
Transcriptome Analysis Identifies Novel Mechanisms Associated with the Antitumor Effect of Chitosan-Stabilized Selenium Nanoparticles

 ,
,  ,
,  and
and 
Abstract
1. Introduction
2. Materials and Methods
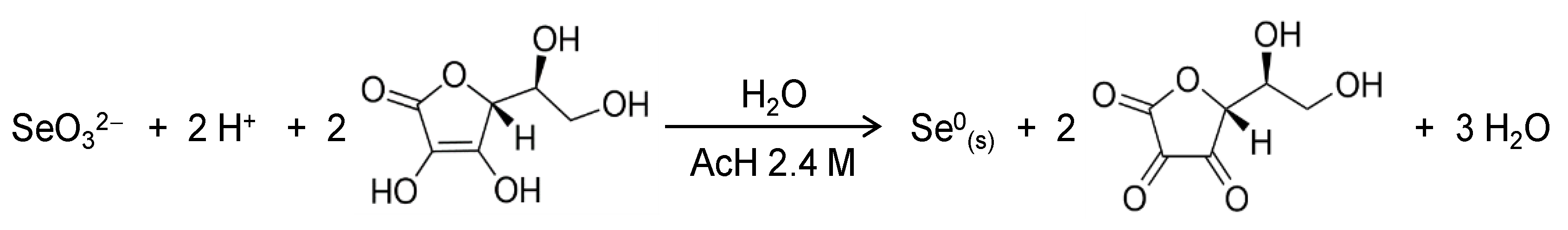
2.1. Synthesis and Characterization of SeNPs
2.2. Cell Culture
2.3. Cytotoxicity Assay
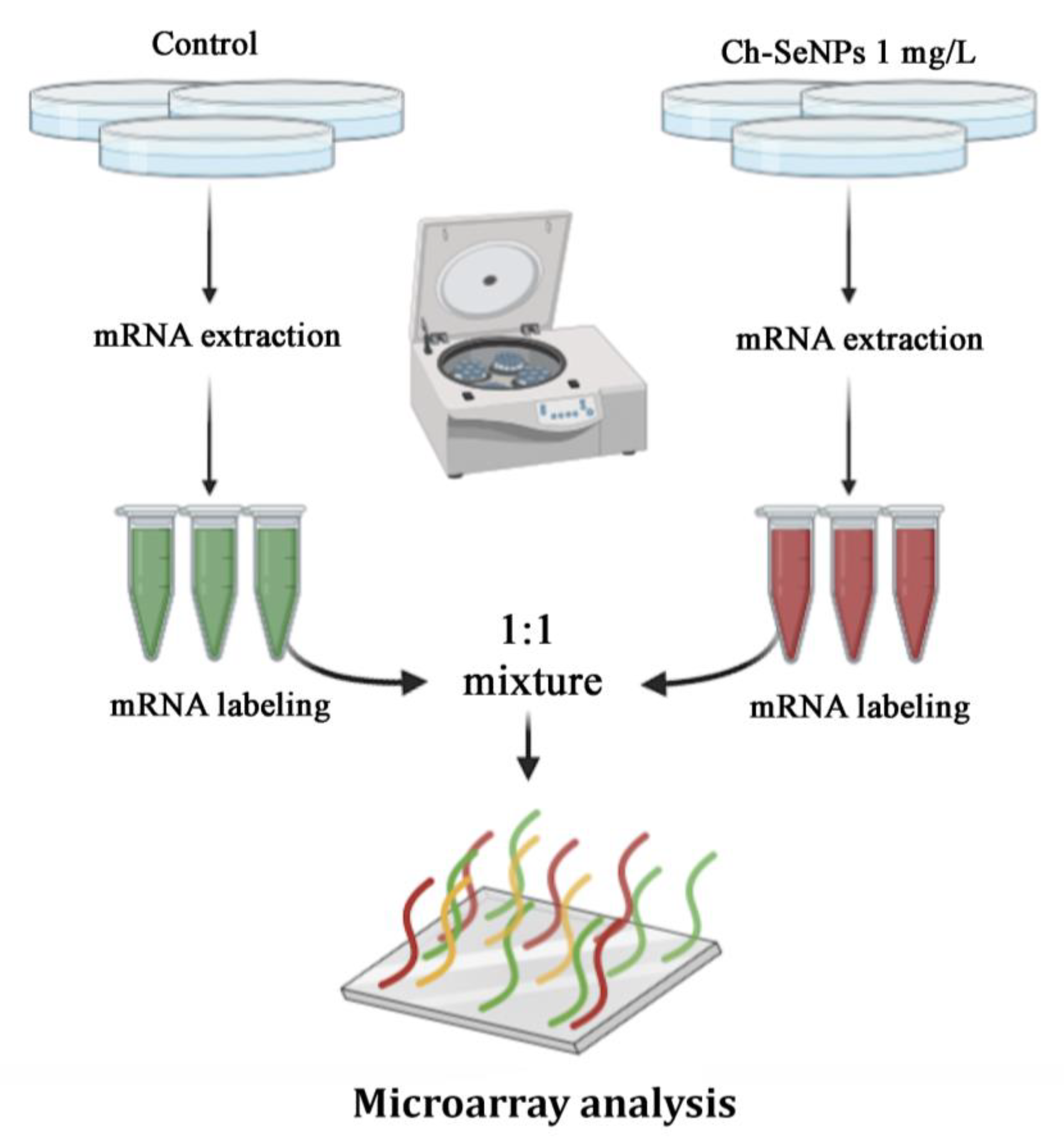
2.4. Transcriptome Analysis
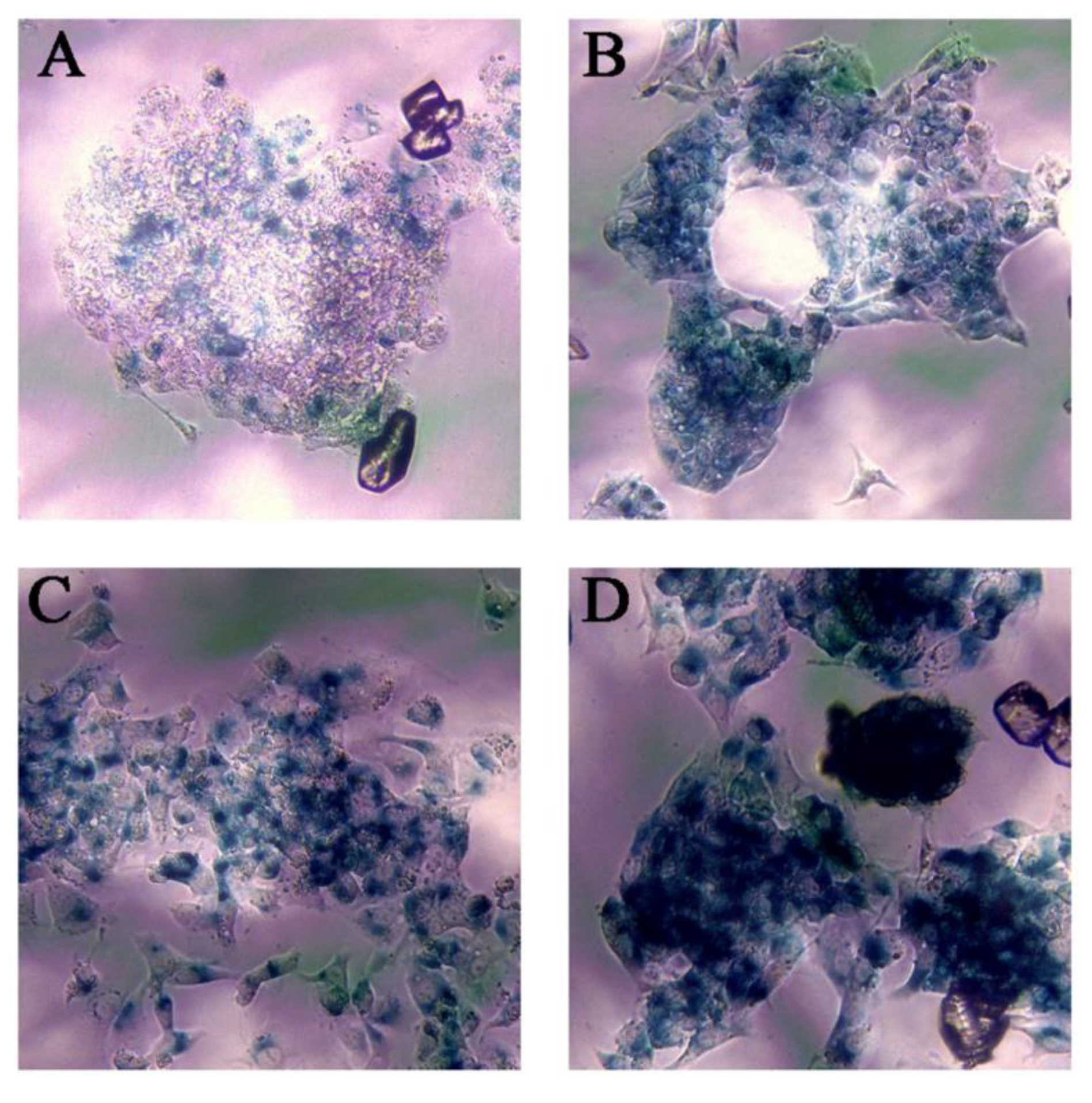
2.5. Senescence Assay
3. Results and Discussion
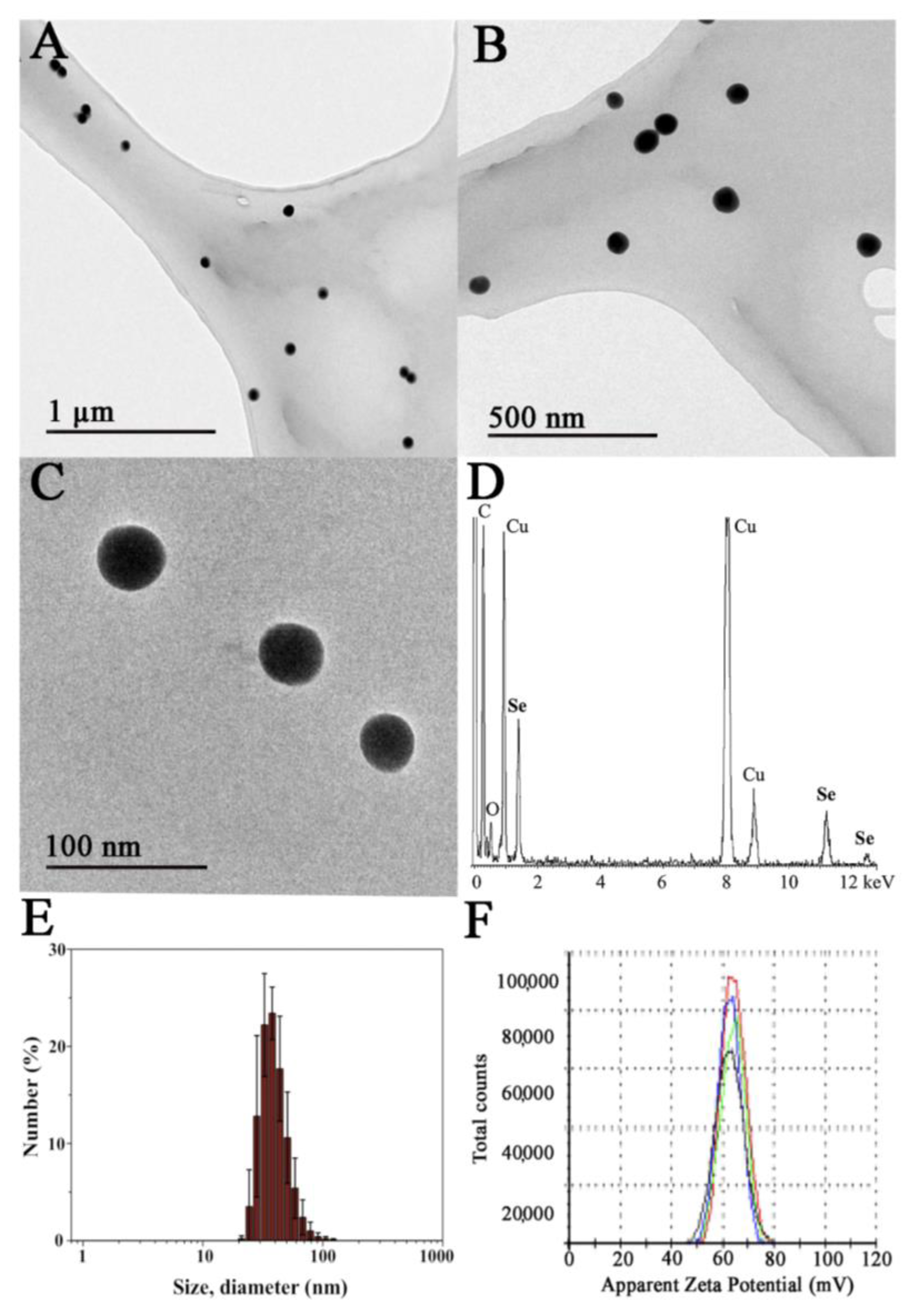
3.1. Synthesis and Characterization of Ch-SeNPs
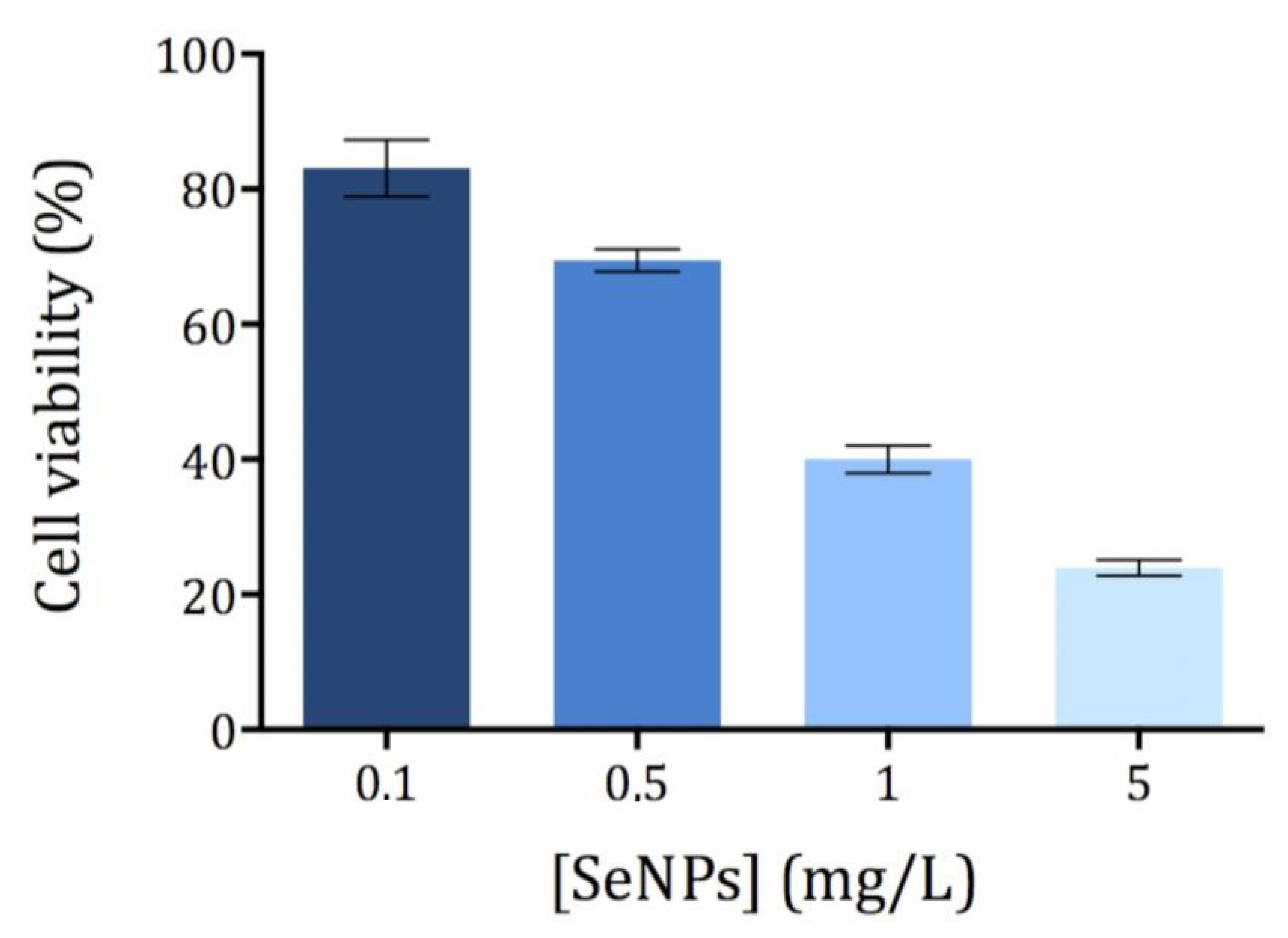
3.2. Cytotoxicity of Ch-SeNPs
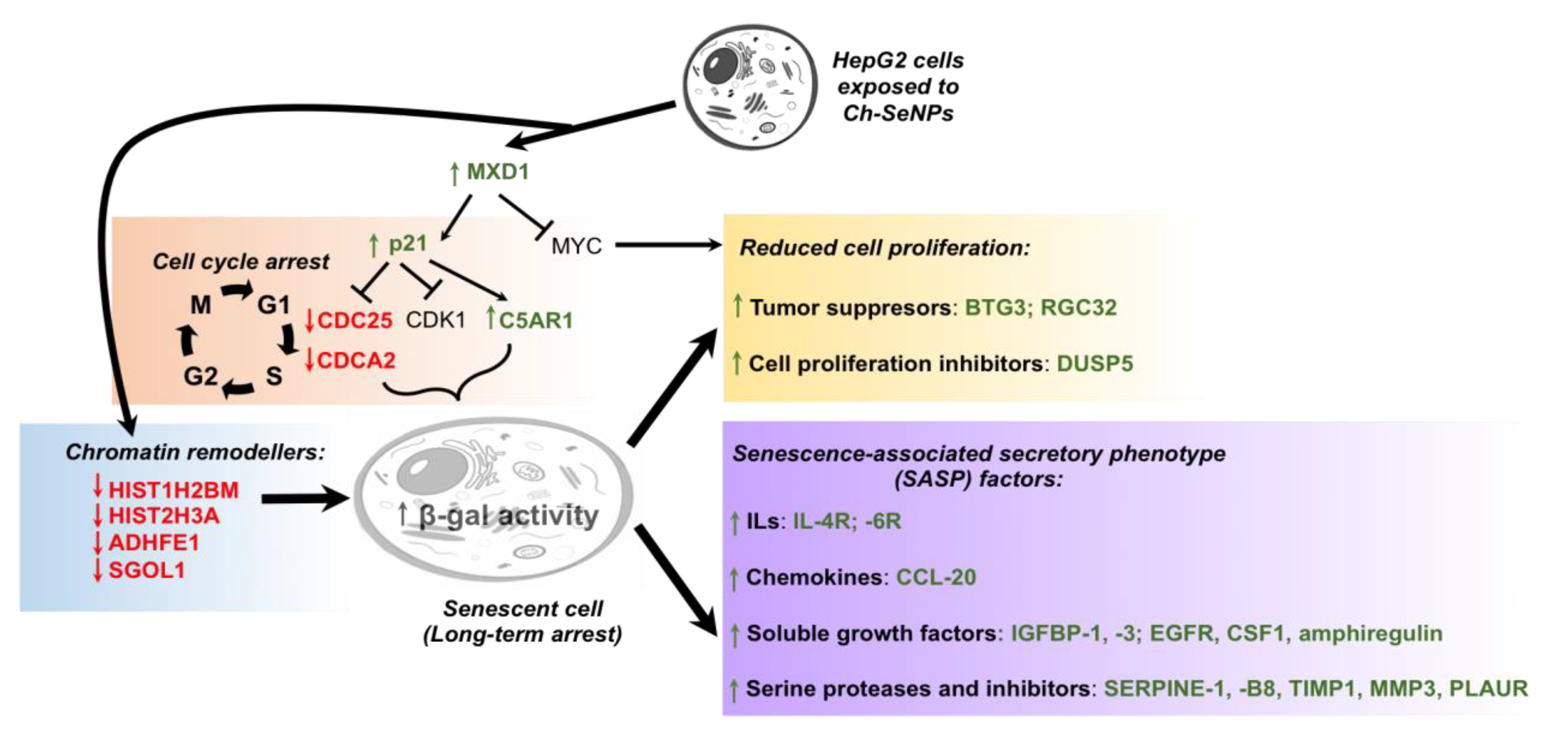
3.3. Revealing the Mechanisms Associated with the Antitumor Effect of Ch-SeNPs by Transcriptomic Analysis
3.4. Ch-SeNPs Induce Senescence in a Preclinical Human Cell Model
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fold Change (FC) | Gene Code | Gene Name |
|---|---|---|
| 14.23 | DUSP5 | dual specificity phosphatase 5 |
| 13.90 | RGCC | regulator of cell cycle |
| 9.70 | PAQR5 | progestin and AdipoQ receptor family member V |
| 9.01 | TMC7 | transmembrane channel-like 7 |
| 8.82 | AQP3 | aquaporin 3 (Gill blood group) |
| 8.11 | AKAP12 | A-kinase (PRKA) anchor protein 12 |
| 7.11 | PALLD | palladin, cytoskeletal associated protein |
| 7.03 | PAI-1 | serpin peptidase inhibitor, clade E (nexin, plasminogen activator inhibitor type 1), member 1 |
| 5.96 | LURAP1L | leucine-rich adaptor protein 1-like |
| 5.90 | CSGALNACT2 | chondroitin sulfate N-acetylgalactosaminyltransferase 2 |
| 5.90 | GPRC5A; MIR614 | G protein-coupled receptor, class C, group 5, member A; microRNA 614 |
| 5.64 | C5AR1 | complement component 5a receptor 1 |
| 5.42 | EMP3 | epithelial membrane protein 3 |
| 5.39 | GPAT3 | glycerol-3-phosphate acyltransferase 3 |
| 5.19 | TM4SF19-TCTEX1D2 | TM4SF19-TCTEX1D2 readthrough (NMD candidate) |
| 5.02 | RBM24 | RNA-binding motif protein 24 |
| 4.88 | TM4SF19 | transmembrane 4 L six family member 19 |
| 4.88 | SERPINB8 | serpin peptidase inhibitor, clade B (ovalbumin), member 8 |
| 4.82 | IER3 | immediate early response 3 |
| 4.75 | PDGFA | platelet-derived growth factor alpha polypeptide |
| 4.65 | MMP3 | matrix metallopeptidase 3 |
| 4.40 | CSTA | cystatin A (stefin A) |
| 4.38 | ITGA2 | integrin, alpha 2 (CD49B, alpha 2 subunit of VLA-2 receptor) |
| 4.31 | CAPN2 | calpain 2, (m/II) large subunit |
| 4.25 | SLC51B | solute carrier family 51, beta subunit |
| 4.22 | EGR1 | early growth response 1 |
| 4.15 | SLC16A6 | solute carrier family 16, member 6 |
| 4.11 | JAG1 | jagged 1 |
| 4.09 | ESAM | endothelial cell adhesion molecule |
| 4.09 | AXL | AXL receptor tyrosine kinase |
| 4.04 | PAEP | progestogen-associated endometrial protein |
| 3.95 | HEY1 | hes-related family bHLH transcription factor with YRPW motif 1 |
| 3.93 | FHL2 | four and a half LIM domains 2 |
| 3.88 | TIMP1 | TIMP metallopeptidase inhibitor 1 |
| 3.85 | IGFBP3 | insulin-like growth factor binding protein 3 |
| 3.69 | SPRY4 | sprouty RTK signaling antagonist 4 |
| 3.65 | F2RL1 | coagulation factor II (thrombin) receptor-like 1 |
| 3.56 | ITPR3 | inositol 1,4,5-trisphosphate receptor, type 3 |
| 3.49 | PLEKHH2 | pleckstrin homology domain containing, family H (with MyTH4 domain) member 2 |
| 3.43 | S100A11 | S100 calcium-binding protein A11 |
| 3.42 | RASD1 | RAS, dexamethasone-induced 1 |
| 3.38 | TUSC3 | tumor suppressor candidate 3 |
| 3.37 | CD55 | CD55 molecule, decay-accelerating factor for complement (Cromer blood group) |
| 3.36 | HKDC1 | hexokinase domain containing 1 |
| 3.35 | SPIRE1 | spire-type actin nucleation factor 1 |
| 3.32 | ARG2 | arginase 2 |
| 3.31 | NKAP | NFKB-activating protein |
| 3.26 | GDF15 | growth differentiation factor 15 |
| 3.25 | ACSL5 | acyl-CoA synthetase long-chain family member 5 |
| 3.24 | AREG | amphiregulin |
| 3.23 | EGFR | epidermal growth factor receptor |
| 3.21 | SLC1A2 | solute carrier family 1 (glial high-affinity glutamate transporter), member 2 |
| 3.21 | ARHGEF2 | Rho/Rac guanine nucleotide exchange factor 2 |
| 3.18 | SFN | stratifin |
| 3.13 | IGFBP1 | insulin-like growth factor-binding protein 1 |
| 3.09 | LGALS3 | lectin, galactoside-binding, soluble, 3 |
| 3.07 | ID1 | inhibitor of DNA binding 1, dominant negative helix–loop–helix protein |
| 3.06 | SRGAP1 | SLIT-ROBO Rho GTPase-activating protein 1 |
| 3.05 | NTSR1 | neurotensin receptor 1 (high affinity) |
| 3.05 | ELK3 | ELK3, ETS-domain protein (SRF accessory protein 2) |
| 3.00 | CACNA2D4 | calcium channel, voltage-dependent, alpha 2/delta subunit 4 |
| 2.99 | HPCAL1 | hippocalcin-like 1 |
| 2.99 | GTPBP2 | GTP-binding protein 2 |
| 2.95 | MCTP1 | multiple C2 domains, transmembrane 1 |
| 2.93 | TCP11L2 | t-complex 11, testis-specific-like 2 |
| 2.92 | SH3RF1 | SH3 domain containing ring finger 1 |
| 2.91 | FOSL1 | FOS-like antigen 1 |
| 2.90 | ATP6V0D2 | ATPase, H+ transporting, lysosomal 38kDa, V0 subunit d2 |
| 2.87 | DYNC2H1 | dynein, cytoplasmic 2, heavy-chain 1 |
| 2.85 | CLIP4 | CAP-GLY domain-containing linker protein family, member 4 |
| 2.85 | RAP1GAP2 | RAP1 GTPase-activating protein 2 |
| 2.83 | SERPINE2 | serpin peptidase inhibitor, clade E (nexin, plasminogen activator inhibitor type 1), member 2 |
| 2.83 | IL6R | interleukin 6 receptor |
| 2.81 | KLF5 | Kruppel-like factor 5 (intestinal) |
| 2.80 | NDE1; MIR484 | nudE neurodevelopment protein 1; microRNA 484 |
| 2.80 | ATF6 | activating transcription factor 6 |
| 2.78 | CCPG1; MIR628 | cell cycle progression 1; microRNA 628 |
| 2.78 | ARAP2 | ArfGAP with RhoGAP domain, ankyrin repeat and PH domain 2 |
| 2.77 | TRIB1 | tribbles pseudokinase 1 |
| 2.74 | ERRFI1 | ERBB receptor feedback inhibitor 1 |
| 2.74 | GCLC | glutamate-cysteine ligase, catalytic subunit |
| 2.73 | SGMS2 | sphingomyelin synthase 2 |
| 2.73 | CD22; MIR5196 | CD22 molecule; microRNA 5196 |
| 2.71 | MT2A | metallothionein 2A |
| 2.69 | GLIPR1 | GLI pathogenesis-related 1 |
| 2.67 | CDKN1A | cyclin-dependent kinase inhibitor 1A (p21, Cip1) |
| 2.67 | CEMIP | cell migration inducing protein, hyaluronan-binding |
| 2.66 | TGFB1 | transforming growth factor beta 1 |
| 2.65 | CD58 | CD58 molecule |
| 2.63 | MEP1A | meprin A, alpha (PABA peptide hydrolase) |
| 2.62 | BHLHE40 | basic helix–loop–helix family, member e40 |
| 2.62 | TIMM9 | translocase of inner mitochondrial membrane 9 homolog (yeast) |
| 2.62 | BMP6 | bone morphogenetic protein 6 |
| 2.62 | IL4R | interleukin 4 receptor |
| 2.60 | TMEM2 | transcript identified by AceView, Entrez Gene ID(s) 23670 |
| 2.58 | DMRTA1 | DMRT-like family A1 |
| 2.58 | ANKRD1 | ankyrin repeat domain 1 (cardiac muscle) |
| 2.55 | MT1B; MT1CP | metallothionein 1B; metallothionein 1C, pseudogene |
| 2.51 | AKR1B10 | aldo-keto reductase family 1, member B10 (aldose reductase) |
| 2.49 | CD109 | CD109 molecule |
| 2.48 | SLC20A1 | solute carrier family 20 (phosphate transporter), member 1 |
| 2.46 | ATF3 | activating transcription factor 3 |
| 2.46 | TAGLN3 | transgelin 3 |
| 2.45 | MOSPD1 | motile sperm domain-containing 1 |
| 2.45 | IL11 | interleukin 11 |
| 2.43 | KDM7A | lysine (K)-specific demethylase 7A |
| 2.42 | CCR7 | chemokine (C-C motif) receptor 7 |
| 2.42 | MXD1 | MAX dimerization protein 1 |
| 2.40 | PITPNC1 | phosphatidylinositol transfer protein, cytoplasmic 1 |
| 2.40 | PLAUR | plasminogen activator, urokinase receptor |
| 2.40 | RABGGTB; SNORD45B; SNORD45A; SNORD45C | Rab geranylgeranyltransferase, beta subunit; small nucleolar RNA, C/D box 45B; small nucleolar RNA, C/D box 45A; small nucleolar RNA, C/D box 45C |
| 2.39 | LETM2 | leucine zipper-EF-hand-containing transmembrane protein 2 |
| 2.36 | C6orf48; SNORD52; SNORD48 | chromosome 6 open reading frame 48; small nucleolar RNA, C/D box 52; small nucleolar RNA, C/D box 48 |
| 2.36 | AP1S3 | adaptor-related protein complex 1 sigma 3 subunit |
| 2.36 | PPP1R15A | protein phosphatase 1, regulatory subunit 15A |
| 2.36 | ARL8A | ADP-ribosylation factor like GTPase 8A |
| 2.36 | PAQR3 | progestin and AdipoQ receptor family member III |
| 2.35 | LCP1 | lymphocyte cytosolic protein 1 (L-plastin) |
| 2.34 | HMGA1 | high-mobility group AT-hook 1 |
| 2.33 | RCL1 | RNA terminal phosphate cyclase-like 1 |
| 2.33 | ANTXR2 | anthrax toxin receptor 2 |
| 2.31 | KPNA5 | karyopherin alpha 5 (importin alpha 6) |
| 2.30 | RRAS2 | related RAS viral (r-ras) oncogene homolog 2 |
| 2.30 | CCL20 | chemokine (C–C motif) ligand 20 |
| 2.30 | TUBE1 | tubulin, epsilon 1 |
| 2.30 | LRRC8B | leucine-rich repeat-containing 8 family, member B |
| 2.29 | DFNA5 | deafness, autosomal dominant 5 |
| 2.28 | ERN1 | endoplasmic reticulum to nucleus signaling 1 |
| 2.28 | ZADH2 | zinc binding alcohol dehydrogenase domain-containing 2 |
| 2.27 | PPP1R18 | protein phosphatase 1, regulatory subunit 18 |
| 2.26 | ASB2 | ankyrin repeat and SOCS box-containing 2 |
| 2.26 | IDS | iduronate 2-sulfatase |
| 2.25 | MAGI2 | membrane-associated guanylate kinase, WW and PDZ domain-containing 2 |
| 2.25 | MT1A | metallothionein 1A |
| 2.25 | ANXA3 | annexin A3 |
| 2.25 | ZAK | sterile alpha motif and leucine zipper-containing kinase |
| 2.24 | CSF1 | colony-stimulating factor 1 (macrophage) |
| 2.24 | PIM1 | Pim-1 proto-oncogene, serine/threonine kinase |
| 2.23 | MSANTD3 | Myb/SANT-like DNA-binding domain containing 3 |
| 2.22 | NAGS | N-acetylglutamate synthase |
| 2.22 | REXO2 | RNA exonuclease 2 |
| 2.22 | VASP | vasodilator-stimulated phosphoprotein |
| 2.21 | CREB5 | cAMP responsive element-binding protein 5 |
| 2.21 | STEAP2 | STEAP family member 2, metalloreductase |
| 2.21 | MRGPRX4 | MAS-related GPR, member X4 |
| 2.20 | ITPRIP | inositol 1,4,5-trisphosphate receptor-interacting protein |
| 2.20 | DIEXF | digestive organ expansion factor homolog (zebrafish) |
| 2.20 | MT1X | metallothionein 1X |
| 2.19 | PTGR1 | prostaglandin reductase 1 |
| 2.19 | C9orf72 | chromosome 9 open reading frame 72 |
| 2.19 | ABR | active BCR-related |
| 2.18 | HIVEP2 | human immunodeficiency virus type I enhancer-binding protein 2 |
| 2.18 | KLF11 | Kruppel-like factor 11 |
| 2.18 | VNN1 | Vanin 1 |
| 2.17 | LAPTM5 | lysosomal protein transmembrane 5 |
| 2.17 | MT1IP | metallothionein 1I, pseudogene |
| 2.17 | CAMSAP2 | calmodulin regulated spectrin-associated protein family, member 2 |
| 2.16 | DPY19L4 | dpy-19-like 4 (C. elegans) |
| 2.16 | SLC22A15 | solute carrier family 22, member 15 |
| 2.16 | PTPN3 | protein tyrosine phosphatase, non-receptor type 3 |
| 2.16 | SPP1 | secreted phosphoprotein 1 |
| 2.16 | NLRC4 | NLR family, CARD domain-containing 4 |
| 2.16 | CMSS1 | cms1 ribosomal small subunit homolog (yeast) |
| 2.15 | HBEGF | heparin-binding EGF-like growth factor |
| 2.15 | TGM2 | transglutaminase 2 |
| 2.15 | NLN | neurolysin (metallopeptidase M3 family) |
| 2.14 | APOPT1 | apoptogenic 1, mitochondrial |
| 2.14 | UPP1 | uridine phosphorylase 1 |
| 2.13 | LAMP3 | lysosomal-associated membrane protein 3 |
| 2.13 | C5orf28 | chromosome 5 open reading frame 28 |
| 2.13 | P2RX5-TAX1BP3 | P2RX5-TAX1BP3 readthrough (NMD candidate) |
| 2.13 | PHF21A | PHD finger protein 21A |
| 2.12 | CTSB | cathepsin B |
| 2.12 | S100P | S100 calcium-binding protein P |
| 2.11 | ANKRA2 | ankyrin repeat, family A (RFXANK-like), 2 |
| 2.11 | FLOT1 | transcript identified by AceView, Entrez Gene ID(s) 10211 |
| 2.11 | CEP290 | centrosomal protein 290kDa |
| 2.11 | MT1H | metallothionein 1H |
| 2.10 | RIT1 | Ras-like without CAAX 1 |
| 2.09 | CEP295NL; TIMP2 | CEP295 N-terminal like; TIMP metallopeptidase inhibitor 2 |
| 2.09 | CES1 | carboxylesterase 1 |
| 2.09 | ASF1A | anti-silencing function 1A histone chaperone |
| 2.09 | OPTN | optineurin |
| 2.09 | GRB10 | growth factor receptor-bound protein 10 |
| 2.09 | AKR1B15 | aldo-keto reductase family 1, member B15 |
| 2.08 | MTMR6 | myotubularin-related protein 6 |
| 2.08 | FAN1 | FANCD2/FANCI-associated nuclease 1 |
| 2.08 | ACTR10 | actin-related protein 10 homolog (S. cerevisiae) |
| 2.08 | RNF19B | ring finger protein 19B |
| 2.08 | NFIL3 | nuclear factor, interleukin 3-regulated |
| 2.08 | NQO1 | NAD(P)H dehydrogenase, quinone 1 |
| 2.08 | DHRS7 | dehydrogenase/reductase (SDR family) member 7 |
| 2.08 | MT1L | metallothionein 1L (gene/pseudogene) |
| 2.07 | LIF | leukemia inhibitory factor |
| 2.07 | DUSP12 | dual specificity phosphatase 12 |
| 2.07 | KCNMB3 | potassium channel subfamily M regulatory beta subunit 3 |
| 2.06 | PXK | PX domain-containing serine/threonine kinase |
| 2.06 | CD9 | CD9 molecule |
| 2.06 | H1F0 | H1 histone family, member 0 |
| 2.06 | ADORA2B | adenosine A2b receptor |
| 2.06 | KRT23 | keratin 23, type I |
| 2.06 | BTG3 | BTG family, member 3 |
| 2.05 | AOX1 | aldehyde oxidase 1 |
| 2.04 | SNORA17A; SNORA17B; SNHG7 | small nucleolar RNA, H/ACA box 17A; small nucleolar RNA, H/ACA box 17B; small nucleolar RNA host gene 7 |
| 2.04 | CPNE8 | copine VIII |
| 2.04 | EIF4A2; SNORA63; SNORD2; SNORA4; SNORA81; MIR1248 | eukaryotic translation initiation factor 4A2; small nucleolar RNA, H/ACA box 63; small nucleolar RNA, C/D box 2; small nucleolar RNA, H/ACA box 4; small nucleolar RNA, H/ACA box 81; microRNA 1248 |
| 2.03 | SMAD6 | SMAD family member 6 |
| 2.03 | YIPF4 | Yip1 domain family member 4 |
| 2.03 | FASTKD1 | FAST kinase domains 1 |
| 2.03 | TMTC3 | transmembrane and tetratricopeptide repeat containing 3 |
| 2.02 | ADAT2 | adenosine deaminase, tRNA-specific 2 |
| 2.02 | ago-02 | argonaute RISC catalytic component 2 |
| 2.02 | TMEM167B | transmembrane protein 167B |
| 2.01 | DCAF10 | DDB1 and CUL4 associated factor 10 |
| 2.01 | RAB3GAP1 | RAB3 GTPase-activating protein subunit 1 (catalytic) |
| 2.00 | GBE1 | glucan (1,4-alpha-), branching enzyme 1 |
| 2.00 | CLIP2 | CAP-GLY domain-containing linker protein 2 |
| 2.00 | SOWAHC | sosondowah ankyrin repeat domain family member C |
| 2.00 | NEK3 | NIMA-related kinase 3 |
| 2.00 | IFRD1 | interferon-related developmental regulator 1 |
| 2.00 | TBPL1 | TBP-like 1 |
| 0.50 | ROR1 | receptor tyrosine kinase-like orphan receptor 1 |
| 0.50 | FAM111B | family with sequence similarity 111, member B |
| 0.50 | CDCA2 | cell division cycle-associated 2 |
| 0.50 | DIO1 | deiodinase, iodothyronine, type I |
| 0.50 | HIST1H1B | histone cluster 1, H1b |
| 0.50 | HOOK2 | hook microtubule-tethering protein 2 |
| 0.50 | CYP7A1 | cytochrome P450, family 7, subfamily A, polypeptide 1 |
| 0.50 | CYP4F2 | cytochrome P450, family 4, subfamily F, polypeptide 2 |
| 0.50 | PNPLA3 | patatin-like phospholipase domain-containing 3 |
| 0.49 | TSACC | TSSK6-activating co-chaperone |
| 0.49 | HIST1H4A | histone cluster 1, H4a |
| 0.49 | ANGPTL3 | angiopoietin-like 3 |
| 0.49 | ADHFE1; C8orf46 | alcohol dehydrogenase, iron-containing 1; chromosome 8 open reading frame 46 |
| 0.49 | GINS2 | GINS complex subunit 2 (Psf2 homolog) |
| 0.49 | ARHGEF39 | Rho guanine nucleotide exchange factor 39 |
| 0.49 | POTEF | POTE ankyrin domain family, member F |
| 0.49 | HIST1H2AG | histone cluster 1, H2ag |
| 0.49 | ZNF341 | zinc finger protein 341 |
| 0.49 | NAT6 | N-acetyltransferase 6 (GCN5-related) |
| 0.48 | FRY | FRY microtubule-binding protein |
| 0.48 | SOX6; MIR6073 | SRY box 6; microRNA 6073 |
| 0.48 | SGOL2 | shugoshin-like 2 (S. pombe) |
| 0.48 | MTHFR | methylenetetrahydrofolate reductase (NAD(P)H) |
| 0.48 | CD24 | CD24 molecule |
| 0.47 | ITIH1 | inter-alpha-trypsin inhibitor heavy-chain 1 |
| 0.47 | SCARA3 | scavenger receptor class A, member 3 |
| 0.47 | TMEM143 | transmembrane protein 143 |
| 0.47 | CDC25C | cell division cycle 25C |
| 0.46 | LEAP2 | liver-expressed antimicrobial peptide 2 |
| 0.46 | ZNF565 | zinc finger protein 565 |
| 0.46 | E2F8 | E2F transcription factor 8 |
| 0.46 | CYP3A7; CYP3A7-CYP3A51P | cytochrome P450, family 3, subfamily A, polypeptide 7; CYP3A7-CYP3A51P readthrough |
| 0.45 | ESCO2 | establishment of sister chromatid cohesion N-acetyltransferase 2 |
| 0.45 | ODAM | odontogenic, ameloblast-associated |
| 0.45 | C1orf116 | chromosome 1 open reading frame 116 |
| 0.44 | HIST2H3A | histone cluster 2, H3a |
| 0.44 | CHGB | chromogranin B |
| 0.44 | PRLR | prolactin receptor |
| 0.44 | HIST2H3A; HIST2H3C | histone cluster 2, H3a; histone cluster 2, H3c |
| 0.44 | CYP3A5 | cytochrome P450, family 3, subfamily A, polypeptide 5 |
| 0.43 | HIST1H2BM | histone cluster 1, H2bm |
| 0.43 | SNAI2 | snail family zinc finger 2 |
| 0.43 | PRRG2 | proline-rich Gla (G-carboxyglutamic acid) 2 |
| 0.42 | SGOL1 | shugoshin-like 1 (S. pombe) |
| 0.42 | SLC22A7 | solute carrier family 22 (organic anion transporter), member 7 |
| 0.41 | CYP3A43 | cytochrome P450, family 3, subfamily A, polypeptide 43 |
| 0.41 | LINC00612 | long intergenic non-protein coding RNA 612 |
| 0.41 | IFIT3 | interferon-induced protein with tetratricopeptide repeats 3 |
| 0.40 | HMMR | hyaluronan-mediated motility receptor (RHAMM) |
| 0.39 | CDH1 | cadherin 1, type 1 |
| 0.39 | HIST1H3B | histone cluster 1, H3b |
| 0.38 | AFM | afamin |
| 0.36 | DEPDC4 | DEP domain-containing 4 |
| 0.36 | YPEL2 | yippee like 2 |
| 0.32 | G6PC | glucose-6-phosphatase, catalytic subunit |
| 0.30 | ANGPTL8 | angiopoietin like 8 |
| 0.29 | CENPI | centromere protein I |
| 0.20 | ALDOB | aldolase B, fructose-bisphosphate |
References
- Sreevalsan, S.; Safe, S. Reactive oxygen species and colorectal cancer. Curr. Color. Cancer Rep. 2013, 9, 350–357. [Google Scholar] [CrossRef]
- Marinescu, S.; Anghel, R.; Gruia, M.I.; Beuran, M. Involvement of reactive oxygen species in the mechanisms associated with cervical cancer specific treatment. Chirurgia 2015, 109, 806–811. [Google Scholar]
- Thyagarajan-Sahu, A.; Sahu, R.P. Potential contributions of antioxidants to cancer therapy: Immunomodulation and radiosensitization. Integr. Cancer Ther. 2018, 17, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Nunes, R.; Das Neves, J.; Sarmento, B. Nanoparticles for the regulation of intestinal inflammation: Opportunities and challenges. Nanomedicine 2019, 14, 2631–2644. [Google Scholar] [CrossRef]
- Tapeinos, C.; Battaglini, M.; Ciofani, G. Advances in the design of solid lipid nanoparticles and nanostructured lipid carriers for targeting brain diseases. J. Control. Release 2017, 264, 306–332. [Google Scholar] [CrossRef]
- Machuca, A.; Garcia-Calvo, E.; Anunciação, D.S.; Garcia, J.L.L. Rhodium nanoparticles as a novel photosensitizing agent in photodynamic therapy against cancer. Chem. A Eur. J. 2020, 26, 7685–7691. [Google Scholar] [CrossRef]
- Montalvo-Quiros, S.; Aragoneses-Cazorla, G.; Garcia-Alcalde, L.; Vallet-Regí, M.; González, B.; Luque-Garcia, J.L. Cancer cell targeting and therapeutic delivery of silver nanoparticles by mesoporous silica nanocarriers: Insights into the action mechanisms using quantitative proteomics. Nanoscale 2019, 11, 4531–4545. [Google Scholar] [CrossRef] [PubMed]
- Oueslati, M.H.; Ben Tahar, L.; Harrath, A.H. Catalytic, antioxidant and anticancer activities of gold nanoparticles synthesized by kaempferol glucoside from Lotus leguminosae. Arab. J. Chem. 2020, 13, 3112–3122. [Google Scholar] [CrossRef]
- Vinceti, M.; Filippini, T.; Del Giovane, C.; Dennert, G.; Zwahlen, M.; Brinkman, M.; Zeegers, M.P.; Horneber, M.; D’Amico, R.; Crespi, C.M. Selenium for preventing cancer. Cochrane Database Syst. Rev. 2018, 1, CD005195. [Google Scholar] [CrossRef] [PubMed]
- Aboul-Fadl, T. Selenium derivatives as cancer preventive agents. Curr. Med. Chem. Agents 2005, 5, 637–652. [Google Scholar] [CrossRef] [PubMed]
- Geoffrion, L.D.; Hesabizadeh, T.; Medina-Cruz, D.; Kusper, M.; Taylor, P.; Vernet-Crua, A.; Chen, J.; Ajo, A.; Webster, T.J.; Guisbiers, G. Naked selenium nanoparticles for antibacterial and anticancer treatments. ACS Omega 2020, 5, 2660–2669. [Google Scholar] [CrossRef]
- Estevez, H.; Garcia-Lidon, J.C.; Luque-Garcia, J.L.; Camara, C. Effects of chitosan-stabilized selenium nanoparticles on cell proliferation, apoptosis and cell cycle pattern in HepG2 cells: Comparison with other selenospecies. Colloids Surf. B Biointerfaces 2014, 122, 184–193. [Google Scholar] [CrossRef]
- Malekifard, F.; Tavassoli, M.; Vaziri, K. In vitro assessment antiparasitic effect of selenium and copper nanoparticles on Giardia deodenalis cyst. Iran. J. Parasitol. 2020, 15, 411–417. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; He, Y.; Liu, L.; Tao, W.; Wang, G.; Sun, W.; Pei, X.; Xiao, Z.; Jin, Y.; Wang, M. Prooxidation and cytotoxicity of selenium nanoparticles at nonlethal level in Sprague-Dawley rats and Buffalo rat liver cells. Oxidative Med. Cell. Longev. 2020, 2020. [Google Scholar] [CrossRef] [PubMed]
- Toubhans, B.; Gazze, S.A.; Bissardon, C.; Bohic, S.; Gourlan, A.T.; Gonzalez, D.; Charlet, L.; Conlan, R.S.; Francis, L.W. Selenium nanoparticles trigger alterations in ovarian cancer cell biomechanics. Nanomed. Nanotechnol. Biol. Med. 2020, 29, 102258. [Google Scholar] [CrossRef]
- Zhao, G.; Dong, R.; Teng, J.; Yang, L.; Liu, T.; Wu, X.; He, Y.; Wang, Z.; Pu, H.; Wang, Y. N-acetyl-l-cysteine enhances the effect of selenium nanoparticles on cancer cytotoxicity by increasing the production of selenium-induced reactive oxygen species. ACS Omega 2020, 5, 11710–11720. [Google Scholar] [CrossRef]
- Hussein, H.-A.A.; Darwesh, O.M.; Mekki, B.B.; El-Hallouty, S.M. Evaluation of cytotoxicity, biochemical profile and yield components of groundnut plants treated with nano-selenium. Biotechnol. Rep. 2019, 24, e00377. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Heras, I.; Sanchez-Diaz, R.; Anunciacao, D.S.; Madrid, Y.; Luque-Garcia, J.L.; Camara, C. Effect of chitosan-stabilized selenium nanoparticles on cell cycle arrest and invasiveness in hepatocarcinoma cells revealed by quantitative proteomics. J. Nanomed. Nanotechnol. 2014, 5. [Google Scholar] [CrossRef]
- Neagu, M.; Boda, D. Transcriptomics in cancer—Stages toward patents in biomarkers? Recent Pat. Biomark. 2012, 2, 75–82. [Google Scholar] [CrossRef]
- Alkhateeb, A.; Rezaeian, I.; Singireddy, S.; Cavallo-Medved, D.; Porter, L.A.; Rueda, L. transcriptomics signature from next-generation sequencing data reveals new transcriptomic biomarkers related to prostate cancer. Cancer Inform. 2019, 18. [Google Scholar] [CrossRef]
- Bai, Y.; Wang, Y.; Zhou, Y.; Li, W.; Zheng, W. Modification and modulation of saccharides on elemental selenium nanoparticles in liquid phase. Mater. Lett. 2008, 62, 2311–2314. [Google Scholar] [CrossRef]
- Mees, D.R.; Pysto, W.; Tarcha, P.J. Formation of selenium colloids using sodium ascorbate as the reducing agent. J. Colloid Interface Sci. 1995, 170, 254–260. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, J.; Zhang, L. Creation of highly stable selenium nanoparticles capped with hyperbranched polysaccharide in water. Langmuir 2010, 26, 17617–17623. [Google Scholar] [CrossRef]
- Zhang, S.; Luo, Y.; Zeng, H.; Wang, Q.; Tian, F.; Song, J.; Cheng, W.-H. Encapsulation of selenium in chitosan nanoparticles improves selenium availability and protects cells from selenium-induced DNA damage response. J. Nutr. Biochem. 2011, 22, 1137–1142. [Google Scholar] [CrossRef] [PubMed]
- Zhai, X.; Zhang, C.; Zhao, G.; Stoll, S.; Ren, F.; Leng, X. Antioxidant capacities of the selenium nanoparticles stabilized by chitosan. J. Nanobiotechnol. 2017, 15, 4. [Google Scholar] [CrossRef] [PubMed]
- Estevez, H.; Palacios, A.; Gil, D.; Anguita, J.; Vallet-Regi, M.; González, B.; Prados-Rosales, R.; Luque-Garcia, J.L. Antimycobacterial effect of selenium nanoparticles on mycobacterium tuberculosis. Front. Microbiol. 2020, 11, 800. [Google Scholar] [CrossRef] [PubMed]
- Charrier-Savournin, F.B.; Château, M.-T.; Gire, V.; Sedivy, J.; Piette, J.; Dulić, V. p21-mediated nuclear retention of cyclin B1-Cdk1 in response to genotoxic stress. Mol. Biol. Cell 2004, 15, 3965–3976. [Google Scholar] [CrossRef] [PubMed]
- Abbas, T.; Dutta, A. p21 in cancer: Intricate networks and multiple activities. Nat. Rev. Cancer 2009, 9, 400–414. [Google Scholar] [CrossRef] [PubMed]
- Saha, P.; Eichbaum, Q.; Silberman, E.D.; Mayer, B.J.; Dutta, A. p21CIP1 and Cdc25A: Competition between an inhibitor and an activator of cyclin-dependent kinases. Mol. Cell. Biol. 1997, 17, 4338–4345. [Google Scholar] [CrossRef]
- Feng, Y.; Qian, W.; Zhang, Y.; Peng, W.; Li, J.; Gu, Q.; Ji, D.; Zhang, Z.; Wang, Q.; Zhang, D.; et al. CDCA2 promotes the proliferation of colorectal cancer cells by activating the AKT/CCND1 pathway in vitro and in vivo. BMC Cancer 2019, 19, 576. [Google Scholar] [CrossRef]
- Uchida, F.; Uzawa, K.; Kasamatsu, A.; Takatori, H.; Sakamoto, Y.; Ogawara, K.; Shiiba, M.; Bukawa, H.; Tanzawa, H. Overexpression of CDCA2 in human squamous cell carcinoma: Correlation with prevention of G1 phase arrest and apoptosis. PLoS ONE 2013, 8, e56381. [Google Scholar] [CrossRef]
- Wiese, K.E.; Walz, S.; Von Eyss, B.; Wolf, E.; Athineos, D.; Sansom, O.; Eilers, M. The role of MIZ-1 in MYC-dependent tumorigenesis. Cold Spring Harb. Perspect. Med. 2013, 3, a014290. [Google Scholar] [CrossRef]
- Koyama, T.; Ogawara, K.; Kasamatsu, A.; Okamoto, A.; Kasama, H.; Minakawa, Y.; Shimada, K.; Yokoe, H.; Shiiba, M.; Tanzawa, H.; et al. ANGPTL3 is a novel biomarker as it activates ERK/MAPK pathway in oral cancer. Cancer Med. 2015, 4, 759–769. [Google Scholar] [CrossRef]
- Sun, H.; Liu, K.; Huang, J.; Sun, Q.; Shao, C.; Luo, J.; Xu, L.; Shen, Y.; Ren, B. FAM111B, a direct target of p53, promotes the malignant process of lung adenocarcinoma. OncoTargets Ther. 2019, 12, 2829–2842. [Google Scholar] [CrossRef] [PubMed]
- Regulski, M.J. Cellular senescence: What, why, and how. Wounds 2017, 29, 168–174. [Google Scholar] [PubMed]
- Cruickshanks, H.A.; McBryan, T.; Nelson, D.M.; VanderKraats, N.D.; Shah, P.P.; Van Tuyn, J.; Rai, T.S.; Brock, C.; Donahue, G.; Dunican, D.S.; et al. Senescent cells harbour features of the cancer epigenome. Nat. Cell Biol. 2013, 15, 1495–1506. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.W.; Lee, S.K.; Lee, Y.W.; Lee, J.O.; Kim, N.; Lee, H.J.; Seo, J.S.; Kim, J.; Kim, H.S.; Park, S.-H. Alcohol induces cell proliferation via hypermethylation of ADHFE1 in colorectal cancer cells. BMC Cancer 2014, 14, 377. [Google Scholar] [CrossRef]
- Murakami-Tonami, Y.; Ikeda, H.; Yamagishi, R.; Inayoshi, M.; Inagaki, S.; Kishida, S.; Komata, Y.; Koster, J.; Takeuchi, I.; Kondo, Y.; et al. SGO1 is involved in the DNA damage response in MYCN-amplified neuroblastoma cells. Sci. Rep. 2016, 6, 31615. [Google Scholar] [CrossRef]
- Hernandez, M.X.; Jiang, S.; Cole, T.A.; Chu, S.-H.; Fonseca, M.I.; Fang, M.J.; Hohsfield, L.A.; Torres, M.D.; Green, K.N.; Wetsel, R.A.; et al. Prevention of C5aR1 signaling delays microglial inflammatory polarization, favors clearance pathways and suppresses cognitive loss. Mol. Neurodegener. 2017, 12, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Castellano, G.; Franzin, R.; Sallustio, F.; Stasi, A.; Banelli, B.; Romani, M.; De Palma, G.; Lucarelli, G.; Divella, C.; Battaglia, M.; et al. Complement component C5a induces aberrant epigenetic modifications in renal tubular epithelial cells accelerating senescence by Wnt4/βcatenin signaling after ischemia/reperfusion injury. Aging 2019, 11, 4382–4406. [Google Scholar] [CrossRef]
- Gao, S.; Jin, L.; Liu, G.; Wang, P.; Sun, Z.; Cao, Y.; Shi, H.; Liu, X.; Shi, Q.; Zhou, X.; et al. Overexpression of RASD1 inhibits glioma cell migration/invasion and inactivates the AKT/mTOR signaling pathway. Sci. Rep. 2017, 7, 3202. [Google Scholar] [CrossRef]
- Krones-Herzig, A.; Mittal, S.; Yule, K.; Liang, H.; English, C.; Urcis, R.; Soni, T.; Adamson, E.D.; Mercola, D. Early growth response 1 acts as a tumor suppressor in vivo and in vitro via regulation of p53. Cancer Res. 2005, 65, 5133–5143. [Google Scholar] [CrossRef]
- Ruoslahti, E. Fibronectin and its integrin receptors in cancer. Adv. Cancer Res. 1999, 76, 1–20. [Google Scholar] [CrossRef]
- Ma, Y.; Chen, Y.; Li, Y.; Grün, K.; Berndt, A.; Zhou, Z.; Petersen, I. Cystatin A suppresses tumor cell growth through inhibiting epithelial to mesenchymal transition in human lung cancer. Oncotarget 2017, 9, 14084–14098. [Google Scholar] [CrossRef]
- Kidger, A.M.; Keyse, S.M. The regulation of oncogenic Ras/ERK signalling by dual-specificity mitogen activated protein kinase phosphatases (MKPs). Semin. Cell Dev. Biol. 2016, 50, 125–132. [Google Scholar] [CrossRef]
- Goeppert, B.; Schmezer, P.; Dutruel, C.; Oakes, C.; Renner, M.; Breinig, M.; Warth, A.; Vogel, M.N.; Mittelbronn, M.; Mehrabi, A.; et al. Down-regulation of tumor suppressor a kinase anchor protein 12 in human hepatocarcinogenesis by epigenetic mechanisms. Hepatology 2010, 52, 2023–2033. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Xu, X.; Wu, M.; Guan, Z.; Su, X.; Chen, S.; Wang, H.; Teng, L. GPRC5A: An emerging biomarker in human cancer. BioMed Res. Int. 2018, 2018. [Google Scholar] [CrossRef] [PubMed]
- Cetin, M.; Odabas, G.; Douglas, L.R.; Duriez, P.J.; Balcik-Ercin, P.; Yalim-Camci, I.; Sayan, A.E.; Yagci, T. ROR1 expression and its functional significance in hepatocellular carcinoma cells. Cells 2019, 8, 210. [Google Scholar] [CrossRef] [PubMed]
- Asad, A.S.; Candia, A.J.N.; Gonzalez, N.; Zuccato, C.F.; Abt, A.; Orrillo, S.J.; Lastra, Y.; De Simone, E.; Boutillon, F.; Goffin, V.; et al. Prolactin and its receptor as therapeutic targets in glioblastoma multiforme. Sci. Rep. 2019, 9, 19578. [Google Scholar] [CrossRef]
- Tilghman, J.; Wu, H.; Sang, Y.; Shi, X.; Guerrero-Cazares, H.; Quinones-Hinojosa, A.; Eberhart, C.G.; Laterra, J.; Ying, M. HMMR maintains the stemness and tumorigenicity of glioblastoma stem-like cells. Cancer Res. 2014, 74, 3168–3179. [Google Scholar] [CrossRef]
- Thangavelu, P.U.; Lin, C.-Y.; Vaidyanathan, S.; Nguyen, T.H.; Dray, E.; Duijf, P.H. Overexpression of the E2F target gene CENPI promotes chromosome instability and predicts poor prognosis in estrogen receptor-positive breast cancer. Oncotarget 2017, 8, 62167–62182. [Google Scholar] [CrossRef]
- Bu, P.; Chen, K.-Y.; Xiang, K.; Johnson, C.; Crown, S.B.; Rakhilin, N.; Ai, Y.; Wang, L.; Xi, R.; Astapova, I.; et al. Aldolase B-mediated fructose metabolism drives metabolic reprogramming of colon cancer liver metastasis. Cell Metab. 2018, 27, 1249–1262.e4. [Google Scholar] [CrossRef] [PubMed]
- Liou, G.Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 5, 479–496. [Google Scholar] [CrossRef] [PubMed]
- Paardekooper, L.M.; Vos, W.; Van Der Bogaart, G. Oxygen in the tumor microenvironment: Effects on dendritic cell function. Oncotarget 2019, 10, 883–896. [Google Scholar] [CrossRef] [PubMed]
- Holmannova, D.; Borska, L.; Andrys, C.; Borsky, P.; Kremlacek, J.; Hamakova, K.; Rehacek, V.; Malkova, A.; Svadlakova, T.; Palicka, V.; et al. The impact of psoriasis and metabolic syndrome on the systemic inflammation and oxidative damage to nucleic acids. J. Immunol. Res. 2020, 2020, 7352637. [Google Scholar] [CrossRef] [PubMed]
- Bensimon, J.; Biard, D.; Paget, V.; Goislard, M.; Morel-Altmeyer, S.; Konge, J.; Chevillard, S.; Lebeau, J. Forced extinction of CD24 stem-like breast cancer marker alone promotes radiation resistance through the control of oxidative stress. Mol. Carcinog. 2015, 55, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Hrycay, E.G.; Bandiera, S.M. Involvement of cytochrome P450 in reactive oxygen species formation and cancer. Adv. Pharmacol. 2015, 74, 35–84. [Google Scholar] [PubMed]
- Veith, A.; Moorthy, B. Role of cytochrome P450s in the generation and metabolism of reactive oxygen species. Curr. Opin. Toxicol. 2018, 7, 44–51. [Google Scholar] [CrossRef]
- Mishra, P.; Tang, W.; Ambs, S. ADHFE1 is a MYC-linked oncogene that induces metabolic reprogramming and cellular de-differentiation in breast cancer. Mol. Cell. Oncol. 2018, 5, e1432260. [Google Scholar] [CrossRef] [PubMed]
- Marchetto, A.; Ohmura, S.; Orth, M.F.; Knott, M.M.L.; Colombo, M.V.; Arrigoni, C.; Bardinet, V.; Saucier, D.; Wehweck, F.S.; Li, J.; et al. Oncogenic hijacking of a developmental transcription factor evokes vulnerability toward oxidative stress in Ewing sarcoma. Nat. Commun. 2020, 11, 2423. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Estevez, H.; Garcia-Calvo, E.; Rivera-Torres, J.; Vallet-Regí, M.; González, B.; Luque-Garcia, J.L. Transcriptome Analysis Identifies Novel Mechanisms Associated with the Antitumor Effect of Chitosan-Stabilized Selenium Nanoparticles. Pharmaceutics 2021, 13, 356. https://doi.org/10.3390/pharmaceutics13030356
Estevez H, Garcia-Calvo E, Rivera-Torres J, Vallet-Regí M, González B, Luque-Garcia JL. Transcriptome Analysis Identifies Novel Mechanisms Associated with the Antitumor Effect of Chitosan-Stabilized Selenium Nanoparticles. Pharmaceutics. 2021; 13(3):356. https://doi.org/10.3390/pharmaceutics13030356
Chicago/Turabian StyleEstevez, Hector, Estefania Garcia-Calvo, Jose Rivera-Torres, María Vallet-Regí, Blanca González, and Jose L. Luque-Garcia. 2021. "Transcriptome Analysis Identifies Novel Mechanisms Associated with the Antitumor Effect of Chitosan-Stabilized Selenium Nanoparticles" Pharmaceutics 13, no. 3: 356. https://doi.org/10.3390/pharmaceutics13030356
APA StyleEstevez, H., Garcia-Calvo, E., Rivera-Torres, J., Vallet-Regí, M., González, B., & Luque-Garcia, J. L. (2021). Transcriptome Analysis Identifies Novel Mechanisms Associated with the Antitumor Effect of Chitosan-Stabilized Selenium Nanoparticles. Pharmaceutics, 13(3), 356. https://doi.org/10.3390/pharmaceutics13030356