
A Strategy for Selective Deletion of Autoimmunity-Related T Cells by pMHC-Targeted Delivery

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Construction of Plasmids
2.3. Expression and Purification of Fusion Proteins
2.4. Biotin Conjugation
2.5. MMAF Conjugation
2.6. Protein Characterization Methods
2.7. 3DNA Synthesis
2.8. Generation of T cell Hybridomas
2.9. Tetramer Formation and FACS
2.10. Cytotoxicity Assays
3. Results and Discussion
3.1. Design of pMHC Fc Fusion Proteins
3.2. Optimization of Conjugation Scheme
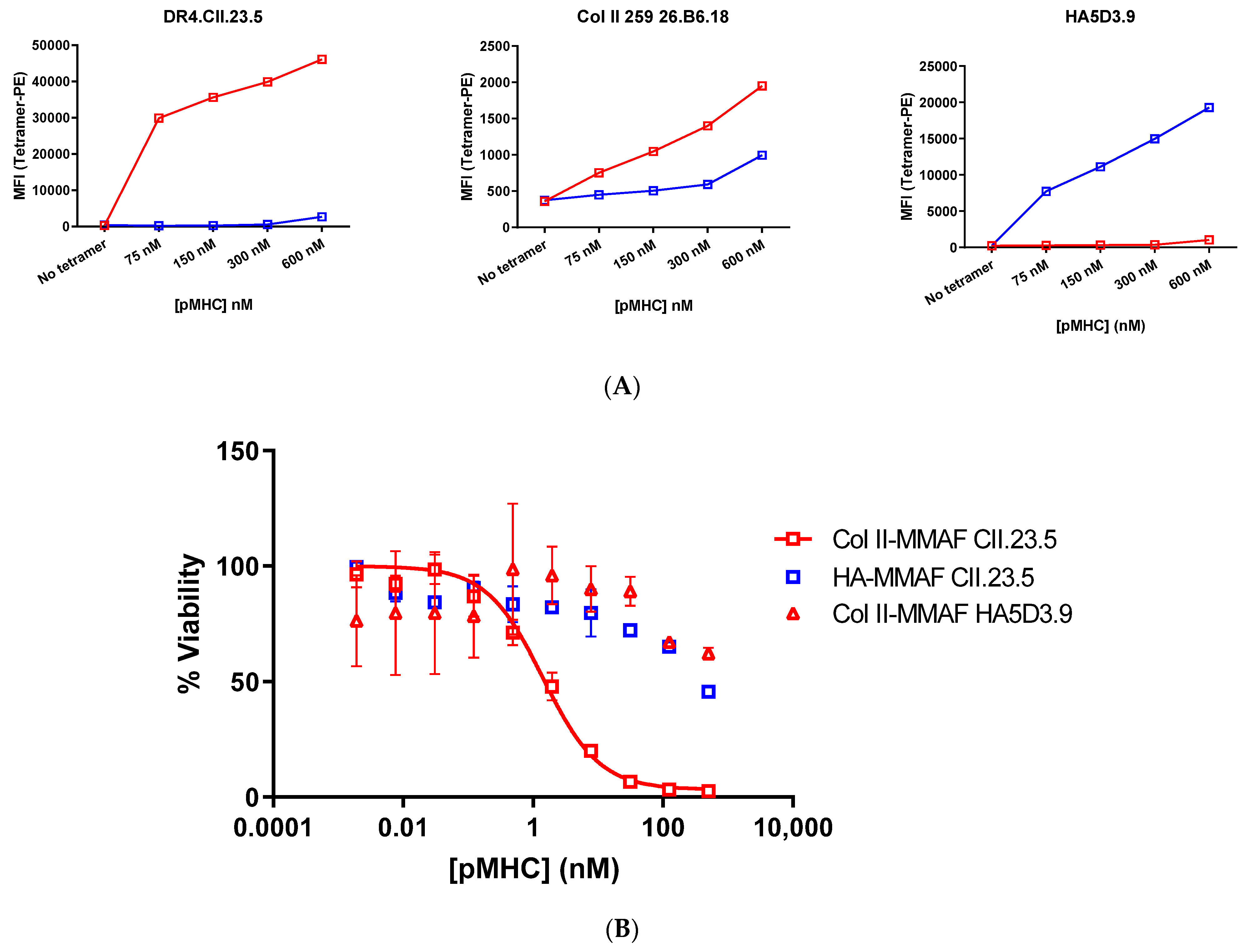
3.3. pMHC-Tetramer Cell Killing Assays
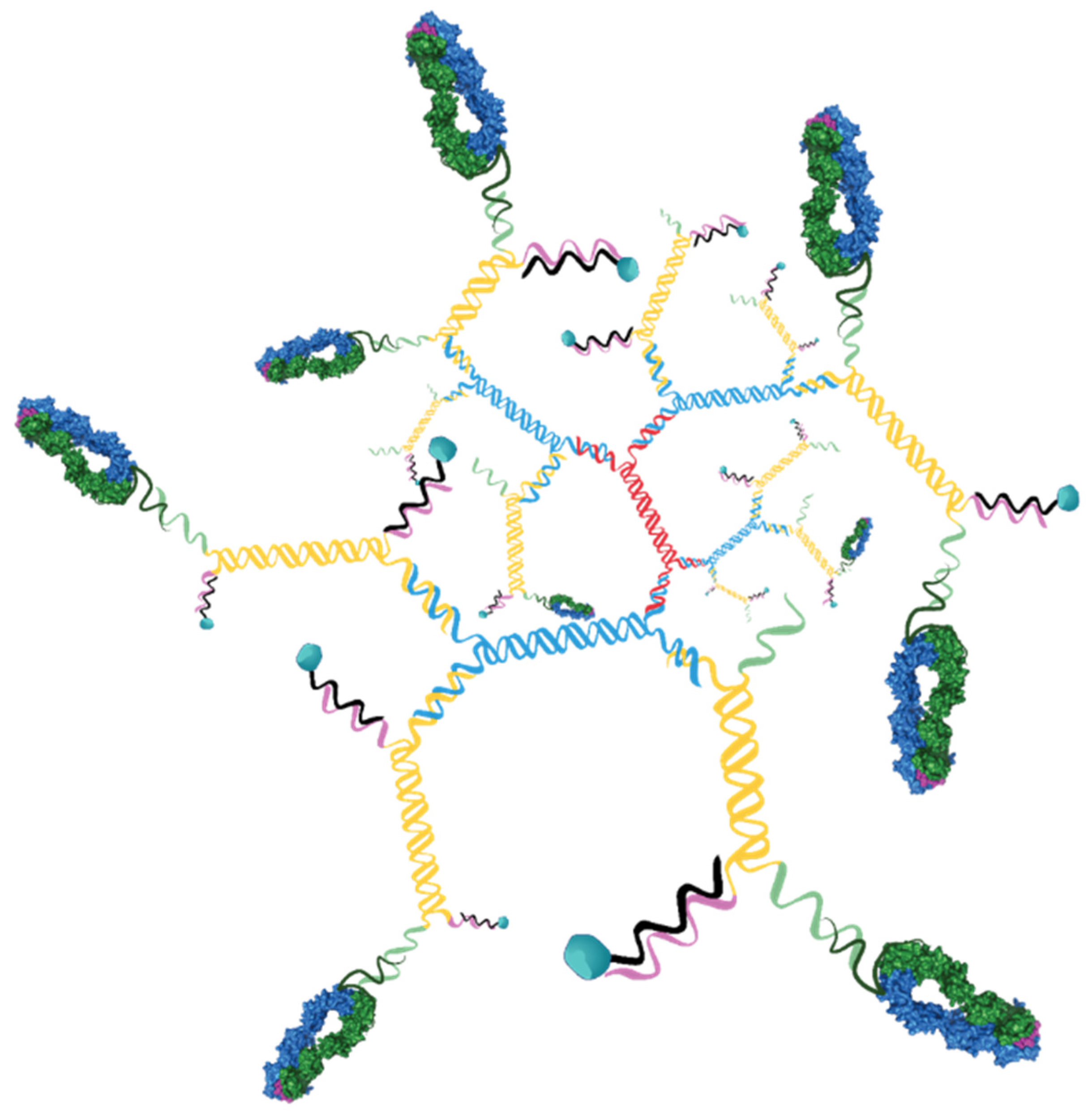
3.4. Production of 3DNA-pMHC Drug Loaded Assemblies
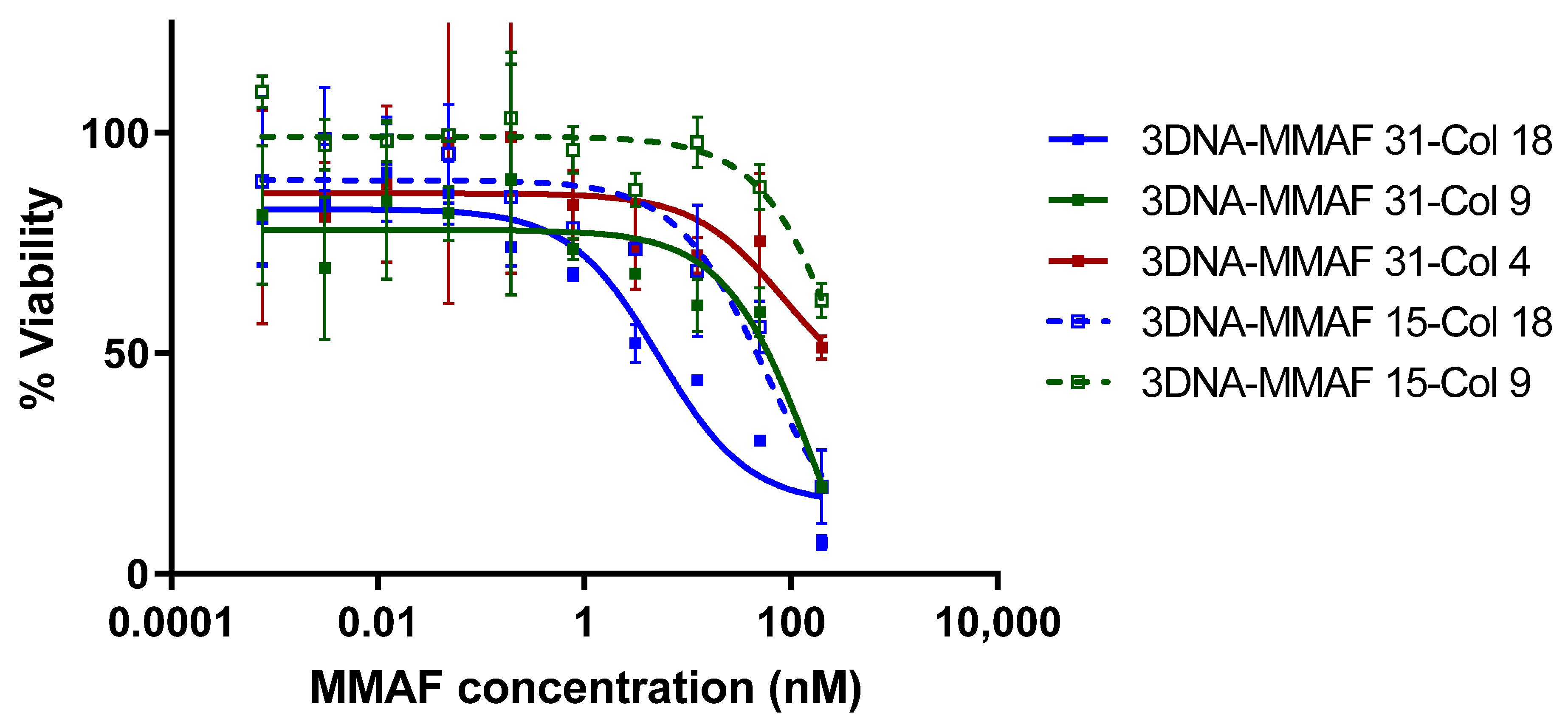
3.5. 3DNA Cell Killing Assays
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Polakis, P. Antibody Drug Conjugates for Cancer Therapy. Pharmacol. Rev. 2015, 68, 3–19. [Google Scholar] [CrossRef]
- Lim, R.K.V.; Yu, S.; Cheng, B.; Li, S.; Kim, N.-J.; Cao, Y.; Chi, V.; Kim, J.Y.; Chatterjee, A.K.; Schultz, P.G.; et al. Targeted Delivery of LXR Agonist Using a Site-Specific Antibody–Drug Conjugate. Bioconj. Chem. 2015, 26, 2216–2222. [Google Scholar] [CrossRef]
- Zhou, C.; Cai, H.; Baruch, A.; Lewin-Koh, N.; Yang, M.; Guo, F.; Xu, D.; Deng, R.; Hazenbos, W.; Kamath, A.V. Sustained activity of novel THIOMAB antibody-antibiotic conjugate against Staphylococcus aureus in a mouse model: Longitudinal pharmacodynamic assessment by bioluminescence imaging. PLoS ONE 2019, 14, e0224096. [Google Scholar] [CrossRef]
- Wang, R.; Liu, T.; Wang, Y.; Cao, Y.; Du, J.; Luo, X.; Deshmukh, V.; Kim, C.H.; Lawson, B.R.; Tremblay, M.S.; et al. An Immunosuppressive Antibody–Drug Conjugate. J. Am. Chem. Soc. 2015, 137, 3229–3232. [Google Scholar] [CrossRef]
- Klareskog, L.; Catrina, A.I.; Paget, S. Rheumatoid arthritis. Lancet 2009, 373, 659–672. [Google Scholar] [CrossRef]
- Fugger, L.; Svejgaard, A. Association of MHC and rheumatoid arthritis. HLA-DR4 and rheumatoid arthritis: Studies in mice and men. Arthritis Res. 2000, 2, 208–211. [Google Scholar] [CrossRef][Green Version]
- Rosloniec, E.F.; Whittington, K.B.; Zaller, D.M.; Kang, A.H. HLA-DR1 (DRB1*0101) and DR4 (DRB1*0401) Use the Same Anchor Residues for Binding an Immunodominant Peptide Derived from Human Type II Collagen. J. Immunol. 2002, 168, 253–259. [Google Scholar] [CrossRef]
- Ria, F.; Penitente, R.; De Santis, M.; Nicolò, C.; Di Sante, G.; Orsini, M.; Arzani, D.; Fattorossi, A.; Battaglia, A.; Ferraccioli, G. Collagen-specific T-cell repertoire in blood and synovial fluid varies with disease activity in early rheumatoid arthritis. Arthritis Res. Ther. 2008, 10, R135. [Google Scholar] [CrossRef] [PubMed]
- Bilal, J.; Berlinberg, A.; Riaz, I.B.; Faridi, W.; Bhattacharjee, S.; Ortega, G.; Murad, M.H.; Wang, Z.; Prokop, L.J.; Alhifany, A.A.; et al. Risk of Infections and Cancer in Patients With Rheumatologic Diseases Receiving Interleukin Inhibitors: A Systematic Review and Meta-analysis. JAMA Netw Open 2019, 2, e1913102. [Google Scholar] [CrossRef] [PubMed]
- Aleksic, M.; Liddy, N.; Molloy, P.; Pumphrey, N.; Vuidepot, A.; Chang, K.-M.; Jakobsen, B.K. Different affinity windows for virus and cancer-specific T-cell receptors: Implications for therapeutic strategies. Eur. J. Immunol. 2012, 42, 3174–3179. [Google Scholar] [CrossRef]
- Altman, J.D.; Moss, P.A.H.; Goulder, P.J.R.; Barouch, D.H.; McHeyzer-Williams, M.G.; Bell, J.I.; McMichael, A.J.; Davis, M.M. Phenotypic Analysis of Antigen-Specific T Lymphocytes. Science 1996, 274, 94–96. [Google Scholar] [CrossRef] [PubMed]
- Yuan, R.R.; Wong, P.; McDevitt, M.R.; Doubrovina, E.; Leiner, I.; Bornmann, W.; O’Reilly, R.; Pamer, E.G.; Scheinberg, D.A. Targeted deletion of T-cell clones using alpha-emitting suicide MHC tetramers. Blood 2004, 104, 2397–2402. [Google Scholar] [CrossRef] [PubMed]
- Vincent, B.G.; Young, E.F.; Buntzman, A.; Stevens, R.; Kepler, T.B.; Tisch, R.M.; Frelinger, J.A.; Hess, P.R. Toxin-Coupled MHC Class I Tetramers Can Specifically Ablate Autoreactive CD8+ T Cells and Delay Diabetes in Nonobese Diabetic Mice. J. Immunol. 2010, 184, 4196–4204. [Google Scholar] [CrossRef] [PubMed]
- Clemente-Casares, X.; Blanco, J.; Ambalavanan, P.; Yamanouchi, J.; Singha, S.; Fandos, C.; Tsai, S.; Wang, J.; Garabatos, N.; Izquierdo, C.; et al. Expanding antigen-specific regulatory networks to treat autoimmunity. Nature 2016, 530, 434–440. [Google Scholar] [CrossRef]
- Serra, P.; Santamaria, P. Nanoparticle-based approaches to immune tolerance for the treatment of autoimmune diseases. Eur. J. Immunol. 2018, 48, 751–756. [Google Scholar] [CrossRef]
- Goldenberg, D.M.; Sharkey, R.M.; Paganelli, G.; Barbet, J.; Chatal, J.-F. Antibody Pretargeting Advances Cancer Radioimmunodetection and Radioimmunotherapy. J. Clin. Oncol. 2006, 24, 823–834. [Google Scholar] [CrossRef]
- Tam, S.H.; McCarthy, S.G.; Armstrong, A.A.; Somani, S.; Wu, S.-J.; Liu, X.; Gervais, A.; Ernst, R.; Saro, D.; Decker, R.; et al. Functional, Biophysical, and Structural Characterization of Human IgG1 and IgG4 Fc Variants with Ablated Immune Functionality. Antibodies 2017, 6, 12. [Google Scholar] [CrossRef]
- Chen, I.; Dorr, B.M.; Liu, D.R. A general strategy for the evolution of bond-forming enzymes using yeast display. Proc. Natl. Acad. Sci. USA 2011, 108, 11399–11404. [Google Scholar] [CrossRef]
- Nilsen, T.W.; Grayzel, J.; Prensky, W. Dendritic Nucleic Acid Structures. J. Theor. Biol. 1997, 187, 273–284. [Google Scholar] [CrossRef]
- Roki, N.; Tsinas, Z.; Solomon, M.; Bowers, J.; Getts, R.C.; Muro, S. Unprecedently high targeting specificity toward lung ICAM-1 using 3DNA nanocarriers. J. Control. Release 2019, 305, 41–49. [Google Scholar] [CrossRef]
- Felix, N.J.; Donermeyer, D.L.; Horvath, S.; Walters, J.J.; Gross, M.L.; Suri, A.; Allen, P.M. Alloreactive T cells respond specifically to multiple distinct peptide-MHC complexes. Nat. Immunol. 2007, 8, 388–397. [Google Scholar] [CrossRef] [PubMed]
- Leisner, C.; Loeth, N.; Lamberth, K.; Justesen, S.; Sylvester-Hvid, C.; Schmidt, E.G.; Claesson, M.; Buus, S.; Stryhn, A. One-Pot, Mix-and-Read Peptide-MHC Tetramers. PLoS ONE 2008, 3, e1678. [Google Scholar] [CrossRef] [PubMed]
- Cunliffe, S.L.; Wyer, J.R.; Sutton, J.K.; Lucas, M.; Harcourt, G.; Klenerman, P.; McMichael, A.J.; Kelleher, A.D. Optimization of peptide linker length in production of MHC class II/peptide tetrameric complexes increases yield and stability, and allows identification of antigen-specific CD4+T cells in peripheral blood mononuclear cells. Eur. J. Immunol. 2002, 32, 3366–3375. [Google Scholar] [CrossRef]
- Popp, M.W.-L.; Ploegh, H.L. Making and Breaking Peptide Bonds: Protein Engineering Using Sortase. Angew. Chem. Int. Ed. 2011, 50, 5024–5032. [Google Scholar] [CrossRef] [PubMed]
- Ooi, J.D.; Petersen, J.; Tan, Y.H.; Huynh, M.; Willett, Z.J.; Ramarathinam, S.; Eggenhuizen, P.J.; Loh, K.L.; Watson, K.A.; Gan, P.Y.; et al. Dominant protection from HLA-linked autoimmunity by antigen-specific regulatory T cells. Nature 2017, 545, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Moiani, D.; Salvalaglio, M.; Cavallotti, C.; Bujacz, A.; Redzynia, I.; Bujacz, G.; Dinon, F.; Pengo, P.; Fassina, G. Structural Characterization of a Protein A Mimetic Peptide Dendrimer Bound to Human IgG. J. Phys. Chem. B 2009, 113, 16268–16275. [Google Scholar] [CrossRef]
- Doronina, S.O.; Mendelsohn, B.A.; Bovee, T.D.; Cerveny, C.G.; Alley, S.C.; Meyer, D.L.; Oflazoglu, E.; Toki, B.E.; Sanderson, R.J.; Zabinski, R.F.; et al. Enhanced Activity of Monomethylauristatin F through Monoclonal Antibody Delivery: Effects of Linker Technology on Efficacy and Toxicity. Bioconj. Chem. 2005, 17, 114–124. [Google Scholar] [CrossRef]
- Dubowchik, G.M.; Firestone, R.A.; Padilla, L.; Willner, D.; Hofstead, S.J.; Mosure, K.; Knipe, J.O.; Lasch, S.J.; Trail, P.A. Cathepsin B-Labile Dipeptide Linkers for Lysosomal Release of Doxorubicin from Internalizing Immunoconjugates: Model Studies of Enzymatic Drug Release and Antigen-Specific In Vitro Anticancer Activity. Bioconj. Chem. 2002, 13, 855–869. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Drugs per 3DNA | pMHCs per 3DNA | Z-Avg (nm) | PDI |
|---|---|---|---|---|
| 3DNA-MMAF 15—pMHC CII 18 | 15 | 18 | 81.17 +/− 0.67 | 0.145 +/− 0.008 |
| 3DNA-MMAF 15—pMHC CII 9 | 15 | 9 | 80.56 +/− 2.71 | 0.201 +/− 0.020 |
| 3DNA-MMAF 15—pMHC CII 4 | 15 | 4 | 75.98 +/− 1.58 | 0.201 +/− 0.016 |
| 3DNA-MMAF 15—pMHC HA 18 | 15 | 18 | 81.68 +/− 0.94 | 0.155 +/− 0.007 |
| 3DNA-MMAF 15—pMHC HA 9 | 15 | 9 | 75.67 +/− 1.09 | 0.169 +/− 0.014 |
| 3DNA-MMAF 15—pMHC HA 4 | 15 | 4 | 78.81 +/− 1.97 | 0.218 +/− 0.014 |
| 3DNA-MMAF 31—pMHC CII 18 | 31 | 18 | 75.35 +/− 0.36 | 0.134 +/− 0.012 |
| 3DNA-MMAF 31—pMHC CII 9 | 31 | 9 | 67.92 +/− 0.75 | 0.132 +/− 0.014 |
| 3DNA-MMAF 31—pMHC CII 4 | 31 | 4 | 67.25 +/− 3.51 | 0.18 +/− 0.029 |
| 3DNA-MMAF 31—pMHC HA 18 | 31 | 18 | 83.15 +/− 1.19 | 0.212 +/− 0.008 |
| 3DNA-MMAF 31—pMHC HA 9 | 31 | 9 | 69.43 +/− 1.38 | 0.152 +/− 0.024 |
| 3DNA-MMAF 31—pMHC HA 4 | 31 | 4 | 65.29 +/− 1.55 | 0.184 +/− 0.020 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goldberg, S.D.; Felix, N.; McCauley, M.; Eberwine, R.; Casta, L.; Haskell, K.; Lin, T.; Palovick, E.; Klein, D.; Getts, L.; et al. A Strategy for Selective Deletion of Autoimmunity-Related T Cells by pMHC-Targeted Delivery. Pharmaceutics 2021, 13, 1669. https://doi.org/10.3390/pharmaceutics13101669
Goldberg SD, Felix N, McCauley M, Eberwine R, Casta L, Haskell K, Lin T, Palovick E, Klein D, Getts L, et al. A Strategy for Selective Deletion of Autoimmunity-Related T Cells by pMHC-Targeted Delivery. Pharmaceutics. 2021; 13(10):1669. https://doi.org/10.3390/pharmaceutics13101669
Chicago/Turabian StyleGoldberg, Shalom D., Nathan Felix, Michael McCauley, Ryan Eberwine, Lou Casta, Kathleen Haskell, Tricia Lin, Elizabeth Palovick, Donna Klein, Lori Getts, and et al. 2021. "A Strategy for Selective Deletion of Autoimmunity-Related T Cells by pMHC-Targeted Delivery" Pharmaceutics 13, no. 10: 1669. https://doi.org/10.3390/pharmaceutics13101669
APA StyleGoldberg, S. D., Felix, N., McCauley, M., Eberwine, R., Casta, L., Haskell, K., Lin, T., Palovick, E., Klein, D., Getts, L., Getts, R., Zhou, M., Bansal-Pakala, P., & Dudkin, V. (2021). A Strategy for Selective Deletion of Autoimmunity-Related T Cells by pMHC-Targeted Delivery. Pharmaceutics, 13(10), 1669. https://doi.org/10.3390/pharmaceutics13101669