Caffeic Acid Phenethyl Ester Assisted by Reversible Electroporation—In Vitro Study on Human Melanoma Cells



Abstract
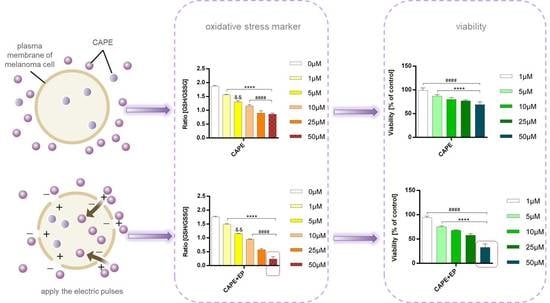
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Electroporation
2.3. Cellular Viability—MTT Assay and IC50 Determination
2.4. Cloning Efficacy Test
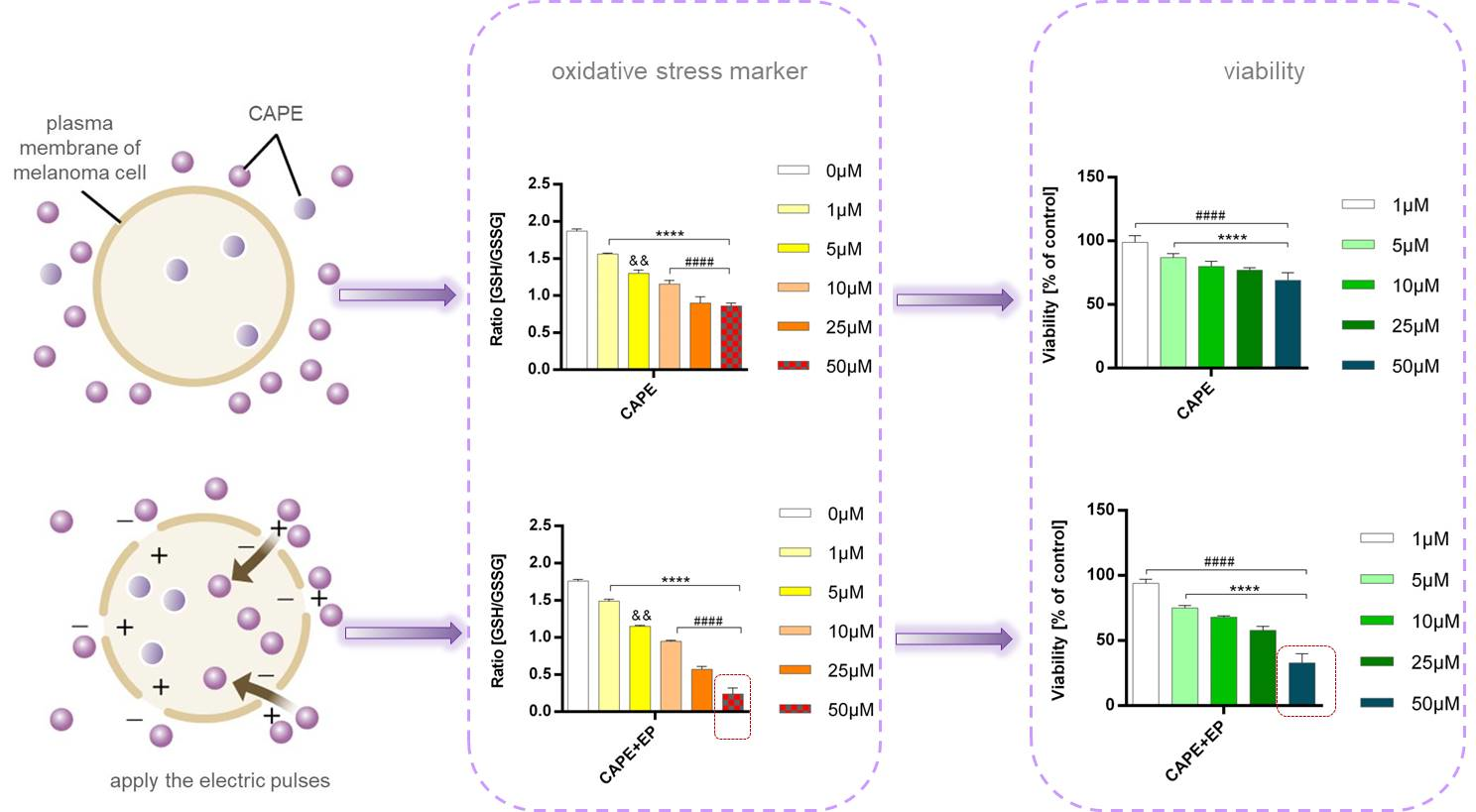
2.5. GSH/GSSG Assay
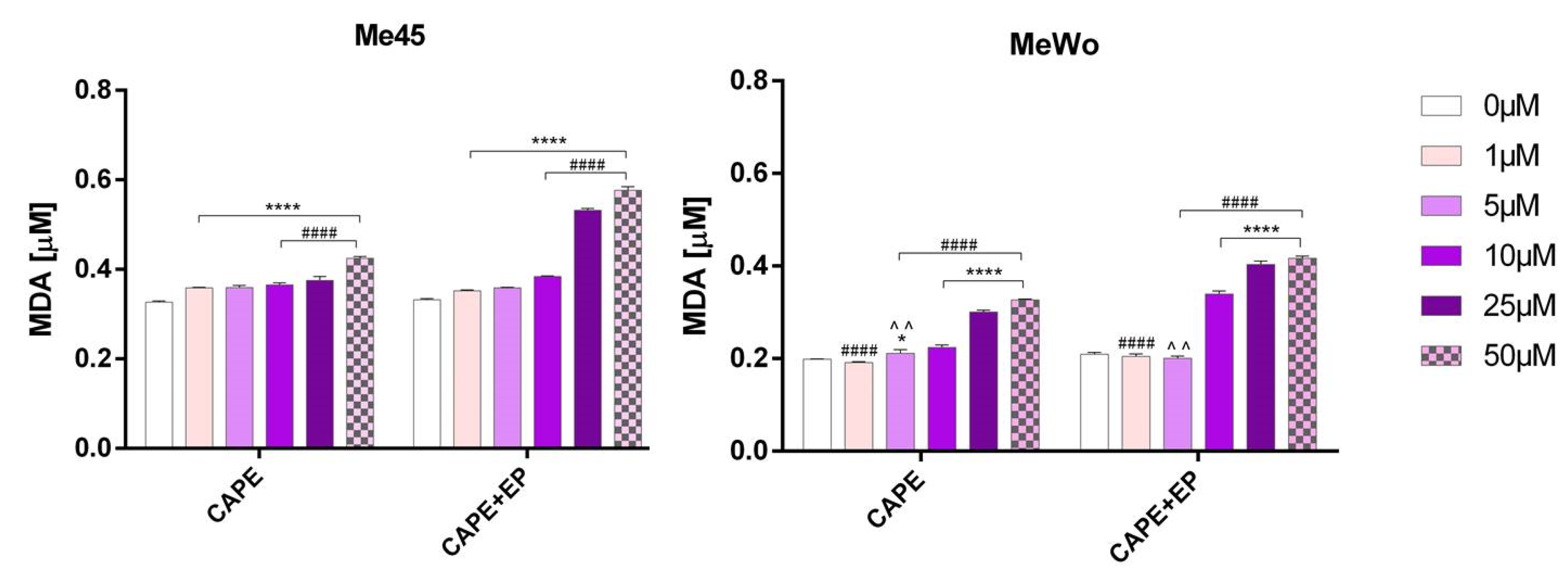
2.6. Lipid Peroxidation
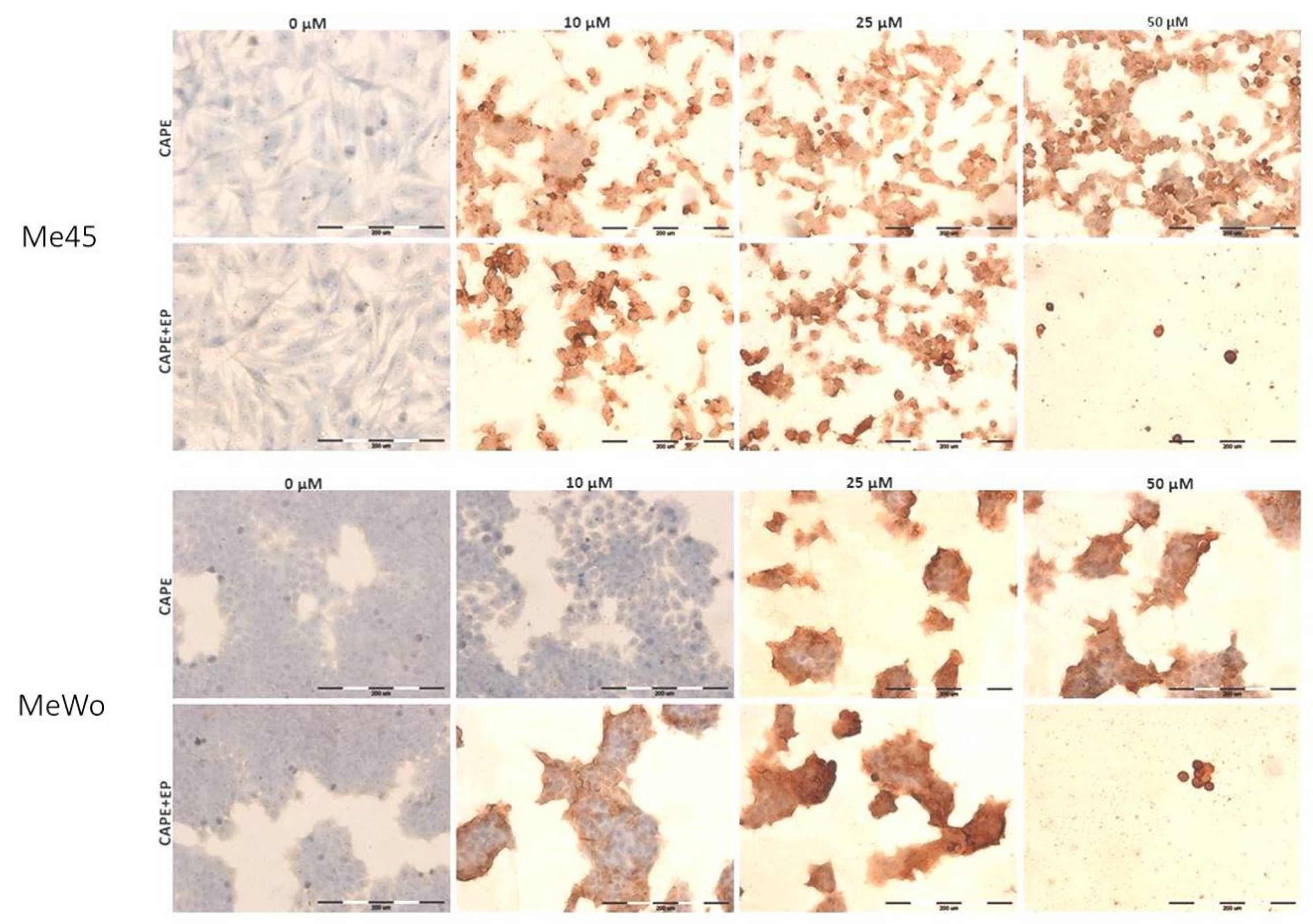
2.7. Immunocytochemical Cleaved PARP-1 Protein Evaluation
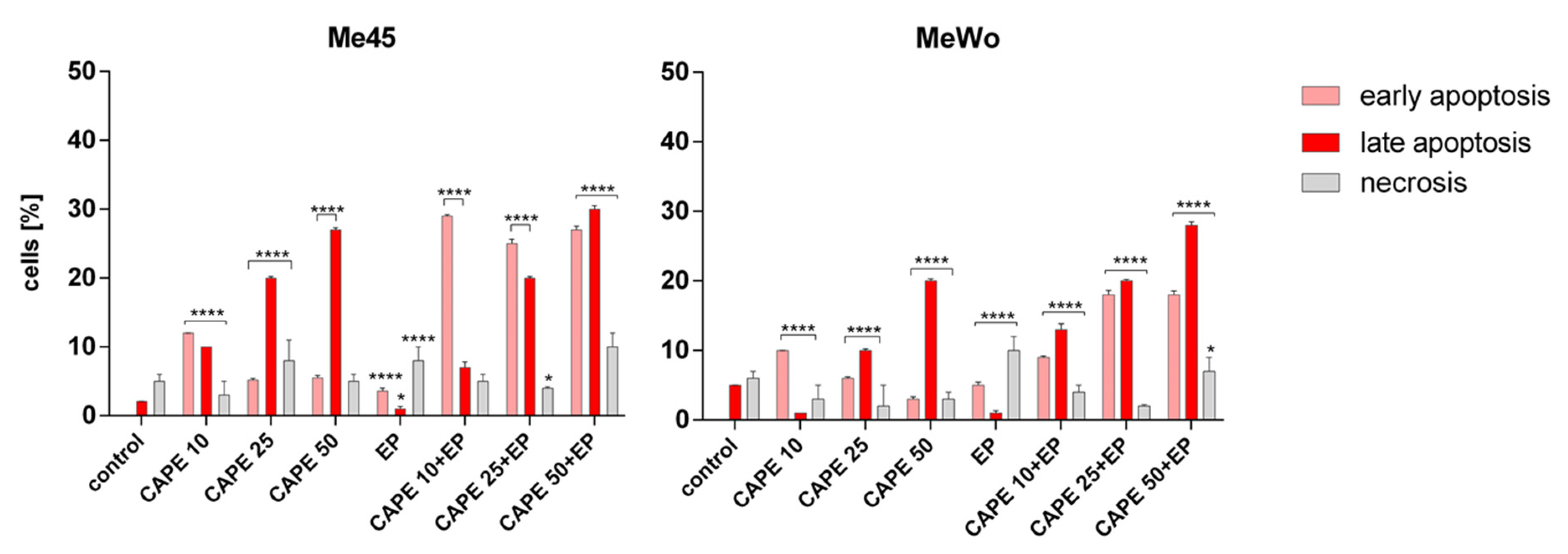
2.8. Apoptosis and Necrosis Evaluation
2.9. Statistical Analysis
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Paul, M.; Ma, J.K. Plant-made pharmaceuticals: Leading products and production platforms. Biotechnol. Appl. Biochem. 2011, 58, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Issa, N.; Wathieu, H.; Ojo, A.; Byers, S.; Dakshanamurthy, S. Drug metabolism in preclinical drug development: A survey of the discovery process, toxicology, and computational tools. Curr. Drug Metab. 2017, 18, 556–565. [Google Scholar] [CrossRef] [PubMed]
- Murtaza, G.; Karim, S.; Akram, M.R.; Khan, S.A.; Azhar, S.; Mumtaz, A.; Asad, M.H. Caffeic acid phenethyl ester and therapeutic potentials. BioMed. Res. Int. 2014, 2014, 145342. [Google Scholar] [CrossRef] [PubMed]
- Kumazawa, S.; Ahn, M.-R.; Fujimoto, T.; Kato, M. Radical-scavenging activity and phenolic constituents of propolis from different regions of Argentina. Nat. Prod. Res. 2010, 24, 804–812. [Google Scholar] [CrossRef] [PubMed]
- Kabała-Dzik, A.; Rzepecka-Stojko, A.; Kubina, R.; Jastrzębska-Stojko, Ż.; Stojko, R.; Wojtyczka, R.D.; Stojko, J. Migration rate inhibition of breast cancer cells treated by caffeic acid and caffeic acid phenethyl ester: An In Vitro comparison study. Nutrients. 2017, 19, 1144. [Google Scholar] [CrossRef]
- Ishida, Y.; Gao, R.; Shah, N.; Bhargava, P.; Furune, T.; Kaul, S.C.; Terao, K.; Wadhwa, R. Anticancer activity in honeybee propolis: Functional insights to the role of caffeic acid phenethyl ester and its complex with γ-cyclodextrin. Integr. Cancer Ther. 2018, 17, 867–873. [Google Scholar] [CrossRef] [PubMed]
- Fırat, F.; Özgül, M.; Türköz Uluer, E.; Inan, S. Effects of caffeic acid phenethyl ester (CAPE) on angiogenesis, apoptosis and oxidatıve stress ın various cancer cell lines. Biotech. Histochem. 2019, 94, 491–497. [Google Scholar] [CrossRef]
- Morin, P.; St-Coeur, P.D.; Doiron, J.A.; Cormier, M.; Poitras, J.J.; Surette, M.E.; Touaibia, M. Substituted caffeic and ferulic acid phenethyl esters: Synthesis, leukotrienes biosynthesis inhibition, and cytotoxic activity. Molecules. 2017, 22, 1124. [Google Scholar] [CrossRef]
- Grunberger, D.; Banerjee, R.; Eisinger, K.; Oltz, E.M.; Efros, L.; Caldwell, M.; Estevez, V.; Nakanishi, K. Preferential cytotoxicity on tumor cells by caffeic acid phenethyl ester isolated from propolis. Experientia 1988, 44, 230–232. [Google Scholar] [CrossRef]
- Chen, H.-C.; Chen, J.-H.; Chang, C.; Shieh, C.-J. Optimization of ultrasound-accelerated synthesis of enzymatic caffeic acid phenethyl ester by response surface methodology. Ultrason. Sonochem. 2011, 18, 455–459. [Google Scholar] [CrossRef]
- Ozturk, G.; Ginis, Z.; Akyol, S.; Erden, G.; Gurel, A.; Akyol, O. The anticancer mechanism of caffeic acid phenethyl ester (CAPE): Review of melanomas, lung and prostate cancers. Eur. Rev. Med. Phar. Sci. 2012, 16, 2064–2068. [Google Scholar]
- Saleem, M.; Maddodi, N.; Zaid, A.M.; Khan, N.; bin Hafeez, B.; Asim, M.; Suh, Y.; Yun, J.M.; Setaluri, V.; Mukhtar, H. Lupeol inhibits growth of highly aggressive human metastatic melanoma cells In Vitro and In Vivo by inducing apoptosis. Clin. Cancer Res. 2008, 14, 2119–2127. [Google Scholar] [CrossRef] [PubMed]
- Liao, H.-F.; Chen, Y.-Y.; Liu, J.-J.; Hsu, M.L.; Shieh, H.J.; Liao, H.J.; Shieh, C.J.; Shiao, M.S.; Chen, Y.J. Inhibitory effect of caffeic acid phenethyl ester on angiogenesis, tumor invasion, and metastasis. J. Agric. Food Chem. 2003, 51, 7907–7912. [Google Scholar] [CrossRef] [PubMed]
- Bhimani, R.S.; Troll, W.; Grunberger, D.; Frenkel, K. Inhibition of oxidative stress in HeLa cells by chemopreventive agents. Cancer Res. 1993, 53, 4528–4533. [Google Scholar] [PubMed]
- Kudugunti, S.K.; Vad, N.M.; Ekogbo, E.; Moridani, M.Y. Efficacy of caffeic acid phenethyl ester (CAPE) in skin B16-F0 melanoma tumor bearing C57BL/6 mice. Invest. New Drugs 2011, 29, 52–62. [Google Scholar] [CrossRef] [PubMed]
- De Luca, A.; Parker, L.J.; Ang, W.H.; Rodolfo, C.; Gabbarini, V.; Hancock, N.C.; Palone, F.; Mazzetti, A.P.; Menin, L.; Morton, C.J.; et al. A structure-based mechanism of cisplatin resistance mediated by glutathione transferase P1-1. Proc. Natl. Acad. Sci. USA 2019, 116, 13943–13951. [Google Scholar] [CrossRef]
- Bocedi, A.; Noce, A.; Marrone, G.; Noce, G.; Cattani, G.; Gambardella, G.; Di Lauro, M.; Di Daniele, N.; Ricci, G. Glutathione transferase P1-1 an enzyme useful in biomedicine and as biomarker in clinical practice and in environmental pollution. Nutrients 2019, 11, 1741. [Google Scholar] [CrossRef]
- Gou, J.; Yao, X.; Tang, H.; Zou, K.; Liu, Y.; Zuo, H.; Zhao, X.; Li, Z. Absorption properties and effects of caffeic acid phenethyl ester and its p-nitro-derivative on P-glycoprotein in Caco-2 cells and rats. Pharm. Biol. 2016, 54, 2960–2967. [Google Scholar] [CrossRef][Green Version]
- Hong, Y.J.; Yang, S.Y.; Nam, M.H.; Koo, Y.C.; Lee, K.W. Caffeic acid inhibits the uptake of 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP) by inducing the efflux transporters expression in Caco-2 cells. Biol. Pharm. Bull. 2015, 38, 201–207. [Google Scholar] [CrossRef]
- Colone, M.; Calcabrini, A.; Toccacieli, L.; Bozzuto, G.; Stringaro, A.; Gentile, M.; Cianfriglia, M.; Ciervo, A.; Caraglia, M.; Budillon, A.; et al. The multidrug transporter P-glycoprotein: A mediator of melanoma invasion? J. Invest. Dermatol. 2008, 128, 957–971. [Google Scholar] [CrossRef]
- Gehl, J. Electroporation: Theory and methods, perspectives for drug delivery, gene therapy and research. Acta Physiol. Scand. 2003, 177, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Choromanska, A.; Kulbacka, J.; Rembialkowska, N.; Pilat, J.; Drag-Zalesinska, M.; Wysocka, T.; Garbiec, A.; Kotulska, M.; Saczko, J. Effects of electrophotodynamic therapy In Vitro on human melanoma cells-melanotic (MeWo) and amelanotic (C32). Melanoma Res. 2015, 25, 210–224. [Google Scholar] [CrossRef] [PubMed]
- Choromanska, A.; Lubinska, S.; Szewczyk, A.; Saczko, J.; Kulbacka, J. Mechanisms of antimelanoma effect of oat β-glucan supported by electroporation. Bioelectrochemistry 2018, 123, 255–259. [Google Scholar] [CrossRef] [PubMed]
- Wadhawa, R.; Nigam, N.; Bhargava, P.; Dhanjal, J.K.; Goyal, S.; Grover, A.; Sundar, D.; Ishida, Y.; Terao, K.; Kaul, S.C. Molecular characterization and enhancement of anticancer activity of caffeic acid phenethyl ester by γ-cycldextrin. J. Cancer 2016, 13, 1755–1771. [Google Scholar] [CrossRef]
- Zhao, W.-X.; Wang, L.; Yang, J.-L.; Li, L.-Z.; Xu, W.-M.; Li, T. Caffeic acid phenethyl ester attenuates pro-inflammatory and fibrogenic phenotypes of LPS-stimulated hepatic stellate cells through the inhibition of NF-κB signaling. J. Pharmacol. 2014, 33, 687–694. [Google Scholar] [CrossRef]
- Ayla, S.; Tunalı, G.; Bilgiç, B.E.; Sofuoğlu, K.; Özdemir, A.A.; Tanrıverdi, G.; Özdemir, S.; Soner, B.C.; Öztürk, B.; Karahüseyinoğlu, S.; et al. Antioxidant activity of CAPE (caffeic acid phenethyl ester) In Vitro can protect human sperm deoxyribonucleic acid from oxidative damage. Acta Histochem. 2018, 120, 117–121. [Google Scholar] [CrossRef]
- Tolba, M.F.; Omar, H.A.; Azab, S.S.; Khalifa, A.E.; Abdel-Naim, A.B.; Abdel-Rahman, S.Z. Caffeic acid phenethyl ester: A review of its antioxidant activity, protective effects against ischemia-reperfusion injury and drug adverse reactions. Crit. Rev. Food Sci. Nutr. 2016, 56, 2183–2190. [Google Scholar] [CrossRef]
- Kudugunti, S.K.; Vad, N.M.; Whiteside, A.J.; Naik, B.U.; Yusuf, M.A.; Sirvenugpol, K.S.; Moridani, M.Y. Biochemical mechanism of caffeic acid phenylethyl ester (CAPE) selective toxicity towards melanoma cell lines. Chem. Biol. Interact. 2010, 188, 1–14. [Google Scholar] [CrossRef]
- Slominski, A.; Zbytek, B.; Slominski, R. Inhibitors of melanogenesis increase toxicity of cyclophosphamide and lymphocytes against melanoma cells. Int. J. Cancer 2009, 124, 1470–1477. [Google Scholar] [CrossRef]
- Sawers, L.; Ferguson, M.J.; Ihrig, B.R.; Young, H.C.; Chakravarty, P.; Wolf, C.R.; Smith, G. Glutathione S-transferase P1 (GSTP1) directly influences platinum drug chemosensitivity in ovarian tumour cell lines. Br. J. Cancer 2014, 111, 1150–1158. [Google Scholar] [CrossRef]
- Chen, M.J.; Chang, W.H.; Lin, C.C.; Liu, C.Y.; Wang, T.E.; Chu, C.H.; Shih, S.C.; Chen, Y.J. Caffeic acid phenethyl ester induces apoptosis of human pancreatic cancer cells involving caspase and mitochondrial dysfunction. Pancreatology 2008, 8, 566–576. [Google Scholar] [CrossRef] [PubMed]
- Pramanik, K.C.; Kudugunti, S.K.; Fofaria, N.M.; Moridani, M.Y.; Srivastava, S.K. Caffeic acid phenethyl ester suppresses melanoma tumor growth by inhibiting PI3K/AKT/XIAP pathway. Carcinogenesis 2013, 34, 2061–2070. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Tang, H.; Ren, Q.; Zhao, X.; Zuo, H.; Li, Z. Inhibited effects of CAPE-pNO2 on cervical carcinoma In Vivo and In Vitro and its detected metabolites. Oncotarget 2017, 8, 94197–94209. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| The Intensity of Staining | Positively Stained Cells [%] | |||
|---|---|---|---|---|
| Me45 | CAPE | CONTROL | - | 0 |
| 10 μM | ++/+++ | 100 | ||
| 25 μM | ++/+++ | 100 | ||
| 50 μM | +++ | 100 | ||
| CAPE+EP | CONTROL | - | 0 | |
| 10 μM | +++ | 100 | ||
| 25 μM | +++ | 100 | ||
| 50 μM | +++ | 100 | ||
| MeWo | CAPE | CONTROL | - | 0 |
| 10 μM | - | 0 | ||
| 25 μM | +++ | 100 | ||
| 50 μM | +++ | 100 | ||
| CAPE+EP | CONTROL | - | 0 | |
| 10 μM | ++/+++ | 100 | ||
| 25 μM | +++ | 100 | ||
| 50 μM | +++ | 100 | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choromanska, A.; Saczko, J.; Kulbacka, J. Caffeic Acid Phenethyl Ester Assisted by Reversible Electroporation—In Vitro Study on Human Melanoma Cells. Pharmaceutics 2020, 12, 478. https://doi.org/10.3390/pharmaceutics12050478
Choromanska A, Saczko J, Kulbacka J. Caffeic Acid Phenethyl Ester Assisted by Reversible Electroporation—In Vitro Study on Human Melanoma Cells. Pharmaceutics. 2020; 12(5):478. https://doi.org/10.3390/pharmaceutics12050478
Chicago/Turabian StyleChoromanska, Anna, Jolanta Saczko, and Julita Kulbacka. 2020. "Caffeic Acid Phenethyl Ester Assisted by Reversible Electroporation—In Vitro Study on Human Melanoma Cells" Pharmaceutics 12, no. 5: 478. https://doi.org/10.3390/pharmaceutics12050478
APA StyleChoromanska, A., Saczko, J., & Kulbacka, J. (2020). Caffeic Acid Phenethyl Ester Assisted by Reversible Electroporation—In Vitro Study on Human Melanoma Cells. Pharmaceutics, 12(5), 478. https://doi.org/10.3390/pharmaceutics12050478