Endoplasmic Reticulum-Associated Degradation-Dependent Processing in Cross-Presentation and Its Potential for Dendritic Cell Vaccinations: A Review

Abstract

1. Introduction
- (i)
- Restricted lysosomal degradation of extracellular proteins
- (ii)
- Recruitment of ERAD-related molecules into endocytotic compartments
- (iii)
- Retro-transport of extracellular proteins into the cytosol
2. Dendritic Cell (DC) Vaccination
2.1. DC Subsets
2.2. DC Vaccination
3. ERAD-Dependent Processing in CP
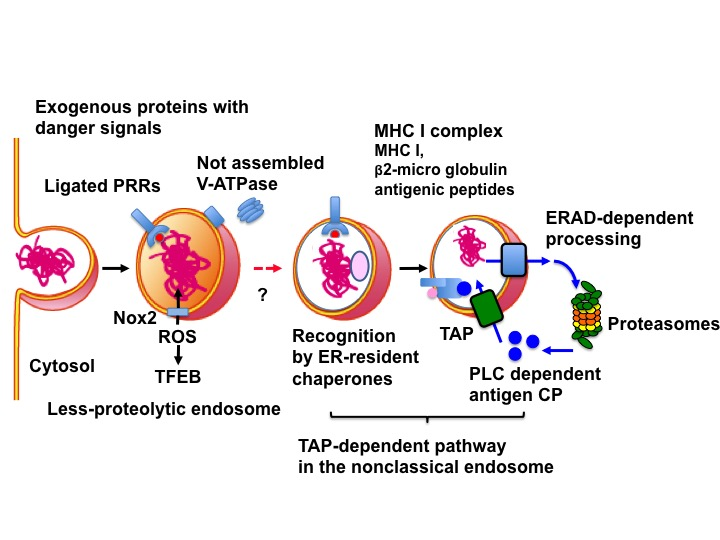
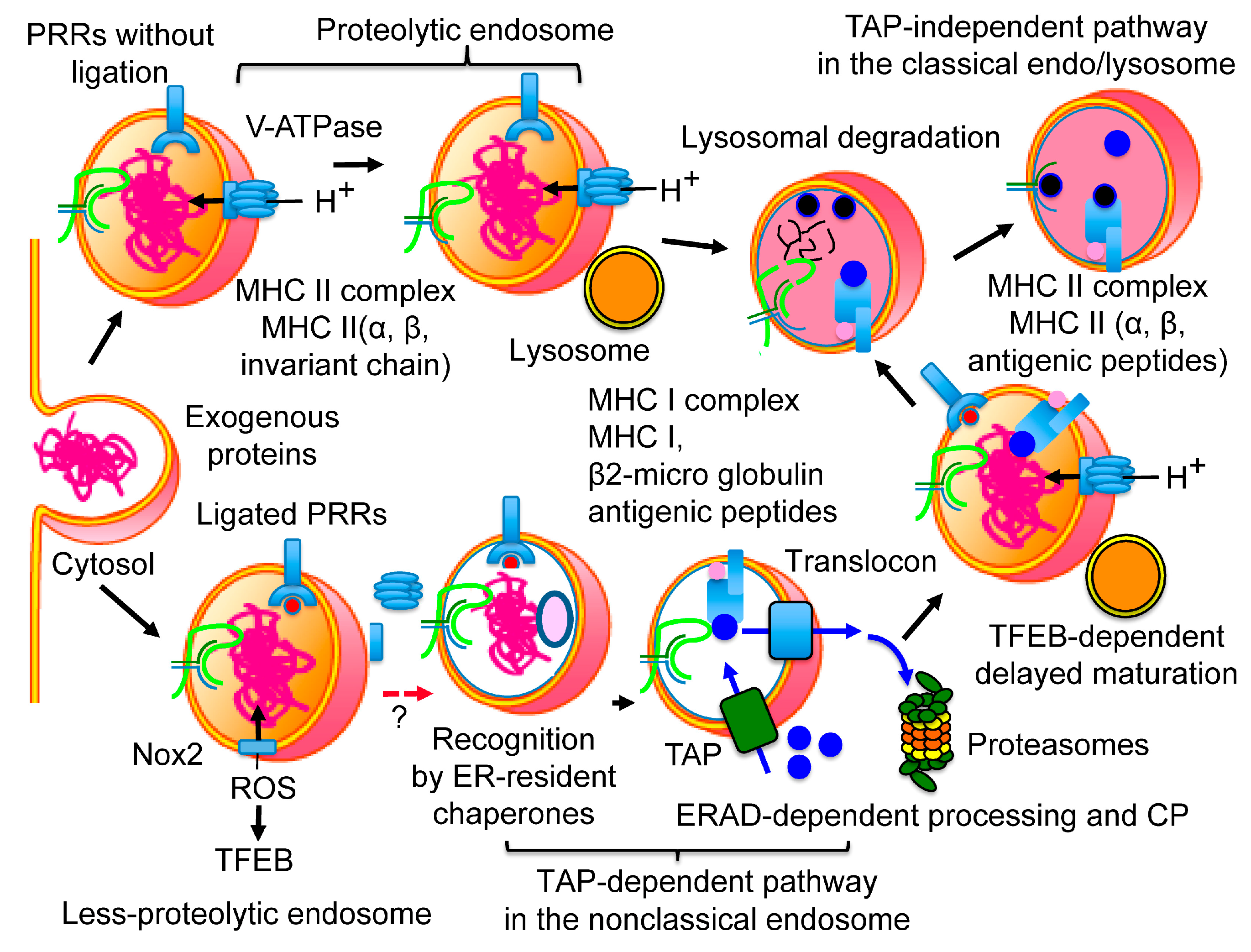
3.1. Two Pathways for Cross-Presentation (CP)
3.2. Molecular Mechanism of the TAP-Dependent Pathway
3.2.1. Protection of Extracellular Proteins from Lysosomal Degradation
3.2.2. The Non-Classical Endosome
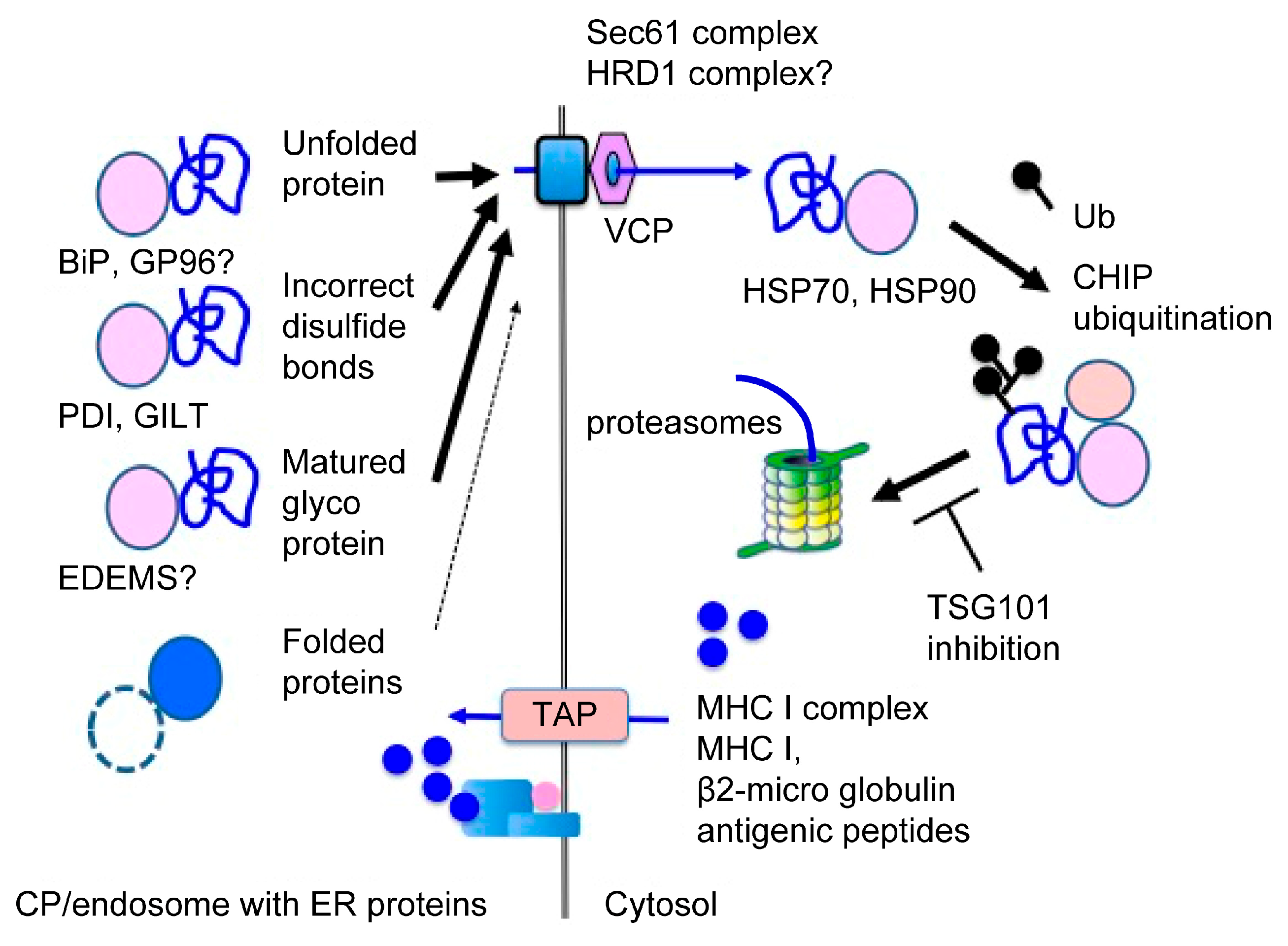
3.2.3. Recognition of Extracellular Proteins as ERAD Substrates
3.2.4. The Translocon in CP
4. Peptide Loading onto MHC I
5. Improvements in CP Efficiency by ERAD-Dependent Processing
6. DC Maturation
7. Conclusions
Funding
Conflicts of Interest
References
- Finn, O.J. Human tumor antigens yesterday, today, and tomorrow. Cancer Immunol. Res. 2017, 5, 347–354. [Google Scholar] [CrossRef]
- Stanley, M. Tumour virus vaccines: Hepatitis B virus and human papillomavirus. Philos. Trans. R. Soc. B Biol. Sci. 2016, 372, 1732. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer-immune set point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef]
- Sigal, L.J.; Crotty, S.; Andino, R.; Rock, K.L. Cytotoxic T cell immunity to virus-infected non-haematopoietic cells requires presentation of exogenous antigen. Nature 1999, 398, 77–80. [Google Scholar] [CrossRef]
- Bonifaz, L.; Bonnyay, D.; Mahnke, K.; Rivera, M.; Nussenzweig, M.C.; Steinman, R.M. Efficient targeting of protein antigen to the dendritic cell receptor DEC-205 in the steady state leads to antigen presentation on major histocompatibility complex class I products and peripheral CD8+ T cell tolerance. J. Exp. Med. 2002, 196, 1627–1638. [Google Scholar] [CrossRef]
- Den Haan, J.M.; Bevan, M.J. Antigen presentation to CD8+ T cells: Cross-priming in infectious diseases. Curr. Opin. Immunol. 2001, 13, 437–441. [Google Scholar] [CrossRef]
- Heath, W.R.; Carbone, F.R. Cross-presentation in viral immunity and self-tolerance. Nat. Rev. Immunol. 2001, 1, 126–134. [Google Scholar] [CrossRef]
- Mittal, D.; Gubin, M.M.; Schreiber, R.D.; Smyth, M.J. New insights into cancer immunoediting and its three components phases-elimination, equilibrium and escape. Curr. Opin. Immunol. 2014, 27, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Lennerz, V.; Fatho, M.; Gentilini, C.; Frye, R.A.; Lifke, A.; Ferel, D.; Wölfel, C.; Huber, C.; Wölfel, T. The response of autologous T cells to a human melanoma is dominated by mutated neoantigens. Proc. Natl. Acad. Sci. USA 2005, 102, 16013–16018. [Google Scholar] [CrossRef] [PubMed]
- Baurain, J.F.; Colau, D.; van Baren, N.; Landry, C.; Martelange, V.; Vikkula, M.; Boon, T.; Coulie, P.G. High frequency of autologous anti-melanoma CTL directed against an antigen generated by a point mutation in a new helicase gene. J. Immunol. 2000, 164, 6057–6066. [Google Scholar] [CrossRef] [PubMed]
- Salmon, H.; Idoyaga, J.; Rahman, A.; Leboeuf, M.; Remark, R.; Jordan, S.; Casanova-Acebes, M.; Khudoynazarova, M.; Agudo, J.; Tung, N.; et al. Expansion and Activation of CD103(+) Dendritic Cell Progenitors at the Tumor Site Enhances Tumor Responses to Therapeutic PD-L1 and BRAF Inhibition. Immunity 2016, 44, 924–938. [Google Scholar] [CrossRef] [PubMed]
- Spranger, S.; Dai, D.; Horton, B.; Gajewski, T.F. Tumor-Residing Batf3 Dendritic Cells Are Required for Effector T Cell Trafficking and Adoptive T Cell Therapy. Cancer Cell 2017, 31, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Broz, M.L.; Binnewies, M.; Boldajipour, B.; Nelson, A.E.; Pollack, J.L.; Erle, D.J.; Barczak, A.; Rosenblum, M.D.; Daud, A.; Barber, D.L.; et al. Dissecting the tumor myeloid compartment reveals rare activating antigen-presenting cells critical for T cell immunity. Cancer Cell 2014, 26, 638–652. [Google Scholar] [CrossRef] [PubMed]
- Hampton, R.Y. ER-associated degradation in protein quality control and cellular regulation. Curr. Opin. Cell Biol. 2002, 14, 476–482. [Google Scholar] [CrossRef]
- Tsai, B.; Ye, Y.; Rapoport, T.A. Retro-translocation of proteins from the endoplasmic reticulum into the cytosol. Nat. Rev. Mol. Cell Biol. 2002, 3, 246–255. [Google Scholar] [CrossRef]
- Preston, G.M.; Brodsky, J.L. The evolving role of ubiquitin modification in endoplasmic reticulum-associated degradation. Biochem. J. 2017, 474, 445–469. [Google Scholar] [CrossRef]
- Tamura, Y.; Hirohashi, Y.; Kutomi, G.; Nakanishi, K.; Kamiguchi, K.; Torigoe, T.; Sato, N. Tumor-produced secreted form of binding of immunoglobulin protein elicits antigen-specific tumor immunity. J. Immunol. 2011, 186, 4325–4330. [Google Scholar] [CrossRef]
- Basu, S.; Srivastava, P.K. Heat shock proteins: The fountainhead of innate and adaptive immune responses. Cell Stress Chaperones 2000, 5, 443–451. [Google Scholar] [CrossRef]
- Srivastava, P. Roles of heat-shock proteins in innate and adaptive immunity. Nat. Rev. Immunol. 2002, 3, 185–194. [Google Scholar] [CrossRef]
- Constantino, J.; Gomes, C.; Falcão, A.; Cruz, M.T.; Neves, B.M. Antitumor dendritic cell-based vaccines: Lessons from 20 years of clinical trials and future perspectives. Transl. Res. 2016, 168, 74–95. [Google Scholar] [CrossRef]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Heery, C.R.; O’Sullivan-Coyne, G.; Madan, R.A.; Cordes, L.; Rajan, A.; Rauckhorst, M.; Lamping, E.; Oyelakin, I.; Marté, J.L.; Lepone, L.M.; et al. Avelumab for metastatic or locally advanced previously treated solid tumours (JAVELIN Solid Tumor): A phase 1a, multicohort, dose-escalation trial. Lancet Oncol. 2017, 18, 587–598. [Google Scholar] [CrossRef]
- Syn, N.L.; Teng, M.W.L.; Mok, T.S.K.; Soo, R.A. De-novo and acquired resistance to immune checkpoint targeting. Lancet Oncol. 2017, 18, e731–e741. [Google Scholar] [CrossRef]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, Activity, and Immune Correlates of Anti-PD-1 Antibody in Cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef]
- Garris, C.S.; Arlauckas, S.P.; Kohler, R.H.; Trefny, M.P.; Garren, S.; Piot, C.; Engblom, C.; Pfirschke, C.; Siwicki, M.; Gungabeesoon, J.; et al. Successful Anti-PD-1 Cancer Immunotherapy Requires T Cell-Dendritic Cell Crosstalk Involving the Cytokines IFN-gamma and IL-12. Immunity 2018, 49, 1148–1161. [Google Scholar] [CrossRef]
- Kreutz, M.; Tacken, P.J.; Figdor, C.G. Targeting dendritic cells—Why bother? Blood 2013, 121, 2836–2844. [Google Scholar] [CrossRef]
- Ofuji, K.; Tada, Y.; Yoshikawa, T.; Shimomura, M.; Yoshimura, M.; Saito, K.; Nakamoto, Y.; Nakatsura, T. A peptide antigen derived from EGFR T790M is immunogenic in non-small cell lung cancer. Int. J. Oncol. 2015, 46, 497–504. [Google Scholar]
- Teitz-Tennenbaum, S.; Li, Q.; Rynkiewicz, S.; Ito, F.; Davis, M.A.; Mcginn, C.J.; Chang, A.E. Radiotherapy Potentiates the Therapeutic Efficacy of Intratumoral Dendritic Cell Administration. Cancer Res. 2003, 63, 8466–8475. [Google Scholar]
- Steinman, R.M. Decisions about dendritic cells: Past, present, and future. Annu. Rev. Immunol. 2011, 30, 1–22. [Google Scholar] [CrossRef]
- Schlitzer, A.; McGovern, N.; Ginhoux, F. Dendritic cells and monocyte-derived cells: Two complementary and integrated functional systems. Semin. Cell Dev. Biol. 2015, 41, 9–22. [Google Scholar] [CrossRef]
- Collin, M.; Bigley, V. Human dendritic cell subsets: An update. Immunology 2008, 154, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Merad, M.; Sathe, P.; Helft, J.; Miller, J.; Mortha, A. The dendritic cell lineage: Ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu. Rev. Immunol. 2013, 31, 563–604. [Google Scholar] [CrossRef] [PubMed]
- Böttcher, J.P.; Reis Sousa, C. The role of type 1 conventional dendritic cells in cancer immunity. Trends Cancer 2018, 4, 784–792. [Google Scholar] [CrossRef] [PubMed]
- Demoulin, S.; Herfs, M.; Delvenne, P.; Hubert, P. Tumor microenvironment converts plasmacytoid dendritic cells into immunosuppressive/tolerogenic cells: Insight into the molecular mechanisms. J. Leukoc. Biol. 2013, 93, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Guilliams, M.; Ginhoux, F.; Jakubzick, C.; Naik, S.H.; Onai, N.; Schraml, B.U.; Segura, E.; Tussiwand, R.; Yona, S. Dendritic cells, monocytes and macrophages: A unified nomenclature based on ontogeny. Nat. Rev. Immunol. 2014, 14, 571–578. [Google Scholar] [CrossRef]
- Segura, E.; Albiston, A.L.; Wicks, I.P.; Chai, S.Y.; Villadangos, J.A. Different cross-presentation pathways in steady-state and inflammatory dendritic cells. Proc. Natl. Acad. Sci. USA 2009, 106, 20377–20381. [Google Scholar] [CrossRef]
- Villani, A.C.; Satija, R.; Reynolds, G.; Sarkizova, S.; Shekhar, K.; Fletcher, J.; Griesbeck, M.; Butler, A.; Zheng, S.; Lazo, S.; et al. Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science 2017, 356, eaah4573. [Google Scholar] [CrossRef]
- Kamphorst, A.O.; Guermonprez, P.; Dudziak, D.; Nussenzweig, M.C. Route of antigen uptake differentially impacts presentation by dendritic cells and activated monocytes. J. Immunol. 2010, 185, 3426–3435. [Google Scholar] [CrossRef]
- Eickhoff, S.; Brewitz, A.; Gerner, M.Y.; Klauschen, F.; Komander, K.; Hemmi, H.; Garbi, N.; Kaisho, T.; Germain, R.N.; Kastenmüller, W. Robust anti-viral immunity requires multiple distinct T cell-dendritic cell interactions. Cell 2015, 162, 1322–1337. [Google Scholar] [CrossRef]
- Hor, J.L.; Whitney, P.G.; Zaid, A.; Brooks, A.G.; Heath, W.R.; Mueller, S.N. Spatiotemporally distinct interactions with dendritic cell subsets facilitates CD4+ and CD8+ T cell activation to localized viral infection. Immunity 2015, 43, 554–565. [Google Scholar] [CrossRef]
- Sun, J.C.; Williams, M.A.; Bevan, M.J. CD4+ T cells are required for the maintenance, not programming, of memory CD8+ T cells after acute infection. Nat. Immunol. 2004, 5, 927–933. [Google Scholar] [CrossRef] [PubMed]
- Hochrein, H.; Shortman, K.; Vremec, D.; Scott, B.; Hertzog, P.; O’Keeffe, M. Differential production of IL-12, IFN-alpha, and IFN-gamma by mouse dendritic cell subsets. J. Immunol. 2001, 166, 5448–5455. [Google Scholar] [CrossRef] [PubMed]
- Mashayekhi, M.; Sandau, M.M.; Dunay, I.R.; Frickel, E.M.; Khan, A.; Goldszmid, R.S.; Sher, A.; Ploegh, H.L.; Murphy, T.L.; Sibley, L.D.; et al. CD8alpha(+) dendritic cells are the critical source of interleukin-12 that controls acute infection by Toxoplasma gondii tachyzoites. Immunity 2011, 35, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Dudziak, D.; Kamphorst, A.O.; Heidkamp, G.F.; Buchholz, V.R.; Trumpfheller, C.; Yamazaki, S.; Cheong, C.; Liu, K.; Lee, H.W.; Park, C.G.; et al. Differential antigen processing by dendritic cell subsets in vivo. Science 2007, 315, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Paulete, A.R.; Cueto, F.J.; Martínez-López, M.; Labiano, S.; Morales-Kastresana, A.; Rodríguez-Ruiz, M.E.; Jure-Kunkel, M.; Azpilikueta, A.; Aznar, M.A.; Quetglas, J.I.; et al. Cancer immunotherapy with immunomodulatory anti-CD137 and anti-PD-1 monoclonal antibodies requires BATF3-dependent dendritic cells. Cancer Discov. 2016, 6, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Barry, K.C.; Hsu, J.; Broz, M.L.; Cueto, F.J.; Binnewies, M.; Combes, A.J.; Nelson, A.E.; Loo, K.; Kumar, R.; Rosenblum, M.D.; et al. A natural killer-dendritic cell axis defines checkpoint therapy-responsive tumor microenvironments. Nat. Med. 2018, 24, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Chiang, M.C.; Chiang, M.C.; Tullett, K.M.; Lee, Y.S.; Idris, A.; Ding, Y.; McDonald, K.J.; Kassianos, A.; Leal Rojas, I.M.; Jeet, V.; et al. Differential uptake and cross- presentation of soluble and necrotic cell antigen by human DC subsets. Eur. J. Immunol. 2016, 46, 329–339. [Google Scholar] [CrossRef]
- Sittig, S.P.; Bakdash, G.; Weiden, J.; Sköld, A.E.; Tel, J.; Figdor, C.G.; de Vries, I.J.; Schreibelt, G. A comparative study of the T cell stimulatory and polarizing capacity of human primary blood dendritic cell subsets. Mediat. Inflamm. 2016, 2016, 3605643. [Google Scholar] [CrossRef]
- Segura, E.; Durand, M.; Amigorena, S. Similar antigen cross-presentation capacity and phagocytic functions in all freshly isolated human lymphoid organ-resident dendritic cells. J. Exp. Med. 2013, 210, 1035–1047. [Google Scholar] [CrossRef]
- Yin, X.; Yu, H.; Jin, X.; Li, J.; Guo, H.; Shi, Q.; Yin, Z.; Xu, Y.; Wang, X.; Liu, R.; et al. Human blood CD1c+ dendritic cells encompass CD5high and CD5low subsets that differ significantly in phenotype, gene expression, and functions. J. Immunol. 2017, 198, 1553–1564. [Google Scholar] [CrossRef]
- Mitchell, D.; Chintala, S.; Dey, M. Plasmacytoid dendritic cell in immunity and cancer. J. Neuroimmunol. 2018, 322, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Di Pucchio, T.; Chatterjee, B.; Smed-Sörensen, A.; Clayton, S.; Palazzo, A.; Montes, M.; Xue, Y.; Mellman, I.; Banchereau, J.; Connolly, J.E. Direct proteasome-independent cross-presentation of viral antigen by plasmacytoid dendritic cells on major histocompatibility complex class I. Nat. Immunol. 2008, 9, 551–557. [Google Scholar] [CrossRef] [PubMed]
- Leon, B.; Martinez del Hoyo, G.; Parrillas, V.; Vargas, H.H.; Sanchez-Mateos, P.; Longo, N.; Lopez-Bravo, M.; Ardavin, C. Dendritic cell differentiation potential of mouse monocytes: Monocytes represent immediate precursors of CD8- and CD8+ splenic dendritic cells. Blood 2004, 103, 2668–2676. [Google Scholar] [CrossRef] [PubMed]
- Auffray, C.; Fogg, D.K.; Narni-Mancinelli, E.; Senechal, B.; Trouillet, C.; Saederup, N.; Leemput, J.; Bigot, K.; Campisi, L.; Abitbol, M.; et al. CX3CR1+ CD115+ CD135+ common macrophage/DC precursors and the role of CX3CR1 in their response to inflammation. J. Exp. Med. 2009, 206, 595–606. [Google Scholar] [CrossRef]
- Segura, E.; Touzot, M.; Bohineust, A.; Cappuccio, A.; Chiocchia, G.; Hosmalin, A.; Dalod, M.; Soumelis, V.; Amigorena, S. Human inflammatory dendritic cells induce Th17 cell differentiation. Immunity 2013, 38, 336–348. [Google Scholar] [CrossRef]
- Shortman, K.; Liu, Y.J. Mouse and human dendritic cell subtypes. Nat. Rev. Immunol. 2002, 2, 151–161. [Google Scholar] [CrossRef]
- Cheong, C.; Matos, I.; Choi, J.H.; Dandamudi, D.B.; Shrestha, E.; Longhi, M.P.; Jeffrey, K.L.; Anthony, R.M.; Kluger, C.; Nchinda, G.; et al. Microbial stimulation fully differentiates monocytes to DC-SIGN/CD209(+) dendritic cells for immune T cell areas. Cell 2010, 143, 416–429. [Google Scholar] [CrossRef]
- Markov, O.V.; Mironova, N.L.; Vlasov, V.V.; Zenkova, M.A. Molecular and Cellular Mechanisms of Antitumor Immune Response Activation by Dendritic Cells. Acta Nat. 2016, 8, 17–30. [Google Scholar] [CrossRef]
- Medel, B.; Costoya, C.; Fernandez, D.; Pereda, C.; Lladser, A.; Sauma, D.; Pacheco, R.; Iwawaki, T.; Salazar-Onfray, F.; Osorio, F. IRE1alpha Activation in Bone Marrow-Derived Dendritic Cells Modulates Innate Recognition of Melanoma Cells and Favors CD8(+) T Cell Priming. Front. Immunol. 2018, 9, 3050. [Google Scholar] [CrossRef]
- Plantinga, M.; Guilliams, M.; Vanheerswynghels, M.; Deswarte, K.; Branco-Madeira, F.; Toussaint, W.; Vanhoutte, L.; Neyt, K.; Killeen, N.; Malissen, B.; et al. Conventional and monocyte-derived CD11b(+) dendritic cells initiate and maintain T helper 2 cell-mediated immunity to house dust mite allergen. Immunity 2013, 38, 322–335. [Google Scholar] [CrossRef]
- Briseno, C.G.; Haldar, M.; Kretzer, N.M.; Wu, X.; Theisen, D.J.; Kc, W.; Durai, V.; Grajales-Reyes, G.E.; Iwata, A.; Bagadia, P.; et al. Distinct Transcriptional Programs Control Cross-Priming in Classical and Monocyte-Derived Dendritic Cells. Cell Rep. 2016, 15, 2462–2474. [Google Scholar] [CrossRef] [PubMed]
- Mahadevan, N.R.; Anufreichik, V.; Rodvold, J.J.; Chiu, K.T.; Sepulveda, H.; Zanetti, M. Cell-extrinsic effects of tumor ER stress imprint myeloid dendritic cells and impair CD8(+) T cell priming. PLoS ONE 2012, 7, e51845. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, E.; Cao, Y.; Rodriguez, P.C. Endoplasmic reticulum stress regulates tumor growth and anti-tumor immunity: A promising opportunity for cancer immunotherapy. Cancer Immunol. Immunother. 2017, 66, 1069–1078. [Google Scholar] [CrossRef] [PubMed]
- Valladeau, J.; Dezutter-Dambuyant, C.; Saeland, S. Langerin/CD207 sheds light on formation of birbeck granules and their possible function in Langerhans cells. Immunol. Res. 2008, 28, 93–107. [Google Scholar] [CrossRef]
- Feinberg, H.; Taylor, M.E.; Razi, N.; McBride, R.; Knirel, Y.A.; Graham, S.A.; Drickamer, K.; Weis, W.I. Structural basis for langerin recognition of diverse pathogen and mammalian glycans through a single binding site. J. Mol. Biol. 2011, 405, 1027–1039. [Google Scholar] [CrossRef]
- Furio, L.; Briotet, I.; Journeaux, A.; Billard, H.; Peguet-Navarro, J. Human Langerhans cells are more efficient than CD14(-)CD1c(1) dermal dendritic cells at priming naive CD4(1) T cells. J. Investig. Dermatol. 2010, 130, 1345–1354. [Google Scholar] [CrossRef]
- Furio, L.; Billard, H.; Valladeau, J.; Peguet-Navarro, J.; Berthier-Vergnes, O. Poly(I:C)-Treated human Langerhans cells promote the differentiation of CD41 T cells producing IFN-gamma and IL-10. J. Investig. Dermatol. 2009, 129, 1963–1971. [Google Scholar] [CrossRef]
- Igyártó, B.Z.; Haley, K.; Ortner, D.; Bobr, A.; Gerami-Nejad, M.; Edelson, B.T.; Zurawski, S.M.; Malissen, B.; Zurawski, G.; Berman, J.; et al. Skin-resident murine dendritic cell subsets promote distinct and opposing antigen-specific T helper cell responses. Immunity 2011, 35, 260–272. [Google Scholar] [CrossRef]
- Fehres, C.M.; Duinkerken, S.; Bruijns, S.C.; Kalay, H.; van Vliet, S.J.; Ambrosini, M.; de Gruijl, T.D.; Unger, W.W.; Garcia-Vallejo, J.J.; van Kooyk, Y. Langerin-mediated internalization of a modified peptide routes antigens to early endosomes and enhances cross-presentation by human Langerhans cells. Cell. Mol. Immunol. 2017, 14, 360–370. [Google Scholar] [CrossRef]
- O’Keeffe, M.; Mok, W.H.; Radford, K. Human dendritic cell subsets and function in health and disease. Cell. Mol. Life Sci. 2015, 72, 4309–4325. [Google Scholar] [CrossRef]
- Wculek, S.K.; Cueto, F.J.; Mujal, A.M.; Melero, I.; Krummel, M.F.; Sancho, D. Dendritic cells in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2020, 20, 7–24. [Google Scholar] [CrossRef] [PubMed]
- Constantino, J.; Gomes, C.; Falcão, A.; Neves, B.M.; Cruz, M.T. Dendritic cell-based immunotherapy: A basic review and recent advances. Immunol. Res. 2017, 65, 798–810. [Google Scholar] [CrossRef] [PubMed]
- Schreibelt, G.; Bol, K.F.; Westdorp, H.; Wimmers, F.; Aarntzen, E.H.; Duiveman-de Boer, T.; van de Rakt, M.W.; Scharenborg, N.M.; de Boer, A.J.; Pots, J.M.; et al. Effective clinical responses in metastatic melanoma patients after vaccination with primary myeloid dendritic cells. Clin. Cancer Res. 2016, 22, 2155–2166. [Google Scholar] [CrossRef] [PubMed]
- Hildner, K.; Edelson, B.T.; Purtha, W.E.; Diamond, M.; Matsushita, H.; Kohyama, M.; Calderon, B.; Schraml, B.U.; Unanue, E.R.; Diamond, M.S.; et al. Batf3 Deficiency Reveals a Critical Role for CD8+ Dendritic Cells in Cytotoxic T Cell Immunity. Science 2008, 322, 1097–1100. [Google Scholar] [CrossRef] [PubMed]
- Sachamitr, P.; Leishman, A.J.; Davies, T.J.; Fairchild, P.J. Directed Differentiation of Human Induced Pluripotent Stem Cells into Dendritic Cells Displaying Tolerogenic Properties and Resembling the CD141+ Subset. Front. Immunol. 2018, 8, 1935. [Google Scholar] [CrossRef]
- Apter, D.; Wheeler, C.M.; Paavonen, J.; Naud, P.; Salmerón, J.; Wheeler, C.M.; Chow, S.N.; Apter, D.; Kitchener, H.; Castellsague, X.; et al. Efficacy of human papillomavirus (HPV)-16/18 AS04-adjuvanted vaccine against cervical infection and precancer caused by oncogenic HPV types (PATRICIA): Final analysis of a double-blind, randomised study in young women. Lancet 2009, 374, 301–314. [Google Scholar]
- Kantoff, P.W.; Higano, C.S.; Shore, N.D.; Berger, E.R.; Small, E.J.; Penson, D.F.; Redfern, C.H.; Ferrari, A.C.; Dreicer, R.; Sims, R.B.; et al. Sipuleucel-T Immunotherapy for Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2010, 363, 411–422. [Google Scholar] [CrossRef]
- Andersen, M.H. Immune Regulation by Self-Recognition: Novel Possibilities for Anticancer Immunotherapy. JNCI J. Natl. Cancer Inst. 2015, 107, 154. [Google Scholar] [CrossRef]
- Mant, A.; Chinnery, F.; Elliott, T.; Williams, A.P. The pathway of cross-presentation is influenced by the particle size of phagocytosed antigen. Immunology 2012, 136, 163–175. [Google Scholar] [CrossRef]
- Kovacsovics-Bankowski, M.; Rock, K.L. A phagosome-to-cytosol pathway for exogenous antigens presented on MHC class I molecules. Science 1995, 267, 243–246. [Google Scholar] [CrossRef]
- Ackerman, A.L.; Kyritsis, C.; Tampé, R.; Cresswell, P. Early phagosomes in dendritic cells form a cellular compartment sufficient for cross presentation of exogenous antigens. Proc. Natl. Acad. Sci. USA 2003, 100, 12889–12894. [Google Scholar] [CrossRef] [PubMed]
- Palmowski, M.J.; Gileadi, U.; Salio, M.; Gallimore, A.; Millrain, M.; James, E.; Addey, C.; Scott, D.; Dyson, J.; Simpson, E.; et al. Role of immunoproteasomes in cross-presentation. J. Immunol. 2006, 177, 983–990. [Google Scholar] [CrossRef] [PubMed]
- Embgenbroich, M.; Burgdorf, S. Current Concepts of Antigen Cross-Presentation. Front. Immunol. 2018, 9, 1643. [Google Scholar] [CrossRef] [PubMed]
- Gros, M.; Amigorena, S. Regulation of Antigen Export to the Cytosol During Cross-Presentation. Front. Immunol. 2019, 10, 41. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.Y.; Bruce, A.T.; Pardoll, D.M.; Levitsky, H.I. In vivo cross-priming of MHC class I-restricted antigens requires the TAP transporter. Immunity 1996, 4, 349–355. [Google Scholar] [CrossRef]
- Kloetzel, P.M. Antigen processing by the proteasome. Nat. Rev. Mol. Cell Biol. 2001, 2, 179–187. [Google Scholar] [CrossRef]
- Rodriguez, A.; Regnault, A.; Kleijmeer, M.; Ricciardi-Castagnoli, P.; Amigorena, S. Selective transport of internalized antigens to the cytosol for MHC class I presentation in dendritic cells. Nat. Cell Biol. 1999, 1, 362–368. [Google Scholar] [CrossRef]
- Shen, L.; Sigal, L.J.; Boes, M.; Rock, K.L. Important role of cathepsin S in generating peptides for TAP-independent MHC class I crosspresentation in vivo. Immunity 2004, 21, 155–165. [Google Scholar] [CrossRef]
- Jia, Y.; Omri, A.; Krishnan, L.; McCluskie, M.J. Potential applications of nanoparticles in cancer immunotherapy. Hum. Vaccines Immunother. 2017, 13, 63–741. [Google Scholar] [CrossRef]
- Belz, G.T.; Behrens, G.M.; Smith, C.M.; Miller, J.F.; Jones, C.; Lejon, K.; Fathman, C.G.; Mueller, S.N.; Shortman, K.; Carbone, F.R.; et al. The CD8alpha(+) dendritic cell is responsible for inducing peripheral self-tolerance to tissue-associated antigens. J. Exp. Med. 2002, 196, 1099–1104. [Google Scholar] [CrossRef]
- Sengupta, D.; Graham, M.; Liu, X.; Cresswell, P. Proteasomal degradation within endocytic organelles mediates antigen cross-presentation. EMBO J. 2019, 38, e99266. [Google Scholar] [CrossRef] [PubMed]
- Dingjan, I.; Verboogen, D.R.; Paardekooper, L.M.; Revelo, N.H.; Sittig, S.P.; Visser, L.J.; Mollard, G.F.; Henriet, S.S.; Figdor, C.G.; Ter Beest, M.; et al. Lipid peroxidation causes endosomal antigen release for cross-presentation. Sci. Rep. 2016, 6, 22064. [Google Scholar] [CrossRef] [PubMed]
- Imai, J.; Otani, M.; Sakai, T.; Hatta, S. Purification of the subcellular compartment in which exogenous antigens undergo endoplasmic reticulum-associated degradation from dendritic cells. Heliyon 2016, 2, e00151. [Google Scholar] [CrossRef]
- Zehner, M.; Marschall, A.L.; Bos, E.; Schloetel, J.G.; Kreer, C.; Fehrenschild, D.; Limmer, A.; Ossendorp, F.; Lang, T.; Koster, A.J.; et al. The translocon protein Sec61 mediates antigen transport from endosomes in the cytosol for cross-presentation to CD8(+) T cells. Immunity 2015, 42, 850–863. [Google Scholar] [CrossRef] [PubMed]
- Menager, J.; Ebstein, F.; Oger, R.; Hulin, P.; Nedellec, S.; Duverger, E.; Lehmann, A.; Kloetzel, P.M.; Jotereau, F.; Guilloux, Y. Cross-presentation of synthetic long peptides by human dendritic cells: A process dependent on ERAD component p97/VCP but not sec61 and/or Derlin-1. PLoS ONE 2014, 9, e89897. [Google Scholar] [CrossRef] [PubMed]
- Delamarre, L.; Pack, M.; Chang, H.; Mellman, I.; Trombetta, E.S. Differential lysosomal proteolysis in antigen-presenting cells determines antigen fate. Science 2005, 307, 1630–1634. [Google Scholar] [CrossRef]
- Lennon-Dumenil, A.M.; Bakker, A.H.; Wolf-Bryant, P.; Ploegh, H.L.; Lagaudriere-Gesbert, C. A closer look at proteolysis and MHC-class-II-restricted antigen presentation. Curr. Opin. Immunol. 2002, 14, 15–21. [Google Scholar] [CrossRef]
- Savina, A.; Jancic, C.; Hugues, S.; Guermonprez, P.; Vargas, P.; Moura, I.C.; Lennon-Duménil, A.M.; Seabra, M.C.; Raposo, G.; Amigorena, S. NOX2 controls phagosomal pH to regulate antigen processing during crosspresentation by dendritic cells. Cell 2006, 126, 205–218. [Google Scholar] [CrossRef] [PubMed]
- Savina, A.; Peres, A.; Cebrian, I.; Carmo, N.; Moita, C.; Hacohen, N.; Moita, L.F.; Amigorena, S. The small GTPase Rac2 controls phagosomal alkalinization and antigen crosspresentation selectively in CD8(+) dendritic cells. Immunity 2009, 30, 544–555. [Google Scholar] [CrossRef]
- Claus, V.; Jahraus, A.; Tjelle, T.; Berg, T.; Kirschke, H.; Faulstich, H.; Griffiths, G. Lysosomal enzyme trafficking between phagosomes, endosomes, and lysosomes in J774 macrophages. Enrichment of cathepsin H in early endosomes. J. Biol. Chem. 1998, 273, 9842–9851. [Google Scholar] [CrossRef]
- Trombetta, E.S.; Ebersold, M.; Garrett, W.; Pypaert, M.; Mellman, I. Activation of lysosomal function during dendritic cell maturation. Science 2003, 299, 1400–1403. [Google Scholar] [CrossRef]
- Forgac, M. Vacuolar ATPases: Rotary proton pumps in physiology and pathophysiology. Nat. Rev. Mol. Cell Biol. 2007, 8, 917–929. [Google Scholar] [CrossRef] [PubMed]
- Liberman, R.; Bond, S.; Shainheit, M.G.; Stadecker, M.J.; Forgac, M. Regulated assembly of vacuolar ATPase is increased during cluster disruption-induced maturation of dendritic cells through a phosphatidylinositol 3-kinase/mTOR-dependent pathway. J. Biol. Chem. 2014, 289, 1355–1363. [Google Scholar] [CrossRef] [PubMed]
- Gerriets, V.A.; Kishton, R.J.; Johnson, M.O.; Cohen, S.; Siska, P.J.; Nichols, A.G.; Warmoes, M.O.; de Cubas, A.A.; MacIver, N.J.; Locasale, J.W.; et al. Foxp3 and Toll-like receptor signaling balance Treg cell anabolic metabolism for suppression. Nat. Immunol. 2016, 12, 1459–1466. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, G.T.; Green, E.R.; Mecsas, J. Neutrophils to the ROScue: Mechanisms of NADPH Oxidase Activation and Bacterial Resistance. Front. Cell. Infect. Microbiol. 2017, 7, 373. [Google Scholar] [CrossRef]
- Jancic, C.; Savina, A.; Wasmeier, C.; Tolmachova, T.; El-Benna, J.; Dang, P.M.; Pascolo, S.; Gougerot-Pocdalo, M.A.; Raposo, G.; Seabra, M.C.; et al. Rab27a regulates phagosomal pH and NADPH oxidase recruitment to dendritic cell phagosomes. Nat. Cell Biol. 2007, 9, 367–378. [Google Scholar] [CrossRef]
- Paardekooper, L.M.; Dingjan, I.; Linders, P.T.A.; Staal, A.H.J.; Cristescu, S.M.; Verberk, W.; van den Bogaart, G. Human Monocyte-Derived Dendritic Cells Produce Millimolar Concentrations of ROS in Phagosomes Per Second. Front. Immunol. 2019, 10, 1216. [Google Scholar] [CrossRef]
- Dingjan, I.; Paardekooper, L.M.; Verboogen, D.R.J.; von Mollard, G.F.; Ter Beest, M.; van den Bogaart, G. VAMP8-mediated NOX2 recruitment to endosomes is necessary for antigen release. Eur. J. Cell Biol. 2017, 96, 705–714. [Google Scholar] [CrossRef]
- Matheoud, D.; Moradin, N.; Bellemare-Pelletier, A.; Shio, M.T.; Hong, W.J.; Olivier, M.; Gagnon, E.; Desjardins, M.; Descoteaux, A. Leishmania evades host immunity by inhibiting antigen cross-presentation through direct cleavage of the SNARE VAMP8. Cell Host Microbe 2013, 14, 15–25. [Google Scholar] [CrossRef]
- Baptista, M.A.; Keszei, M.; Oliveira, M.; Sunahara, K.K.; Andersson, J.; Dahlberg, C.I.; Worth, A.J.; Lieden, A.; Kuo, I.C.; Wallin, R.P.; et al. Deletion of Wiskott-Aldrich syndrome protein triggers Rac2 activity and increased cross-presentation by dendritic cells. Nat. Commun. 2016, 7, 12175. [Google Scholar] [CrossRef]
- Ding, Y.; Guo, Z.; Liu, Y.; Li, X.; Zhang, Q.; Xu, X.; Gu, Y.; Zhang, Y.; Zhao, D.; Cao, X. The lectin Siglec-G inhibits dendritic cell cross-presentation by impairing MHC class I-peptide complex formation. Nat. Immunol. 2016, 17, 1167–1175. [Google Scholar] [CrossRef] [PubMed]
- Stegmann, T.; Booy, F.P.; Wilschut, J. Effects of low pH on influenza virus. Activation and inactivation of the membrane fusion capacity of the hemagglutinin. J. Biol. Chem. 1987, 262, 17744–17749. [Google Scholar] [PubMed]
- Roche, S.; Gaudin, Y. Characterization of the equilibrium between the native and fusion-inactive conformation of rabies virus glycoprotein indicates that the fusion complex is made of several trimers. Virology 2002, 297, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Komala Sari, T.; Pritchard, S.M.; Cunha, C.W.; Wudiri, G.A.; Laws, E.I.; Aguilar, H.C.; Taus, N.S.; Nicola, A.V. Contributions of herpes simplex virus 1 envelope proteins to entry by endocytosis. J. Virol. 2013, 87, 13922–13926. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gil-Torregrosa, B.C.; Lennon-Duménil, A.M.; Kessler, B.; Guermonprez, P.; Ploegh, H.L.; Fruci, D.; van Endert, P.; Amigorena, S. Control of cross-presentation during dendritic cell maturation. Eur. J. Immunol. 2004, 2, 398–407. [Google Scholar] [CrossRef]
- Belizaire, R.; Unanue, E.R. Targeting proteins to distinct subcellular compartments reveals unique requirements for MHC class I and II presentation. Proc. Natl. Acad. Sci. USA 2009, 106, 17463–17468. [Google Scholar] [CrossRef]
- Accapezzato, D.; Visco, V.; Francavilla, V.; Molette, C.; Donato, T.; Paroli, M.; Mondelli, M.U.; Doria, M.; Torrisi, M.R.; Barnaba, V. Chloroquine enhances human CD8+ T cell responses against soluble antigens in vivo. J. Exp. Med. 2005, 202, 817–828. [Google Scholar] [CrossRef]
- Chatterjee, B.; Smed-Sörensen, A.; Cohn, L.; Chalouni, C.; Vandlen, R.; Lee, B.C.; Widger, J.; Keler, T.; Delamarre, L.; Mellman, I. Internalization and endosomal degradation of receptor-bound antigens regulate the efficiency of cross presentation by human dendritic cells. Blood 2012, 120, 2011–2020. [Google Scholar] [CrossRef]
- Liu, J.; Liu, X.; Han, Y.; Zhang, J.; Liu, D.; Ma, G.; Li, C.; Liu, L.; Kong, D. Nanovaccine Incorporated with Hydroxychloroquine Enhances Antigen Cross-Presentation and Promotes Antitumor Immune Responses. ACS Appl. Mater. Interfaces 2018, 10, 30983–30993. [Google Scholar] [CrossRef]
- Burgdorf, S.; Kautz, A.; Böhnert, V.; Knolle, P.A.; Kurts, C. Distinct pathways of antigen uptake and intracellular routing in CD4 and CD8 T cell activation. Science 2007, 316, 612–616. [Google Scholar] [CrossRef]
- Tacken, P.J.; de Vries, I.J.; Gijzen, K.; Joosten, B.; Wu, D.; Rother, R.P.; Faas, S.J.; Punt, C.J.; Torensma, R.; Adema, G.J.; et al. Effective induction of naive and recall T-cell responses by targeting antigen to human dendritic cells via a humanized anti-DC-SIGN antibody. Blood 2005, 106, 1278–1285. [Google Scholar] [CrossRef] [PubMed]
- Tacken, P.J.; Ginter, W.; Berod, L.; Cruz, L.J.; Joosten, B.; Sparwasser, T.; Figdor, C.G.; Cambi, A. Targeting DC-SIGN via its neck region leads to prolonged antigen residence in early endosomes, delayed lysosomal degradation, and cross-presentation. Blood 2011, 118, 4111–4119. [Google Scholar] [CrossRef]
- Cohn, L.; Chatterjee, B.; Esselborn, F.; Smed-Sörensen, A.; Nakamura, N.; Chalouni, C.; Lee, B.C.; Vandlen, R.; Keler, T.; Lauer, P.; et al. Antigen delivery to early endosomes eliminates the superiority of human blood BDCA3+ dendritic cells at cross presentation. J. Exp. Med. 2013, 210, 1049–1063. [Google Scholar] [CrossRef] [PubMed]
- Gagnon, E.; Duclos, S.; Rondeau, C.; Chevet, E.; Cameron, P.H.; Steele-Mortimer, O.; Paiement, J.; Bergeron, J.J.; Desjardins, M. Endoplasmic reticulum-mediated phagocytosis is a mechanism of entry into macrophages. Cell 2002, 110, 119–131. [Google Scholar] [CrossRef]
- Houde, M.; Bertholet, S.; Gagnon, E.; Brunet, S.; Goyette, G.; Laplante, A.; Princiotta, M.F.; Thibault, P.; Sacks, D.; Desjardins, M. Phagosomes are competent organelles for antigen cross-presentation. Nature 2003, 425, 402–406. [Google Scholar] [CrossRef] [PubMed]
- Guermonprez, P.; Saveanu, L.; Kleijmeer, M.; Davoust, J.; Van Endert, P.; Amigorena, S. ER-phagosome fusion defines an MHC class I cross-presentation compartment in dendritic cells. Nature 2003, 425, 397–402. [Google Scholar] [CrossRef] [PubMed]
- Eden, E.R. The formation and function of ER-endosome membrane contact sites. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2016, 1861, 874–879. [Google Scholar] [CrossRef]
- Cebrian, I.; Visentin, G.; Blanchard, N.; Jouve, M.; Bobard, A.; Moita, C.; Enninga, J.; Moita, L.F.; Amigorena, S.; Savina, A. Sec22b regulates phagosomal maturation and antigen crosspresentation by dendritic cells. Cell 2011, 147, 1355–1368. [Google Scholar] [CrossRef]
- Nair-Gupta, P.; Baccarini, A.; Tung, N.; Seyffer, F.; Florey, O.; Huang, Y.; Banerjee, M.; Overholtzer, M.; Roche, P.A.; Tampe, R.; et al. TLR signals induce phagosomal MHC-I delivery from the endosomal recycling compartment to allow cross-presentation. Cell 2014, 158, 506–521. [Google Scholar] [CrossRef]
- Alloatti, A.; Rookhuizen, D.C.; Joannas, L.; Carpier, J.M.; Iborra, S.; Magalhaes, J.G.; Yatim, N.; Kozik, P.; Sancho, D.; Albert, M.L.; et al. Critical role for Sec22b-dependent antigen cross-presentation in antitumor immunity. J. Exp. Med. 2017, 214, 2231–2241. [Google Scholar] [CrossRef]
- Wu, S.J.; Niknafs, Y.S.; Kim, S.H.; Oravecz-Wilson, K.; Zajac, C.; Toubai, T.; Sun, Y.; Prasad, J.; Peltier, D.; Fujiwara, H.; et al. A critical analysis of the role of SNARE protein SEC22B in antigen cross-presentation. Cell Rep. 2017, 19, 2645–2656. [Google Scholar] [CrossRef] [PubMed]
- Montealegre, S.; van Endert, P. MHC class I cross-presentation: Stage lights on Sec22b. Trends Immunol. 2017, 38, 618–662. [Google Scholar] [CrossRef] [PubMed]
- Giodini, A.; Cresswell, P. Hsp90-mediated cytosolic refolding of exogenous proteins internalized by dendritic cells. EMBO J. 2008, 27, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Imai, J.; Hasegawa, H.; Maruya, M.; Koyasu, S.; Yahara, I. Exogenous antigens are processed through the endoplasmic reticulum-associated degradation (ERAD) in cross-presentation by dendritic cells. Int. Immunol. 2005, 17, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Cresswell, P. Defective cross-presentation of viral antigens in GILT-free mice. Science 2010, 328, 1394–1398. [Google Scholar] [CrossRef]
- Hurst, K.E.; Lawrence, K.A.; Reyes Angeles, L.; Ye, Z.; Zhang, J.; Townsend, D.M.; Dolloff, N.; Thaxton, J.E. Endoplasmic Reticulum protein disulfide isomerase shapes T cell efficacy for adoptive cellular therapy of tumors. Cells 2019, 8, 1514. [Google Scholar] [CrossRef]
- Morito, D.; Nagata, K. ER Stress Proteins in Autoimmune and Inflammatory Diseases. Front. Immunol. 2012, 3, 48. [Google Scholar] [CrossRef]
- Zehner, M.; Chasan, A.I.; Schuette, V.; Embgenbroich, M.; Quast, T.; Kolanus, W.; Burgdorf, S. Mannose receptor polyubiquitination regulates endosomal recruitment of p97 and cytosolic antigen translocation for cross-presentation. Proc. Natl. Acad. Sci. USA 2011, 108, 9933–9938. [Google Scholar] [CrossRef]
- Sancho, D.; Mourão-Sá, D.; Joffre, O.P.; Schulz, O.; Rogers, N.C.; Pennington, D.J.; Carlyle, J.R.; Reis Sousa, C. Tumor therapy in mice via antigen targeting to a novel, DC-restricted C-type lectin. J. Clin. Investig. 2008, 118, 2098–2110. [Google Scholar] [CrossRef]
- Caminschi, I.; Proietto, A.I.; Ahmet, F.; Kitsoulis, S.; Shin The, J.; Lo, J.C.; Rizzitelli, A.; Wu, L.; Vremec, D.; van Dommelen, S.L.; et al. The dendritic cell subtype- restricted C-type lectin Clec9A is a target for vaccine enhancement. Blood 2008, 112, 3264–3273. [Google Scholar] [CrossRef]
- Martinez-Pomares, L. The mannose receptor. J. Leukoc. Biol. 2012, 92, 1177–1186. [Google Scholar] [CrossRef] [PubMed]
- Hosokawa, N.; Kamiya, Y.; Kamiya, D.; Kato, K.; Nagata, K. Human OS-9, a lectin required for glycoprotein endoplasmic reticulum-associated degradation, recognizes mannose-trimmed N-glycans. J. Biol. Chem. 2009, 284, 17061–17068. [Google Scholar] [CrossRef] [PubMed]
- Kreer, C.; Kuepper, J.M.; Zehner, M.; Quast, T.; Kolanus, W.; Schumak, B.; Burgdorf, S. N-glycosylation converts non-glycoproteins into mannose receptor ligands and reveals antigen-specific T cell responses in vivo. Oncotarget 2017, 8, 6857–6872. [Google Scholar] [CrossRef] [PubMed]
- Friedlander, R.; Jarosch, E.; Urban, J.; Volkwein, C.; Sommer, T. A regulatory link between ER-associated protein degradation and the unfolded-protein response. Nat. Cell Biol. 2000, 2, 379–384. [Google Scholar] [CrossRef] [PubMed]
- Greenfield, J.J.; High, S. The Sec61 complex is located in both the ER and the ER-Golgi intermediate compartment. J. Cell Sci. 1999, 112, 1477–1486. [Google Scholar] [PubMed]
- Kalies, K.U.; Rapoport, T.A.; Hartmann, E. The beta subunit of the Sec61 complex facilitates cotranslational protein transport and interacts with the signal peptidase during translocation. J. Cell Biol. 1998, 141, 887–894. [Google Scholar] [CrossRef]
- Koopmann, J.O.; Albring, J.; Hüter, E.; Bulbuc, N.; Spee, P.; Neefjes, J.; Hämmerling, G.J.; Momburg, F. Export of antigenic peptides from the endoplasmic reticulum intersects with retro-grade protein translocation through the Sec61p channel. Immunity 2000, 13, 117–127. [Google Scholar] [CrossRef]
- Wirth, A.; Jung, M.; Bies, C.; Frien, M.; Tyedmers, J.; Zimmermann, R.; Wagner, R. The Sec61p complex is a dynamic precursor activated channel. Mol. Cell 2003, 12, 261–268. [Google Scholar] [CrossRef]
- Schäuble, N.; Cavalié, A.; Zimmermann, R.; Jung, M. Interaction of Pseudomonas aeruginosa Exotoxin A with the human Sec61 complex suppresses passive calcium efflux from the endoplasmic reticulum. Channels 2014, 8, 76–83. [Google Scholar] [CrossRef]
- Ackerman, A.L.; Giodini, A.; Cresswell, P. A role for the endoplasmic reticulum protein retrotranslocation machinery during crosspresentation by dendritic cells. Immunity 2006, 25, 607–617. [Google Scholar] [CrossRef]
- Romisch, K. A Case for Sec61 Channel Involvement in ERAD. Trends Biochem. Sci. 2017, 42, 171–179. [Google Scholar] [CrossRef] [PubMed]
- You, X.; Xu, D.D.; Zhang, D.; Chen, J.; Gao, F.G. PYR-41 and Thalidomide Impair Dendritic Cell Cross-Presentation by Inhibiting Myddosome Formation and Attenuating the Endosomal Recruitments of p97 and Sec61 via NF-kappaB Inactivation. J. Immunol. Res. 2018, 2018, 5070573. [Google Scholar] [CrossRef] [PubMed]
- Burgdorf, S.; Leister, P.; Scheidtmann, K.H. TSG101 interacts with apoptosis-antagonizing transcription factor and enhances androgen receptor-mediated transcription by promoting its monoubiquitination. J. Biol. Chem. 2004, 279, 17524–17534. [Google Scholar] [CrossRef] [PubMed]
- Imai, T.; Kato, Y.; Kajiwara, C.; Mizukami, S.; Ishige, I.; Ichiyanagi, T.; Hikida, M.; Wang, J.Y.; Udono, H. Heat shock protein 90 (HSP90) contributes to cytosolic translocation of extracellular antigen for cross-presentation by dendritic cells. Proc. Natl. Acad. Sci. USA 2011, 108, 16363–16368. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Li, L.; Ye, Y. Regulation of retrotranslocation by p97-associated deubiquitinating enzyme ataxin-3. J. Cell Biol. 2006, 174, 963–971. [Google Scholar] [CrossRef] [PubMed]
- Reits, E.; Neijssen, J.; Herberts, C.; Benckhuijsen, W.; Janssen, L.; Drijfhout, J.W.; Neefjes, J. A major role for TPPII in trimming proteasomal degradation products for MHC class I antigen presentation. Immunity 2004, 20, 495–506. [Google Scholar] [CrossRef]
- Burgdorf, S.; Schölz, C.; Kautz, A.; Tampé, R.; Kurts, C. Spatial and mechanistic separation of cross-presentation and endogenous antigen presentation. Nat. Immunol. 2008, 9, 558–566. [Google Scholar] [CrossRef]
- Saveanu, L.; Carroll, O.; Hassainya, Y.; van Endert, P. Complexity, contradictions, and conundrums: Studying post-proteasomal proteolysis in HLA class I antigen presentation. Immunol. Rev. 2005, 207, 42–59. [Google Scholar] [CrossRef]
- Saveanu, L.; Carroll, O.; Weimershaus, M.; Guermonprez, P.; Firat, E.; Lindo, V.; Greer, F.; Davoust, J.; Kratzer, R.; Keller, S.R.; et al. IRAP identifies an endosomal compartment required for MHC class I cross-presentation. Science 2009, 325, 213–217. [Google Scholar] [CrossRef]
- Subtil, A.; Lampson, M.A.; Keller, S.R.; McGraw, T.E. Characterization of the insulin-regulated endocytic recycling mechanism in 3T3-L1 adipocytes using a novel reporter molecule. J. Biol. Chem. 2000, 275, 4787–4795. [Google Scholar] [CrossRef]
- Firat, E.; Saveanu, L.; Aichele, P.; Staeheli, P.; Huai, J.; Gaedicke, S.; Nil, A.; Besin, G.; Kanzler, B.; van Endert, P.; et al. The role of endoplasmic reticulum-associated aminopeptidase 1 in immunity to infection and in cross-presentation. J. Immunol. 2007, 178, 2241–2248. [Google Scholar] [CrossRef] [PubMed]
- Zagorac, G.B.; Mahmutefendić, H.; Tomaš, M.I.; Kučić, N.; Le Bouteiller, P.; Lučin, P. Early endosomal rerouting of major histocompatibility class I conformers. J. Cell Physiol. 2012, 227, 2953–2964. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Zhang, Y.; Vigneron, N.; Stroobant, V.; Thielemans, K.; van der Bruggen, P.; Van den Eynde, B.J. Long-peptide cross-presentation by human dendritic cells occurs in vacuoles by peptide exchange on nascent MHC class I molecules. J. Immunol. 2016, 196, 1711–1720. [Google Scholar] [CrossRef] [PubMed]
- Osorio, F.; Tavernier, S.J.; Hoffmann, E.; Saeys, Y.; Martens, L.; Vetters, J.; Delrue, I.; De Rycke, R.; Parthoens, E.; Pouliot, P.; et al. The unfolded-protein-response sensor IRE-1alpha regulates the function of CD8alpha+ dendritic cells. Nat. Immunol. 2014, 15, 248–257. [Google Scholar] [CrossRef]
- Cubillos-Ruiz, J.R.; Silberman, P.C.; Rutkowski, M.R.; Chopra, S.; Perales-Puchalt, A.; Song, M.; Zhang, S.; Bettigole, S.E.; Gupta, D.; Holcomb, K.; et al. ER Stress Sensor XBP1 Controls Anti-tumor Immunity by Disrupting Dendritic Cell Homeostasis. Cell 2015, 161, 1527–1538. [Google Scholar] [CrossRef]
- Imai, Y.; Kuba, K.; Neely, G.G.; Yaghubian-Malhami, R.; Perkmann, T.; van Loo, G.; Ermolaeva, M.; Veldhuizen, R.; Leung, Y.H.; Wang, H.; et al. Identification of oxidative stress and Toll-like receptor 4 signaling as a key pathway of acute lung injury. Cell 2008, 133, 235–249. [Google Scholar] [CrossRef]
- Mutoh, A.; Ueda, S. Peroxidized unsaturated fatty acids stimulate Toll-like receptor 4 signaling in endothelial cells. Life Sci. 2013, 92, 984–992. [Google Scholar] [CrossRef]
- Retamal-Diaz, A.; Weiss, K.A.; Tognarelli, E.I.; Freire, M.; Bueno, S.M.; Herold, B.C.; Jacobs, W.R., Jr.; Gonzalez, P.A. US6 Gene Deletion in Herpes Simplex Virus Type 2 Enhances Dendritic Cell Function and T Cell Activation. Front. Immunol. 2017, 8, 1523. [Google Scholar] [CrossRef]
- Rodvold, J.J.; Mahadevan, N.R.; Zanetti, M. Immune modulation by ER stress and inflammation in the tumor microenvironment. Cancer Lett. 2015, 380, 227–236. [Google Scholar] [CrossRef]
- Reading, J.L.; Gálvez-Cancino, F.; Swanton, C.; Lladser, A.; Peggs, K.S.; Quezada, S.A. The function and dysfunction of memory CD8(+) T cells in tumor immunity. Immunol. Rev. 2018, 283, 194–212. [Google Scholar] [CrossRef]
- Gebhardt, T.; Palendira, U.; Tscharke, D.C.; Bedoui, S. Tissue-resident memory T cells in tissue homeostasis, persistent infection, and cancer surveillance. Immunol. Rev. 2018, 283, 54–76. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R. Toll-like receptors and innate immunity. Nat. Rev. Immunol. 2001, 1, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Reverendo, M.; Mendes, A.; Arguello, R.J.; Gatti, E.; Pierre, P. At the crossway of ER-stress and proinflammatory responses. FEBS J. 2019, 286, 297–310. [Google Scholar] [CrossRef] [PubMed]
- Pizzurro, G.A.; Tapia, I.J.; Sganga, L.; Podhajcer, O.L.; Mordoh, J.; Barrio, M.M. Cytokine-enhanced maturation and migration to the lymph nodes of a human dying melanoma cell-loaded dendritic cell vaccine. Cancer Immunol. Immunother. 2015, 64, 1393–1406. [Google Scholar] [CrossRef] [PubMed]
- Alloatti, A.; Kotsias, F.; Magalhaes, J.G.; Amigorena, S. Dendritic cell maturation and cross-presentation: Timing matters! Immunol. Rev. 2016, 272, 97–108. [Google Scholar] [CrossRef]
- Chi, H.; Li, C.; Zhao, F.S.; Zhang, L.; Ng, T.B.; Jin, G.; Sha, O. Anti-tumor Activity of Toll-Like Receptor 7 Agonists. Front. Pharmacol. 2017, 8, 304. [Google Scholar] [CrossRef]
- Vacchelli, E.; Eggermont, A.; Sautès-Fridman, C.; Galon, J.; Zitvogel, L.; Kroemer, G.; Galluzzi, L. Trial Watch: Toll-like receptor agonists for cancer therapy. Oncoimmunology 2013, 8, e25238. [Google Scholar] [CrossRef]
- Bourque, J.; Hawiger, D. Immunomodulatory Bonds of the Partnership between Dendritic Cells and T Cells. Crit. Rev. Immunol. 2018, 38, 379–401. [Google Scholar] [CrossRef]
- Weimershaus, M.; Mauvais, F.X.; Saveanu, L.; Adiko, C.; Babdor, J.; Abramova, A.; Montealegre, S.; Lawand, M.; Evnouchidou, I.; Huber, K.J.; et al. Innate Immune Signals Induce Anterograde Endosome Transport Promoting MHC Class I Cross-Presentation. Cell Rep. 2018, 24, 3568–3581. [Google Scholar] [CrossRef]
- Pauwels, A.M.; Hartlova, A.; Peltier, J.; Driege, Y.; Baudelet, G.; Brodin, P.; Trost, M.; Beyaert, R.; Hoffmann, E. Spatiotemporal Changes of the Phagosomal Proteome in Dendritic Cells in Response to LPS Stimulation. Mol. Cell. Proteom. 2019, 18, 909–922. [Google Scholar] [CrossRef] [PubMed]
- Blander, J.M.; Medzhitov, R. Toll-dependent selection of microbial antigens for presentation by dendritic cells. Nature 2006, 440, 808–812. [Google Scholar] [CrossRef] [PubMed]
- Corridoni, D.; Shiraishi, S.; Chapman, T.; Steevels, T.; Muraro, D.; Thezenas, M.L.; Prota, G.; Chen, J.L.; Gileadi, U.; Ternette, N.; et al. NOD2 and TLR2 Signal via TBK1 and PI31 to Direct Cross-Presentation and CD8 T Cell Responses. Front. Immunol. 2019, 10, 958. [Google Scholar] [CrossRef] [PubMed]
- Jelinek, I.; Leonard, J.N.; Price, G.E.; Brown, K.N.; Meyer-Manlapat, A.; Goldsmith, P.K.; Wang, Y.; Venzon, D.; Epstein, S.L.; Segal, D.M. TLR3-specific double-stranded RNA oligonucleotide adjuvants induce dendritic cell cross-presentation, CTL responses, and antiviral protection. J. Immunol. 2011, 186, 2422–2429. [Google Scholar] [CrossRef] [PubMed]
- Asano, J.; Tada, H.; Onai, N.; Sato, T.; Horie, Y.; Fujimoto, Y.; Fukase, K.; Suzuki, A.; Mak, T.W.; Ohteki, T. Nucleotide oligomerization binding domain-like receptor signaling enhances dendritic cell-mediated cross-priming in vivo. J. Immunol. 2010, 184, 736–745. [Google Scholar] [CrossRef] [PubMed]
- Visvikis, O.; Ihuegbu, N.; Labed, S.A.; Luhachack, L.G.; Alves, A.F.; Wollenberg, A.C.; Stuart, L.M.; Stormo, G.D.; Irazoqui, J.E. Innate Host Defense Requires TFEB-Mediated Transcription of Cytoprotective and Antimicrobial Genes. Immunity 2014, 40, 896–909. [Google Scholar] [CrossRef]
- Ma, J.; Wei, K.; Zhang, H.; Tang, K.; Li, F.; Zhang, T.; Liu, J.; Xu, P.; Yu, Y.; Sun, W.; et al. Mechanisms by which dendritic cells present tumor microparticle antigens to CD8+ T cells. Cancer Immunol. Res. 2018, 9, 1057–1068. [Google Scholar] [CrossRef]
- Faure-André, G.; Vargas, P.; Yuseff, M.; Heuzé, M.; Diaz, J.; Lankar, D.; Steri, V.; Manry, J.; Hugues, S.; Vascotto, F.; et al. Regulation of dendritic cell migration by CD74, the MHC class II-associated invariant chain. Science 2008, 322, 1705–1710. [Google Scholar] [CrossRef]
- Vargas, P.; Maiuri, P.; Bretou, M.; Sáez, P.J.; Pierobon, P.; Maurin, M.; Chabaud, M.; Lankar, D.; Obino, D.; Terriac, E.; et al. Innate control of actin nucleation determines two distinct migration behaviours in dendritic cells. Nat. Cell Biol. 2015, 18, 43–53. [Google Scholar] [CrossRef]
- Dalod, M.; Chelbi, R.; Malissen, B.; Lawrence, T. Dendritic cell maturation: Functional specialization through signaling specificity and transcriptional programming. EMBO J. 2014, 33, 1104–1116. [Google Scholar] [CrossRef]
- Galluzzi, L.; Senovilla, L.; Vacchelli, E.; Eggermont, A.; Fridman, W.H.; Galon, J.; Sautès-Fridman, C.; Tartour, E.; Zitvogel, L.; Kroemer, G. Trial watch: Dendritic cell-based interventions for cancer therapy. Oncoimmunology 2012, 1, 1111–1134. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| DC subsets | Mouse | Human | ||
|---|---|---|---|---|
| Surface Marker | Cytokine Profile | Surface Marker | Cytokine Profile | |
| cDC1 | CD11c, MHC II, CD8α, CD103, CD24, XCR1, CLEC9A, CD205 | IL-12 (high), IFN-III, IFN- λ | CD11c (low), HLA-DR, CD141, CD205, CLEC9A, XCR1, Nec12 | IL-12 (low), IFN-III, IFN-λ |
| cDC2 | CD11c, MHC II, CD11b, CD172a (Sirpα), ESAM | IL-6, TNF | CD11c, HLA-DR, CD1c, CD11b, CD172a (Sirpα), CD1a, CD14, CD5 | IL-12, IL-1β, TNF, IL-6, IL-10, IL-23, IFN-γ |
| pDC | CD11c, MHC II, B220, CD317, SIGLEC-H, CD172a, CD209, CCR2, CCR9, CXCR3 | Type I and III IFN | CD11c (low), HLA-DR (low), CD123, CD303, CD304, CCR2, CXCR3 | Type I and III IFN |
| moDC | CD11c, MHC II, CD11b, CD172, F4/80, Ly6C, CD64 (FcεRI) | IL-12, IL-23, IL-6, IL-10 | CD11c, HLA-DR, CD1c, CD1a, CD11b, CD172a, CD64 (FcεRI), CD14, CD5, CD206 | IL-12, IL-23, IL-6, IL-10 |
| LC | CD11c, CD1d, CD207 (langerin), E-cadherin, MHC II, CD205 | CD11c, CD1a, CD1b, CD1c, CD207 (langerin), E-cadherin, HLA-DR, CD205 | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Imai, J.; Ohashi, S.; Sakai, T. Endoplasmic Reticulum-Associated Degradation-Dependent Processing in Cross-Presentation and Its Potential for Dendritic Cell Vaccinations: A Review. Pharmaceutics 2020, 12, 153. https://doi.org/10.3390/pharmaceutics12020153
Imai J, Ohashi S, Sakai T. Endoplasmic Reticulum-Associated Degradation-Dependent Processing in Cross-Presentation and Its Potential for Dendritic Cell Vaccinations: A Review. Pharmaceutics. 2020; 12(2):153. https://doi.org/10.3390/pharmaceutics12020153
Chicago/Turabian StyleImai, Jun, Sayaka Ohashi, and Takahiro Sakai. 2020. "Endoplasmic Reticulum-Associated Degradation-Dependent Processing in Cross-Presentation and Its Potential for Dendritic Cell Vaccinations: A Review" Pharmaceutics 12, no. 2: 153. https://doi.org/10.3390/pharmaceutics12020153
APA StyleImai, J., Ohashi, S., & Sakai, T. (2020). Endoplasmic Reticulum-Associated Degradation-Dependent Processing in Cross-Presentation and Its Potential for Dendritic Cell Vaccinations: A Review. Pharmaceutics, 12(2), 153. https://doi.org/10.3390/pharmaceutics12020153