Inhaled Antibiotics for Mycobacterial Lung Disease

Abstract
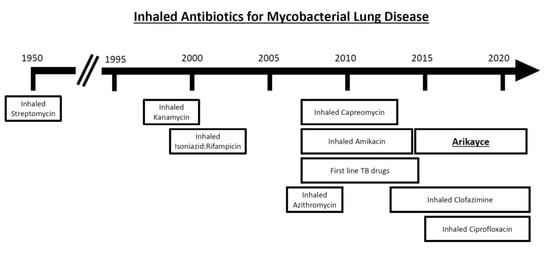
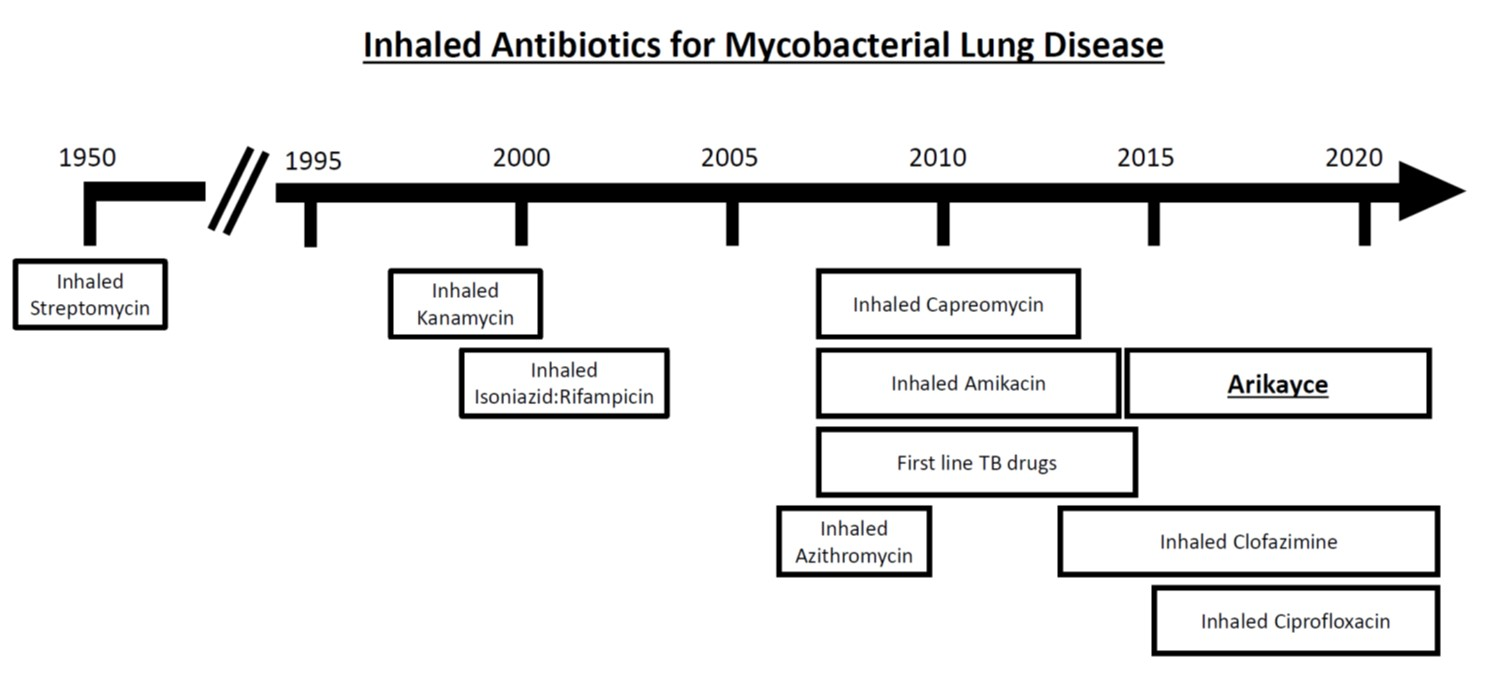{kind=link}
1. Introduction
1.1. Current Treatment Strategies and Limitations
1.2. The Potential for Inhaled Therapies
2. Drug Delivery Strategies for Inhaled Antibiotics
3. Past and Present Inhaled Therapies for Mycobacterial Lung Disease
3.1. Tuberculosis
3.2. Nontuberculous Mycobacteria
4. Ongoing Drug Development Programs
4.1. First Line TB Antibiotics
4.2. Inhaled Clofazimine
4.3. Inhaled Azithromycin
4.4. Inhaled Ciprofloxacin
4.5. Recombinant Human GM-CSF
4.6. Nitric Oxide Gas
5. Regulatory Considerations for Inhaled Drug Programs
6. Cross-Fertilization between TB and NTM Development Programs
7. Barriers to Entry of Inhaled Therapeutics
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jarlier, V.; Nikaido, H. Mycobacterial cell wall: Structure and role in natural resistance to antibiotics. FEMS Microbiol. Lett. 1994, 123, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Glickman, M.S.; Jacobs, W.R., Jr. Microbial Pathogenesis of Mycobacterium tuberculosis: Dawn of a Discipline. Cell 2001, 104, 477–485. [Google Scholar] [CrossRef]
- Griffith, D.E.; Aksamit, T.; Brown-Elliott, B.A.; Catanzaro, A.; Daley, C.; Gordin, F.; Holland, S.M.; Horsburgh, R.; Huitt, G.; Iademarco, M.F.; et al. An official ATS/IDSA statement: Diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am. J. Respir. Crit. Care Med. 2007, 175, 367–416. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M. Leprosy—An overview of clinical features, diagnosis, and treatment. JDDG J. Dtsch. Dermatol. Ges. 2017, 15, 801–827. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. WHO Global Tuberculosis Report 2017; World Health Organization: Geneva, Switzerland, 2017; ISBN 9789241565516. [Google Scholar]
- Prince, D.S.; Peterson, D.D.; Steiner, R.M.; Gottlieb, J.E.; Scott, R.; Israel, H.L.; Figueroa, W.G.; Fish, J.E. Infection with Mycobacterium avium Complex in Patients without Predisposing Conditions. N. Engl. J. Med. 1989, 321, 863–868. [Google Scholar] [CrossRef] [PubMed]
- Prevots, D.R.; Shaw, P.A.; Strickland, D.; Jackson, L.A.; Raebel, M.A.; Blosky, M.A.; De Oca, R.M.; Shea, Y.R.; Seitz, A.E.; Holland, S.M.; et al. Nontuberculous mycobacterial lung disease prevalence at four integrated health care delivery systems. Am. J. Respir. Crit. Care Med. 2010, 182, 970–976. [Google Scholar] [CrossRef] [PubMed]
- Honda, J.R.; Knight, V.; Chan, E.D. Pathogenesis and risk factors for nontuberculous mycobacterial lung disease. Clin. Chest Med. 2015, 36, 1–11. [Google Scholar] [CrossRef]
- Prevots, D.R.; Marras, T.K. Epidemiology of human pulmonary infection with non-tuberculous mycobacteria: A review. Clin. Chest Med. 2015, 36, 13–34. [Google Scholar] [CrossRef] [PubMed]
- Marras, T.K.; Daley, C.L. Epidemiology of human pulmonary infection with nontuberculous mycobacteria. Clin. Chest Med. 2002, 23, 553–567. [Google Scholar] [CrossRef]
- Strollo, S.E.; Adjemian, J.; Adjemian, M.K.; Prevots, D.R. The burden of pulmonary nontuberculous mycobacterial disease in the United States. Ann. Am. Thorac. Soc. 2015, 12, 1458–1464. [Google Scholar] [CrossRef]
- Ringshausen, F.C.; Wagner, D.; de Roux, A.; Diel, R.; Hohmann, D.; Hickstein, L.; Welte, T.; Rademacher, J. Prevalence of nontuberculous mycobacterial pulmonary disease, Germany, 2009–2014. Emerg. Infect. Dis. 2016, 22, 1102–1105. [Google Scholar] [CrossRef] [PubMed]
- Namkoong, H.; Kurashima, A.; Morimoto, K.; Hoshino, Y.; Hasegawa, N.; Ato, M.; Mitarai, S. Epidemiology of Pulmonary Nontuberculous Mycobacterial Disease, Japan. Emerg. Infect. Dis. 2016, 22, 1116–1117. [Google Scholar] [CrossRef] [PubMed]
- Adjemian, J.; Frankland, T.B.; Daida, Y.G.; Honda, J.R.; Olivier, K.N.; Zelazny, A.; Honda, S.; Prevots, D.R. Epidemiology of nontuberculous mycobacterial lung disease and Tuberculosis, Hawaii, USA. Emerg. Infect. Dis. 2017, 23. [Google Scholar] [CrossRef] [PubMed]
- De Mello, K.G.C.; Queiroz Mello, F.C.; Borga, L.; Rolla, V.; Duarte, R.S.; Sampaio, E.P.; Holland, S.M.; Prevots, D.R.; Dalcolmo, M.P. Clinical and therapeutic features of pulmonary nontuberculous mycobacterial disease, Brazil, 1993–2011. Emerg. Infect. Dis. 2013, 19, 393–399. [Google Scholar] [PubMed]
- Raju, R.M.; Raju, S.M.; Zhao, Y.; Rubin, E.J. Leveraging Advances in Tuberculosis Diagnosis and Treatment to Address Nontuberculous Mycobacterial Disease. Emerg. Infect. Dis. 2016, 22, 365–369. [Google Scholar] [CrossRef] [PubMed]
- Appelberg, R. Pathogenesis of Mycobacterium avium infection: Typical responses to an atypical Mycobacterium? Immunol. Res. 2006, 35, 179–190. [Google Scholar] [CrossRef]
- Pai, M.; Behr, M.A.; Dowdy, D.; Dheda, K.; Divangahi, M.; Boehme, C.C.; Ginsberg, A.; Swaminathan, S.; Spigelman, M.; Getahun, H.; et al. Tuberculosis. Nat. Rev. Dis. Primers 2016, 2, 16076. [Google Scholar] [CrossRef]
- Tortoli, E. Clinical manifestations of nontuberculous mycobacteria infections. Clin. Microbiol. Infect. 2009, 15, 906–910. [Google Scholar] [CrossRef]
- Pagan, A.J.; Ramakrishnan, L. The formation and function of granulomas. Annu. Rev. Immunol. 2018, 36, 639–665. [Google Scholar] [CrossRef]
- Orme, I.M.; Robinson, R.T.; Cooper, A.M. The balance between protective and pathogenic immune responses in the TB-infected lung. Nat. Immunol. 2015, 16, 57–63. [Google Scholar] [CrossRef]
- Gadkowski, L.B.; Stout, J.E. Cavitary pulmonary disease. Clin. Microbiol. Rev. 2008, 21, 305–333. [Google Scholar] [CrossRef] [PubMed]
- Haworth, C.S.; Banks, J.; Capstick, T.; Fisher, A.J.; Gorsuch, T.; Laurenson, I.F.; Leitch, A.; Loebinger, M.R.; Milburn, H.J.; Nightingale, M.; et al. British Thoracic Society guidelines for the management of non-tuberculous mycobacterial pulmonary disease (NTM-PD). Thorax 2017, 72, ii1–ii64. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Companion Handbook to the WHO Guidelines for the Programmatic Management of Drug-Resistant Tuberculosis; World Health Organization: Geneva, Switzerland, 2014; ISBN 9789241548441. [Google Scholar]
- Dheda, K.; Barry, C.E.; Maartens, G. Tuberculosis. Lancet 2016, 387, 1211–1226. [Google Scholar] [CrossRef]
- Griffith, D. Treatment of Mycobacterium avium Complex (MAC). Semin. Respir. Crit. Care Med. 2018, 39, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Strnad, L.; Winthrop, K.L. Treatment of Mycobacterium abscessus complex. Semin. Respir. Crit. Care Med. 2018, 39, 362–376. [Google Scholar] [PubMed]
- Basille, D.; Jounieaux, V. Treatment of Other Nontuberculous Mycobacteria. Semin. Respir. Crit. Care Med. 2018, 39, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Koh, W.J.; Moon, S.M.; Kim, S.Y.; Woo, M.A.; Kim, S.; Jhun, B.W.; Park, H.Y.; Jeon, K.; Huh, H.J.; Ki, C.S.; et al. Outcomes of Mycobacterium avium complex lung disease based on clinical phenotype. Eur. Respir. J. 2017, 50, 1602503. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.; Kim, S.Y.; Kim, D.H.; Huh, H.J.; Ki, C.S.; Lee, N.Y.; Lee, S.H.; Shin, S.; Shin, S.J.; Daley, C.L.; et al. Clinical characteristics and treatment outcomes of patients with acquired macrolide-resistant Mycobacterium abscessus lung disease. Antimicrob. Agents Chemother. 2017, 61, e01146-17. [Google Scholar] [CrossRef]
- Koh, W.J.; Jeong, B.H.; Kim, S.Y.; Jeon, K.; Park, K.U.; Jhun, B.W.; Lee, H.; Park, H.Y.; Kim, D.H.; Huh, H.J.; et al. Mycobacterial characteristics and treatment outcomes in Mycobacterium abscessus lung disease. Clin. Infect. Dis. 2017, 64, 309–316. [Google Scholar] [CrossRef]
- Park, J.; Cho, J.; Lee, C.-H.; Han, S.K.; Yim, J.-J. Progression and Treatment Outcomes of Lung Disease Caused by Mycobacterium abscessus and Mycobacterium massiliense. Clin. Infect. Dis. 2017, 64, 301–308. [Google Scholar] [CrossRef]
- Koh, W.J.; Jeon, K.; Lee, N.Y.; Kim, B.J.; Kook, Y.H.; Lee, S.H.; Park, Y.K.; Kim, C.K.; Shin, S.J.; Huitt, G.A.; et al. Clinical significance of differentiation of Mycobacterium massiliense from Mycobacterium abscessus. Am. J. Respir. Crit. Care Med. 2012, 183, 405–410. [Google Scholar] [CrossRef] [PubMed]
- Kotilainen, H.; Valtonen, V.; Tukiainen, P.; Poussa, T.; Eskola, J.; Järvinen, A. Clinical findings in relation to mortality in non-tuberculous mycobacterial infections: Patients with Mycobacterium avium complex have better survival than patients with other mycobacteria. Eur. J. Clin. Microbiol. Infect. Dis. 2015, 34, 1909–1918. [Google Scholar] [CrossRef] [PubMed]
- Ballarino, G.J.; Olivier, K.N.; Claypool, R.J.; Holland, S.M.; Prevots, D.R. Pulmonary nontuberculous mycobacterial infections: Antibiotic treatment and associated costs. Respir. Med. 2009, 103, 1448–1455. [Google Scholar] [CrossRef] [PubMed]
- Collier, S.A.; Stockman, L.J.; Hicks, L.A.; Garrison, L.E.; Zhou, F.J.; Beach, M.J. Direct healthcare costs of selected diseases primarily or partially transmitted by water. Epidemiol. Infect. 2012, 140, 2003–2013. [Google Scholar] [CrossRef]
- Kruk, M.E.; Schwalbe, N.R.; Aguiar, C.A. Timing of default from tuberculosis treatment: A systematic review. Trop. Med. Int. Health 2008, 13, 703–712. [Google Scholar] [CrossRef]
- Balavoine, C.; Blanc, F.-X.; Lanotte, P.; Meurice, J.C.; Andrejak, C.; Marchand-Adam, S. Adverse events during treatment of nontuberculous mycobacterial lung disease: Do they really matter? Eur. Respir. J. 2018, 52, PA2664. [Google Scholar]
- Wallace, R.J.; Brown-Elliott, B.A.; McNulty, S.; Philley, J.V.; Killingley, J.; Wilson, R.W.; York, D.S.; Shepherd, S.; Griffith, D.E. Macrolide/azalide therapy for nodular/bronchiectatic Mycobacterium avium complex lung disease. Chest 2014, 146, 276–282. [Google Scholar] [CrossRef]
- Xu, H.B.; Jiang, R.H.; Li, L. Treatment outcomes for Mycobacterium avium complex: A systematic review and meta-analysis. Eur. J. Clin. Microbiol. Infect. Dis. 2014, 33, 347–358. [Google Scholar] [CrossRef]
- Field, S.K.; Fisher, D.; Cowie, R.L. Mycobacterium avium complex Pulmonary Disease in Patients without HIV Infection. Chest 2004, 126, 566–581. [Google Scholar] [CrossRef]
- Cherkaoui, I.; Sabouni, R.; Ghali, I.; Kizub, D.; Billioux, A.C.; Bennani, K.; Bourkadi, J.E.; Benmamoun, A.; Lahlou, O.; El Aouad, R.; et al. Treatment default amongst patients with tuberculosis in urban Morocco: Predicting and explaining default and post-default sputum smear and drug susceptibility results. PLoS ONE 2014, 9, e93574. [Google Scholar] [CrossRef]
- Chida, N.; Ansari, Z.; Hussain, H.; Jaswal, M.; Symes, S.; Khan, A.J.; Mohammed, S. Determinants of default from tuberculosis treatment among patients with drug-susceptible tuberculosis in Karachi, Pakistan: A mixed methods: Study. PLoS ONE 2015, 10, e0142384. [Google Scholar] [CrossRef]
- Yang, T.W.; Park, H.O.; Jang, H.N.; Yang, J.H.; Kim, S.H.; Moon, S.H.; Byun, J.H.; Lee, C.E.; Kim, J.W.; Kang, D.H. Side effects associated with the treatment of multidrug-resistant tuberculosis at a tuberculosis referral hospital in South Korea. Medicine (Baltimore) 2017, 96, e7482. [Google Scholar] [CrossRef]
- Karumbi, J.; Garner, P. Directly observed therapy for treating tuberculosis. Cochrane Database Syst. Rev. 2015, 29, 1–56. [Google Scholar] [CrossRef] [PubMed]
- Quon, B.S.; Goss, C.H.; Ramsey, B.W. Inhaled antibiotics for lower airway infections. Ann. Am. Thorac. Soc. 2014, 11, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Konstan, M.W.; Flume, P.A.; Kappler, M.; Chiron, R.; Higgins, M.; Brockhaus, F.; Zhang, J.; Angyalosi, G.; He, E.; Geller, D.E. Safety, efficacy and convenience of tobramycin inhalation powder in cystic fibrosis patients: The EAGER trial. J. Cyst. Fibros. 2011, 10, 54–61. [Google Scholar] [CrossRef]
- VanDevanter, D.R.; Geller, D.E. Tobramycin administered by the TOBI® Podhaler® for persons with cystic fibrosis: A review. Med. Devices Evid. Res. 2011, 4, 179–188. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Geller, D.E. Aerosol Antibiotics in Cystic Fibrosis. Respir. Care 2009, 54, 658–670. [Google Scholar] [CrossRef]
- Hickey, A.J.; Durham, P.G.; Dharmadhikari, A.; Nardell, E.A. Inhaled drug treatment for tuberculosis: Past progress and future prospects. J. Control. Release 2016, 240, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Fink, J.B.; Stapleton, K.W. Nebulizers. In ISAM Textbook of Aerosol Medicine; International Society for Aerosols in Medicine: Munich, Germany, 2015; pp. 617–656. [Google Scholar]
- Geller, D.E.; Weers, J.; Heuerding, S. Development of an Inhaled Dry-Powder Formulation of Tobramycin Using PulmoSphereTM Technology. J. Aerosol Med. Pulm. Drug Deliv. 2011, 24, 175–182. [Google Scholar] [CrossRef]
- Hagedoorn, P.; Grasmeijer, F.; Hoppentocht, M.; Akkerman, O.; Frijlink, H.; de Boer, A. In vitro evaluation of the Twincer colistin dry powder inhaler as a non-cough-inducing alternative to Colobreathe. Eur. Respir. J. 2016, 48, PA2561. [Google Scholar]
- Weers, J.; Clark, A.; Challoner, P. High dose inhaled powder delivery: Challenges and techniques. Respir. Drug Deliv. IX 2004, 9, 281–288. [Google Scholar]
- Morton, D.; Barling, D. Development of Dry Powder Inhalation Formulations. In ISAM Textbook of Aerosol Medicine; International Society for Aerosols in Medicine: Munich, Germany, 2015; pp. 651–680. [Google Scholar]
- Drugs.com. Available online: https://www.drugs.com/price-guide/ (accessed on 1 July 2019).
- Hickey, A.J.; Misra, A.; Fourie, P.B. Dry powder antibiotic aerosol product development: Inhaled therapy for tuberculosis. J. Pharm. Sci. 2013, 102, 3900–3907. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.; Abramson, H.; Ratner, B. Aerosol streptomycin treatment of advanced pulmonary tuberculosis in children. Am. J. Dis. Child. 1950, 80, 207–237. [Google Scholar] [CrossRef] [PubMed]
- Isaakidis, P.; Rangan, S.; Pradhan, A.; Ladomirska, J.; Reid, T.; Kielmann, K. “I cry every day”: Experiences of patients co-infected with HIV and multidrug-resistant tuberculosis. Trop. Med. Int. Health 2013, 18, 1128–1133. [Google Scholar] [CrossRef] [PubMed]
- Toczek, A.; Cox, H.; Du Cros, P.; Cooke, G.; Ford, N. Strategies for reducing treatment default in drug-resistant tuberculosis: Systematic review and meta-analysis. Int. J. Tuberc. Lung Dis. 2013, 17, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.T.; Haskal, R.; McGowan, K.; Nardell, E.; Sabbag, R. Inhaled kanamycin in the treatment of multidrug-resistant tuberculosis: A study of five patients. Infect. Dis. Clin. Pract. 1998, 7, 49–53. [Google Scholar] [CrossRef]
- Sacks, L.V.; Pendle, S.; Orlovic, D.; Andre, M.; Popara, M.; Moore, G.; Thonell, L.; Hurwitz, S. Adjunctive Salvage Therapy with Inhaled Aminoglycosides for Patients with Persistent Smear-Positive Pulmonary Tuberculosis. Clin. Infect. Dis. 2001, 32, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Justo, O.R.; Moraes, A.M. Kanamycin incorporation in lipid vesicles prepared by ethanol injection designed for tuberculosis treatment. J. Pharm. Pharmacol. 2005, 57, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Jin, H.E.; Kim, D.D.; Chung, S.J.; Shim, W.S.; Shim, C.K. Chitosan microspheres as an alveolar macrophage delivery system of ofloxacin via pulmonary inhalation. Int. J. Pharm. 2013, 441, 562–569. [Google Scholar] [CrossRef] [PubMed]
- Rojanarat, W.; Nakpheng, T.; Thawithong, E.; Yanyium, N.; Srichana, T. Levofloxacin-Proliposomes: Opportunities for Use in Lung Tuberculosis. Pharmaceutics 2012, 4, 385–412. [Google Scholar] [CrossRef]
- Zhou, Q.; Leung, S.S.Y.; Tang, P.; Parumasivam, T.; Loh, Z.H.; Chan, H.K. Inhaled formulations and pulmonary drug delivery systems for respiratory infections. Adv. Drug Deliv. Rev. 2014, 85, 83–99. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Saxena, D.; Dwivedi, A.K.; Misra, A. Drug combinations to target alveolar macrophages for treatment of pulmonary tuberculosis. Pharm. Res. 2001, 18, 1405–1410. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Treatment of Tuberculosis: Guidelines, 4th ed.; World Health Organization: Geneva, Switzerland, 2010. [Google Scholar]
- Garcia-Contreras, L.; Muttil, P.; Fallon, J.K.; Kabadi, M.; Gerety, R.; Hickey, A.J. Pharmacokinetics of Sequential Doses of Capreomycin Powder for Inhalation in Guinea Pigs. Antimicrob. Agents Chemother. 2012, 56, 2612–2618. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Contreras, L.; Fiegel, J.; Telko, M.J.; Elbert, K.; Hawi, A.; Thomas, M.; VerBerkmoes, J.; Germishuizen, W.A.; Fourie, P.B.; Hickey, A.J.; et al. Inhaled large porous particles of capreomycin for treatment of tuberculosis in a guinea pig model. Antimicrob. Agents Chemother. 2007, 51, 2830–2836. [Google Scholar] [CrossRef] [PubMed]
- Dharmadhikari, A.S.; Kabadi, M.; Gerety, B.; Hickey, A.J.; Fourie, P.B.; Nardell, E. Phase I, Single-Dose, Dose-Escalating Study of Inhaled Dry Powder Capreomycin: A New Approach to Therapy of Drug-Resistant Tuberculosis. Antimicrob. Agents Chemother. 2013, 57, 2613–2619. [Google Scholar] [CrossRef] [PubMed]
- Davis, K.K.; Kao, P.N.; Jacobs, S.S.; Ruoss, S.J. Aerosolized amikacin for treatment of pulmonary Mycobacterium avium infections: An observational case series. BMC Pulm. Med. 2007, 7, 2. [Google Scholar] [CrossRef] [PubMed]
- Yagi, K.; Ishii, M.; Namkoong, H.; Asami, T.; Iketani, O.; Asakura, T.; Suzuki, S.; Sugiura, H.; Yamada, Y.; Nishimura, T.; et al. The efficacy, safety, and feasibility of inhaled amikacin for the treatment of difficult-to-treat non-tuberculous mycobacterial lung diseases. BMC Infect. Dis. 2017, 17, 558. [Google Scholar] [CrossRef]
- Olivier, K.N.; Shaw, P.A.; Glaser, T.S.; Bhattacharyya, D.; Fleshner, M.; Brewer, C.C.; Zalewski, C.K.; Folio, L.R.; Siegelman, J.R.; Shallom, S.; et al. Inhaled amikacin for treatment of refractory pulmonary nontuberculous mycobacterial disease. Ann. Am. Thorac. Soc. 2014, 11, 30–35. [Google Scholar] [CrossRef]
- Rose, S.J.; Neville, M.E.; Gupta, R.; Bermudez, L.E. Delivery of aerosolized liposomal amikacin as a novel approach for the treatment of nontuberculous mycobacteria in an experimental model of pulmonary infection. PLoS ONE 2014, 9, e108703. [Google Scholar] [CrossRef]
- Malinin, V.; Neville, M.; Eagle, G.; Gupta, R.; Perkins, W.R. Pulmonary deposition and elimination of liposomal amikacin for inhalation and effect on macrophage function after administration in rats. Antimicrob. Agents Chemother. 2016, 60, 6540–6549. [Google Scholar] [CrossRef]
- Zhang, J.; Leifer, F.; Rose, S.; Chun, D.Y.; Thaisz, J.; Herr, T.; Nashed, M.; Joseph, J.; Perkins, W.R.; DiPetrillo, K. Amikacin Liposome Inhalation Suspension (ALIS) Penetrates Non-tuberculous Mycobacterial Biofilms and Enhances Amikacin Uptake Into Macrophages. Front. Microbiol. 2018, 9, 915. [Google Scholar] [CrossRef]
- Griffith, D.E.; Eagle, G.; Thomson, R.; Aksamit, T.R.; Hasegawa, N.; Morimoto, K.; Addrizzo-Harris, D.J.; O’Donnell, A.E.; Marras, T.K.; Flume, P.A.; et al. Amikacin liposome inhalation suspension for treatment-refractory lung disease caused by Mycobacterium avium complex (CONVERT) a prospective, open-label, randomized study. Am. J. Respir. Crit. Care Med. 2018, 198, 1559–1569. [Google Scholar] [CrossRef] [PubMed]
- Olivier, K.N.; Griffith, D.E.; Eagle, G.; McGinnis, J.P.; Micioni, L.; Liu, K.; Daley, C.L.; Winthrop, K.L.; Ruoss, S.; Addrizzo-Harris, D.J.; et al. Randomized trial of liposomal amikacin for inhalation in nontuberculous mycobacterial lung disease Supplemental Data. Am. J. Respir. Crit. Care Med. 2017, 195, 814–823. [Google Scholar] [CrossRef] [PubMed]
- Muttil, P.; Kaur, J.; Kumar, K.; Yadav, A.B.; Sharma, R.; Misra, A. Inhalable microparticles containing large payload of anti-tuberculosis drugs. Eur. J. Pharm. Sci. 2007, 32, 140–150. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.G.Y.; Chan, H.K.; Prestidge, C.A.; Denman, J.A.; Young, P.M.; Traini, D. A novel dry powder inhalable formulation incorporating three first-line anti-tubercular antibiotics. Eur. J. Pharm. Biopharm. 2013, 83, 285–292. [Google Scholar] [CrossRef]
- Maretti, E.; Rossi, T.; Bondi, M.; Croce, M.A.; Hanuskova, M.; Leo, E.; Sacchetti, F.; Iannuccelli, V. Inhaled Solid Lipid Microparticles to target alveolar macrophages for tuberculosis. Int. J. Pharm. 2014, 462, 74–82. [Google Scholar] [CrossRef]
- Barry, V.C.; Belton, J.; Conalty, M.L.; Denneny, J.M.; Edward, D.W.; O’Sullivan, J.F.; Twomey, D.; Winder, F. A New Series of Phenazines (Rimino-Compounds) With High Antituberculosis Activity. Nature 1957, 179, 1013–1015. [Google Scholar] [CrossRef]
- Barry, V.C.; Conalty, M.L.; Gaffney, E.E. Antituberculosis activity in the phenazine series; isomeric pigments obtained by oxidation of O-phenylenediamine derivatives. J. Pharm. Pharmacol. 1956, 8, 1089–1096. [Google Scholar] [CrossRef] [PubMed]
- Yawalkar, S.J.; Vischer, W. Lamprene (Clofazimine) in Leprosy. Lepr. Rev. 1979, 50, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Baik, J.; Rosania, G.R. Macrophages Sequester Clofazimine in an Intracellular Liquid Crystal-Like Supramolecular Organization. PLoS ONE 2012, 7, e47494. [Google Scholar] [CrossRef]
- Van Deun, A.; Maug, A.K.J.; Salim, M.A.H.; Das, P.K.; Sarker, M.R.; Daru, P.; Rieder, H.L. Short, highly effective, and inexpensive standardized treatment of multidrug-resistant tuberculosis. Am. J. Respir. Crit. Care Med. 2010, 182, 684–692. [Google Scholar] [CrossRef] [PubMed]
- Aung, K.J.M.; Van Deun, A.; Declercq, E.; Sarker, M.R.; Das, P.K.; Hossain, M.A.; Rieder, H.L. Successful “9-month Bangladesh regimen” for multidrugresistant tuberculosis among over 500 consecutive patients. Int. J. Tuberc. Lung Dis. 2014, 18, 1180–1187. [Google Scholar] [CrossRef] [PubMed]
- Pietersen, E.; Ignatius, E.; Streicher, E.M.; Mastrapa, B.; Padanilam, X.; Pooran, A.; Badri, M.; Lesosky, M.; Van Helden, P.; Sirgel, F.A.; et al. Long-term outcomes of patients with extensively drug-resistant tuberculosis in South Africa: A cohort study. Lancet 2014, 383, 1230–1239. [Google Scholar] [CrossRef]
- Diacon, A.H.; Dawson, R.; Von Groote-Bidlingmaier, F.; Symons, G.; Venter, A.; Donald, P.R.; Van Niekerk, C.; Everitt, D.; Hutchings, J.; Burger, D.A.; et al. Bactericidal activity of pyrazinamide and clofazimine alone and in combinations with pretomanid and bedaquiline. Am. J. Respir. Crit. Care Med. 2015, 191, 943–953. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Yao, L.; Hao, X.; Liu, Y.; Zeng, L.; Liu, G.; Li, M.; Li, F.; Wu, M.; Zhu, Y.; et al. Clofazimine for the treatment of multidrug-resistant tuberculosis: Prospective, multicenter, randomized controlled study in China. Clin. Infect. Dis. 2015, 60, 1361–1367. [Google Scholar] [PubMed]
- Hwang, T.J.; Dotsenko, S.; Jafarov, A.; Weyer, K.; Falzon, D.; Lunte, K.; Nunn, P.; Jaramillo, E.; Keshavjee, S.; Wares, D.F. Safety and availability of clofazimine in the treatment of multidrug and extensively drug-resistant tuberculosis: Analysis of published guidance and meta-analysis of cohort studies. BMJ Open 2014, 4, e004143. [Google Scholar] [CrossRef]
- Xu, H.B.; Jiang, R.H.; Xiao, H.P. Clofazimine in the treatment of multidrug-resistant tuberculosis. Clin. Microbiol. Infect. 2012, 18, 1104–1110. [Google Scholar] [CrossRef]
- Wang, Q.; Pang, Y.; Jing, W.; Liu, Y.; Wang, N.; Yin, H.; Zhang, Q.; Ye, Z.; Zhu, M.; Li, F.; et al. Clofazimine for treatment of extensively drug-resistant pulmonary tuberculosis in China. Antimicrob. Agents Chemother. 2018, 62, e02149-17. [Google Scholar] [CrossRef]
- Dalcolmo, M.; Gayoso, R.; Sotgiu, G.; D’Ambrosio, L.; Rocha, J.L.; Borga, L.; Fandinho, F.; Braga, J.U.; Galesi, V.M.N.; Barreira, D.; et al. Effectiveness and safety of clofazimine in multidrug-resistant tuberculosis: A nationwide report from Brazil. Eur. Respir. J. 2017, 49, 9–13. [Google Scholar] [CrossRef]
- Jarand, J.; Paul Davis, J.; Cowie, R.L.; Field, S.K.; Fisher, D.A. Long-term follow-up of Mycobacterium avium complex lung disease in patients treated with regimens including clofazimine and/or rifampin. Chest 2016, 149, 1285–1293. [Google Scholar] [CrossRef]
- Martiniano, S.L.; Wagner, B.D.; Levin, A.; Nick, J.A.; Sagel, S.D.; Daley, C.L.; Martiniano, S.L. Safety and Effectiveness of Clofazimine for Primary and Refractory Nontuberculous Mycobacterial Infection. Chest 2017, 152, 800–809. [Google Scholar] [CrossRef] [PubMed]
- Martiniano, S.L.; Sontag, M.K.; Daley, C.L.; Nick, J.A.; Sagel, S.D. Clinical significance of a first positive nontuberculous mycobacteria culture in cystic fibrosis. Ann. Am. Thorac. Soc. 2014, 11, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Aznar, M.L.; Brode, S.K.; Mehrabi, M.; Marras, T.K. Safety and effectiveness of clofazimine in nontuberculous mycobacterial lung disease. Can. J. Respir. Crit. Care Sleep Med. 2018, 2, 72–77. [Google Scholar] [CrossRef]
- Henkel, A.G.; Paulson, M.L.; Claypool, R.J.; Prevots, D.R.; Holland, S.M.; Olivier, K.N. Safety and Tolerability of Clofazimine for the Treatment of Nontuberculous Mycobacterial Infections. Am. J. Respir. Crit. Care Med. 2012, 185, A4038. [Google Scholar]
- Baik, J.; Stringer, K.A.; Mane, G.; Rosania, G.R. Multiscale distribution and bioaccumulation analysis of clofazimine reveals a massive immune system-mediated xenobiotic sequestration response. Antimicrob. Agents Chemother. 2013, 57, 1218–1230. [Google Scholar] [CrossRef] [PubMed]
- Swanson, R.V.; Adamson, J.; Moodley, C.; Ngcobo, B.; Ammerman, N.C.; Dorasamy, A.; Moodley, S.; Mgaga, Z.; Tapley, A.; Bester, L.A.; et al. Pharmacokinetics and pharmacodynamics of clofazimine in a mouse model of tuberculosis. Antimicrob. Agents Chemother. 2015, 59, 3042–3051. [Google Scholar] [CrossRef]
- Verma, R.K.; Germishuizen, W.A.; Motheo, M.P.; Agrawal, A.K.; Singh, A.K.; Mohan, M.; Gupta, P.; Gupta, U.D.; Cholo, M.; Anderson, R.; et al. Inhaled microparticles containing clofazimine are efficacious in treatment of experimental tuberculosis in mice. Antimicrob. Agents Chemother. 2013, 57, 1050–1052. [Google Scholar] [CrossRef] [PubMed]
- Brunaugh, A.D.; Jan, S.U.; Ferrati, S.; Smyth, H.D.C. Excipient-Free Pulmonary Delivery and Macrophage Targeting of Clofazimine via Air Jet Micronization. Mol. Pharm. 2017, 14, 4019–4031. [Google Scholar] [CrossRef]
- Murashov, M.D.; Diaz-espinosa, J.; Lalone, V.; Tan, J.W.Y.; Laza, R.; Wang, X.; Stringer, K.A.; Rosania, G.R. Synthesis and Characterization of a Biomimetic Formulation of Clofazimine Hydrochloride Microcrystals for Parenteral Administration. Pharmaceutics 2018, 10, 238. [Google Scholar] [CrossRef]
- Banaschewski, B.; Verma, D.; Pennings, L.J.; Zimmerman, M.; Ye, Q.; Gadawa, J.; Dartois, V.; Ordway, D.; Van Ingen, J.; Ufer, S.; et al. Clofazimine inhalation suspension for the aerosol treatment of pulmonary nontuberculous mycobacterial infections. J. Cyst. Fibros. 2019, in press. [Google Scholar] [CrossRef]
- Stout, J.E.; Floto, R.A. Treatment of Mycobacterium abscessus: All macrolides are equal, but perhaps some are more equal than others. Am. J. Respir. Crit. Care Med. 2012, 186, 822–823. [Google Scholar] [CrossRef] [PubMed]
- Parameswaran, G.I.; Sethi, S. Long-term macrolide therapy in chronic obstructive pulmonary disease. CMAJ 2014, 186, 1127–1128. [Google Scholar] [CrossRef] [PubMed]
- Hickey, A.J.; Lu, D.; Ashley, E.D.; Stout, J. Inhaled Azithromycin Therapy. J. Aerosol Med. 2006, 19, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; You, Y.; Ren, Y.; Zhang, Y.; Tang, X. Formulation, characteristics and aerosolization performance of azithromycin DPI prepared by spray-drying. Powder Technol. 2008, 187, 214–221. [Google Scholar] [CrossRef]
- Serisier, D.J.; Bilton, D.; De Soyza, A.; Thompson, P.J.; Kolbe, J.; Greville, H.W.; Cipolla, D.; Bruinenberg, P.; Gonda, I. Inhaled, dual release liposomal ciprofloxacin in non-cystic fibrosis bronchiectasis (ORBIT-2): A randomised, double-blind, placebo-controlled trial. Thorax 2013, 68, 812–817. [Google Scholar] [CrossRef] [PubMed]
- Haworth, C.S.; Bilton, D.; Chalmers, J.D.; Davis, A.M.; Froehlich, J.; Gonda, I.; Thompson, B.; Wanner, A.; O’Donnell, A.E. Inhaled liposomal ciprofloxacin in patients with non-cystic fibrosis bronchiectasis and chronic lung infection with Pseudomonas aeruginosa (ORBIT-3 and ORBIT-4): Two phase 3, randomised controlled trials. Lancet Respir. Med. 2019, 7, 213–226. [Google Scholar] [CrossRef]
- Blanchard, J.D.; Elias, V.; Cipolla, D.; Gonda, I.; Bermudez, L.E. Effective treatment of Mycobacterium avium subsp. hominissuis and Mycobacterium abscessus species infections in macrophages, biofilm, and mice by using liposomal ciprofloxacin. Animicrobial Agents Chemother. 2018, 62, e00440-18. [Google Scholar] [CrossRef] [PubMed]
- Bermudez, L.E.; Blanchard, J.; Babrak, L.; Gonda, I. Treatment of Mycobacterium avium subsp hominissuis (MAH) lung infection with liposome-encapsulated ciprofloxacin resulted in significant decrease in bacterial load in the lung. Am. J. Respir. Crit. Care Med. 2015, 191, A6293. [Google Scholar]
- Shibata, Y.; Berclaz, P.Y.; Chroneos, Z.C.; Yoshida, M.; Whitsett, J.A.; Trapnell, B.C. GM-CSF regulates alveolar macrophage differentiation and innate immunity in the lung through PU.1. Immunity 2001, 15, 557–567. [Google Scholar] [CrossRef]
- Ballinger, M.N.; Paine, R.; Serezani, C.H.C.; Aronoff, D.M.; Choi, E.S.; Standiford, T.J.; Toews, G.B.; Moore, B.B. Role of granulocyte macrophage colony-stimulating factor during gram-negative lung infection with Pseudomonas aeruginosa. Am. J. Respir. Cell Mol. Biol. 2006, 34, 766–774. [Google Scholar] [CrossRef]
- De Groote, M.A.; Johnson, L.; Podell, B.; Brooks, E.; Basaraba, R.; Gonzalez-Juarrero, M. GM-CSF knockout mice for preclinical testing of agents with antimicrobial activity against Mycobacterium abscessus. J. Antimicrob. Chemother. 2014, 69, 1057–1064. [Google Scholar] [CrossRef] [PubMed]
- Scott, J.; Ji, Y.; Kannan, M.; Wylam, M.E. Inhaled Granulocyte-Macrophage Colony Stimulating Factor for Mycobacterium Abscessus in Cystic Fibrosis. Eur. Respir. J. 2018, 51, 1702127. [Google Scholar] [CrossRef] [PubMed]
- Moser, C.; Jensen, P.Ø.; Pressler, T.; Frederiksen, B.; Lanng, S.; Kharazmi, A.; Koch, C.; Høiby, N. Serum concentrations of GM-CSF and G-CSF correlate with the Th1/Th2 cytokine response in cystic fibrosis patients with chronic Pseudomonas aeruginosa lung infection. Apmis 2005, 113, 400–409. [Google Scholar] [CrossRef] [PubMed]
- Heslet, L.; Bay, C.; Nepper-Christensen, S. The immunomodulatory effect of inhaled granulocyte-macrophage colony-stimulating factor in cystic fibrosis. A new treatment paradigm. J. Inflamm. Res. 2012, 5, 19–27. [Google Scholar] [PubMed]
- Wylam, M.E.; Ten, R.; Prakash, U.B.S.; Nadrous, H.F.; Clawson, M.L.; Anderson, P.M. Aerosol granulocyte-macrophage colony-stimulating factor for pulmonary alveolar proteinosis. Eur. Respir. J. 2006, 27, 585–593. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, K.; Sato, A.; Takada, T.; Arai, T.; Nei, T.; Kasahara, Y.; Motoi, N.; Hojo, M.; Urano, S.; Ishii, H.; et al. Direct evidence that GM-CSF inhalation improves lung clearance in pulmonary alveolar proteinosis. Respir. Med. 2012, 106, 284–293. [Google Scholar] [CrossRef]
- Tazawa, R.; Hamano, E.; Arai, T.; Ohta, H.; Ishimoto, O.; Uchida, K.; Watanabe, M.; Saito, J.; Takeshita, M.; Hirabayashi, Y.; et al. Granulocyte-macrophage colony-stimulating factor and lung immunity in pulmonary alveolar proteinosis. Am. J. Respir. Crit. Care Med. 2005, 171, 1142–1149. [Google Scholar] [CrossRef]
- Anderson, P.M.; Markovic, S.N.; Sloan, J.A.; Clawson, M.L.; Wylam, M.; Arndt, C.A.S.; Smithson, W.A.; Burch, P.; Gornet, M.; Rahman, E. Aerosol granulocyte macrophage-colony stimulating factor: A low toxicity, lung-specific biological therapy in patients with lung metastases. Clin. Cancer Res. 1999, 5, 2316–2323. [Google Scholar]
- Xu, J.; Lucas, R.; Schuchmann, M.; Kuhnle, S.; Meergans, T.; Barreiros, A.P.; Lohse, A.W.; Otto, G.; Wendel, A. GM-CSF Restores Innate, But Not Adaptive, Immune Responses in Glucocorticoid-Immunosuppressed Human Blood In Vitro. J. Immunol. 2014, 171, 938–947. [Google Scholar] [CrossRef]
- Moncada, S.; Palmer, R.M.; Higgs, E.A. Nitric oxide: Physiology, pathophysiology, and pharmacology. Pharmacol. Rev. 1991, 43, 109–142. [Google Scholar]
- Griffiths, M.J.; Evans, T.W. Inhaled Nitric Oxide Therapy in Adults. N. Engl. J. Med. 2005, 353, 2683–2695. [Google Scholar] [CrossRef] [PubMed]
- Ghaffari, A.; Neil, D.H.; Ardakani, A.; Road, J.; Ghahary, A.; Miller, C.C. A direct nitric oxide gas delivery system for bacterial and mammalian cell cultures. Nitric Oxide Biol. Chem. 2005, 12, 129–140. [Google Scholar] [CrossRef] [PubMed]
- McMullin, B.B.; Chittock, D.R.; Roscoe, D.L.; Garcha, H.; Wang, L.; Miller, C.C. The antimicrobial effect of nitric oxide on the bacteria that cause nosocomial pneumonia in mechanically ventilated patients in the intensive care unit. Respir. Care 2005, 50, 1451–1456. [Google Scholar] [PubMed]
- Miller, C.C.; Rawat, M.; Johnson, T.; Av-Gay, Y. Innate protection of Mycobacterium smegmatis against the antimicrobial activity of nitric oxide is provided by mycothiol. Antimicrob. Agents Chemother. 2007, 51, 3364–3366. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Miller, C.; Miller, M.; McMullin, B.; Regev, G.; Serghides, L.; Kain, K.; Road, J.; Av-Gay, Y. A phase I clinical study of inhaled nitric oxide in healthy adults. J. Cyst. Fibros. 2012, 11, 324–331. [Google Scholar] [CrossRef]
- Deppisch, C.; Herrmann, G.; Graepler-Mainka, U.; Wirtz, H.; Heyder, S.; Engel, C.; Marschal, M.; Miller, C.C.; Riethmüller, J. Gaseous nitric oxide to treat antibiotic resistant bacterial and fungal lung infections in patients with cystic fibrosis: A phase I clinical study. Infection 2016, 44, 513–520. [Google Scholar] [CrossRef]
- Yaacoby-Bianu, K.; Gur, M.; Toukan, Y.; Nir, V.; Hakim, F.; Geffen, Y.; Bentur, L. Compassionate Nitric Oxide Adjuvant Treatment of Persistent Mycobacterium Infection in Cystic Fibrosis Patients. Pediatr. Infect. Dis. J. 2018, 37, 336–338. [Google Scholar]
- Riccio, D.A.; Schoenfisch, M.H. Nitric oxide release: Part I. Macromolecular scaffolds. Chem. Soc. Rev. 2012, 41, 3731–3741. [Google Scholar] [CrossRef]
- Carpenter, A.W.; Schoenfisch, M.H. Nitric oxide release: Part II. Therapeutic applications. Chem. Soc. Rev. 2012, 41, 3742–3752. [Google Scholar] [CrossRef]
- U.S. Department of Health and Human Services; Food and Drug Administration; Center for Drug Evaluation and Research (CDER); Center for Biologics Evaluation and Research (CBER). Guidance for Industry Expedited Programs for Serious Conditions—Drugs and Biologics; The Food and Drug Administration: Rockville, MD, USA, 2013; pp. 1–40.
- Hou, S.; Wu, J.; Li, X.; Shu, H. Practical, regulatory and clinical considerations for development of inhalation drug products. Asian J. Pharm. Sci. 2015, 10, 490–500. [Google Scholar] [CrossRef]
- Quittner, A.L.; Buu, A.; Messer, M.A.; Modi, A.C.; Watrous, M. Development and validation of the cystic fibrosis questionnaire in the United States: A health-related quality-of-life measure for cystic fibrosis. Chest 2005, 128, 2347–2354. [Google Scholar] [CrossRef] [PubMed]
- Gopal, M.; Padayatchi, N.; Metcalfe, J.Z.; O’Donnell, M.R. Systematic review of clofazimine for the treatment of drug-resistant tuberculosis. Int. J. Tuberc. Lung Dis. 2013, 17, 1001–1007. [Google Scholar] [CrossRef] [PubMed]
- Padayatchi, N.; Gopal, M.; Naidoo, R.; Werner, L.; Naidoo, K.; Master, I.; O’Donnell, M.R. Clofazimine in the treatment of extensively drug-resistant tuberculosis with HIV coinfection in South Africa: A retrospective cohort study. J. Antimicrob. Chemother. 2014, 69, 3103–3107. [Google Scholar] [CrossRef] [PubMed]
- Saini, V.; Ammerman, N.C.; Chang, Y.S.; Tasneen, R.; Chaisson, R.E.; Jain, S.; Nuermberger, E.; Grosset, J.H. Treatment-shortening effect of a novel regimen combining high-dose rifapentine and clofazimine in pathologically distinct mouse models of tuberculosis. Antimicrob. Agents Chemother. 2019, 63, e00388-19. [Google Scholar] [CrossRef] [PubMed]
- Lamprecht, D.A.; Finin, P.M.; Rahman, M.A.; Cumming, B.M.; Russell, S.L.; Jonnala, S.R.; Adamson, J.H.; Steyn, A.J.C. Turning the respiratory flexibility of Mycobacterium tuberculosis against itself. Nat. Commun. 2016, 7, 12393. [Google Scholar] [CrossRef] [PubMed]
- Van Ingen, J.; Totten, S.E.; Helstrom, N.K.; Heifets, L.B.; Boeree, M.J.; Daley, C.L. In vitro synergy between clofazimine and amikacin in treatment of nontuberculous mycobacterial disease. Antimicrob. Agents Chemother. 2012, 56, 6324–6327. [Google Scholar] [CrossRef]
- Ruth, M.M.; Sangen, J.J.N.; Remmers, K.; Pennings, L.J.; Svensson, E.; Aarnoutse, R.E.; Zweijpfenning, S.M.H.; Hoefsloot, W.; Kuipers, S.; Magis-Escurra, C.; et al. A bedaquiline/clofazimine combination regimen might add activity to the treatment of clinically relevant non-tuberculous mycobacteria. J. Antimicrob. Chemother. 2019, 74, 935–942. [Google Scholar] [CrossRef]
- Chau, T.; da Silva, J.; Ghaffari, A.; Zelazny, A.; Olivier, K.N. Synergistic Effect of Nitric Oxide with Antibiotics Against Mycobacterium Abscessus In Vitro. Am. J. Respir. Crit. Care Med. 2019, 199, A2656. [Google Scholar]
- Andries, K.; Verhasselt, P.; Guillemont, J.; Gohlmann, H.W.; Neefs, J.-M.; Winkler, H.; Van Gestel, J.; Timmerman, P.; Zhu, M.; Lee, E.; et al. A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science 2005, 307, 223–227. [Google Scholar] [CrossRef]
- Brown-Elliott, B.A.; Philley, J.V.; Griffith, D.E.; Thakkar, F.; Wallace, R.J. In vitro susceptibility testing of bedaquiline against Mycobacterium avium complex. Antimicrob. Agents Chemother. 2017, 61, e01798. [Google Scholar] [CrossRef]
- Pang, Y.; Zheng, H.; Tan, Y.; Song, Y.; Zhao, Y. In Vitro activity of bedaquiline against nontuberculous mycobacteria in China. Antimicrob. Agents Chemother. 2017, 61, e02627-16. [Google Scholar] [CrossRef] [PubMed]
- Dupont, C.; Viljoen, A.; Thomas, S.; Roquet-Banères, F.; Herrmann, J.-L.; Pethe, K.; Kremer, L. Bedaquiline Inhibits the ATP Synthase in Mycobacterium abscessus and Is Effective in Infected Zebrafish. Antimicrob. Agents Chemother. 2017, 61, e01225-17. [Google Scholar] [CrossRef] [PubMed]
- Obregón-Henao, A.; Arnett, K.A.; Henao-Tamayo, M.; Massoudi, L.; Creissen, E.; Andries, K.; Lenaerts, A.J.; Ordway, D.J. Susceptibility of Mycobacterium abscessus to antimycobacterial drugs in preclinical models. Antimicrob. Agents Chemother. 2015, 59, 6904–6912. [Google Scholar] [CrossRef] [PubMed]
- Bald, D.; Villellas, C.; Lu, P.; Koul, A. Targeting energy metabolism in Mycobacterium tuberculosis, a new paradigm in antimycobacterial drug discovery. MBio 2017, 8, e00272-17. [Google Scholar] [CrossRef] [PubMed]
- Soni, I.; De Groote, M.A.; Dasgupta, A.; Chopra, S. Challenges facing the drug discovery pipeline for non-tuberculous mycobacteria. J. Med. Microbiol. 2016, 65, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Khosravi, A.D.; Mirsaeidi, M.; Farahani, A.; Tabandeh, M.R.; Mohajeri, P.; Shoja, S.; Khosroshahi, S.R.H.L. Prevalence of nontuberculous mycobacteria and high efficacy of D-cycloserine and its synergistic effect with clarithromycin against Mycobacterium fortuitum and Mycobacterium abscessus. Infect. Drug Resist. 2018, 7, 2521–2532. [Google Scholar] [CrossRef] [PubMed]
- Heinrichs, M.; May, R.; Heider, F.; Reimers, T.; Sy, S.K.B.; Peloquin, C.; Derendorf, H. Mycobacterium tuberculosis Strains H37ra and H37rv have equivalent minimum inhibitory concentrations to most antituberculosis drugs. Int. J. Mycobacteriol. 2018, 7, 156–161. [Google Scholar] [CrossRef]
- Bruhn, D.F.; Scherman, M.S.; Liu, J.; Scherbakov, D.; Meibohm, B.; Böttger, E.C.; Lenaerts, A.J.; Lee, R.E. In vitro and in vivo evaluation of synergism between anti-tubercular spectinamides and non-classical tuberculosis antibiotics. Sci. Rep. 2015, 14. [Google Scholar] [CrossRef]
- Finch, C.K.; Chrisman, C.R.; Baciewicz, A.M.; Self, T.H. Rifampin and rifabutin drug interactions: An update. Arch. Intern. Med. 2002, 162, 985–992. [Google Scholar] [CrossRef]
- Strydom, N.; Gupta, S.V.; Fox, W.S.; Via, L.E.; Bang, H.; Lee, M.; Eum, S.; Shim, T.; Barry, C.E.; Zimmerman, M.; et al. Tuberculosis drugs’ distribution and emergence of resistance in patient’s lung lesions: A mechanistic model and tool for regimen and dose optimization. PLoS Med. 2019, 16, e1002773. [Google Scholar] [CrossRef]
- Prideaux, B.; Via, L.E.; Zimmerman, M.D.; Eum, S.; Sarathy, J.; O’Brien, P.; Chen, C.; Kaya, F.; Weiner, D.M.; Chen, P.Y.; et al. The association between sterilizing activity and drug distribution into tuberculosis lesions. Nat. Med. 2015, 21, 1223–1227. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, M.; Hashizume, H.; Tomishige, T.; Kawasaki, M.; Tsubouchi, H.; Sasaki, H.; Shimokawa, Y.; Komatsu, M. OPC-67683, a nitro-dihydro-imidazooxazole derivative with promising action against tuberculosis in vitro and in mice. PLoS Med. 2006, 3, e466. [Google Scholar] [CrossRef] [PubMed]
- Lenaerts, A.J.; Gruppo, V.; Marietta, K.S.; Johnson, C.M.; Driscoll, D.K.; Tompkins, N.M.; Rose, J.D.; Reynolds, R.C.; Orme, I.M. Preclinical testing of the nitroimidazopyran PA-824 for activity against Mycobacterium tuberculosis in a series of in vitro and in vivo models. Antimicrob. Agents Chemother. 2005, 49, 2294–2301. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.-S. Clinical Implications of New Drugs and Regimens for the Treatment of Drug-resistant Tuberculosis. Chonnam Med. J. 2017, 53, 103. [Google Scholar] [CrossRef] [PubMed]
- Schaaf, H.S.; Thee, S.; van der Laan, L.; Hesseling, A.C.; Garcia-Prats, A.J. Adverse effects of oral second-line antituberculosis drugs in children. Expert Opin. Drug Saf. 2016, 15, 1369–1381. [Google Scholar] [CrossRef] [PubMed]
- Ismail, N.A.; Omar, S.V.; Joseph, L.; Govender, N.; Blows, L.; Ismail, F.; Koornhof, H.; Dreyer, A.W.; Kaniga, K.; Ndjeka, N. Defining Bedaquiline Susceptibility, Resistance, Cross-Resistance and Associated Genetic Determinants: A Retrospective Cohort Study. EBioMedicine 2018, 28, 136–142. [Google Scholar] [CrossRef]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Banaschewski, B.; Hofmann, T. Inhaled Antibiotics for Mycobacterial Lung Disease. Pharmaceutics 2019, 11, 352. https://doi.org/10.3390/pharmaceutics11070352
Banaschewski B, Hofmann T. Inhaled Antibiotics for Mycobacterial Lung Disease. Pharmaceutics. 2019; 11(7):352. https://doi.org/10.3390/pharmaceutics11070352
Chicago/Turabian StyleBanaschewski, Brandon, and Thomas Hofmann. 2019. "Inhaled Antibiotics for Mycobacterial Lung Disease" Pharmaceutics 11, no. 7: 352. https://doi.org/10.3390/pharmaceutics11070352
APA StyleBanaschewski, B., & Hofmann, T. (2019). Inhaled Antibiotics for Mycobacterial Lung Disease. Pharmaceutics, 11(7), 352. https://doi.org/10.3390/pharmaceutics11070352