylmethyl)amino)-6-((4-fluorobenzyl)amino)-1,3,5-triazin-2-yl)amino)phenyl)acetic acid (5)
2.1.5. General Procedure for Synthesis of 2-(4-((4-((Cyclohexylmethyl)amino)-6-((4-fluorobenzyl)amino)-1,3,5-triazin-2-yl)amino)phenyl)acetohydrazide (6)
2.1.6. General Procedure for Synthesis of Target Compounds (8 and 8a–j)
2.1.7. General Procedure for Synthesis of Target Compounds (9a–n)
2.1.8. General Procedure for Synthesis of Target Compounds (10a–o)
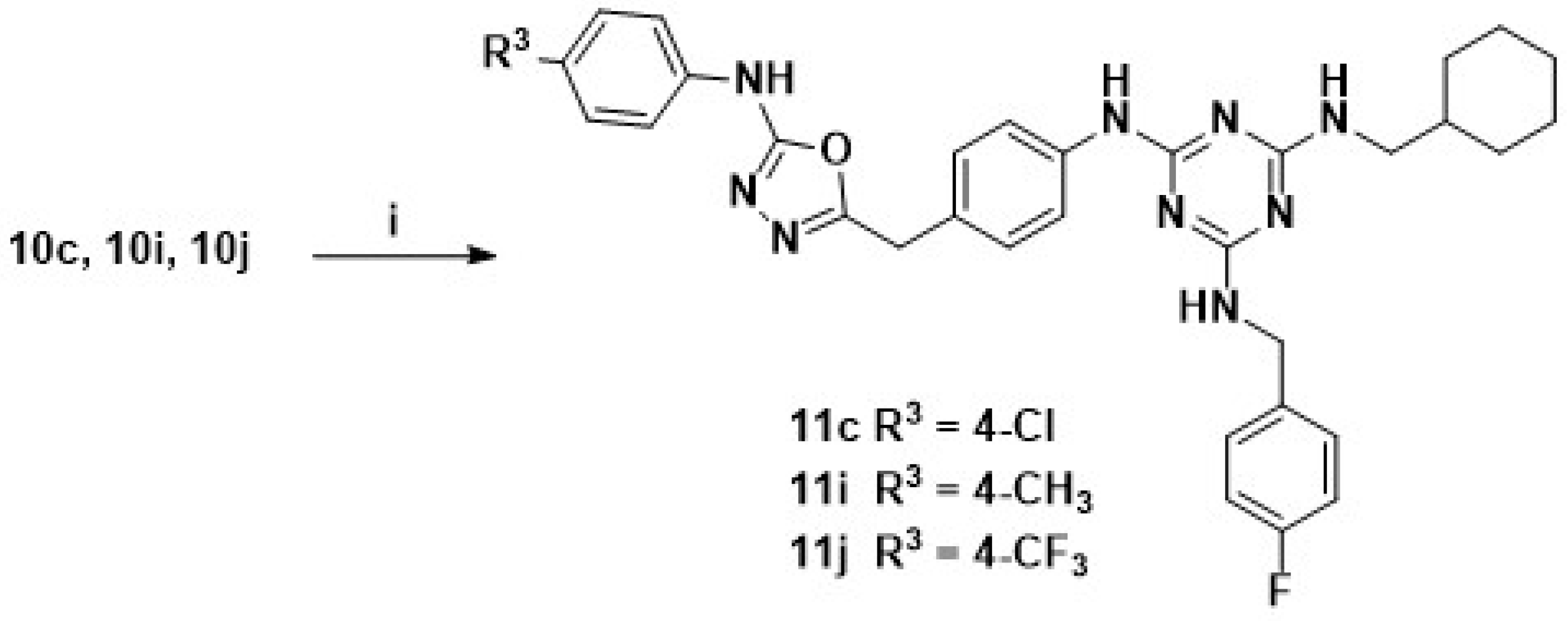
2.1.9. General Procedure for Synthesis of Target Compounds (11c, 11i and 11j)
2.2. Biological Activity
2.2.1. Strains and Culture Conditions
2.2.2. Antifungal Susceptibility Testing
2.2.3. Biofilm Formation Assay
2.2.4. Cytotoxicity Assays
3. Results and Discussion
3.1. Chemistry
3.2. In Vitro Antifungal Evaluation and Structure–Activity Relationships Study
3.3. Checkerboard Microdilution Assay
3.4. Antifungal Spectrum Investigation of Compound 10l
3.5. Inhibitory Effects of Compounds 10l and 10m against the Formation of C. albicans Biofilms
3.6. In Vitro Cytotoxicity Assay
3.7. In Silico Predictive Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Brown, G.D.; Denning, D.W.; Gow, N.A.; Levitz, S.M.; Netea, M.G.; White, T.C. Hidden killers: Human fungal infections. Sci. Transl. Med. 2012, 4, 165rv13. [Google Scholar] [CrossRef] [PubMed]
- Enoch, D.A.; Yang, H.; Aliyu, S.H.; Micallef, C. The Changing Epidemiology of Invasive Fungal Infections. Methods Mol. Biol. 2017, 1508, 17–65. [Google Scholar] [PubMed]
- Rodrigues, M.E.; Silva, S.; Azeredo, J.; Henriques, M. Novel strategies to fight Candida species infection. Crit. Rev. Microbiol. 2016, 42, 594–606. [Google Scholar] [CrossRef] [PubMed]
- Calderone, R.; Sun, N.; Gay-Andrieu, F.; Groutas, W.; Weerawarna, P.; Prasad, S.; Alex, D.; Li, D. Antifungal drug discovery: The process and outcomes. Future Microbiol. 2014, 9, 791–805. [Google Scholar] [CrossRef]
- Denning, D.W.; Bromley, M.J. Infectious Disease. How to bolster the antifungal pipeline. Science 2015, 347, 1414–1416. [Google Scholar] [CrossRef]
- Stewart, A.G.; Paterson, D.L. How urgent is the need for new antifungals? Expert Opin. Pharm. 2021, 22, 1857–1870. [Google Scholar] [CrossRef]
- Maddy, A.J.; Sanchez, N.; Shukla, B.S.; Maderal, A.D. Dermatological manifestations of fungal infection in patients with febrile neutropaenia: A review of the literature. Mycoses 2019, 62, 826–834. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Tu, J.; Ji, C.; Li, Z.; Han, G.; Liu, N.; Li, J.; Sheng, C. Discovery of piperidol derivatives for combinational treatment of azole-resistant candidiasis. ACS Infect. Dis. 2021, 7, 650–660. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Cao, Y.Y.; Dai, B.D.; Sun, X.R.; Zhu, Z.Y.; Cao, Y.B.; Wang, Y.; Gao, P.H.; Jiang, Y.Y. In vitro synergism of fluconazole and baicalein against clinical isolates of Candida albicans resistant to fluconazole. Biol. Pharm. Bull. 2008, 31, 2234–2236. [Google Scholar] [CrossRef]
- Zhao, F.; Dong, H.H.; Wang, Y.H.; Wang, T.Y.; Yan, Z.H.; Yan, F.; Zhang, D.Z.; Cao, Y.Y.; Jin, Y.S. Synthesis and synergistic antifungal effects of monoketone derivatives of curcumin against fluconazole-resistant Candida spp. Medchemcomm 2017, 8, 1093–1102. [Google Scholar] [CrossRef] [PubMed]
- Quan, H.; Cao, Y.Y.; Xu, Z.; Zhao, J.X.; Gao, P.H.; Qin, X.F.; Jiang, Y.Y. Potent in vitro synergism of fluconazole and berberine chloride against clinical isolates of Candida albicans resistant to fluconazole. Antimicrob. Agents Chemother. 2006, 50, 1096–1099. [Google Scholar] [CrossRef]
- Dai, L.; Zang, C.; Tian, S.; Liu, W.; Tan, S.; Cai, Z.; Ni, T.; An, M.; Li, R.; Gao, Y.; et al. Design, synthesis, and evaluation of caffeic acid amides as synergists to sensitize fluconazole-resistant Candida albicans to fluconazole. Bioorganic Med. Chem. Lett. 2015, 25, 34–37. [Google Scholar] [CrossRef]
- Liu, H.; Wang, L.; Li, Y.; Liu, J.; An, M.; Zhu, S.; Cao, Y.; Jiang, Z.; Zhao, M.; Cai, Z.; et al. Structural optimization of berberine as a synergist to restore antifungal activity of fluconazole against drug-resistant Candida albicans. ChemMedChem 2014, 9, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Ding, Z.; Hao, Y.; Ni, T.; Xie, F.; Zhao, J.; Li, R.; Yu, S.; Wang, T.; Chai, X.; et al. Design, synthesis, and SAR study of 3-(benzo[d][1,3]dioxol-5-yl)-N-benzylpropanamide as novel potent synergists against fluconazole-resistant Candida albicans. Bioorganic Med. Chem. Lett. 2017, 27, 4571–4575. [Google Scholar] [CrossRef] [PubMed]
- Mondal, J.; Sivaramakrishna, A. Functionalized triazines and tetrazines: Synthesis and applications. Top. Curr. Chem. (Cham) 2022, 380, 34. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Long, S.; Rakesh, K.P.; Zha, G.F. Structure-activity relationships (SAR) of triazine derivatives: Promising antimicrobial agents. Eur. J. Med. Chem. 2020, 185, 111804. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Mandal, M.K.; Masih, A.; Saha, A.; Ghosh, S.K.; Bhat, H.R.; Singh, U.P. 1,3,5-Triazine: A versatile pharmacophore with diverse biological activities. Arch. Pharm. (Weinh.) 2021, 354, e2000363. [Google Scholar] [CrossRef] [PubMed]
- Bozorov, K.; Zhao, J.; Aisa, H.A. 1,2,3-Triazole-containing hybrids as leads in medicinal chemistry: A recent overview. Bioorganic Med. Chem. 2019, 27, 3511–3531. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, A.; Srivastava, K.; Puri, S.K.; Chauhan, P.M. Syntheses of 2,4,6-trisubstituted triazines as antimalarial agents. Bioorganic Med. Chem. Lett. 2005, 15, 531–533. [Google Scholar] [CrossRef] [PubMed]
- Lazewska, D.; Wiecek, M.; Ner, J.; Kaminska, K.; Kottke, T.; Schwed, J.S.; Zygmunt, M.; Karcz, T.; Olejarz, A.; Kuder, K.; et al. Aryl-1,3,5-triazine derivatives as histamine H4 receptor ligands. Eur. J. Med. Chem. 2014, 83, 534–546. [Google Scholar] [CrossRef] [PubMed]
- Singh, U.P.; Bhat, H.R.; Gahtori, P. Antifungal activity, SAR and physicochemical correlation of some thiazole-1,3,5-triazine derivatives. J. Mycol. Med. 2012, 22, 134–141. [Google Scholar] [CrossRef]
- Alhameed, R.A.; Almarhoon, Z.; Sholkamy, E.N.; Khan, S.A.; Ul-Haq, Z.; Sharma, A.; de la Torre, B.G.; Albericio, F.; El-Faham, A. Novel 4,6-disubstituted s-triazin-2-yl amino acid derivatives as promising antifungal agents. J. Fungi 2020, 6, 237. [Google Scholar] [CrossRef] [PubMed]
- Haiba, N.S.; Khalil, H.H.; Moniem, M.A.; El-Wakil, M.H.; Bekhit, A.A.; Khattab, S.N. Design, synthesis and molecular modeling studies of new series of s-triazine derivatives as antimicrobial agents against multi-drug resistant clinical isolates. Bioorg. Chem. 2019, 89, 103013. [Google Scholar] [CrossRef] [PubMed]
- Al-Zaydi, K.M.; Khalil, H.H.; El-Faham, A.; Khattab, S.N. Synthesis, characterization and evaluation of 1,3,5-triazine aminobenzoic acid derivatives for their antimicrobial activity. Chem. Cent. J. 2017, 11, 39. [Google Scholar] [CrossRef]
- Dong, H.H.; Wang, Y.H.; Peng, X.M.; Zhou, H.Y.; Zhao, F.; Jiang, Y.Y.; Zhang, D.Z.; Jin, Y.S. Synergistic antifungal effects of curcumin derivatives as fungal biofilm inhibitors with fluconazole. Chem. Biol. Drug Des. 2021, 97, 1079. [Google Scholar] [CrossRef]
- Li, L.; Zhang, T.; Xu, J.; Wu, J.; Wang, Y.; Qiu, X.; Zhang, Y.; Hou, W.; Yan, L.; An, M.; et al. The synergism of the small molecule ENOblock and fluconazole against fluconazole-resistant Candida albicans. Front. Microbiol. 2019, 10, 2071. [Google Scholar] [CrossRef]
- M38eA2; Clinical and Laboratory Standards Institute/National Committee for Clinical Laboratory Standards, Reference Method for Broth Dilution Antifungal Susceptibility Testing of Yeast. 2nd ed. Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2008.
- Verma, A.K.; Majid, A.; Hossain, M.S.; Ahmed, S.F.; Ashid, M.; Bhojiya, A.A.; Upadhyay, S.K.; Vishvakarma, N.K.; Alam, M. Identification of 1,2,4-triazine and its derivatives against lanosterol 14-demethylase (CYP51) property of Candida albicans: Influence on the development of new antifungal therapeutic strategies. Front. Med. Technol. 2022, 4, 845322. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Li, Z.; Yin, H.; Hu, J.; Xue, Y.; Zhang, G.; Zheng, X.; Chen, W.; Hu, X. Synergistic antibiofilm effects of pseudolaric acid A combined with fluconazole against Candida albicans via inhibition of adhesion and yeast-to-hypha transition. Microbiol. Spectr. 2022, 10, e01478-21. [Google Scholar] [CrossRef] [PubMed]
- Ramage, G.; Walle, K.V.; Wickes, B.L.; Lopez-Ribot, J.L. Standardized method for in vitro antifungal susceptibility testing of Candida albicans biofilms. Antimicrob. Agents Chemother. 2001, 45, 2475–2479. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compd. | Strains C. albicans. 901 | FICI | Strains C. albicans. 904 | FICI | ||
|---|---|---|---|---|---|---|
| Alone | with FCZ | Alone | with FCZ | |||
| 5 | >64.0 | >64.0 | 1.125 | >64.0 | >64.0 | 1.125 |
| 6 | >64.0 | >64.0 | 1.125 | >64.0 | >64.0 | 1.125 |
| 8 | 32.0 | 16.0 | 0.625 | 32.0 | 8.0 | 0.375 |
| 8b | >64.0 | >64.0 | 1.125 | >64.0 | >64.0 | 1.125 |
| 8c | >64.0 | >64.0 | 1.125 | >64.0 | >64.0 | 1.125 |
| 8d | >64.0 | >64.0 | 1.125 | >64.0 | >64.0 | 1.125 |
| 8e | >64.0 | >64.0 | 1.125 | >64.0 | >64.0 | 1.125 |
| 8f | >64.0 | >64.0 | 1.125 | >64.0 | >64.0 | 1.125 |
| 8g | >64.0 | >64.0 | 1.125 | >64.0 | >64.0 | 1.125 |
| 8h | >64.0 | >64.0 | 1.125 | >64.0 | >64.0 | 1.125 |
| 8i | >64.0 | >64.0 | 1.125 | >64.0 | >64.0 | 1.125 |
| 8j | >64.0 | >64.0 | 1.125 | >64.0 | >64.0 | 1.125 |
| 9a | >64.0 | >64.0 | 1.125 | >64.0 | >64.0 | 1.125 |
| 9b | >64.0 | >64.0 | 1.125 | >64.0 | >64.0 | 1.125 |
| 9c | >64.0 | >64.0 | 1.125 | >64.0 | >64.0 | 1.125 |
| 9d | >64.0 | >64.0 | 1.125 | >64.0 | >64.0 | 1.125 |
| 9e | >64.0 | >64.0 | 1.125 | >64.0 | >64.0 | 1.125 |
| 9f | >64.0 | >64.0 | 1.125 | >64.0 | >64.0 | 1.125 |
| 9g | >64.0 | >64.0 | 1.125 | >64.0 | >64.0 | 1.125 |
| 9h | >64.0 | >64.0 | 1.125 | >64.0 | >64.0 | 1.125 |
| 9i | >64.0 | >64.0 | 1.125 | >64.0 | >64.0 | 1.125 |
| 9j | >64.0 | >64.0 | 1.125 | >64.0 | >64.0 | 1.125 |
| 9k | >64.0 | >64.0 | 1.125 | >64.0 | >64.0 | 1.125 |
| 9l | >64.0 | >64.0 | 1.125 | >64.0 | >64.0 | 1.125 |
| 9m | >64.0 | >64.0 | 1.125 | >64.0 | >64.0 | 1.125 |
| 9n | >64.0 | >64.0 | 1.125 | >64.0 | >64.0 | 1.125 |
| 10a | >64.0 | 0.25 | 0.129 | >64.0 | 0.25 | 0.129 |
| 10b | >64.0 | 0.25 | 0.129 | >64.0 | 1.0 | 0.141 |
| 10c | >64.0 | 0.25 | 0.129 | >64.0 | 2.0 | 0.156 |
| 10d | >64.0 | 0.25 | 0.129 | >64.0 | 1.0 | 0.141 |
| 10e | >64.0 | 0.25 | 0.129 | >64.0 | 1.0 | 0.141 |
| 10f | >64.0 | 0.5 | 0.133 | >64.0 | 2.0 | 0.156 |
| 10g | >64.0 | 1.0 | 0.141 | >64.0 | 1.0 | 0.141 |
| 10h | >64.0 | 0.25 | 0.129 | >64.0 | 1.0 | 0.141 |
| 10i | >64.0 | 0.25 | 0.129 | >64.0 | 2.0 | 0.156 |
| 10j | >64.0 | 0.5 | 0.133 | >64.0 | 0.25 | 0.129 |
| 10k | >64.0 | 0.5 | 0.133 | >64.0 | 0.5 | 0.133 |
| 10l | 4.0 | 0.5 | 0.25 | 4.0 | 0.5 | 0.25 |
| 10m | >64.0 | 0.125 | 0.127 | >64.0 | 0.125 | 0.127 |
| 10n | >64.0 | 1.0 | 0.141 | >64.0 | 1.0 | 0.141 |
| 10o | >64.0 | 2.0 | 0.156 | >64.0 | 2.0 | 0.156 |
| 11c | >64.0 | >64.0 | 1.125 | >64.0 | >64.0 | 1.125 |
| 11i | >64.0 | >64.0 | 1.125 | >64.0 | >64.0 | 1.125 |
| 11j | >64.0 | >64.0 | 1.125 | >64.0 | >64.0 | 1.125 |
| FCZ | >64.0 | / | / | >64.0 | / | / |
| Compd. | Strains C. albicans. 901 | Lowest FICI | Strains C. albicans. 904 | Lowest FICI | ||
|---|---|---|---|---|---|---|
| Alone | with FCZ a | Alone | with FCZ a | |||
| 10a | >64.0 | 1.0(1.0) | 0.031 | >64.0 | 0.5(1.0) | 0.023 |
| 10j | >64.0 | 2.0(1.0) | 0.047 | >64.0 | 0.5(2.0) | 0.039 |
| 10l | 4.0 | 0.5(1.0) | 0.141 | 4.0 | 0.5(1.0) | 0.141 |
| 10m | >64.0 | 0.5(0.5) | 0.016 | >64.0 | 0.5(0.5) | 0.016 |
| 8 | 32.0 | 16.0(0.5) | 0.508 | 32.0 | 16.0(0.5) | 0.508 |
| Species | Strains | FCZ | 8 | 10l |
|---|---|---|---|---|
| C. albicans. | 10,231 | 0.5 | 32.0 | >64.0 |
| C. albicans. | 911 | >64.0 | 16.0 | >64.0 |
| C. neoformans. | 32,609 | 0.5 | 16.0 | ≤0.125 |
| C. parapsilosis. | 22,019 | 1.0 | 16.0 | >64.0 |
| C. glabrata. | 537 | 1.0 | 32.0 | ≤0.125 |
| C. tropicalis. | 8915 | >64.0 | 32.0 | >64.0 |
| C. auris. | 891 | >64.0 | 32.0 | >64.0 |
| C. auris. | 918 | >64.0 | 32.0 | >64.0 |
| C. auris. | 919 | >64.0 | 32.0 | >64.0 |
| A. fumigatus. | 7544 | >64.0 | 32.0 | >64.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xie, F.; Hao, Y.; Liu, J.; Bao, J.; Ni, T.; Liu, Y.; Chi, X.; Wang, T.; Yu, S.; Jin, Y.; et al. Discovery of Novel Thiosemicarbazides Containing 1,3,5-Triazines Derivatives as Potential Synergists against Fluconazole-Resistant Candida albicans. Pharmaceutics 2022, 14, 2334. https://doi.org/10.3390/pharmaceutics14112334
Xie F, Hao Y, Liu J, Bao J, Ni T, Liu Y, Chi X, Wang T, Yu S, Jin Y, et al. Discovery of Novel Thiosemicarbazides Containing 1,3,5-Triazines Derivatives as Potential Synergists against Fluconazole-Resistant Candida albicans. Pharmaceutics. 2022; 14(11):2334. https://doi.org/10.3390/pharmaceutics14112334
Chicago/Turabian StyleXie, Fei, Yumeng Hao, Jiacun Liu, Junhe Bao, Tingjunhong Ni, Yu Liu, Xiaochen Chi, Ting Wang, Shichong Yu, Yongsheng Jin, and et al. 2022. "Discovery of Novel Thiosemicarbazides Containing 1,3,5-Triazines Derivatives as Potential Synergists against Fluconazole-Resistant Candida albicans" Pharmaceutics 14, no. 11: 2334. https://doi.org/10.3390/pharmaceutics14112334
APA StyleXie, F., Hao, Y., Liu, J., Bao, J., Ni, T., Liu, Y., Chi, X., Wang, T., Yu, S., Jin, Y., Li, L., Zhang, D., & Yan, L. (2022). Discovery of Novel Thiosemicarbazides Containing 1,3,5-Triazines Derivatives as Potential Synergists against Fluconazole-Resistant Candida albicans. Pharmaceutics, 14(11), 2334. https://doi.org/10.3390/pharmaceutics14112334